


Abstract
RNA-seq is a key technology for understanding the biology of the cell because of its ability to profile transcriptional and post-transcriptional regulation at single nucleotide resolutions. Compared to DNA sequencing alignment algorithms, RNA-seq alignment algorithms have a diminished ability to accurately detect and map base pair substitutions, gaps, discordant pairs and repetitive regions. These shortcomings adversely affect experiments that require a high degree of accuracy, notably the ability to detect RNA editing. We have developed RNASequel, a software package that runs as a post-processing step in conjunction with an RNA-seq aligner and systematically corrects common alignment artifacts. Its key innovations are a two-pass splice junction alignment system that includes de novo splice junctions and the use of an empirically determined estimate of the fragment size distribution when resolving read pairs. We demonstrate that RNASequel produces improved alignments when used in conjunction with STAR or Tophat2 using two simulated datasets. We then show that RNASequel improves the identification of adenosine to inosine RNA editing sites on biological datasets. This software will be useful in applications requiring the accurate identification of variants in RNA sequencing data, the discovery of RNA editing sites and the analysis of alternative splicing.
INTRODUCTION
RNA-seq is a key technology for understanding the biology of the cell. By enabling the global profiling of the RNA content of a sample at single nucleotide resolutions (1), RNA-seq makes it possible to reveal the details of transcriptional and post-transcriptional regulation (1–3). For instance, a single RNA-seq experiment can simultaneously profile transcript isoforms, gene fusions, alternative splicing, RNA editing and allelic imbalance (4–8). Unfortunately, the current generation of RNA-seq paired-end aligners suffers from shortcomings that obscure biologically important signals or which give rise to false signals. For example, the initial identification of putative non-canonical RNA editing has more recently been demonstrated to arise from false positives derived from sequencing and alignment artifacts (9).
A typical RNA-seq experiment consists of sequencing both ends of a cDNA fragment to generate two reads (a read pair) separated by a variable length of sequence. The accurate alignment of these read pairs is essential to the downstream analysis of an RNA-seq experiment, but RNA-seq read alignment is challenging due to the non-contiguous nature of mRNA transcripts (10). Critically, RNA-seq aligners must be able to identify exonic alignments in regions that can be interspersed with introns that can reach hundreds of thousands of kilobases in length (11). To solve this issue paired-end RNA-seq alignment methods typically apply a distance cutoff to exclude discordantly mapped pairs. However, these cutoffs tend to be arbitrary and very liberal. For example, many algorithms consider mapped pairs to be concordant up to a maximum distance of 500 kb, which is sufficiently high to catch the rare very long intron, but also is prone to incorrectly classifying the more common case of discordant reads that are mapped incorrectly.
To facilitate the mapping of spliced reads while attempting to minimize common systematic errors, various RNA-seq alignment methodologies have been developed. These methods include tools that are dependent on, and optimized for, a specific short read alignment tool such as Bowtie or BWA (12–19). Other tools implement their own alignment algorithms that may not be as accurate as traditional short read alignment tools, or which are less tolerant to gaps and mismatches (20,21). RNA-seq alignment methods also differ in their usage of pre-existing splice junction databases. Most methods perform better when a splice junction database is provided, but this hinders the identification of novel splice junctions, and may not be feasible for less well-characterized species (18,22). In addition, few splice junction aware RNA-seq aligners are able to recognize and handle transcripts that span more than one splice junction or contain a novel combination of existing junctions.
Other common artifacts that lead to issues with spliced alignments include (i) the identification of false positive splice junction alignments due to short alignment overlaps on one side of the splice junction, which is compounded by the reduction of base quality at read ends; (ii) false positive splice junctions due to reverse transcriptase template switching and splicing noise; (iii) splice junctions that are missed because the read has been incorrectly aligned to an intron sequence rather than across a splice junction (18,22–24). These artifacts contribute to false positives for calling insertions, deletions, splice junctions and mismatches. For example, many false positive sites in predicted RNA edits tend to be located near splice sites due to incorrectly spliced alignments (8,9). These are compounded by issues relating to library preparation such as errors generated by reverse transcription and random hexamer priming (25). In general, RNA-seq aligners have a low default tolerance for insertions, deletions and mismatches, which together increase the number of unmapped bases (soft clipping) at read ends and miss alignments to regions with a high mismatch rate. Finally, poor repeat tolerance can also lead to false positive mismatch calls by aligning a read pair to one paralogous gene while missing the alignment to another.
One common method to compensate for spliced alignment artifacts is to execute a two-pass alignment scheme (15,18). A two-pass alignment consists of two steps: (i) the alignment of the reads to known splice junctions and the reference genome for the detection of novel splice junctions; (ii) the generation of a new index file including all, or a subset of, high confidence novel splice junctions. This can drastically improve the spliced alignment of reads with low short exonic overlaps.
To address the common causes of systematic artifacts in RNA-seq library preparation, sequencing and alignment we have constructed an RNA-seq realignment program called RNASequel. RNASequel utilizes the spliced-read output of any read mapper and de novo splice junction detection algorithm to perform an error-tolerant realignment (Figure 1). It takes advantage of an empirically determined fragment size distribution and annotated and novel splice junctions to predict if a read pair maps concordantly. We have tested RNASequel against STAR (21) and Tophat2 (26) for de novo splice junction prediction using real and simulated datasets, and find increases in sensitivity and decreases in false positive predictions. We also show that RNASequel has improved repeat alignment sensitivity that improves the detection of potential single nucleotide variants and RNA editing sites.
Figure 1.
