


Abstract
In contrast to bacteria that have two release factors, RF1 and RF2, eukaryotes only possess one unrelated release factor eRF1, which recognizes all three stop codons of the mRNA and hydrolyses the peptidyl-tRNA bond. While the molecular basis for bacterial termination has been elucidated, high-resolution structures of eukaryotic termination complexes have been lacking. Here we present a 3.8 Å structure of a human translation termination complex with eRF1 decoding a UAA(A) stop codon. The complex was formed using the human cytomegalovirus (hCMV) stalling peptide, which perturbs the peptidyltransferase center (PTC) to silence the hydrolysis activity of eRF1. Moreover, unlike sense codons or bacterial stop codons, the UAA stop codon adopts a U-turn-like conformation within a pocket formed by eRF1 and the ribosome. Inducing the U-turn conformation for stop codon recognition rationalizes how decoding by eRF1 includes monitoring geometry in order to discriminate against sense codons.
INTRODUCTION
Sense and stop codons are recognized in the decoding center of the small ribosomal subunit (SSU) by transfer-RNAs (tRNAs) and release factors (RFs), respectively. For peptide elongation, the tRNA is delivered by eEF1A (EF-Tu in bacteria) to the aminoacyl-tRNA acceptor site (A-site). There, base pairing between the codon of the messenger RNA (mRNA) and the anti-codon of a cognate tRNA at least in the first two positions results in the formation of a short A-helix. Its correct geometry is monitored by the evolutionary conserved ribosomal nucleotides G530, A1492 and A1493 (1,2) according to E. coli (Ec) numbering.
However, for translation termination prokaryotes and eukaryotes diverged and employ evolutionary unrelated release factors and most likely distinct decoding mechanisms. Bacteria possess two class-I release factors, RF1 for recognition of UAA and UAG stop codons and RF2 for decoding UAA and UGA stop codons (3–5). Both factors consist of four domains and adopt a very similar conformation when bound to the ribosome (6–9). Domains 2 and 4 form the stop codon “reading head” comprising conserved amino acids of the so-called recognition loop (PxT-motif in RF1, SPF-motif in RF2), whereas domain 3 possesses the highly conserved GGQ-motif that triggers release of the nascent polypeptide chain in the PTC of the large ribosomal subunit (LSU). Notably, in contrast to A-helix forming sense codons, stop codons adopt a different conformation when decoded by RFs in bacteria. The third (Wobble) base is not stacked on the first two bases and instead interacts with G530 of the 16S rRNA in a distinct conformation. In this geometry, the identity of the codon is recognized by the reading head of the RF via a network of hydrogen bonds between the three individual bases of the stop codon and specific RF amino acid side chains.
In contrast to bacteria, eukaryotes contain one structurally unrelated class-I termination factor, termed eRF1, which reads all three stop codons. eRF1 consists of an N-terminal domain (N domain) necessary for stop-codon recognition (10), the M domain carrying the conserved GGQ-motif (11) and a C-terminal domain (C domain), which mediates the interaction with the class-II termination factor eRF3 and the ribosome recycling factor ABCE1 (12–15). eRF1 is delivered to the ribosomal A-site in form of a ternary complex with the GTPase eRF3. Following stop-codon recognition by eRF1, eRF3 hydrolyses GTP to GDP and dissociates from the ribosome (15–17). Subsequently, eRF1 accommodates into the A-site of the ribosome in an extended state, positioning its GGQ-motif of the M domain in the PTC for peptide release (13,18,19). Subsequently, the ATPase ABCE1 can bind to the ribosome-eRF1 complex further stimulating peptide release (15) and eventually triggering ribosome recycling by dissociating the ribosome into subunits (14,20,21). So far, structures have not been of sufficiently high resolution in order to obtain information about the stop decoding mechanism by eRF1 (13,18,22). However, a wealth of biochemical and mutational studies suggest that the highly conserved TAS-NIKS motif (residues 58–64), the GTS-loop (residues 31–33) and the YxCxxxF motif (residues 125–131) of the eRF1 N domain are important for stop-codon recognition (23–26).
Here, we used cryo-EM to report on the structure of an hCMV stalled human 80S ribosome bound to eRF1. The hCMV stalling peptide perturbs the PTC to silence the peptide hydrolysis activity of eRF1. Moreover, the UAA(A) stop codon adopts a unique U-turn-like conformation within a pocket formed by eRF1 and the ribosome revealing an important contribution of mRNA geometry recognition for stop codon decoding in eukaryotes.
MATERIALS AND METHODS
Plasmid constructions
The hCMV-stalling construct depicted in Figures 2A and 3B was cloned into the pT7CFE1 backbone via the MegaWhop cloning method (27). For the mutational analysis, point mutations to codons encoding for alanine or (only for selected residues) for other amino acids were introduced with the same cloning method. The CMV-truncated control construct that lacks the stop codon (truncated after the Pro22 codon) was generated in the same way. The expression plasmid for human eRF1 was obtained by cloning isoform 1 of the human eRF1 encoding sequence into the pETDuet-1 backbone via MegaWhop cloning. The construct also contained an N-terminal (His)6-tag for protein purification and an HRV 3C protease cleavage-site for tag removal. The Jmjd4-expression plasmid (pET-28a containing (His)6-tagged Jmjd4) was kindly provided by Sarah Wilkins/Mathew Coleman (Oxford University) (28).
