


Abstract
Type I CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)–Cas (CRISPR-associated) systems exist in bacterial and archaeal organisms and provide immunity against foreign DNA. The Cas protein content of the DNA interference complexes (termed Cascade) varies between different CRISPR-Cas subtypes. A minimal variant of the Type I-F system was identified in proteobacterial species including Shewanella putrefaciens CN-32. This variant lacks a large subunit (Csy1), Csy2 and Csy3 and contains two unclassified cas genes. The genome of S. putrefaciens CN-32 contains only five Cas proteins (Cas1, Cas3, Cas6f, Cas1821 and Cas1822) and a single CRISPR array with 81 spacers. RNA-Seq analyses revealed the transcription of this array and the maturation of crRNAs (CRISPR RNAs). Interference assays based on plasmid conjugation demonstrated that this CRISPR-Cas system is active in vivo and that activity is dependent on the recognition of the dinucleotide GG PAM (Protospacer Adjacent Motif) sequence and crRNA abundance. The deletion of cas1821 and cas1822 reduced the cellular crRNA pool. Recombinant Cas1821 was shown to form helical filaments bound to RNA molecules, which suggests its role as the Cascade backbone protein. A Cascade complex was isolated which contained multiple Cas1821 copies, Cas1822, Cas6f and mature crRNAs.
INTRODUCTION
The arms race between viruses on one end and bacteria and archaea on the other end resulted in the evolution of diversified prokaryotic immune systems and viral countermeasures (1,2). Prokaryotic genomes can contain adaptive immune systems, termed CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)–Cas (CRISPR-associated) (3,4), which utilize small RNAs to target foreign genetic elements (5,6). Genomic CRISPR elements consist of an array of short repeat sequences that are interspersed with unique spacer sequences that can be derived from viral genomes and conjugative plasmids (7–9). CRISPR arrays are often located adjacent to a set of cas genes that are essential for conferring immunity (10,11). The CRISPR–Cas immune response can be divided into three stages. First, upon infection of the host by a virus, a segment of a viral genome, i.e. the protospacer, is integrated as a new spacer into an expanding CRISPR array (12). This stage of the immune response, termed acquisition, relies on the recognition of a short signature sequence, the protospacer adjacent motif (PAM) in the viral DNA sequence (13,14). Next, the CRISPR array is transcribed and processed into short CRISPR RNAs (crRNAs) that contain a spacer sequence flanked by parts of the repeats at their termini (15–19). The crRNAs are integrated into CRISPR ribonucleoprotein (crRNP) surveillance complexes that are formed by multiple Cas proteins. These complexes utilize the crRNA guidance to recognize and degrade foreign genetic material during a recurring infection (6,17,20–22). The recognition of the foreign genetic material is achieved via Watson–Crick base pairing between the crRNA and the unwound target DNA strand or target RNA sequence (23–25).
CRISPR–Cas systems are classified into three main types based on the presence of the signature proteins Cas3 (Type I), Cas9 (Type II) and Cas10 (Type III), respectively (10,26). Only Cas1 and Cas2 are conserved in the vast majority of the CRISPR–Cas systems and have been found to be essential for the acquisition of new spacers (13,27,28). Type I systems utilize a crRNP complex termed Cascade (or CRISPR-associated complex for antiviral defense) to identify targets (6,21,29), and Type II systems are characterized by the standalone nuclease Cas9 (30–32). In both cases, the recognition of targets is dependent on the presence of a PAM sequence (25,33,34). In contrast, Type III systems use crRNP complexes that facilitate co-transcriptional ssRNA and DNA cleavage in a PAM sequence-independent manner (20,35,36).
The Type I Cascade crRNP complex has been studied biochemically and structurally for subtype I-E (17,23,24), and cryo-EM structures are available for Cascade complexes of subtypes I-A, I-C and I-F (37–39). The 405 kDa Type I-E Cascade from Escherichia coli has a characteristic ‘seahorse-shape’ architecture and is formed by Cas5e, Cas6e, Cas7 and two subtype-specific proteins Cse1 (large subunit) and Cse2 (small subunit) (23,24,37). The complex has an uneven protein stoichiometry of (Cas7)6–(Cse2)2–(Cse1)1–(Cas5e)1–(Cas6e)1 and binds to a 61 nt-long crRNA, which contains a 32 nt-long spacer flanked by 8 nt- and 21 nt-long repeat sequences, respectively. Cse1, the large subunit protein of the Cascade complex, is involved in PAM recognition and recruits Cas3 after target recognition (17,37,40), whereas the small subunit protein Cse2 functions in stabilizing the R-loop by binding to the displaced DNA strand (17,23,24). The Type I-E Cascade has a helical backbone formed by six copies of Cas7 and contains a groove in which the crRNA is bound and protected. Additionally, the 3′- and 5′-ends of the crRNA are protected by Cas6e and Cas5e, respectively (17,41). This general architecture of the Cascade crRNPs appears to be conserved for all Type I subtypes, even though their Cas protein compositions can differ considerably (42).
One apparent variation of this Cascade architecture is evident for a computationally identified subtype I-F variant present in few beta- and gammaproteobacteria. This variant was first described for Photobacterium profundum SS9 (PBPRB1995-PBPRB1991) and was suggested to rely on a minimal set of five Cas proteins (26). The system contains Cas1, an integrase that mediates spacer acquisition (43,44), Cas2–3, the target DNA nuclease, Cas6f, the endonuclease that generates mature crRNAs (18), and two additional genes that show no apparent sequence similarity to any known Cas protein families. Furthermore, a large subunit could not be detected (26). We identified this minimal CRISPR-Cas subtype in the Shewanella putrefaciens strain CN-32 and experimentally verified crRNA maturation and DNA interference activity in vivo. Recombinant production of the Cas proteins revealed the presence of a Cascade complex and allowed for the assignment of one unclassified Cas protein into the Cas7 group. The two unclassified proteins were shown to be required for a stable crRNA pool in S. putrefaciens CN-32. Our results provide insights into the reductive evolution of CRISPR–Cas systems and demonstrate that a Cascade complex can function without a large subunit.
MATERIALS AND METHODS
Bacterial strains and growth conditions
Bacterial strains and plasmids used in this study are listed in Supplementary Table S1. E. coli strains DH5α, DH5α λpir and WM3064 were grown in LB medium at 37°C. Media for the growth of the 2,6-diamino-pimelic acid (DAP)-auxotroph E. coli WM3064 strain were supplemented with 300 μM of DAP. E. coli strain BL21(DE3) pLysS was grown either in LB or in NZ-amine (1% NZ-amine, 0.5% yeast extract and 1% NaCl) media at 37°C until an OD600 of ∼0.6 was reached. Protein expression was induced by addition of 1 mM Isopropyl β-d-thiogalactopyranoside (IPTG) and continuous growth at 18°C. S. putrefaciens CN-32 was cultured in LB medium at 30°C. Media were supplemented with 50 μg/ml spectinomycin, 50 μg/ml kanamycin, 50 μg/ml ampicillin or 10% (w/v) sucrose.
Construction of cas gene deletion strains
S. putrefaciens CN-32 mutants with markerless in-frame deletions or integrations were constructed using the primers listed in Supplementary Table S2 and the suicide vector pNPTS138-R6KT (KanR) following the procedure reported in (45). Briefly, appropriate ∼500 bp homologous fragments of the up- and downstream regions of the target gene were amplified. For deletions, the codons encoding the first and last four amino acids were retained. The resulting PCR-derived DNA fragments were joined via overlap PCR, treated with appropriate restriction enzymes and ligated with the equally digested pNPTS138-R6KT vector. The resulting plasmids were sequenced and transferred to S. putrefaciens CN-32 by conjugation from E. coli WM3064. Target gene regions were replaced by the corresponding modified version via sequential homologous crossover and correct deletions were identified via colony PCR using appropriate primers.