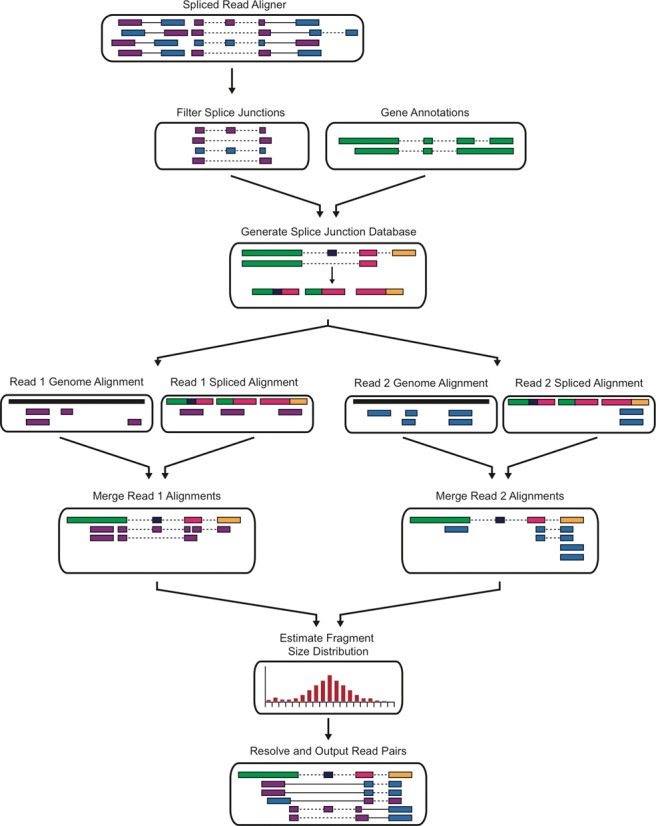
RNA-seq realignment schematic. A spliced read aligner is used to identify sample specific novel splice junctions that are used to generate a splice junction index. Read 1 and read2 from each read pair are independently mapped to the genome and splice junction index using a contiguous read aligner. Low quality alignments are removed, the genomic and splice junction alignments are merged and the read pairs are resolved using an empirically determined fragment size distribution.
RNASequel implemented in C++ is available under the GNU Public License from: https://github.com/GWW/RNASequel.
MATERIALS AND METHODS
Splice junction definitions and alignment scoring
We defined a canonical splice junction as any splice junction with the following motifs: GTAG, GCAG, GCTG, GCAA, GCGG, GTTG, GTAA, ATAC, ATAA and ATAG. The strand of a splice junction was inferred based on gene annotations and the aforementioned splicing motifs. Alignments were scored using the following scoring penalties: gap open = −8, gap extension = −1, splice junction = −4, match = 3, mismatch = −3. For spliced alignments an extra alignment penalty was added for each splice junction. A penalty of −3 was applied for GTAG splice junctions, −6 for other canonical splice motifs and −9 for all other splice motifs. To reduce the chances of choosing an alignment with a long intron over an alignment with a shorter intron and a lower score we applied an additional penalty for splice junctions with introns over a pre-defined length (arbitrarily set at 64 kb by default). For these long introns we applied a penalty of 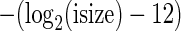 , where isize is the size of the intron.
, where isize is the size of the intron.
Splice junction discovery and splice junction index generation
The splice junction databases combined reference annotations (if available) and novel splice junction predictions from Tophat2 or STAR (if used). Only the novel splice junctions meeting the following criteria were retained (used for analysis): (i) the splice junction must be observed at least 8 bp away from the ends of at least one read; (ii) there are at least two different alignment positions mapping across the pair; (iii) the predicted intron size is at least 21 bp and no more than 500 kb in length. For each novel junction we added to the database N base pairs of flanking sequence on each side of the junction, where N should be chosen based on the size of a sequencing read, for our case we used 76 or 90. To handle cases in which a read could span multiple splice junctions, we supplemented our index by including multiple splice junctions on the same annotated strand if a sequence of length N could span one or more downstream junctions. Splice junctions with an ambiguous strand were considered on both strands. Finally, redundant sets of spanning splice junctions were removed to minimize the database size. The splice junction index can then be used with any contiguous read mapper.
Contiguous and spliced read alignment
For mapping reads to the GRCh37 reference genome (contiguous alignments) and splice junction indexes, we chose BWA-mem version 0.7.8 for its speed and accuracy (19). However any read mapper can be used. Read 1 and read 2 from each pair were independently mapped to the reference genome and the splice junction index. For each splice junction alignment, we resolved the alignments back to the genomic co-ordinates and removed contiguous alignments. To avoid alignment artifacts that occur due to reads improperly aligning to intronic sequences, alignments were trimmed if they overlapped a splice site within six base pairs of the end of the alignment. For each alignment the score was calculated as described above and we defined the minimum alignment score to be 2 × (aligned bases); any alignments with a score less than this were discarded. The retained alignments for read 1 and read 2 were then paired by identifying every potential alignment combination that matched the following criteria: (i) the alignments were on the same chromosome; (ii) the alignments were in the correct orientation and (iii) the distance between the read pairs was <1 Mb.
Estimating the empirical fragment size distribution
As noted earlier, the current generation of RNA-seq aligners uses an arbitrary cutoff to remove read pairs that map too far away from each other. RNASequel uses two different methods to solve this problem in a more disciplined manner. Only read-pairs that mapped uniquely after discarding alignments that had a score less than (the highest alignment score)—12 were used for fragment size estimation. We used a score difference of 12, which equates to four mismatches with our default mismatch penalty of three. This number can be adjusted if an increased repeat sensitivity is desired. In the case in which a gene annotation file is available for the organism under study, we estimated the expected fragment size distribution from the annotated gene model introns. For organisms with gene annotations we identified pairs that mapped to long exons (>250 bp) that should be larger than the insert size of the library or pairs that mapped to single isoform genes (7). In the case in which gene annotations were unavailable, we used maximum distance criteria of 1500 bp between the read pairs. In both cases we set a size cutoff to 1500 bp and required at least 100 000 fragment size observations. Both methods for estimating the fragment size distribution may lead to rare cases where an intron is included and the fragment size is overestimated. To compensate, the empirical distribution was normalized and a confidence interval retaining the smallest 99% of the observations was applied.