Figure 2.
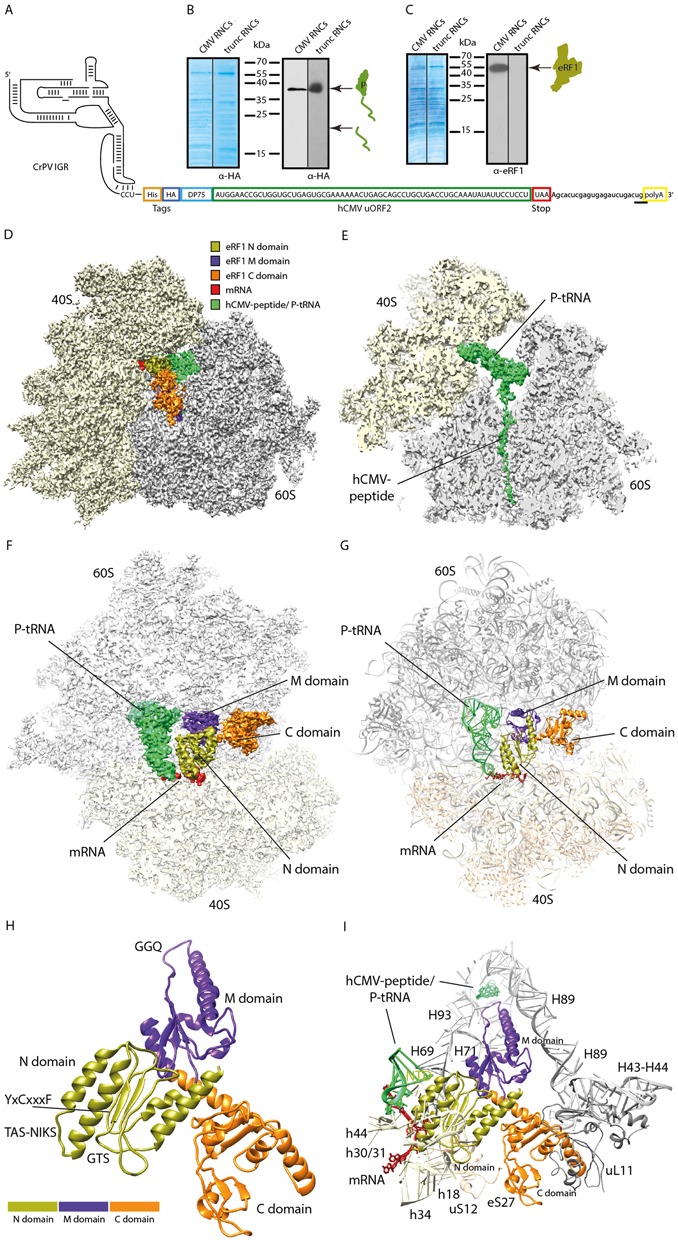
Isolation and cryo-EM structure of a stalled human 80S ribosome bound to eRF1. (A) Schematic representation of the hCMV stalling mRNA construct used in the human translation system for 80S-CMV-RNC generation. The final mRNA construct encoded a CrPV IGR IRES sequence for translation initiation, an N-terminal HA- and (His)6-tag, the well characterized DP75 dipeptidyl-aminopeptidase B (DPAPB) (30), the hCMV uORF2 stalling sequence and a polyA-tail. The termination site relevant for the Stop23Ala mutated construct is underlined. (B, C) Western blots of purified human RNCs programmed with the hCMV mRNA shown in (A) or programmed with truncated mRNA (harbouring no stop codon in the A-site). Signal detection was performed with anti-HA antibody for detection of (B) peptidyl-tRNA or (C) anti-eRF1 antibody. (D) Cryo-EM structure of the eRF1-bound human 80S-CMV-RNC at 3.8 Å resolution. The colour code for mRNA, tRNA and the eRF1 domains is given. (E) Section of (D) focusing on the hCMV peptidyl-tRNA. (F) Top view section of the human eRF1 bound 80S-CMV-RNC cryo-EM structure. (G) Molecular models of (F). For docking, the model of the human ribosome POST structure (pdb code: 5AJ0) and the crystal/NMR structures of human eRF1 (pdb codes: 3E1Y-A, 2KTV) were used. (H, I) Molecular models displaying (H) all 3 eRF1 domains and (I) ribosomal contacts of human eRF1.
Figure 3.