Point mutations were introduced into the S. putrefaciens CN-32 gene Sputcn32_1822. To this end, the wild-type gene sequence, along with 500 bp of upstream and downstream sequences was cloned into pNPTS138-R6KT. Point mutations were introduced into this plasmid by QuickChange site-directed mutagenesis (Stratagene) using primers that were designed using the Agilent QuickChange Primer Design Tool. The plasmids carrying the point mutations were applied for in-frame insertions in S. putrefaciens CN-32 as described above.
RNA isolation, sequencing and Northern blot analysis
Nucleic acids were extracted from an overnight culture of S. putrefaciens CN-32 using phenol/chloroform (1:1 phenol pH 5 for RNA and pH 8 for DNA). The mirVana RNA extraction kit (Ambion) was used to isolate small RNAs (<200 nt) from total RNA. To ensure proper termini for adapter ligation, 1 μg of the small RNA preparation was incubated with 10 U of T4 polynucleotide kinase (T4 PNK, Ambion) at 37°C overnight, followed by 1 h incubation at 37°C in presence of 1 mM ATP. This treatment ensures 5′-monophosphate termini suitable for RNA-Seq library preparations. RNA libraries were sequenced by Illumina HiSeq2000 sequencing at the Max-Planck Genome Centre, Cologne.
Northern blot analyses required the extraction of 10 μg of total RNA from wild-type and cas gene-knockout strains of S. putrefaciens CN-32. The RNA preparations were denatured (95°C for 5 min) in formamide loading buffer and separated via electrophoresis on a 10% TBE–8 M urea polyacrylamide gels at 200 V for 1 h. The separated bands were transferred to a nylon membrane (Roth) and subjected to UV-crosslinking at 25 V for 2 h. DNA probes were radiolabeled with 5′-γ[32P]-ATP using T4 PNK and incubated with the membrane, which was pre-incubated at 42°C for 30 min with ULTRAhyb-Oligo buffer (Ambion). After overnight hybridization at 42°C, the membranes were washed two times for 15 min with 2× SSC buffer and 0.1% SDS and with 0.1× SSC and 0.1% SDS. Radioactive signals were detected by phosphorimaging.
Conjugation-assays for DNA interference analysis
Target plasmids for conjugation assays were designed with the primers listed in Supplementary Table S2. Individual primer pairs were hybridized generating the sticky ends of EcoRI and BamHI restriction sites that flanked the protospacer sequences. The primer pairs were phosphorylated at the 5′-termini by T4 PNK, mixed and hybridized (95°C for 5 min, followed by slow cooling to room temperature). The obtained DNA fragments were ligated into a linearized pBBR1MCS2 (KanR) vector. Insertion of the fragment was confirmed via sequencing.
The obtained plasmids were transformed into the donor strain E. coli WM3064. Equal amounts of E. coli and S. putrefaciens CN-32 strains overnight cultures were harvested. The strains were suspended in 100 μl of DAP-supplemented LB medium and mixed before spotting a single drop on nonselective LB agar supplemented with DAP. After overnight incubation at 30°C, the cells were washed from the plate using 2 ml of nonselective LB. The mating mixture was then washed three times with 1 ml of nonselective LB and screened for conjugated S. putrefaciens CN-32 by plating 100 μl of the mating mixture on LB plates supplemented with Kan but lacking DAP. This procedure was performed in triplicate and in parallel for the control plasmid without protospacer sequences. The number of obtained colonies was counted for conjugation of the plasmid with protospacer (pT) and the control plasmid (pNT). Relative conjugation efficiency was calculated as pT/pNT ratio and the transconjugant counts are detailed in Supplementary Table S3.
Production and purification of recombinant proteins
The cas genes of S. putrefaciens CN-32 were amplified from genomic DNA and cloned into gene expression vectors. The cas6f gene was cloned into pET20b (AmpR) to generate Cas6f with a C-terminal His-tag. The gene cassette cas1821, cas1822, cas6f was cloned into pRSFDuet; this construct allows the simultaneous production of all three proteins with only Cas1821 having an N-terminal His-tag fusion. The individual cas1821 gene was cloned to produce a His-Sumo-tag fusion that was processed as previously described for Thermoproteus tenax Cas proteins (22). Cas6f production was induced by addition of 1 mM IPTG followed by overnight growth at 18°C of the E. coli BL21(DE3)pLysS host strain. Cell lysates were pepared by sonication in a buffer containing 50 mM Tris-HCl pH 8.0, 500 mM NaCl, 5% glycerol, 1 mM dithiothreitol (DTT), 0.01% Triton X100 and 20 mM imidazole. Proteins were purified on a HiTrap Ni-NTA column (GE Healthcare) in a linear imidazole gradient ranging from 100 to 500 mM imidazole. The Cas1821 protein was subjected to Ni-NTA chromatography in a buffer containing 100 mM potassium phosphate pH 7.5, 500 mM NaCl, 10% glycerol. A high-salt washing step with a buffer including 1 M NaCl was added before protein elution to remove nucleic acid contaminants. The SUMO tag was cleaved using SUMO protease during overnight dialysis at 4°C in a buffer containing 50 mM Tris–HCl pH 7.0, 100 mM NaCl and 10% glycerol. The protein was further purified over a HiTrap Heparin Sepharose HP column and size exclusion chromatography on a Superdex 200 column.
The production of recombinant Cascade required the presence of the pRSFDuet vector containing the genes cas1821, cas1822, cas6f and a pUC19 vector containing the repeat-spacer4-repeat sequence from the CRISPR array cloned downstream of a T7 RNA polymerase promoter. The 100 nt transcript is processed by Cas6f into a 60 nt crRNA. BL21(DE3)pLysS cells with both plasmids were grown in the presence of ampicillin, kanamycin and spectinomycin. Protein and pre-crRNA production was induced at an OD600 of 0.6 by addition of 1 mM IPTG, followed by overnight growth at 18°C. Cascade crRNP complexes were purified via Ni-NTA chromatography in a buffer containing 50 mM Tris-HCl pH 7.0, 500 mM NaCl, 10% glycerol, 1 mM DTT and 20 to 500 mM imidazole. Size exclusion chromatography was performed on a Superdex 200 column (calibrated with a molecular weight standard kit (12 000–200 000 Da, Sigma-Aldrich)) in a buffer containing 50 mM HEPES-NaOH pH 7.0, 150 mM NaCl and 1 mM DTT.
Transmission electron microscopy of Cas1821
Purified protein samples were negatively stained with 2% (w/v) uranyl acetate as described previously (22,46,47). Subsequent electron microscopy was carried out with a JEOL JEM-2100 transmission electron microscope equipped with a LaB6 cathode at 120 kV (JEOL, Tokyo, Japan). Electron micrographs were taken with a 2k × 2k fast-scan CCD camera F214 combined with EM-Menu 4 (TVIPS, Gauting, Germany).
RESULTS
Identification of a minimal CRISPR-Cas subtype in S. putrefaciens CN-32
A minimal Type I-F variant CRISPR–Cas subtype was first described for Photobacterium profundum (PBPRB1995-PBPRB1991) (26). The activity of this unusual system has not been evaluated experimentally. We identified a similar system in S. putrefaciens CN-32 (Figure 1). S. putrefaciens CN-32 harbors a single CRISPR array with 81 spacers and only five cas genes: cas1 (Sputcn32_1819), cas3 (Sputcn32_1820), cas6f (Sputcn32_1823), cas1821 and cas1822. The comparison of this system with the subtype I-F system present in the Shewanella putrefaciens strain W3–18–1 illustrates the loss of the large subunit and the possible diversification of the Csy2 and Csy3 proteins (Figure 1A). These CRISPR-Cas systems share 99–100% amino acid identity for the flanking Cas1 and a CRISPR-array adjacent DinG-helicase. In addition, the Cas6f (Csy4) endonuclease and the N-terminal Cas2-like domain of the Cas2–3 nuclease show significant sequence identity. Homology searches of Cas1821 and Cas1822 revealed the presence of similar I-F variant systems in few other beta- and gammaproteobacteria. These include the human pathogens Legionella pneumophila (strain 2300/99 Alcoy) and Vibrio cholerae (strain TM 11079–80).