Resolving read pair alignments
To identify potential concordant read pairs we examined all of the potential combinations between the alignments for read 1 and read 2 that were correctly oriented, mapped to the same chromosome and were <1 Mbp apart. For each of these potential pairs every potential fragment size using different combination of splice junctions between the pairs was compared to the empirically determined fragment size distribution. Each potential fragment was then assigned a score of 10 × |(normalized fragment distribution score)/(maximum fragment distribution score)| + (read 1 alignment score) + (read 2 alignment score). The highest scoring pair was marked as primary; any pair with a score difference of <12 was marked as secondary and the remaining alignments were discarded. The score difference when calling repeat alignments should be carefully chosen based on the desired repeat tolerance, for our purpose we found that 12, which is equivalent to a difference of four mismatches, was reasonable. If no valid pairs were found using the fragment size distribution and the potential read pair was uniquely aligned it was outputted and marked as discordant. Furthermore, we implemented two different fallback methods depending on whether or not gene annotations were provided. Both of these methods are optional and deactivated by default. In the case where gene annotations were provided if both pairs mapped within the same annotated gene and were less than a user-defined distance apart they were considered concordant. If no gene annotations were provided we considered a pair concordant if the distance between the pair was at least a user-defined distance apart. For alignments where there were no valid alignments for one of the reads in a pair we reduced the score difference threshold to six, since we are only examining a single read rather than both reads in a pair. The highest scoring singleton alignment was marked as primary and the remaining alignments were marked as secondary.
Simulated dataset benchmarking
The simulated datasets were downloaded from ArrayExpress using the accession number E-MTAB-1728 (22) and alignments that mapped to ‘random’ and ‘NA’ chromosomes were removed. To simplify the comparison of alignment pipeline outputs to the ‘ground truth’ of the simulated datasets, we removed read pairs if either read had an edit distance of 25 or more. We left-shifted gaps, trimmed spliced alignments with less than eight base pairs of exonic overlap at the read ends and converted spliced alignments into deletions for predicted introns with a length <21 bp. For repeat mapped alignments we considered only the primary alignment. An alignment was considered perfect if the paired alignment exactly matched the true alignment. Partial alignments overlapped the true alignment but may have been soft clipped or included alternate insertions or deletions. Singleton alignments were classified as paired alignments in which either read 1 or 2 was unmapped. For spliced read alignment comparisons we counted a junction as correct only if the junction was present in the true alignment. A spliced alignment was considered partially correct if it contained at least one of the correct junctions and no incorrect junctions (this also encompasses the case in which the alignment contained all of the correct junctions but some of them were lost due to soft clipping). Finally, alignments that were mapped but did not meet the criteria for a perfect or partial alignment were marked as failed.
Identifying putative adenosine to inosine RNA editing events
The reads from the poly(A)-depleted YH lymphoblastoid cell line were mapped with the same alignment algorithm combinations as the benchmarking datasets. The alignments were retained if they had no more than two aligned ambiguous bases and no more than 10 soft clipped bases at either end. The retained alignments were then searched for potential edits using the following criteria to discard low quality calls: (i) positions mapping to tandem repeats using trf (27) or low complexity and simple regions according to RepeatMasker were discarded, (ii) for positions overlapping an inverted repeat annotated by einverted (28) or a repeat element identified by RepeatMasker we used a less stringent coverage criteria and required at least 10× coverage and a 10% alternative allele frequency, for positions with no repeat overlap we required 16× coverage and a 20% alternative allele frequency, (iii) at least one of the reads supporting the alternative base was outside of the first and last eight base pairs of the read ends, (iv) potential changes for which more than 90% of the supporting reads contained an insertion or deletion were removed, (v) potential sites where more than 70% of the supporting alignments contained different kinds of mismatches were discarded. After removing low quality calls, we also discarded changes found in the UCSC Genome Browser ‘Common SNP’ track, which is derived from dbSNP v141 if no genome sequence was available. For the GM12878 and YH datasets SNPs that were called from genome sequencing data were discarded (see Supplementary Methods) (29,30).
RESULTS
Developing an accurate RNA-seq realignment tool
We have developed RNASequel, an accurate and error-tolerant paired-end RNA-seq realignment tool, which functions as a post-processing step attached to an RNA-seq alignment algorithm. Our implementation allows the user to utilize his or her preferred aligner and future-proofs the tool: it can be used to improve the accuracy of any current or future RNA-seq alignment software that emits its results in standard BAM format. The tool refines the splice junction predictions prior to realignment by removing junctions that experience has shown are likely to be false positives, for example junctions found only in the end of reads or junctions found within repeat alignments. To improve paired-end alignment accuracy the reads from each pair are independently mapped to the genome sequence (genomic index) and a database of splice junctions (splice junction index) (Figure 1). An advantage of aligning the reads independently to the genome and splice junction index is the reduction of indexing time, since indexing the reference sequence can take a long time while indexing the RNASequel-generated splice junction database is comparatively fast. These four alignments can be performed in parallel using a computational cluster. The genomic and splice junction database alignments for each read are merged and alignments are discarded based on user-configured filtering parameters. Lastly, we refine paired-end read analysis by validating that each potential read pair alignment falls within an empirically determined fragment size distribution. This is in contrast to most spliced alignment methods that consider a read pair concordant if it aligns within a preset distance.