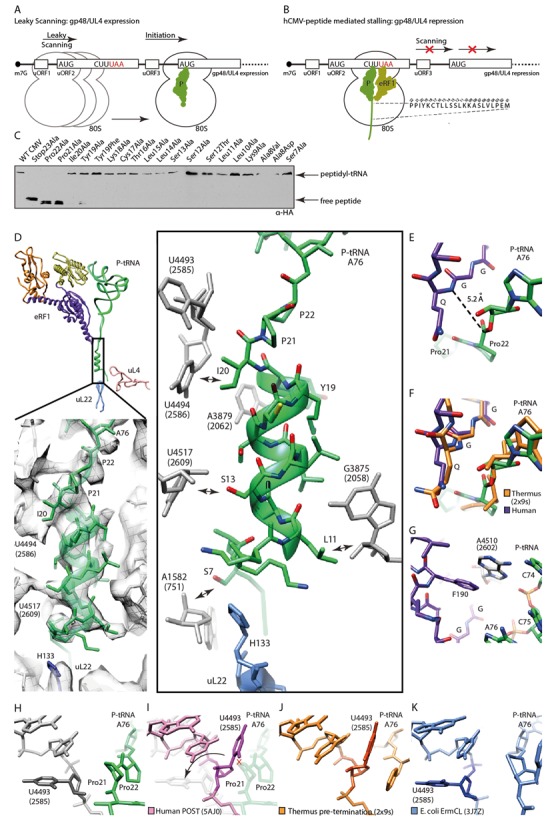
Mechanism of termination silencing by nascent hCMV peptide. (A) Schematic representation of the gp48/UL4 mRNA illustrating expression regulation of gp48/UL4 by leaky scanning of uORF2. (B) Ribosome stalling on the hCMV uORF2 leads to gp48/UL4 repression. (C) Identification of arrest-defective amino acid substitutions within the hCMV stalling peptide indicated by decreased amount of hCMV peptidyl-tRNA and accumulation of free peptide detected by Western blotting. (D, upper left) Overview of eRF1, peptidyl-tRNA and ribosomal proteins uL22 and uL4 labelled and coloured distinctively. (D, lower left) EM density section revealing the helical structure which is formed by the nascent hCMV peptide within the upper part of the ribosomal tunnel. (D, right) Same section, showing H133 of uL22 and nucleotides of the ribosomal tunnel wall which interact with the helical part of the nascent chain (arrows). (E) Position of the eRF1 GGQ-motif and the peptidyl-tRNA within the PTCs. (F) Comparison of the position of the bacterial RF2 and the human eRF1 GGQ-motifs and the peptidyl-tRNAs within the PTC. (G) eRF1-stabilizing position of Hs A4510 (Ec A2602). (H-K) Comparison of the position of Hs U4493 (Ec U2585) in (H) the hCMV stalled ribosome complex, (I) the human ribosome in the POST state, (J) the prokaryotic pre-termination complex and (K) the ErmCL stalled ribosomal complex.
Human in vitro translation extract
The translation extract was prepared on the basis of Mikami et al. (29) with significant adjustments (Figure 1A). HeLa S3 cells were grown to a density of 3.0–5.5 × 105 in SMEM (Sigma), supplemented with 10% heat-inactivated FCS (Gibco), Penicillin (100 U/ml)/Streptomycin 100 μg/ml (Gibco) and 1x GlutaMAX (Gibco) at 37°C, 5% CO2 using a spinner flask (40 rpm). Subsequently, cells were harvested (2 min, 650x g) and the resulting pellet was washed 3x with Washing Buffer (35 mM HEPES/KOH pH 7.5, 140 mM NaCl, 11 mM Glucose) (1 min, 650x g) and 1x with Extraction Buffer (20 mM HEPES/KOH pH 7.5/4°C, 45 mM KOAc, 45 mM KCl, 1.8 mM Mg(OAc)2, 1 mM DTT). Further, the cell pellet was resuspended in Extraction Buffer (1.2 × 109 cells/ml) and disrupted by nitrogen pressure (300 psi, 30 min, 4°C) in a cell disruption vessel (Parr Instrument). The lysate was mixed with 1/29 volume High Potassium Buffer (20 mM HEPES/KOH pH 7.5/4°C, 945 mM KOAc, 945 mM KCl, 1.8 mM Mg(OAc)2, 1 mM DTT), incubated at 4°C for 5 min and centrifuged briefly (20 s, 14 000 rpm, 4°C). Aliquots of the resulting supernatant were frozen in liquid nitrogen and stored at −80°C.
Figure 1.
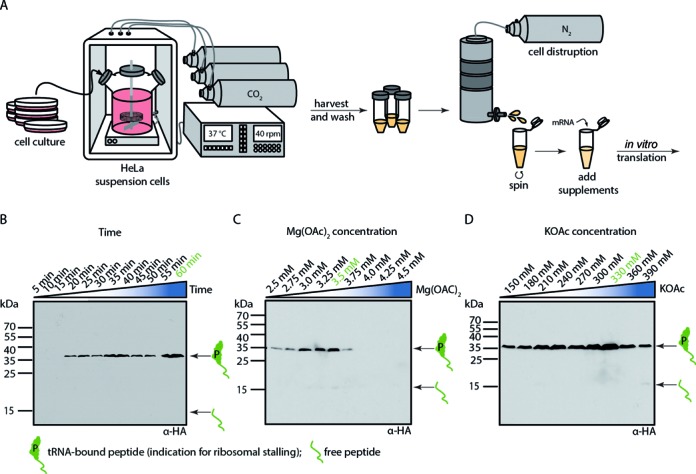
Translation in human cell-free extract. (A) Schematic representation of the individual steps from human HeLa cell culture to a cell-free human in vitro translation extract. Cells were grown in a large spinner flask. After harvesting and washing of the cells, they were subjected to nitrogen pressure and disrupted by sudden pressure release. A quick centrifugation step resulted in a crude extract which can be supplemented for optimal performance of mRNA in vitro translation. Example of in vitro translational stalling optimization for (B) time (C) magnesium acetate concentration and (D) potassium acetate concentration using Western blotting with anti-HA antibody for detection of tRNA-bound hCMV peptide or free hCMV peptide. Optimal conditions vary for each extract.