Figure 1.
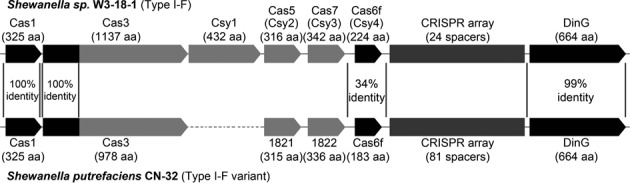
Comparison of CRISPR-Cas systems in S. putrefaciens strains. Schematic diagrams show the single CRISPR-Cas systems from S. putrefaciens W3–18–1 (Sputw3181_2191- Sputw3181_2185, Type I-F) and S. putrefaciens CN-32 (Sputcn32_1819 – Sputcn32_1824, Type I-F variant). The N-terminal 94 amino acids of the Cas3 proteins share 100% aa identity. The C-terminal Cas3 portion and the Cascade components do not reveal significant protein similarity.
The CRISPR array in S. putrefaciens CN-32 is transcribed and processed in vivo
First, we aimed to show crRNA production of the minimal Type I-F variant system. The small RNA fraction was isolated from S. putrefaciens CN-32 and subjected to RNA-Seq analyses. Sequencing of crRNAs required T4 PNK treatment of the isolated small RNA molecules during Illumina Hiseq2000 library preparation which indicates the presence of crRNA 5′ OH termini. More than 11 million sequence reads were mapped to the S. putrefaciens reference genome. The presence of all 81 crRNA transcripts could be verified and precursor transcript processing generated the expected 8 nt repeat tags at the crRNAs’ 5′-ends (5′-CUUAGAAA-3′) and a 20 nt repeat tag at the 3′-termini (Figure 2). 73 377 reads mapped to the CRISPR locus, indicating a lower crRNA abundance in comparison to very similar RNA-Seq studies for type I-A and type I-B systems (16,22). A minimum free-energy five base pair stem-loop was predicted in the 3′-repeat tags of all crRNAs, which could explain the absence of additional 3′-terminal degradation for most crRNAs (Figure 2). The abundance of the individual crRNAs varied significantly.
Figure 2.
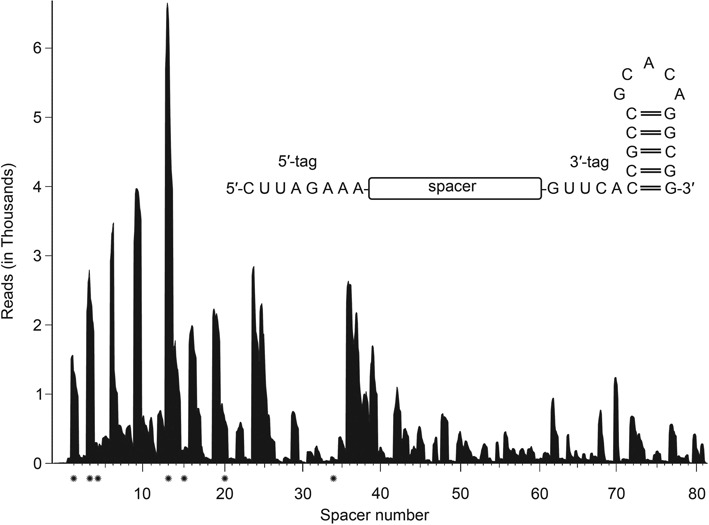
Analysis of CRISPR RNA abundance and termini. The coverage plot of S. putrefaciens CN-32 sequence reads (Illumina HiSeq2000) indicates the variable abundance of individual crRNAs. Mature crRNAs contained 5′-terminal 8 nt tags (5′-CUUAGAAA-3′) and 20 nt 3′-ends without further trimming events (inset). Spacers that are marked with an asterisk were tested for interference activity (see Figure 4)
We verified that the crRNAs are matured by Cas6f. Sputcn32_1823 encodes a protein with 37% amino acid identity to Csy4 (Cas6f) from Pseudomonas aeruginosa. A sequence alignment of the two proteins verified a conserved histidine residue at position 29 that was previously reported to be essential for Cas6f activity (18). The purified recombinant S. putrefaciens Cas6f protein showed endonuclease activity and cleaved a 100 nt-long spacer-repeat–spacer transcript. This endonuclease activity was dependent on the catalytic His29 residue, and a His29Ala point mutation abolished the activity (Supplementary Figure S1).
The S. putrefaciens CRISPR–Cas systems shows in vivo interference activity
Next, we assayed CRISPR-Cas-mediated DNA interference activity in S. putrefaciens CN-32. The low abundance of crRNAs required the establishment of a sensitive conjugation-based in vivo assay (Figure 3A). Two conjugative plasmids were used: (i) a non-target control plasmid (pNT = pBBR1MCS2) and (ii) a target plasmid (pT) containing the 13th spacer of the S. putrefaciens CN-32 CRISPR array (spacer13) and the PAM sequence ‘GG’ at the 3′ end of the protospacer. The PAM sequence was selected based on computational predictions by Mojica et al. (14). In agreement, we could identify potential prophage targets for spacer33 (∼97% identity between spacer and target) and spacer34 (100% identity between spacer and target) using the tool CRISPRtarget (48). In both cases, the di-nucleotide GG was found at the 3′ end of target strand match. The two plasmids pNT and pT were conjugated from E. coli WM 3064 into S. putrefaciens CN-32. In addition, markerless gene deletions were constructed for the genes cas1, cas3, cas1821 and cas1822 and the two plasmids were conjugated into these deletion strains. The colonies carrying pNT or pT were counted and the relative conjugation efficiency was calculated as pT over pNT. Relative conjugation efficiency (pT/pNT) below 1 indicates interference activity. Interference was observed in wild-type S. putrefaciens CN-32 cells (Figure 3B) and the Δcas1 strain, which is in agreement with an exclusive role of Cas1 in spacer acquisition. In contrast, interference activity was not apparent in the Δcas1821 and Δcas1822 strains, and was significantly reduced in the Δcas3 strain and a strain carrying a Cas3 mutant that lacks the HD nuclease motif. To investigate the reason for this loss of interference activity, we assayed the crRNA pool in these deletion strains. Total RNA was isolated from the different S. putrefaciens CN-32 cells and Northern blot analyses were performed using a radioactively labeled probe against the crRNA repeat sequence (Figure 3C). The production of crRNAs was observed in wild-type cells and the Δcas1- and Δcas3-deletion strains. However, crRNAs were absent in the Δcas1821 and Δcas1822 strains. These results are consistent with Cas6f generating mature crRNAs, which are then bound by Cas1821 and Cas1822 to maintain a stable crRNA pool in the cell. Thus, the absence of interference activity for the Δcas1821 and Δcas1822 strains correlates with the absence of crRNAs in these cells.
Figure 3.