RNASequel realignment leads to improved alignment accuracy
To benchmark RNASequel realignment we tested two different de novo splice junction prediction tools, Tophat2 and STAR with gene model annotations (Tophat2 Ann. and STAR Ann) and without annotations (Tophat2 and STAR). The novel splice junctions identified from each of these tools were used for realignment with RNASequel. We also compared RNASequel realignment against STAR with two passes where the splice junctions predicted in the first pass are used to generate a new index for a second pass (STAR Two Pass and STAR Ann. Two Pass). Finally, to benchmark RNASequel without de novo splice junctions RNASequel was used with gene annotations alone in a single pass alignment (Annotation Only). We chose Tophat2 because of its popularity as one of the first RNA-seq alignment tools and STAR for its use within the ENCODE project, its high accuracy and its fast alignment rate (22). Two 2 × 76 bp simulated datasets used by each dataset have roughly 3.7 × 107 read pairs (22). The second of the two simulated datasets was generated with a higher mismatch (∼3× more), gap (∼5× more) and novel splice junction rate (1.5× more). Overall, RNASequel improved the number of reads that perfectly recapitulated the simulated alignment; this was especially the case for the second simulated dataset (Supplementary Tables S1 and S2, Figure 2A and B). For the first simulated dataset RNASequel alignments produced the highest number of perfect alignments, ∼90 versus 80–87% for the other methods, however, on the second simulated dataset RNASequel identified 12–20% more perfect alignments. The performance of the algorithms with and without gene annotations was similar for both simulated datasets. Finally, Tophat2 had the fewest number of partial alignments and the highest number of singleton alignments, likely due to one read in the pair having more mismatches than Tophat2's default cutoff. For both simulated datasets RNASequel realignment demonstrated an increased repeat sensitivity, the number of correct alignments to repetitive elements was typically ∼4× higher for the first simulated dataset and ∼2× higher for the second dataset. This improved alignment accuracy is also reflected in regions in both simulated datasets (Supplementary Figures S1–S4).
Figure 2.
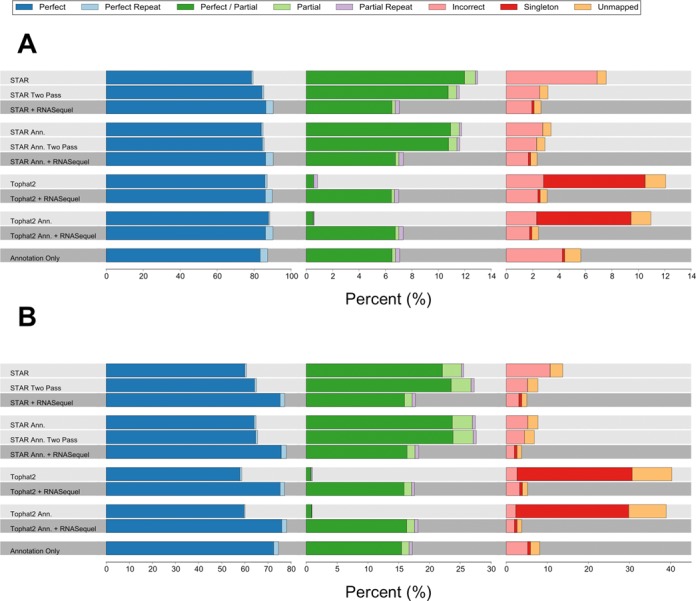
Alignment rates as percentages of the total number of pairs for the first (A) and second (B) simulated datasets with the indicated alignment methods. For a description of the alignment types see the benchmarking methods description.
Realignment to a splice junction database improves spliced read accuracy
A major challenge for de novo splice junction identification is that a single pass alignment scheme may incorrectly align reads with short exonic alignments because the true splice junction has not been discovered. To mitigate this issue we applied a filtering scheme to identify and remove false positives that occurred due to repetitive region mappings, splice junctions occurring exclusively in the ends of a read and/or non-canonical splice motifs. To maximize our ability to align reads across multiple splice junctions we supplemented sample-specific splice junction index with groups of novel and annotated splice junctions that could be spanned by a single sequencing read. For both simulated datasets, realignment with RNASequel or STAR with two passes increased the number of perfectly mapped spliced reads by 2–10% (Supplementary Figure S5). When gene annotations were present the number of perfect alignments increased by 4–10%. This was particularly evident for reads that spanned multiple splice junctions, which demonstrates the usefulness of our splice index alignment (Supplementary Figure S6). RNASequel realignment had the lowest number of incorrect spliced alignments and the highest number of perfect alignments compared to STAR. The rate of incorrect alignments was higher when using Tophat2 for de novo splice junction predictions. This may be due to Tophat2's higher false negative rate. The importance of including de novo splice junctions for alignment is highlighted by examining RNASequel using only gene annotations which had the highest number of incorrect spliced reads. The number of perfect spliced reads was more pronounced for the second simulated dataset where the number was increased by ∼10% and the number of failed alignments decreased by 5% without annotations and 2% with annotations for RNASequel realignment versus STAR with two passes. Overall, RNASequel realignment had the highest precision for both annotated and novel splice junctions (Figure 3A and B, Supplementary Figure S7A and B, Supplementary Tables S1 and S2). For annotated splice junctions RNASequel realignment had the highest recall for both simulated datasets and comparable precision. The increase was small for the first simulated dataset, but 7–30% higher for the second simulated dataset. As expected, the recall and precision were highest when gene model annotations were supplied.
Figure 3.