In vitro transcription
Plasmids were linearized with SpeI-HF (NEB) in CutSmart (NEB) buffer at 37°C overnight and purified (QIAquick PCR purification kit (Qiagen)) before in vitro transcription (tc) to ensure the right length of the mRNA product. The final mRNA construct encoded a CrPV IGR IRES sequence for translation initiation, an N-terminal HA- and (His)6-tag, the well characterized DP75 dipeptidyl-aminopeptidase B (DPAPB) (30), the hCMV uORF2 stalling sequence and a polyA-tail. The reaction was performed in Buffer tc (40 mM Tris/HCl pH 7.9/4°C, 20 mM MgCl2, 0.01% Triton X-100, 2.5 mM Spermidine, 5 mM DTT, 6.25 mM ATP, 6.25 mM CTP, 6.25 mM GTP, 6.25 mM UTP, 0.2 U/ml RNasin (Ambion)) with home-made T7 RNA polymerase and 1.5 μg mRNA/100 μl reaction. The tc reaction was incubated for 4 h at 37°C. Additional T7 RNA polymerase was added during incubation. The resulting mRNA was purified by LiCl precipitation and stored at −80°C.
In vitro translation
In vitro translation (tl) was performed in Buffer tl (24 mM HEPES/KOH pH 7.5/4°C, 112.5 mM KOAc, 20.1 mM KCl, 2.15 mM Mg(OAc)2, 2.5 mM DTT, 0.25 mM GTP, 1.56 mM ATP, 16 mM Creatine Phosphate (Roche), 0.45 μg/μl Creatine Kinase (Roche), 50 μg/ml yeast tRNA, 0.4 mM Spermidine) with 50% (v/v) extract. This mixture was supplemented with 0.12 mM amino-acid mixture, complete (Promega) and 0.885 U/μl RNasin (Ambion). 100 μg mRNA/600 μl translation reactions was added before incubation for 20–60 min (depending on the extract) at 30°C. Translation time, KOAc and Mg(OAc)2 concentrations were optimized for each extract (Figure 1B–D).
Preparation of eRF1-containing RNCs
Human ribosomes were stalled via the nascent hCMV-peptide and the resulting ribosome nascent chain complexes (RNCs) were purified essentially as described before (31). Briefly, the translation reactions were loaded onto 3 ml sucrose cushion (750 mM sucrose, 50 mM Tris/HCl pH 7.0/4°C, 500 mM KOAc, 125 mM Mg(OAc)2, 0.1% Nikkol, 5 mM β-Mercaptoethanol, 1/1000 complete protease inhibitor (Roche)) for centrifugation (Beckman Coulter TLA 120.2, 100 000 rpm, 45 min, 4°C). The resulting pellet was resuspended in 2 ml Buffer A (50 mM Tris/HCl pH 7.0/4°C, 250 mM KOAc, 25 mM Mg(OAc)2, 0.1% Nikkol, 250 mM sucrose, 5 mM β-Mercaptoethanol, 1/1000 EDTA-free complete protease inhibitor (Roche), 0.2 U/ml RNasin (Ambion)) and subjected to Co2+-affinity purification. 1 ml TALON Metal Affinity Resin (Macherey & Nagel) was pre-equilibrated and pre-exposed to a yeast tRNA mixture (10 μg/ml) in Buffer A. The beads were washed 3x with 5 column volume (CV) Buffer A and 1x with 3 CV Buffer B (50 mM Tris/HCl pH 7.0/4°C, 500 mM KOAc, 25 mM Mg(OAc)2, 0.1% Nikkol, 250 mM sucrose, 5 mM β-Mercaptoethanol, 1/1000 EDTA-free complete protease inhibitor (Roche)). Elution of the His-CMV-RNCs was performed with 3 ml Buffer A containing 150 mM imidazole. The eluate was again loaded onto a sucrose cushion and centrifuged (Beckman Coulter TLA 110, 100 000 rpm, 60 min, 4°C). The pellet was resuspended on ice in 25 μl Buffer C (50 mM Tris/HCl pH 7.0/4°C, 100 mM KOAc, 6 mM Mg(OAc)2, 1 mM DTT, 1/200 EDTA-free complete protease inhibitor (Roche), 0.2 U/ml RNasin (Ambion)), briefly centrifuged (13 000 rpm, 1 min, 4°C), aliquoted, frozen in liquid nitrogen and stored at −80°C. Stalling efficiency and eRF1 co-purification was monitored in the final sample via SDS-PAGE and Western blotting. CMV-truncated control RNCs were prepared accordingly.