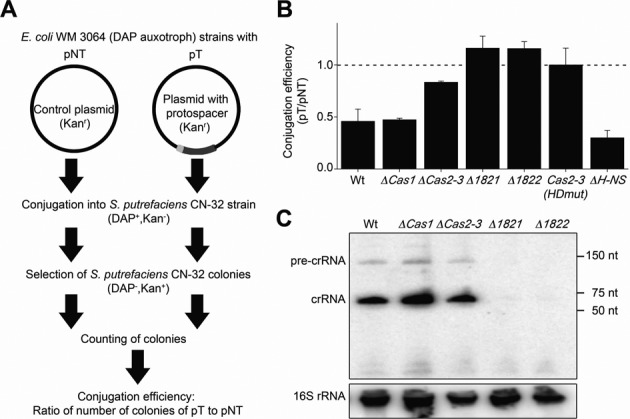
Conjugation assays reveal DNA interference activity of the S. putrefaciens CN-32 CRISPR–Cas system. (A) The experimental set-up of the conjugation assay is depicted. Two plasmids were used: (I) a control plasmid (pNT, plasmid non-target) and (II) a plasmid containing a PAM sequence (light grey) and a sequence matching spacer 13 (dark grey) of the S. putrefaciens CN-32 CRISPR array (pT, plasmid target). The number of S. putrefaciens CN-32 colonies carrying each plasmid was counted and the conjugation efficiency was determined. Interference is observed if pT/pNT is lower than 1 in triplicate assays. (B) The pT/pNT ratio is calculated for conjugation assays into S. putrefaciens CN-32 wild type cells and strains containing deletions of genes encoding Cas1, Cas3, Cas1821, Cas1822 and H-NS. Additionally, a Cas3 active site variant (HDmut, H156A/D157A) was tested. (C) Northern blot analyses were performed with extracted total RNA from S. putrefaciens CN-32 wild-type and cas gene-knockout strains using a 5′-γ-[32P]-ATP labeled probe complementary to the repeat sequence. A stable crRNA pool was absent in the S. putrefaciens CN-32 Δcas1821 and Δcas1822 strains.
Previous studies on the type I-E CRISPR-Cas system in E. coli revealed that the heat-stable nucleoid-structuring (H-NS) protein, a global transcriptional repressor, can regulate the expression of CRISPR-Cas systems (49). We constructed a S. putrefaciens CN-32 Δhns strain and observed slightly increased interference activity (Figure 3B). A two-fold increase in cas gene transcripts was detected via RT-qPCR (Supplementary Figure S2), which could explain increased interference.
The minimal subtype I-F variant CRISPR-Cas system recognizes PAM sequences and targets both DNA strands
Conjugation between the donor and the recipient cell requires the transfer of one strand of plasmid DNA through an intercellular cytoplasmic bridge, beginning at the origin of transfer and progressing in 5′ to 3′ direction. The transferred strand is converted into circular double-stranded plasmid DNA in the recipient and a new strand is synthesized in the donor to replace the transferred strand. In our experimental setup, the transferred strand can be directly targeted by the crRNA as it contains the sequence complementary to spacer13. To address if the minimal I-F variant system targets both DNA strands, a plasmid was constructed with the sequence of spacer13 on the transferred strand. Thus, the complementary target of crRNA 13 exists only after synthesis of the second strand in the recipient cell. The relative conjugation efficiency (pT/pNT) for this plasmid in both wild-type and ΔH-NS strains was comparable to the previous construct (Figure 4). This suggests that the I-F variant CRISPR–Cas system targets dsDNA.
Figure 4.
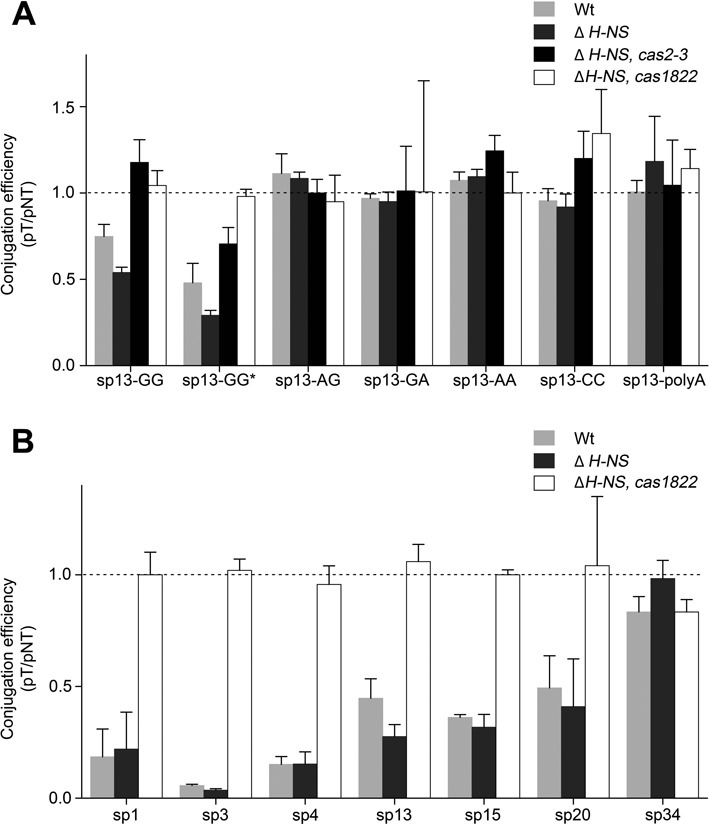
Analysis of the DNA target specificity of the S. putrefaciens CN-32 CRISPR–Cas system. Conjugation assays (see Figure 3) were used to analyze plasmid DNA target variants for DNA interference in S. putrefaciens CN-32 cells. (A) Interference was observed if the transferred DNA strand contained a sequence complementary to the crRNA13 (sp13-GG) or a sequence identical with spacer13 (sp13-GG*). Interference was evident for the wild-type and ΔH-NS strains, which contained all cas genes. However, the deletion of cas3 (Sputcn32_1820) or cas1822 abolished interference activity. The presence of the PAM sequence GG at the 3′ end of the DNA target (5′-protospacer-PAM-3′) was essential for the interference activity and PAM mutants (sp13-AG, sp13-GA, sp13-AA or sp13-CC) and a 10 nt poly-A sequence disrupting crRNA/DNA complementarity (sp13-polyA) were not targeted. (B) DNA interference activity was dependent on spacer sequence or crRNA abundance (spacers 1,3,4,13,15,20,34, see Figure 2).
Next, we aimed to investigate if the absence of a large subunit has an effect on the recognition of PAM sequences or the necessity for perfect crRNA/target complementarity. Variants of the targeted plasmid were constructed that contained PAM sequence mutations. These variants contained the dinucleotides AG, GA, AA or CC at the 3′ end of the spacer13 match on the target strand. In addition, a plasmid was constructed with the PAM-adjacent 10 nt of the protospacer (including the seed sequence) being replaced by a poly-A stretch. The relative conjugation efficiency (pT/pNT) of these constructs highlighted a loss of interference activity and demonstrated that PAM sequences are recognized in S. putrefaciens CN-32 (Figure 4A).
The RNA-Seq analysis of crRNA production revealed a highly variable crRNA abundance profile (Figure 2). We investigated if the abundance of individual crRNAs is correlated with the interference activity. Thus, we generated targeted plasmids with GG PAM sequences in which the spacer13 target was replaced with spacer1, spacer3 (abundant crRNAs) or spacer4, spacer15, spacer20 and spacer34 targets (low crRNA abundance). We performed conjugation assays and verified that highly abundant crRNAs (spacers1,3,13) yield efficient interference activity (Figure 4B). In agreement, the observed absence of stable crRNAs with spacer34 in S. putrefaciens CN-32 correlated with a complete loss of interference activity. However, interference activity was observed for crRNAs with spacer4, spacer15 and spacer20 even though only minimal amounts of these crRNAs were detected in our RNA-Seq analysis (Figure 4B). In general, crRNAs with promoter-adjacent spacers showed the highest interference activity. These results indicate that variable crRNA abundance is a factor that can potentially influence the efficiency of DNA targeting.