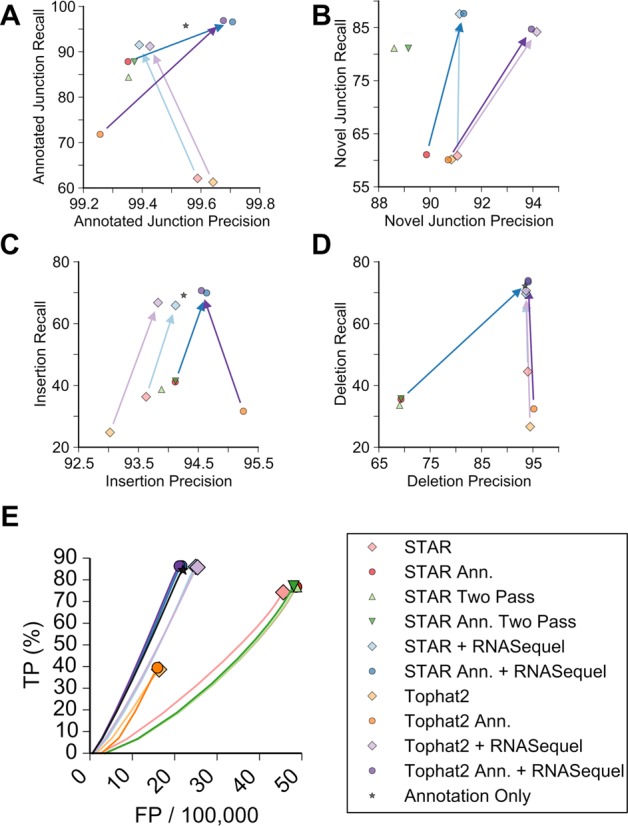
Alignment characteristics for the second simulated dataset. The recall and precision as a percentage of the number of correctly aligned reads for annotated junctions (A), novel junctions (B), insertions (C) and deletions (D). The alignment algorithms used are indicated according to the legend and the arrows indicate the improvement by RNASequel and are colored according to the legend. (E) Receiver-operator curve demonstrating the relationship of correctly called sequence variants (Y axis) to the number of falsely called variants (X axis) for each read pair across each of the alignment methods. Note that the X-axis scale is false positive variant calls per 100 000 reads.
For the identification of novel splice junctions, RNASequel had a slightly lower recall rate due to our filtering scheme, but a ∼3–5% higher precision than STAR for the first simulated dataset. The slight decrease in recall and the increase in precision demonstrates the tradeoff when applying a filtering scheme to novel splice junctions prior to realignment. For the second simulated dataset, RNASequel realignment increased the recall by 6–23% and the precision by 2–4%. We examined the false negative splice junction alignments and observed that majority of them (23–60%) were within 15 bp of the 3′ end of the read sequence. These may have been missed due to the simulated read quality degradation near the 3′ ends (Supplementary Figures S8 and S9).
In summary, by generating a splice junction database and mapping the reads with an accurate error-tolerant realignment we have increased the splice junction accuracy, especially in the case of datasets with high error rates.
RNASequel realignment improves alignments with insertions and deletions
Gapped alignments are a challenge for RNA-seq alignment. For example, a higher gap tolerance threshold can result in additional false positive splice junction predictions by inserting a gap to bridge an alignment to an incorrect splice junction. Furthermore, false positive gaps can be inserted within an alignment that incorrectly aligns to an intronic sequence. To overcome this we have combined RNASequel's accurate splice junction indexing strategy with a gap tolerant alignment using BWA mem followed by a trimming of alignments that map to intron sequences. Using this approach, RNASequel increased the gap recall by ∼20% compared to STAR and Tophat2 (Figure 3C and D, Supplementary Figure S7C and D, Supplementary Tables S1 and S2). The insertion precision was comparable between all of the methods used while the deletion precision after RNASequel realignment was ∼20–25% higher compared to STAR. For each of the alignment algorithms the false negatives for insertions and deletions tended to occur in the first and last 10 bp of each read where aligners are more likely to soft clip the alignment rather than insert a gap (Supplementary Figures S8 and S9). Intriguingly, STAR alignments produced a higher percentage of false positive deletions through the middle of the read compared to Tophat2 and RNASequel realignment. Tophat2 had a slightly higher false positive rate near the read ends due to using an underlying global rather than local alignment algorithm.
The effect of RNASequel's increased gap tolerance is to reduce read artifacts such as mismatches and incorrect splice junction calls due to incorrect gapped alignment.
RNASequel realignment increases mismatch tolerance and accuracy
High mismatch tolerance for RNA-seq alignment can lead to an increase in accuracy, but it can also lead to more false positive splice junction alignments or alignments that should be spliced but are contiguously aligned into an intron sequence. The RNASequel splice junction filtering step helps reduce some of these false positives while our attempt to trim alignments that overlap splice sites near the read ends reduces many false positives. The simulated datasets are dominated by alignments with low numbers of mismatches and to assess the performance of the tools and RNASequel on read pairs with high and low levels of mismatches, we plotted the number of true positive and false positive mismatches stratified by the true number of mismatches in each read pair (Figure 3E, Supplementary Figure S7E). RNASequel realignment had the highest mismatch recall and precision compared to the other tools (Supplementary Tables S1 and S2). Tophat2 had the lowest mismatch accuracy due to a low mismatch tolerance by default. As observed in the splice junction and gap tests, the majority of the false negative and false positive mismatches were near the ends of reads, particularly the 3-prime end of the read (Supplementary Figure S10A and B). This is due to the higher number of mismatches near the 3-prime end of the read from the simulated read quality degradation. It should be noted that we could have improved the other tools’ accuracy by hand-optimizing their alignment parameters, but we felt that the default parameters represented a typical laboratory use case. Furthermore, adjusting the alignment tools mismatch parameters may lead to undesirable alignment artifacts, for example, a higher false positive spliced read alignment rate.
RNASequel execution speed and memory requirements
RNASequel realignment is reasonably fast. The splice junction index generation takes <15 min. The reference and splice junction alignment steps are dependent on the chosen alignment tool, for BWA-mem this takes 2–3 h per 100M reads with 16 threads for the reference alignment and 1 h per 100M reads for the splice junction index alignment. BWA-mem uses 40GB of memory for both alignment types. The merge step processes ∼35M pairs per hour with eight threads and uses 20GB of memory. It should be noted that all four of the BWA-mem alignments could be parallelized on a computational cluster decreasing the RNASequel processing time substantially. As a comparison STAR processes roughly 50M pairs per hour with eight threads and ∼60GB of memory. Tophat2 processes roughly 8M pairs per hour with eight threads and <20GB of memory.