Mutational-screening
Mutated hCMV-(stalling) constructs were in vitro transcribed and translated as described above. Incubation of the translation reaction was terminated by the addition of 1x SDS Sample Buffer. Stalling efficiency after 10, 20, 30 and 40 min at 37°C was analysed via SDS-PAGE and subsequent Western blotting.
eRF1 expression, purification and in vivo hydroxylation
Co-expression of eRF1 and Jmjd4 was performed in E. coli BL21 (DE3) for in vivo hydroxylation of eRF1 at K63. The cells were grown to an OD600 = 0.6 at 37°C and temperature-adjusted for subsequent expression at 18°C overnight. After 20 h, cells were harvested, frozen in liquid nitrogen and stored at −80°C. Lysis Buffer (50 mM HEPES/KOH pH 7.5/4°C, 500 mM NaCl, 5 mM imidazole, 5 mM β-Mercaptoethanol) was added to the pellet and cells were disrupted in a Microfluidizer (model 110I lab homogenizer, Microfluidics) at 3x 15 000 psi. The lysate was cleared (SORVALL, SS-34, 13 500 rpm, 30 min, 4°C). The resulting supernatant was incubated with TALON Metal Affinity Resin (Macherey & Nagel) beads for 1.5 h at 4°C. Beads were washed with 40x CV WashBuffer (50 mM HEPES/KOH pH 7.5/4°C, 500 mM NaCl, 20 mM imidazole, 5 mM β-Mercaptoethanol), before incubation with homemade HRV 3C protease in Buffer E (50 mM HEPES/KOH pH 7.5/4°C, 200 mM NaCl, 1 mM β-Mercaptoethanol, 2.5 mM MgCl2) for 1 h at 4°C was performed for elution of the tag-free human eRF1 protein. The protein was aliquoted, frozen in liquid nitrogen and stored at −80°C. Samples were taken throughout the purification procedure for analysis via SDS-PAGE and Western blotting. In vivo hydroxylation of eRF1 at C4 of K63 was monitored via MS/MS.
Cryo-grid preparation
0.1 pmol/μl eRF1-CMV-RNCs were incubated with 5x molar excess of purified and hydroxylated eRF1 to prevent ligand-dissociation from the ribosome. This sample (20 mM HEPES pH 7.5/4°C, 100 mM KOAc, 2.5 mM Mg(OAc)2, 0.25 mM spermidine, 2 mM DTT, 0.06 U/μl RNasin (Ambion), 1/625 EDTA-free complete protease inhibitor (Roche)) was applied to 2 nm pre-coated Quantifoil R3/3 holey carbon supported grids and vitrified using a Vitrobot Mark IV (FEI Company).
Cryo-electron microscopy and single particle reconstruction
Data collection was performed on a Titan Krios TEM (FEI Company) equipped with a Falcon II direct electron detector, operated at 300 keV using the EPU software (FEI). The magnification settings resulted in a pixel size of 1.062 Å/pixel. The data set was provided with the semi-automatic software EPU (FEI Company) with a dose of 5 e−/Å2 per frame for frames 0–3 and 10 e−/Å−2 per frame for frames 4–6 at defocus values between 1.0 and 2.7 μm. Data were collected in a series of 7 frames (numbered 0–6) whereof frames 3–6 were excluded for the final reconstruction for limitation of the effective dose. The three remaining frames were aligned and summed up using Motion Correction software (32). Automatic particle detection was performed by the program SIGNATURE (33). Initial alignment of the obtained 255 253 particles was done using a human 80S ribosome as reference structure. Data processing and single particle analysis were done using the SPIDER software package (34). Noise and non-ribosomal particles (35,062; 14%) were removed from the data set by further unsupervised 3D sorting (35). Particles lacking ligand or P-tRNA density were excluded from the final data set. The remaining particles (33 165; 13%) were refined to an average resolution of 3.8 Å (FSC0.143) and a local resolution extending to 3.6 Å for the core of the ribosome as computed using ResMap (36) (Supplementary Figure S1). Throughout the refinement, a frequency-limited (truncation of frequencies higher than 8 Å) refinement protocol was used to prevent potential over-fitting (37). The final volume was subjected to B-factor sharpening using the program EM-BFACTOR (38).
Molecular model building
The initial model of human eRF1 was based on the crystal structure by Cheng et al. (12) (pdb code: 3E1Y-A). The missing minidomain herein was added from the NMR structure of the eRF1 C domain (pdb code: 2KTV). The initial models for the ribosomal rRNA and proteins were based on the cryo-EM reconstruction of the human ribosome in the POST state (19) (pdb code: 5AJ0). All models were initially fitted to the electron density using UCSF Chimera (39) and adjusted according to the features of the electron-density using Coot (40). Modelling was followed by refinement using the real-space refine tool of the PHENIX software package (41). Here, default settings of the PHENIX command phenix.real_space_refine model.pdb map.ccp4 were used.