Characterization of recombinant Cas1821 as a helical Cascade backbone protein
The absence of observable crRNAs in the Δcas1821 and Δcas1822 strains suggests that the encoded Cas proteins are required for maintaining a stable crRNA pool in the cell. Thus, these two proteins, Cas1821 and Cas1822, could fulfill the roles of Cas7 and Cas5 proteins even though sequence similarity calculations did not indicate them as members of these Cas protein families (26). We attempted to individually produce recombinant S. putrefaciens CN-32 Cas1821 and Cas1822 proteins in E. coli. The production of Cas1822 did not yield soluble protein in various expression conditions. However, soluble recombinant Cas1821 was produced as a SUMO-tag fusion construct. The SUMO-Cas1821 was purified via Ni-NTA chromatography and co-eluted with bound nucleic acid contaminants. Therefore, a high-salt washing step with a buffer including 1 M NaCl was added before protein elution from the Ni-NTA column, and a second cation exchange chromatography step was included in the purification protocol. This procedure yielded Cas1821 with a purity of >95% and without nucleic acid contaminants. Gel-elution chromatography of Cas1821 revealed a ∼35 kDa monomer (Supplementary Figure S3). Electrophoretic mobility shift assays of Cas1821 with radiolabeled crRNA transcripts revealed a slow migrating band indicating the crRNA-binding potential of Cas1821 (Figure 5A). This band disappeared upon addition of 1 μg of competitor yeast RNA, suggesting a non-specific interaction. This behavior is in agreement with the observed non-specific binding of E. coli contaminant nucleic acids. Recombinant Cas1821 bound to these contaminants eluted near the void volume during gel-elution chromatography (Supplementary Figure S3). These fractions were visualized by electron microscopy and revealed the formation of long helical filament-structures of varying length (Figure 5B). These observations suggest that Cas1821 could fulfil the role of Cas7 which was shown to form the helical Cascade backbone filament after unspecific RNA binding in other CRISPR-Cas subtypes (e.g. Type I-A, Type I-E).
Figure 5.
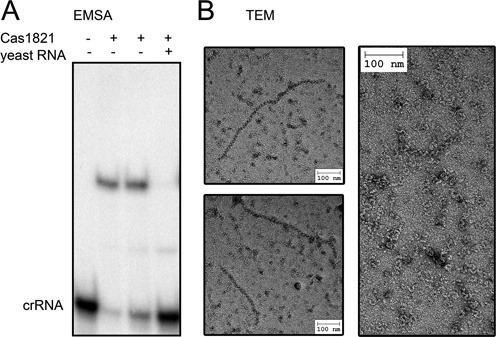
Characterization of recombinant Cas1821 protein. (A) EMSA assays indicate the binding of 5′-γ[32P]-ATP labeled crRNA by 250 nM Cas1821 (lane 2) or 500 nM Cas1821 (lane 3). Bands were separated by 6% native PAGE. The RNA binding is unspecific and outcompeted by yeast total RNA. (B) Transmission electron micrographs of negatively stained helical structures formed by RNA-bound recombinant Cas1821.
In vivo characterization of potential DNA target interaction partners
The absence of a large subunit implies that other Cas proteins can substitute for its role(s) during DNA target interaction. However, even for previously investigated and related subtype I-F CRISPR–Cas systems, the role of the large subunit (Csy1) is not clear, and it is not known which Cas protein is responsible for PAM recognition (44). The S. putrefaciens CN-32 CRISPR-Cas system is flanked by a homolog of the DinG gene (Sputcn32_1824), an ATP-dependent DNA helicase that was predicted to be a functional component of Type I-U CRISPR-Cas systems (26). To test if this protein plays a role in DNA interference, we constructed a ΔDinG strain. We did not observe a loss of interference activity in our conjugation assays indicating that this DinG homolog does not promote DNA interference and is not a component of subtype I-F variant Cascade (Supplementary Figure S4). This is in agreement with the observation that this DinG homolog is conserved in S. putrefaciens strains without CRISPR-arrays or other CRISPR-Cas subtypes.
It is possible that Cas1822 interacts with the DNA target, as arginine residues are known to mediate DNA recognition in Cas proteins (50). A multiple alignment of Cas1822 proteins revealed conserved basic amino acids and a potential Asp/Glu motif (Supplementary Figure S5). Therefore, we assayed if mutations of conserved arginine residues in Cas1822 have an effect on interference activity. We constructed point mutations of the cas1822 gene at the appropriate positions to generate corresponding Ala substitutions in Cas1822 in vivo, and these variants were tested for interference activity. Interference was observed for Δhns/R170A, Δhns/R225A and Δhns/E86A,D87A strains (Figure 6A). A Northern blot analysis verified the presence of mature crRNAs in these strains (Figure 6B). In contrast, the two strains Δhns/R60A and Δhns/R66A lacked interference activity and stable crRNAs. We propose that these arginine residues are required for Cascade formation and crRNA protection. Finally, the two strains Δhns/R29A and Δhns/R258A did not show interference activity even though a stable crRNA pool was observed in the cell. Thus, these arginine residues are possibly involved in either the direct interaction with target DNA or with the Cas component that recognizes the PAM motif. Both scenarios are discussed below.
Figure 6.
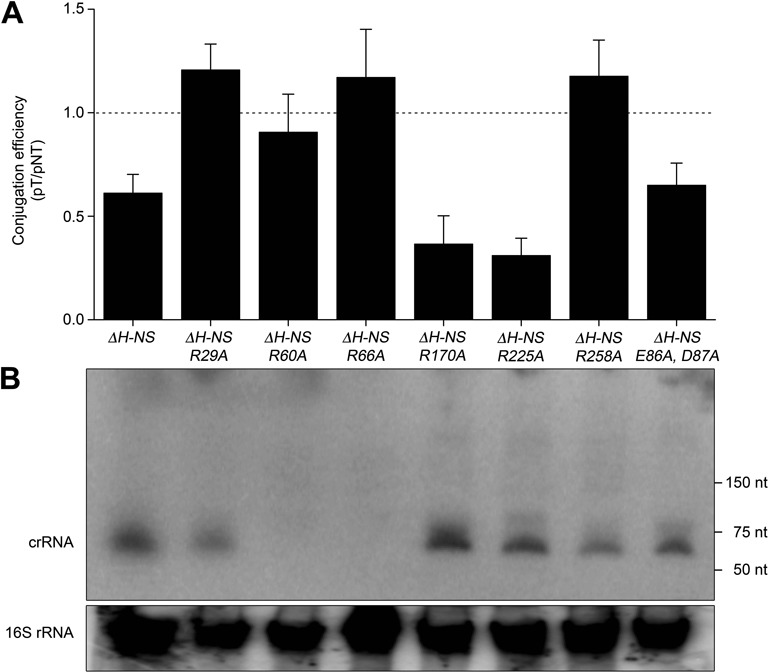
In vivo characterization of Cas1822 point-mutants. (A) S. putrefaciens CN-32 strains carrying Cas1822 proteins with the indicated point mutations were tested for DNA interference using the conjugation assay (see Figure 3). (B) Northern Blot analyses with anti-repeat probes revealed the cellular crRNA pool of these strains.
Production of recombinant subtype I-F variant Cascade
Finally, we aimed to verify that Cas1821 and Cas1822 are assembled into a Cascade complex. Thus, we transformed two plasmids into E. coli to facilitate the T7 RNA polymerase-driven production of (i) a pre-crRNA transcript and (ii) the Cas proteins Cas1821, Cas1822 and Cas6f. Only Cas1821 contains a His-Tag, which was successfully used to purify a Cascade ribonucleoprotein complex from E. coli. This complex was subjected to gel elution chromatography and the RNA and protein content of the fractions was analyzed (Figure 7). A distinct peak revealed the formation of a stable complex with an estimated size of approximately 215 kDa. The Cas1821 protein was overrepresented in the complex which supports its role as Cascade backbone. Cas1822 and Cas6f were co-purified and are suggested to be the proteins that cap the 3′ hairpin tag (Cas6f) and the 8 nt 5′ tag (Cas1822) of the crRNA. Indeed, mature crRNA bands were co-purified in the Cascade fraction. This verified that Cas6f processed the pre-crRNA transcript in E. coli and likely loaded the Cascade complex. In addition, Cas1821-Cas1822 dimers were detected which suggests that these units are similar to the Cas5–Cas7 dimers observed for the type I-A Cascade.