RNASequel realignment improves alignment characteristics on biological datasets
Simulated datasets do not capture all of the potential sources of errors present in a biological RNA-seq library. For example, there may be reads derived from spurious transcripts in non-coding regions of the genome such as pseudogenes. There are also other sequencing errors unique to a biological dataset such as reverse transcriptase template switching (23,24). To compare the alignment accuracy of RNASequel to Tophat2 and STAR, we applied our program to three biological datasets, one derived from a lymphoblastoid cell line (YH) and two replicates derived from the lymphoblastoid cell line GM12878 (31,32). The YH RNA-seq sample used a library that was poly(A) and ribosomal RNA depleted and was deeply sequenced to a depth of ∼400M 2 × 90 bp pairs. The GM12878 samples were sequenced to a depth of ∼100M 2 × 75 bp poly(A) selected pairs. For all three samples RNASequel realignment lead to the concordant mapping of more read pairs. For the YH sample, realignment with RNASequel realignment leads to the concordant mapping of ∼90% of the read pairs while Tophat2 mapped ∼60% and STAR mapped ∼84% (Supplementary Figure S12A, Table S3). For GM12878-1 the paired alignment rates were ∼80% for STAR and RNASequel while Tophat2 mapped ∼48% (Supplementary Figure S12B, Table S4). Finally, for the GM1278-2 sample RNASequel mapped ∼80% of the pairs, while star mapped ∼70% and Tophat2 mapped ∼45% (Supplementary Figure S12C, Table S5). In all three of the cases RNASequel identified 0.3–6% more pairs as repeat mapping compared to STAR and Tophat2. For the YH dataset STAR with two passes mapped a similar number of repeat pairs to RNASequel while mapping 2–3 times less for the GM12878-1 dataset. To further investigate the read mapping improvements conferred by RNASequel we compared STAR Ann. plus RNASequel to STAR Ann. with two passes with an additional 25 poly(A) RNA-seq samples from the ENCODE project (Supplementary Figure S13, Table S6). On average RNASequel mapped 2.75% more pairs and identified an average of 5.5% more repeat mapped pairs.
RNASequel realignment attempts to predict whether a read pair is concordant using the empirically determined fragment size distribution, splice junction predictions and gene annotations. To compare the effect of this on paired alignment we used our fragment size determination algorithm on the alignments produced by STAR and Tophat2 to predict whether the paired alignments have a valid fragment size using the junctions predicted by the tool and gene annotations. We found that ∼1–2% of the pairs uniquely mapped by STAR and Tophat2 had a fragment size outside of the empirical range determined by our algorithm (Supplementary Figure S11). It should be noted that Tophat2 does take advantage of a user-provided fragment size mean and standard deviation. These numbers were also similar for repeat alignments where all or a subset of the alignments had a fragment size that was not within the empirically determined distribution. These represent a small proportion of the alignments that include cases where the fragment size was outside of the tail of fragment size cases with missing splice junction annotations and false positive alignments. For STAR ∼60–80% for unique pairs and ∼20–40% for repeat pairs fall within 50 bp of our confidence interval (data not shown). However, these alignments can contribute to artifacts in downstream analysis, especially when identifying variant or RNA editing calls.
RNASequel realignment generates more robust RNA editing calls
In vertebrates, the ADAR family of enzymes is responsible for the conversion of adenosine to inosine (A-to-I) in RNA (33). This type of RNA editing is thought to be used as a regulatory mechanism (34). In RNA-sequencing, A-to-I edits manifest either as A-to-G or T-to-C substitutions depending on the strand of the transcript. The identification of RNA editing sites using RNA-seq is difficult due to a number of sequencing and alignment artifacts. To demonstrate the degree to which RNASequel realignment improves RNA editing calls we compared our realignment algorithm with Tophat2 and STAR with and without gene model annotations. The potential nucleotide changes were then filtered to remove common sources of false positives including alignments to tandem repeats and changes biased to the ends of reads. We removed somatic polymorphisms (if available) or common polymorphisms in dbSNP (if genome annotations were not available). Prior to filtering the YH and GM12878 datasets RNAsequel realignment yielded comparable numbers (+/-1–3%) of A-to-I changes as STAR and Tophat2 RNASequel yielded 20% more A-to-I changes for the YH dataset and 8–10% fewer changes for the GM12878 datasets (Supplementary Figures S14, S15, S16). For non-A-to-I changes RNASequel yielded 4–11% fewer changes compared to STAR and 23–40% fewer compared to Tophat2. We also compared the total SNV calls between STAR Ann. with RNASequel and STAR Ann. with two passes for 25 additional ENCODE RNA-seq samples. We found an average decrease in the number of A-to-I calls by 0.52% and a decrease in non-A-to-I calls by 3.7% (Supplementary Figures S17 and S18). These results suggest that RNASequel realignment leads to fewer potential false positives prior to filtering than STAR and Tophat2. These results are also consistent with our simulated dataset results that demonstrated the reduction in false positive mismatch calls facilitated by RNASequel realignment compared to Tophat2 and STAR.