Further, we validated the quality of the eRF1-fit by calculating the cross-correlation (CC) between the extracted density and our model using UCSF Chimera (39). The CC-values increased when comparing the initial models (individual domains of the human eRF1 crystal structure docked into the isolated density) with the refined model (0.73–0.76 for the N domain; 0.66–0.68 for the M domain, 0.39–0.70 for the C domain). The significant increase for the CTD CC-value is explained by the lack of the minidomain and the C-terminal helix in the crystal structure.
Figure preparations
Figures depicting atomic models with and without density were prepared using UCSF Chimera (39).
RESULTS AND DISCUSSION
Isolation and cryo-EM of a stalled human 80S ribosome bound to eRF1
We employed the stalling sequence of the hCMV gp48 upstream open reading frame 2 (uORF2) (13,42,43) in order to obtain a ribosome-eRF1 complex suitable for structural analysis using cryo-electron microscopy (cryo-EM). The nascent hCMV-peptide stalls the ribosome in a pre-termination state with a UAA stop codon positioned in the ribosomal A-site. Moreover, hCMV-stalling prevents peptide release and thus leads to an accumulation of ribosome-bound eRF1 (44). For the purification of stable human termination complexes, we developed and optimized a robust human cell-free translation system based on HeLa cell extracts (Figure 1). Notably, we used the cap-independent cricket paralysis virus intergenic region internal ribosomal entry site (CrPV IGR IRES) for translation initiation since this IRES does not require any initiation factors (45). The cell-free system was programmed with mRNA encoding the hCMV stalling peptide and an additional (His)6 affinity tag at the N-terminus (Figure 2A) for purification of the stalled RNCs carrying the UAA stop codon in the A-site. The isolated RNCs displayed a single band for the hCMV-stalled peptidyl-tRNA in Western blots, indicating the homogeneity of the preparation (Figure 2B). Moreover, in contrast to RNCs that were stalled using a truncated mRNA (without a stop codon in the A-site), the hCMV-RNCs co-purified with stably bound endogenous eRF1 (Figure 2C).
For structure determination this preparation was subjected to cryo-EM and single particle analysis in the presence of excess recombinant human eRF1 in order to maintain a high ligand occupancy on the RNCs. Excess eRF1 was in vivo hydroxylated at Lys63 by co-expression with Jmjd4, a 2-oxogluterate and Fe(II)-dependent oxygenase. This post-translational modification by Jmjd4 was previously shown to be endogenously present and required for optimal translation termination efficiency (28). After in silico sorting of the data set into homogeneous subpopulations a final sub-data set of 33 165 particles was obtained, yielding a 3D reconstruction with an average resolution of 3.8 Å (Supplementary Figure S1A). As expected, the electron density map displayed the typical appearance of a human ribosome as reported previously (19,46,47), but also showed additional density for the peptidyl-tRNA in the P-site and in the exit tunnel of the ribosome, as well as for eRF1 bound in the A-site (Figure 2D and E). The overall conformation of the ribosome in the pre-termination state resembles the post-translocation (POST) state, in which the ribosomal subunits are not rotated with respect to each other (19), however, with the stalk base (rRNA helices H43/H44) moved inwards as observed previously for eRF1 and eRF1/ABCE1 containing complexes (13,19).
eRF1 adopts an extended conformation, reaching into the A-site decoding center of the SSU with the N domain while the M domain is oriented towards the LSU positioning the GGQ-motif in the PTC (Figure 2F) (13,18). For molecular analysis, we docked the molecular model for the human ribosome (19) into our density (Supplementary Figures S2A, S2B) and built a model for ribosome-bound eRF1 based on the crystal structure of eRF1 in complex with eRF3 (Figure 2G and H and Supplementary Figure S3A–3D) (12). The main contacts of eRF1 with the ribosome involve all three domains and predominantly involve ribosomal RNA, with the exception of a few contacts to the ribosomal proteins eS27 and uL11 via the α-helix forming C-terminal tail of eRF1 (Figure 2I, Supplementary Figure S3E–3G).
Termination silencing by the nascent hCMV peptide
The hCMV peptide is encoded in its native mRNA as a small ORF upstream of the gp48 ORF and when translated it induces ribosome stalling at its own termination, which in turn prevents ribosome scanning and thereby represses expression of the downstream gp48 ORF translation (Figure 3A and B). We investigated the contribution of the single amino acid residues of the hCMV peptide to the stalling activity in our human cell-free system using scanning mutagenesis. In agreement with previous data (43,48) the most dramatic stalling compromising effects were observed for the mutation of the two C-terminal prolines 21/22, validating their importance for the stalling mechanism in our system (Figure 3C).