Figure 7.
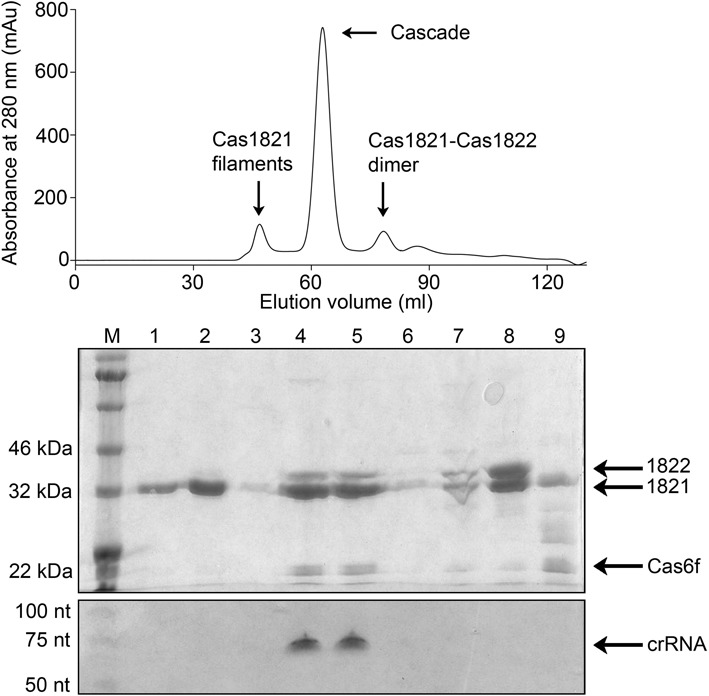
Co-purification of a recombinant subtype I-F variant Cascade complex. His-tagged Cas1821 co-eluted with Cas1822, Cas6f and mature crRNA during gel-elution chromatography (fractions 4 and 5) which verified Cascade complex formation. Cas1821 filaments were observed in the void volume (fractions 1 and 2). Dimers of Cas1821 and Cas1822 (fraction 8) were identified. SDS-PAGE (middle) and 8M urea PAGE (bottom) was used to separate the protein and RNA content of the fraction indicated in the gel-elution chromatogram (top).
DISCUSSION
The Cas protein content of CRISPR-Cas systems varies between different subtypes. The Type I systems target DNA and are characterized by a multi-protein DNA interference complex termed Cascade. We analyzed the interference activity of the S. putrefaciens CN-32 CRISPR-Cas system, which contains a minimal Type I set of five Cas proteins. Three of these proteins can be classified into subtype I-F protein families. One of these proteins is Cas6, the endonuclease responsible for pre-crRNA cleavage reaction. Cas6 variants of the different CRISPR-Cas subtypes show different RNA recognition and cleavage features. The S. putrefaciens Cas6 enzyme is a Cas6f variant. A catalytic histidine residues at position 29 was found to be essential for cleavage activity, similar to the well-characterized Cas6f of P. aeruginosa (18). The mature crRNAs contain a standard 8 nt 5′ tag and a stable hairpin formed by the repeat sequence at the 3′ end, which prevents further 3′-terminal trimming. The 3′-terminal hairpin structures have also been found in other subtypes and are e.g. described for crRNAs found in E. coli and P. aeruginosa (6,18). In these systems, the corresponding Cas6 enzymes remain stably associated with the hairpin after cleavage and protect the crRNA from further degradation (18,37,51). This stable interaction of Cas6f to the crRNA is suggested to serve as a starting point for the assembly of the Cascade backbone by several Cas7 copies (17,23,24). This is suggested to allow Cas5 to bind and cap the 5′ tag of the crRNA. Thus, these three proteins bind and protect crRNA in Cascade and guarantee their stability and their ability to form base pairs with target DNA.
We showed that the deletion of Cas1821 or Cas1822 abolished interference activity in conjugation assays, which corresponded with the loss of an observable cellular crRNA pool. Analyses of E. coli and Haloferax volcanii Cascade structures and Pseudomonas aeruginosa Type I-F system activity showed that Cas5 and Cas7 proteins are essential for crRNA maintenance and stability (52–54). Thus, we propose that Cas1821 and Cas1822 can fulfill the roles of these two proteins. Accordingly, recombinant Cas1821, Cas1822 and Cas6f could be co-purified in Cascade assemblies with mature crRNAs. The non-specific RNA binding of Cas1821 led to the formation of oligomeric structures that could be visualized as helical filaments using electron microscopy. The formation of helical filaments was previously reported for Cas7 from Thermoproteus tenax, a sub-complex of Cas7 and Cas5a subunits from Sulfolobus solfataricus and for Type I-E Cascade in E. coli (22,39). Thus, we conclude that Cas1821 proteins build the helical backbone of the Cascade structures and serve as Cas7 proteins. The observed formation of Cas1821 and Cas1822 dimers suggest that these could be the initial building blocks of Cascade, similar to the Cas7 and Cas5a subunit dimers found in S. solfataricus.
Our RNA-Seq analyses indicated highly variable crRNA abundance patterns. The general crRNA abundance was lower than found for other subtypes, e.g. subtype I-B (M. maripaludis) or subtype I-A (T. tenax) (16,22). Usually, the most abundant crRNAs are the ones closest to the promoter in the CRISPR leader regions (16,22,55). These crRNAs contain the most recently acquired spacers. The variability of crRNA abundance could be explained by the presence of anti-crRNA sequences and internal promoters, which were observed in other cases (56). These scenarios are not apparent for the S. putrefaciens CRISPR array. It is plausible that the Cascade loading efficiency differs for different crRNAs. Additionally, pre-crRNA could form internal structures that would influence Cas6f cleavage site availability.
The assembly of crRNAs into Cascade complexes containing multiple Cas1821 copies, Cas1822 and Cas6f highlights the universality of this architecture in Type I systems. It is apparent that small and large subunits are missing in this subtype I-F variant CRISPR-Cas system. We utilized a conjugation assay to study if the loss of these subunits has an influence on known features of Type I-mediated DNA interference. However, we observed that this system targets dsDNA and depends on the presence of a PAM sequence as expected for a Type I CRISPR-Cas activity. The PAM sequence was found to be GG and mutations in this sequence resulted in the loss of interference activity. The PAM sequence is shared with subtype I-F CRISPR-Cas systems (14,57). We asked if undetected Cas proteins exist in the genomes of organisms that contain the subtype I-F variant, which could fulfill the function of small and large subunits. It should be noted that the small subunit is also missing for subtype I-F CRISPR-Cas systems. The subtype I-F variant CRISPR-Cas system was identified in nine mesophilic beta- and gamma-proteobacteria: S. putrefaciens (strains CN-32 & 200), Oligella ureolytica (strain DSM 18253), Pseudoalteromonas tunicata (strain D2), Legionella pneumophila (strain 2300/99 Alcoy), Oxalobacter formigenes (strain OXCC13), P. profundum (strain SS9), Vibrio cholerae (strain TM 11079–80) and Methylophaga nitratireducenticrescens. Phylogenetic profiling did not reveal additional Cas proteins that co-evolved with the unusual Cascade assemblies. A homolog of a DinG helicase was identified downstream of the CRISPR array, which was reported to be part of Type I-U CRISPR-Cas system (26). However, these enzymes are also found in related Shewanella strains without CRISPR systems or with different CRISPR-Cas subtypes. A deletion of the dinG gene did not abolish the interference activity indicating that the encoded protein is not required for subtype I-F variant Cascade activity. In conclusion, we do not see any evidence for additional Cascade components that are not encoded in the minimal cas gene operon. To date, RNase III and PNPase are the only non-Cas proteins that can be involved in conferring CRISPR-Cas based immunity (58,59). Both proteins play a key role in crRNA generation and are not involved in DNA interactions. The large subunit of the subtype I-E E. coli Cascade has been shown to interact with the PAM motif and to recruit Cas3. In addition, both small and large subunits are required for the stabilization of the crRNA-target hybrid (23,60). The role of the large subunit (Csy1) of subtype I-F CRISPR-Cas systems is not fully understood yet. We hypothesize that either Cas1822 and/or Cas3 are involved in DNA target PAM recognition. We showed that the mutation of conserved arginine residues in Cas1822 influenced DNA interference activity even in the presence of a stable cellular crRNA pool. Cas3 interacts with the DNA and could provide additional DNA target selectivity. It is interesting to note that the subtype I-F and subtype I-F variant Cas3 enzymes contain an N-terminal portion that resembles Cas2 (44). Only this region is conserved between Cas3 enzymes found in different Shewanella strains, while the larger C-terminal portion shows no apparent homology. Thus, unexpected Cas3 functions could be encoded in this part. The soluble recombinant production of S. putrefaciens Cas1822 and Cas3 was not successful and we rely on future studies of macromolecular Cascade structures to pinpoint the molecular details of PAM recognition mechanisms in subtype I-F and subtype I-F variant CRISPR-Cas systems. The loss of large and small subunits might coincide with the evolution of specialized minimal Type I Cascade systems that still rely on PAM motif recognition for DNA target specificity.