After filtering potential false positives we observed that RNASequel and STAR found similar somatic SNV calls (∼1% difference) (Supplementary Figures S14–S16). For Tophat2 alignments RNASequel realignment yielded 20–40% more somatic SNV or dbSNP calls. We also observed an average 3.1% reduction in dbSNP calls for ENCODE samples (Supplementary Figures S17 and S18A). For A-to-I calls we observed a comparable number of changes between STAR and RNASequel for the YH dataset (∼0.1–1% increase after realignment) and ∼4–10% fewer changes for the GM12878 datasets and for Tophat2 alignments we found 2–3 times as many A-to-I calls. For non-A-to-I changes we observed a 15–25% decrease in the number of calls after RNASequel realignment compared to STAR and a 1.4–3 times as many compared to Tophat2. For the 25 ENCODE datasets we found an average of 7.3% fewer A-to-I changes and 10.4% fewer non-A-to-I changes. Combined together these results suggest that RNASequel realignment yields fewer false positive SNV calls compared to STAR due to RNASequel realignment reducing the number of non-A-to-I changes. Furthermore, for the YH-1 dataset we found more somatic SNV's suggesting an improved false negative score compared to STAR and Tophat. Tophat2 uses a global alignment algorithm and low mismatch tolerance that leads to a higher false negative rate for reads with more than two mismatches and a higher false positive rate at the read end for reads with few mismatches. In conclusion, we feel that RNASequel realignment shows that the false positive is reduced with minimal effect on the false negative rate.
Finally, to explore the features of RNASequel realignment that leads to improved SNV and RNA editing calls we assessed the impact of RNASequel's improved repeat detection and fragment size estimation algorithms. To assess the impact of repeat mapped reads on calling of RNA editing sites, we collected the union of pairs that mapped across any of the variant sites by either the base alignment program or the alignment program with RNASequel. Pairs that were multi-mapped by one tool and uniquely mapped by the other were discarded and the edit sites were called and filtered again. To assess the impact of our fragment size determination algorithm on identifying concordant read pairs we removed uniquely mapped reads that did not have a valid fragment size as determined by our algorithm. We found that within the union of alignments RNASequel marked 4–10% of the reads as multi-mapping, compared to STAR and Tophat2, which marked ∼1% as multi-mapping with the exception of STAR with two passes which had 2.7% for the YH sample (Figure 4, Supplementary Table S7). We also identified more alignments marked as singleton compared to STAR (1–10 versus 0%) and fewer than Tophat2 (1–9 versus 17–35%). For the 25 ENCODE samples we observed an average of 13% multi-mapped reads with RNASequel versus 1.3% with STAR with two passes (Supplementary Figure S19, Table S8). RNASequel realignments mapped more pairs where 0.6–4% of the reads were unmapped compared to STAR and Tophat2 where 4–25% of the pairs were unmapped. RNASequel also mapped more of the alignments than STAR for the 25 ENCODE datasets 1 versus 4%. A portion of the alignments identified by STAR as concordant pairs were marked as discordant pairs by RNASequel (0.1–0.8% of the alignments). Tophat2 marked the highest proportion of reads as discordant but this was also the case for the simulated and full set of reads for the biological datasets. Finally, our fragment size estimation algorithm identified ∼1% of the reads mapped as unique by STAR or Tophat2 as being discordant. After removing the alignments that were marked as unique by STAR and reads marked as discordant with our fragment size determination algorithm the difference in the number of calls was lessened or increased in favor of RNASequel (Supplementary Figures S14–S16 and S18). For example, the number of non-A-to-I edits is reduced after removing reads that were uniquely mapped by STAR but were repeat mapped by RNASequel. Collectively, these results imply that the improvements in alignment characteristics, particularly increased repeat sensitivity and improved identification of concordantly mapped read pairs leads to an improved alignment for the purposes of calling SNVs and RNA edits.
Figure 4.
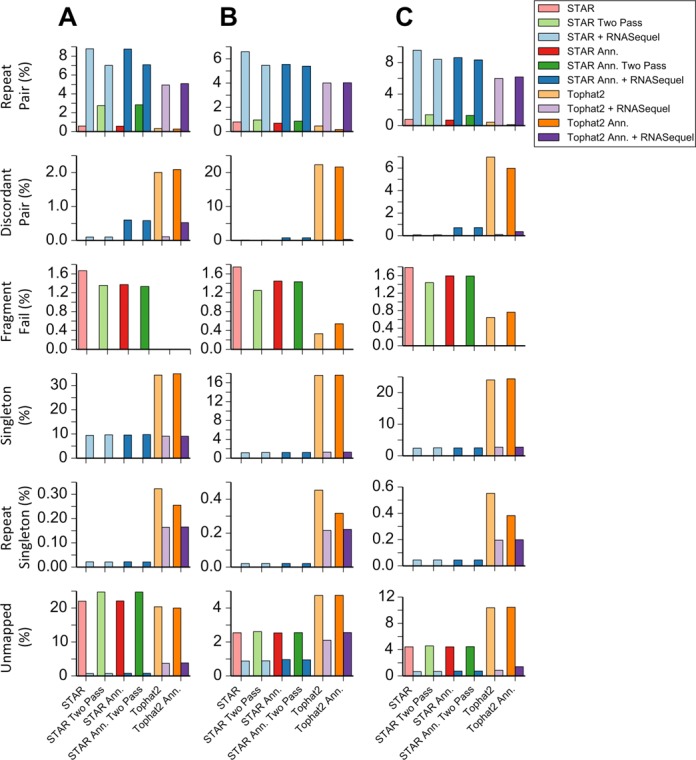
Comparision of the alignment type between the union of all reads that support a genomic SNV, dbSNP entry, retained A-to-I change or retained non-A-to-I change for YH (A) GM12878–1 (B) and GM12878–2 (C). The bar on the left indicates the percentage of alignment types for the labeled tool, the bar on the right indicates the alignment rate for the tool with RNASequel realignment. For STAR with two passes, the alignment rate for RNASequel with STAR as a single pass is used for comparison.