The hCMV peptide is clearly resolved in our structure from the PTC to the tunnel constriction (formed by uL22 and uL4 ∼30 Å from the PTC), thereby encompassing all residues that contribute to the stalling activity (43,44,48); from the most C-terminal Pro22 to Ser7 near ribosomal protein uL22 (Figure 3D). The density revealed that the largest part of the peptide adopts an α-helical conformation and engages in several interactions with the ribosomal tunnel (Figure 3D). Several nucleotides of the tunnel wall (numbering of Homo sapiens (Hs) is according to Behrmann et al. (19)) participate in these interactions (Hs U4494, Ec U2586; Hs A3879, Ec A2062; Hs U4517, Ec 2609; Hs G3875, Ec G2058) and at the N-terminal end of the α-helix Ser7 reaches the most distal His133 of ribosomal uL22 (Figure 3D).
The GGQ-motif of eRF1 was found in a position indistinguishable from the position observed in the bacterial RF-ribosome complexes (Figure 3E and F and Supplementary Figure S2C) (9). Given that the GGQ-mediated release mechanism is indeed conserved between eukaryotes and prokaryotes, we conclude that eRF1 is properly positioned in its active conformation also when termination is stalled by the hCMV peptide, as observed before at lower resolution (13). Compared to the bacterial pre-termination complex (9), the ester bond between A76 of the tRNA and the C-terminal Pro22 also appears to be in its canonical position. In bacteria, it has been shown that two rRNA nucleotides, Ec A2602 and Ec U2585, within the highly conserved PTC are required for release activity (49). Ec A2602 stabilizes the GGQ containing loop of bacterial release factors (8) and, analogously, we find an equivalent situation in the human complex where Hs A4510 (Ec 2602) stacks upon the side chain of Phe190 of eRF1 (Figure 3G, Supplementary Figure S2D).
In contrast, we observed a drastically changed conformation for Hs U4493 (Ec U2585) (Figure 3H) when comparing to its canonical position in another human ribosome structure (Figure 3I) (19) or in a bacterial pre-termination complex (Figure 3J) (9). Specifically, Hs U4493 is flipped by approximately 90° most likely due to a steric clash with the penultimate Pro21 of the hCMV peptide. This unusual conformation has been observed before for U2585 in the context of the bacterial drug-dependent stalling peptide ErmCL (Figure 3K) (50), where the C-terminal residues of the ErmCL nascent peptide chain adopts a similar path compared to the hCMV peptide. It is very unlikely that Hs U4493 in this conformation can still participate in the release reaction. Therefore, we suggest that the rotation of Hs U4493 contributes silencing of the PTC release activity. This would also rationalize the lack of hCMV mediated stalling when replacing the stop codon with a sense codon (Figure 3C) since mutations of Ec U2585 have severe effects on termination but not on peptide bond formation (49). Taken together, α-helix formation of the hCMV peptide combined with the restricted geometry of the residues Pro21 and Pro22 positions the peptide such that the PTC is perturbed and loses its ability to support release activity of an otherwise productively positioned GGQ-loop of eRF1.
The UAA(A) stop codon resembles a U-turn conformation
The N domain of eRF1 reaches into the decoding center of the SSU and creates a tight pocket to accommodate the UAA stop codon as well as the fourth nucleotide (A4) immediately following the stop codon (Figure 4A, Supplementary Figure S4). Notably, compared to the geometry that was observed for sense codon and bacterial stop codon decoding, the eukaryotic stop codon adopts a dramatically different conformation, namely, that resembling the geometry of a classical RNA U-turn motif of the UNR-type (Figure 4A and B) (51). Formation of the U-turn results in shortening of the mRNA, which is in agreement with a smaller toeprinting signal observed upon eRF1 binding (16).
Figure 4.
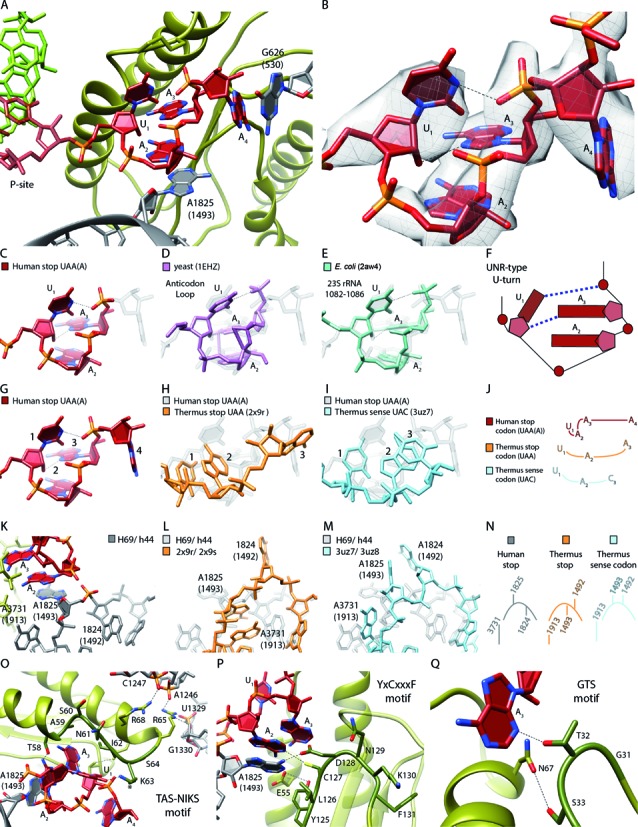
U-turn-like geometry of the UAA stop-codon bound by eRF1 and the ribosome. (A) Human UAA(A) stop-codon (red) positioned in a cavity between eRF1 (green) and the ribosomal bases Hs G626 (Ec G530) and Hs A1825 (Ec A1493) (gray). (B) EM density revealing the geometry of the mRNA sequence UAA(A) during stop codon recognition. (C–F) Comparison of the human stop codon mRNA geometry with the known UNR-type U-turn structures (D) yeast anticodon loop (purple) and (E) Ec 23S rRNA (1082–1086) (turquois). (F) Schematic representation of (C–E). (G–J) Comparison of the A-site mRNA geometry during (G) human UAA stop codon recognition (H), Thermus thermophilus (Tt) UAA stop codon recognition (light orange) and (I) Tt UAC sense decoding (light blue). (J) Schematic representation of (G–I). (K–N) Comparison of the h44 (Hs A1824 (Ec A1492), Hs A1825 (Ec A1493))/H69 (Hs A3731 (Ec A1913)) states during (K) human UAA stop codon recognition, (L) Tt UAA stop codon recognition and (M) Tt UAC decoding. (N) Schematic representation of (K–M). (O) Interactions of the eRF1 TAS-NIKS motif (residues 58–64) with the mRNA/rRNA. The hydroxylation site of K63 is located at C4 (*). (P) Interactions of the eRF1 YxCxxxF (residues 125–131) motif with the mRNA/rRNA. (Q) Interactions of the eRF1 GTS (residues 31–33) motif with the mRNA.