Supplementary Material
Acknowledgments
The authors are grateful to Prof. Uwe-G. Maier for allocation of the electron microscopic facilities.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Max Planck Society; Deutsche Forschungsgemeinschaft [DFG FOR1680 to L.R.]; International Max Planck Research School (to S.B.); Deutsche Forschungsgemeinschaft [DFG 831/4-1 to K.M.T.]. Funding for open access charge: Max Planck Society.
Conflict of interest statement. None declared.
REFERENCES
- 1.Goldfarb T., Sberro H., Weinstock E., Cohen O., Doron S., Charpak-Amikam Y., Afik S., Ofir G., Sorek R. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 2015;34:169–183. doi: 10.15252/embj.201489455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stern A., Sorek R. The phage-host arms race: shaping the evolution of microbes. Bioessays. 2011;33:43–51. doi: 10.1002/bies.201000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horvath P., Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- 4.Westra E.R., Swarts D.C., Staals R.H., Jore M.M., Brouns S.J., van der Oost J. The CRISPRs, they are a-changin’: how prokaryotes generate adaptive immunity. Annu. Rev. Genet. 2012;46:311–339. doi: 10.1146/annurev-genet-110711-155447. [DOI] [PubMed] [Google Scholar]
- 5.Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 6.Brouns S.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J., Snijders A.P., Dickman M.J., Makarova K.S., Koonin E.V., Van Der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pourcel C., Salvignol G., Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151:653–663. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- 8.Mojica F.J., García-Martínez J., Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005;60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- 9.Grissa I., Vergnaud G., Pourcel C. Molecular Epidemiology of Microorganisms. Springer; 2009. pp. 105–116. [Google Scholar]
- 10.Makarova K.S., Haft D.H., Barrangou R., Brouns S.J., Charpentier E., Horvath P., Moineau S., Mojica F.J., Wolf Y.I., Yakunin A.F. Evolution and classification of the CRISPR–Cas systems. Nat. Rev. Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haft D.H., Selengut J., Mongodin E.F., Nelson K.E. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 2005;1:e60. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savitskaya E., Semenova E., Dedkov V., Metlitskaya A., Severinov K. High-throughput analysis of type IE CRISPR/Cas spacer acquisition in E. coli. RNA Biol. 2013;10:716–725. doi: 10.4161/rna.24325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nuñez J.K., Lee A.S., Engelman A., Doudna J.A. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature. 2015;519:193–198. doi: 10.1038/nature14237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mojica F., Diez-Villasenor C., Garcia-Martinez J., Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–740. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- 15.Gesner E.M., Schellenberg M.J., Garside E.L., George M.M., MacMillan A.M. Recognition and maturation of effector RNAs in a CRISPR interference pathway. Nat. Struct. Mol. Biol. 2011;18:688–692. doi: 10.1038/nsmb.2042. [DOI] [PubMed] [Google Scholar]
- 16.Richter H., Zoephel J., Schermuly J., Maticzka D., Backofen R., Randau L. Characterization of CRISPR RNA processing in Clostridium thermocellum and Methanococcus maripaludis. Nucleic Acids Res. 2012;40:9887–9896. doi: 10.1093/nar/gks737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jore M.M., Lundgren M., van Duijn E., Bultema J.B., Westra E.R., Waghmare S.P., Wiedenheft B., Pul Ü., Wurm R., Wagner R. Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat. Struct. Mol. Biol. 2011;18:529–536. doi: 10.1038/nsmb.2019. [DOI] [PubMed] [Google Scholar]
- 18.Haurwitz R.E., Jinek M., Wiedenheft B., Zhou K., Doudna J.A. Sequence-and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010;329:1355–1358. doi: 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carte J., Pfister N.T., Compton M.M., Terns R.M., Terns M.P. Binding and cleavage of CRISPR RNA by Cas6. RNA. 2010;16:2181–2188. doi: 10.1261/rna.2230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rouillon C., Zhou M., Zhang J., Politis A., Beilsten-Edmands V., Cannone G., Graham S., Robinson C.V., Spagnolo L., White M.F. Structure of the CRISPR interference complex CSM reveals key similarities with cascade. Mol. Cell. 2013;52:124–134. doi: 10.1016/j.molcel.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van der Oost J., Westra E.R., Jackson R.N., Wiedenheft B. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat. Rev. Microbiol. 2014;12:479–492. doi: 10.1038/nrmicro3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plagens A., Tripp V., Daume M., Sharma K., Klingl A., Hrle A., Conti E., Urlaub H., Randau L. In vitro assembly and activity of an archaeal CRISPR-Cas type IA Cascade interference complex. Nucleic Acids Res. 2014;42:5125–5138. doi: 10.1093/nar/gku120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mulepati S., Héroux A., Bailey S. Crystal structure of a CRISPR RNA–guided surveillance complex bound to a ssDNA target. Science. 2014;345:1479–1484. doi: 10.1126/science.1256996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson R.N., Golden S.M., van Erp P.B., Carter J., Westra E.R., Brouns S.J., van der Oost J., Terwilliger T.C., Read R.J., Wiedenheft B. Crystal structure of the CRISPR RNA–guided surveillance complex from Escherichia coli. Science. 2014;345:1473–1479. doi: 10.1126/science.1256328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maier L.-K., Lange S.J., Stoll B., Haas K.A., Fischer S.M., Fischer E., Duchardt-Ferner E., Wöhnert J., Backofen R., Marchfelder A. Essential requirements for the detection and degradation of invaders by the Haloferax volcanii CRISPR/Cas system IB. RNA Biol. 2013;10:865–874. doi: 10.4161/rna.24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Makarova K.S., Aravind L., Wolf Y.I., Koonin E.V. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol. Direct. 2011;6:38. doi: 10.1186/1745-6150-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nuñez J.K., Kranzusch P.J., Noeske J., Wright A.V., Davies C.W., Doudna J.A. Cas1–Cas2 complex formation mediates spacer acquisition during CRISPR–Cas adaptive immunity. Nature Struct. Mol. Biol. 2014;21:528–534. doi: 10.1038/nsmb.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richter C., Dy R.L., McKenzie R.E., Watson B.N., Taylor C., Chang J.T., McNeil M.B., Staals R.H., Fineran P.C. Priming in the Type IF CRISPR-Cas system triggers strand-independent spacer acquisition, bi-directionally from the primed protospacer. Nucleic Acids Res. 2014;42:8516–8526. doi: 10.1093/nar/gku527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garneau J.E., Dupuis M.-È., Villion M., Romero D.A., Barrangou R., Boyaval P., Fremaux C., Horvath P., Magadán A.H., Moineau S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 30.Jinek M., Jiang F., Taylor D.W., Sternberg S.H., Kaya E., Ma E., Anders C., Hauer M., Zhou K., Lin S. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343:1247997. doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chylinski K., Le Rhun A., Charpentier E. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol. 2013;10:726–737. doi: 10.4161/rna.24321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Westra E.R., Semenova E., Datsenko K.A., Jackson R.N., Wiedenheft B., Severinov K., Brouns S.J. Type IE CRISPR-cas systems discriminate target from non-target DNA through base pairing-independent PAM recognition. PLoS Genet. 2013;9:e1003742. doi: 10.1371/journal.pgen.1003742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anders C., Niewoehner O., Duerst A., Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513:569–573. doi: 10.1038/nature13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Staals R.H., Agari Y., Maki-Yonekura S., Zhu Y., Taylor D.W., van Duijn E., Barendregt A., Vlot M., Koehorst J.J., Sakamoto K. Structure and activity of the RNA-targeting Type III-B CRISPR-Cas complex of Thermus thermophilus. Mol. Cell. 2013;52:135–145. doi: 10.1016/j.molcel.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Samai P., Pyenson N., Jiang W., Goldberg G.W., Hatoum-Aslan A., Marraffini L.A. Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell. 2015;161:1164–1174. doi: 10.1016/j.cell.2015.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiedenheft B., van Duijn E., Bultema J.B., Waghmare S.P., Zhou K., Barendregt A., Westphal W., Heck A.J., Boekema E.J., Dickman M.J. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc. Natl. Acad. Sci. U.S.A. 2011;108:10092–10097. doi: 10.1073/pnas.1102716108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nam K.H., Haitjema C., Liu X., Ding F., Wang H., DeLisa M.P., Ke A. Cas5d protein processes pre-crRNA and assembles into a cascade-like interference complex in subtype IC/Dvulg CRISPR-Cas system. Structure. 2012;20:1574–1584. doi: 10.1016/j.str.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lintner N.G., Kerou M., Brumfield S.K., Graham S., Liu H., Naismith J.H., Sdano M., Peng N., She Q., Copié V. Structural and functional characterization of an archaeal clustered regularly interspaced short palindromic repeat (CRISPR)-associated complex for antiviral defense (CASCADE) J. Biol. Chem. 2011;286:21643–21656. doi: 10.1074/jbc.M111.238485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sashital D.G., Wiedenheft B., Doudna J.A. Mechanism of foreign DNA selection in a bacterial adaptive immune system. Mol. Cell. 2012;46:606–615. doi: 10.1016/j.molcel.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niewoehner O., Jinek M., Doudna J.A. Evolution of CRISPR RNA recognition and processing by Cas6 endonucleases. Nucleic Acids Res. 2014;42:1341–1353. doi: 10.1093/nar/gkt922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Judith R., James H.N., Malcolm F.W. CRISPR interference: a structural perspective. Biochem. J. 2013;453:155–166. doi: 10.1042/BJ20130316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nunez J.K., Lee A.S., Engelman A., Doudna J.A. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature. 2015;519:193–198. doi: 10.1038/nature14237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richter C., Gristwood T., Clulow J.S., Fineran P.C. In vivo protein interactions and complex formation in the Pectobacterium atrosepticum subtype IF CRISPR/Cas System. PLoS One. 2012;7:e49549. doi: 10.1371/journal.pone.0049549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lassak J., Henche A.-L., Binnenkade L., Thormann K.M. ArcS, the cognate sensor kinase in an atypical Arc system of Shewanella oneidensis MR-1. Appl. Environ. Microbiol. 2010;76:3263–3274. doi: 10.1128/AEM.00512-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klingl A., Moissl-Eichinger C., Wanner G., Zweck J., Huber H., Thomm M., Rachel R. Analysis of the surface proteins of Acidithiobacillus ferrooxidans strain SP5/1 and the new, pyrite-oxidizing Acidithiobacillus isolate HV2/2, and their possible involvement in pyrite oxidation. Arch. Microbiol. 2011;193:867–882. doi: 10.1007/s00203-011-0720-y. [DOI] [PubMed] [Google Scholar]
- 47.Rachel R., Meyer C., Klingl A., Gurster S., Heimerl T., Wasserburger N., Burghardt T., Kuper U., Bellack A., Schopf S., et al. Analysis of the ultrastructure of archaea by electron microscopy. Methods Cell Biol. 2010;96:47–69. doi: 10.1016/S0091-679X(10)96003-2. [DOI] [PubMed] [Google Scholar]
- 48.Biswas A., Gagnon J.N., Brouns S.J., Fineran P.C., Brown C.M. CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 2013;10:817–827. doi: 10.4161/rna.24046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pul Ü., Wurm R., Arslan Z., Geißen R., Hofmann N., Wagner R. Identification and characterization of E. coli CRISPR-cas promoters and their silencing by H-NS. Mol. Microbiol. 2010;75:1495–1512. doi: 10.1111/j.1365-2958.2010.07073.x. [DOI] [PubMed] [Google Scholar]
- 50.Luscombe N.M., Laskowski R.A., Thornton J.M. Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang R., Preamplume G., Terns M.P., Terns R.M., Li H. Interaction of the Cas6 riboendonuclease with CRISPR RNAs: recognition and cleavage. Structure. 2011;19:257–264. doi: 10.1016/j.str.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brendel J., Stoll B., Lange S.J., Sharma K., Lenz C., Stachler A.-E., Maier L.-K., Richter H., Nickel L., Schmitz R.A. A complex of Cas proteins 5, 6, and 7 is required for the biogenesis and stability of clustered regularly interspaced short palindromic repeats (crispr)-derived rnas (crrnas) in Haloferax volcanii. J. Biol. Chem. 2014;289:7164–7177. doi: 10.1074/jbc.M113.508184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pougach K., Semenova E., Bogdanova E., Datsenko K.A., Djordjevic M., Wanner B.L., Severinov K. Transcription, processing and function of CRISPR cassettes in Escherichia coli. Mol. Microbiol. 2010;77:1367–1379. doi: 10.1111/j.1365-2958.2010.07265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cady K.C., O'Toole G.A. Non-identity-mediated CRISPR-bacteriophage interaction mediated via the Csy and Cas3 proteins. J. Bacteriol. 2011;193:3433–3445. doi: 10.1128/JB.01411-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Su A.A., Tripp V., Randau L. RNA-Seq analyses reveal the order of tRNA processing events and the maturation of C/D box and CRISPR RNAs in the hyperthermophile Methanopyrus kandleri. Nucleic Acids Res. 2013;41:6250–6258. doi: 10.1093/nar/gkt317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zoephel J., Randau L. RNA-Seq analyses reveal CRISPR RNA processing and regulation patterns. Biochem. Soc. Trans. 2013;41:1459–1463. doi: 10.1042/BST20130129. [DOI] [PubMed] [Google Scholar]
- 57.Cady K.C., Bondy-Denomy J., Heussler G.E., Davidson A.R., O'Toole G.A. The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J. Bacteriol. 2012;194:5728–5738. doi: 10.1128/JB.01184-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sesto N., Touchon M., Andrade J.M., Kondo J., Rocha E.P., Arraiano C.M., Archambaud C., Westhof É., Romby P., Cossart P. A PNPase dependent CRISPR system in Listeria. PLoS Genet. 2014;10:e1004065. doi: 10.1371/journal.pgen.1004065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Deltcheva E., Chylinski K., Sharma C.M., Gonzales K., Chao Y., Pirzada Z.A., Eckert M.R., Vogel J., Charpentier E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hochstrasser M.L., Taylor D.W., Bhat P., Guegler C.K., Sternberg S.H., Nogales E., Doudna J.A. CasA mediates Cas3-catalyzed target degradation during CRISPR RNA-guided interference. Proc. Natl. Acad. Sci. U.S.A. 2014;111:6618–6623. doi: 10.1073/pnas.1405079111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.