DISCUSSION
By systematically mitigating common artifacts that occur during RNA-seq library preparation and alignment, RNASequel increases the accuracy of splice junction, gap and mismatch calling while decreasing the false discovery rate. When applied to the challenging problem of RNA editing detection, the RNASequel post-processing method reduces the number of apparent false positives without adversely affecting sensitivity. We have found that using RNASequel in combination with STAR provides the best accuracy metrics. Crucially, we show that despite our higher error tolerance, we identify fewer non-canonical edits compared to STAR on a biological dataset. This implies that many potential RNA editing calls are due to systematic alignment errors that can be mitigated with RNASequel realignment thereby strengthening the interpretation of biological datasets. STAR is also preferred because it has better performance characteristics than Tophat2. RNASequel realignment is agnostic to the underlying aligners used for splice junction prediction and contiguous read alignment leading to an adaptable RNA-seq alignment tool that can take advantage of new alignment methods. In the future, we are investigating methods to improve the performance and disk space usage of RNASequel by calling the underlying contiguous aligner as a library. We are also investigating methods to capture aligned pairs that fall within the tail of the fragment size distribution to increase the number of concordantly mapped pairs. The improvements facilitated by RNASequel realignment are useful for the analysis of alternative splicing, gene and isoform expression, sequence variant calling and RNA editing.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We would like to thank Michael Brudno, Ewan Gibb and Paul Krzyzankowski for helpful discussion regarding the manuscript and the design of RNASequel.
FUNDING
Ministry of Economic Development and Innovation, Ontario; Frederick Banting and Charles Best Canada Graduate Scholarship (to G.W.W., in part). Funding for open access charge: Ontario Ministry of Research and Innovation.
Conflict of interest statement. None declared.
REFERENCES
- 1.Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lister R., O'Malley R.C., Tonti-Filippini J., Gregory B.D., Berry C.C., Millar A.H., Ecker J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 4.Pan Q., Shai O., Lee L.J., Frey B.J., Blencowe B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 5.Maher C.A., Kumar-Sinha C., Cao X., Kalyana-Sundaram S., Han B., Jing X., Sam L., Barrette T., Palanisamy N., Chinnaiyan A.M. Transcriptome sequencing to detect gene fusions in cancer. Nature. 2009;458:97–101. doi: 10.1038/nature07638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang E.T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S.F., Schroth G.P., Burge C.B. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;29:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramaswami G., Lin W., Piskol R., Tan M.H., Davis C., Li J.B. Accurate identification of human Alu and non-Alu RNA editing sites. Nat. Method. 2012;9:579–581. doi: 10.1038/nmeth.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piskol R., Peng Z., Wang J., Li J.B. Lack of evidence for existence of noncanonical RNA editing. Nat. Biotechnol. 2013;31:19–20. doi: 10.1038/nbt.2472. [DOI] [PubMed] [Google Scholar]
- 10.Hastings M.L., Krainer A.R. Pre-mRNA splicing in the new millennium. Curr. Opin. Cell Biol. 2001;13:302–309. doi: 10.1016/s0955-0674(00)00212-x. [DOI] [PubMed] [Google Scholar]
- 11.Garber M., Grabherr M.G., Guttman M., Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat. Methods. 2011;8:469–477. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- 12.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K., Singh D., Zeng Z., Coleman S.J., Huang Y., Savich G.L., He X., Mieczkowski P., Grimm S.A., Perou C.M. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Au K.F., Jiang H., Lin L., Xing Y., Wong W.H. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res. 2010;38:4570–4578. doi: 10.1093/nar/gkq211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grant G.R., Farkas M.H., Pizarro A., Lahens N., Schug J., Brunk B., Stoeckert C.J., Hogenesch J.B., Pierce E.A. Comparative analysis of RNA-seq alignment algorithms and the RNA-Seq Unified Mapper (RUM) Bioinformatics. 2011;27:2518–2528. doi: 10.1093/bioinformatics/btr427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics. 2014;30:2843–2851. doi: 10.1093/bioinformatics/btu356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu T.D., Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012;29:21–22. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engström P.G., Steijger T., Sipos B., Grant G.R., Kahles A., Alioto T., Behr J., Bertone P., Bohnert R., Campagna D., et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat. Methods. 2013;10:1185–1191. doi: 10.1038/nmeth.2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cocquet J., Chong A., Zhang G., Veitia R.A. Reverse transcriptase template switching and false alternative transcripts. Genomics. 2006;88:127–131. doi: 10.1016/j.ygeno.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 24.Houseley J., Tollervey D. Apparent non-canonical trans-splicing is generated by reverse transcriptase in vitro. PLoS One. 2010;5:e12271. doi: 10.1371/journal.pone.0012271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Gurp T.P., McIntyre L.M., Verhoeven K.J.F. Consistent errors in first strand cDNA due to random hexamer mispriming. PLoS One. 2013;8:e85583. doi: 10.1371/journal.pone.0085583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes inthe presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rice P., Longden I., Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 29.Consortium, T.1.G.P., The 1000 Genomes Consortium Participants are arranged by project role, T.B.I.A.A.F.A.W.I.E.F.P.I.A.P.L.A.I., author, C., committee, S., Medicine, P.G.B.C.O., BGI-Shenzhen, Broad Institute of MIT and Harvard, European Bioinformatics Institute, Illumina, Max Planck Institute for Molecular Genetics. An integrated map of genetic variation from 1,092 human genomes. Nature. 2013;490:56–65. [Google Scholar]
- 30.Wang J., Wang W., Li R., Li Y., Tian G., Goodman L., Fan W., Zhang J., Li J., Zhang J., et al. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng Z., Cheng Y., Tan B.C.-M., Kang L., Tian Z., Zhu Y., Zhang W., Liang Y., Hu X., Tan X. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nature Biotechnol. 2012;30:253–260. doi: 10.1038/nbt.2122. [DOI] [PubMed] [Google Scholar]
- 32.Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F., et al. Landscape of transcription in human cells. Nature. 2013;488:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gott J.M., Emeson R.B. Functions and mechanisms of RNA editing. Annu. Rev. Genet. 2000;34:499–531. doi: 10.1146/annurev.genet.34.1.499. [DOI] [PubMed] [Google Scholar]
- 34.Nishikura K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010;79:321–349. doi: 10.1146/annurev-biochem-060208-105251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.