The observed stop codon geometry is stabilized by the characteristic H-bond pattern of an UNR-type U-turn between the first U (U1) and the third phosphate group as well as between the 2′-hydroxyl group of the first ribose and the N7 of the third base that is always a purine (A3 in our case) in this class of turns (Figure 4C–F). This geometry is very similar to other U-turns of the same type that have been found, for example in tRNA anticodon loops as well as in the 23S ribosomal RNA (Figure 4D and E). This distinct conformation of the eukaryotic stop codon also results in a unique mode of interaction with the ribosome and the release factor, when compared to sense codons or bacterial stop codons during decoding (Figure 4G–J). In bacteria the ribosome stabilizes the third base of the stop codon by a stacking interaction with Ec G530 (6–9). In case of the eukaryotic stop codon Hs G626 (Ec G530) stacks onto the fourth base (A4) allowing for formation of the U-turn between U1 and A3 (Figure 4A). Moreover, the eukaryotic stop codon stacks onto the flipped-out Hs A1825 (Ec A1493), while Hs A1824 (Ec A1492) remains within helix44 (h44) of the SSU rRNA (Figure 4K–N). Conversely, in bacterial stop codon recognition A1493 remains in h44 and only A1492 flips out (6,7,9), where it contributes to the positioning of A3 of the stop codon on Ec G530, but does not engage in any stacking interaction (Figure 4L). For sense codons, both A1492 and A1493 flip out of h44 to insert into the minor groove of the codon-anticodon helix, forming an A-minor interaction (Figure 4M).
In addition to the ribosome, all three previously identified conserved motifs of the eRF1 N domain contribute to the formation of the stop codon binding pocket as well as establishing interactions with the ribosome (Figure 4O–Q): Almost the entire TAS-NIKS motif (residues 58–64) lines the stop codon on top from Thr58 to Lys63, while residues Ala59, Ile62 and Ser64 rather stabilize the loop conformation of eRF1, and the following Arg65 and Arg68 interact with ribosomal RNA (52). The YxCxxxF motif (residues 125–131) of eRF1 forms the middle part of the binding pocket and contributes to the stabilization of the flipped out Hs A1825 which is in agreement with a role of eRF1 in influencing the conformation of this ribosomal base (53). The region of eRF1 from Tyr125 to Asp128 is positioned in direct proximity to the stop codon, whereas Asn129, Lys130 and Phe131 are oriented away. The GTS motif from Gly31 to Ser33, in particular Thr32, closes the binding pocket near A3 of the stop codon.
Recognition of the stop codon and discrimination against sense codons
How are the three stop codons UAA, UGA and UAG distinguished from sense codons? A large contribution is apparently provided by the geometry resembling the UNR-like U-turn motif (51), which is required to optimally fit into the pocket formed between the ribosome and eRF1. Having the base U in the first position is mandatory for stabilizing this geometry, and, in addition U1 is found in hydrogen bonding distance to Lys63 within the TAS-NIKS motif of eRF1 (Figure 5A). Consistently, a direct contact between U1 and Lys63 has been suggested before based on chemical cross-linking data (54). At this resolution, modifications like the hydroxylation of Lys63 (C4) cannot be assigned. However, since this modification improves translation termination efficiency, we speculate its involvement in the proper orientation of the K63 side chain to improve base discrimination efficiency at position 1 of the stop codon. Purines in position 1 would be too large for the pocket and, like cytosine in position 1, would not allow for a stable UNR-turn geometry. Moreover, cytosine would not allow the equivalent hydrogen bonding interactions with Lys63 of eRF1.
Figure 5.
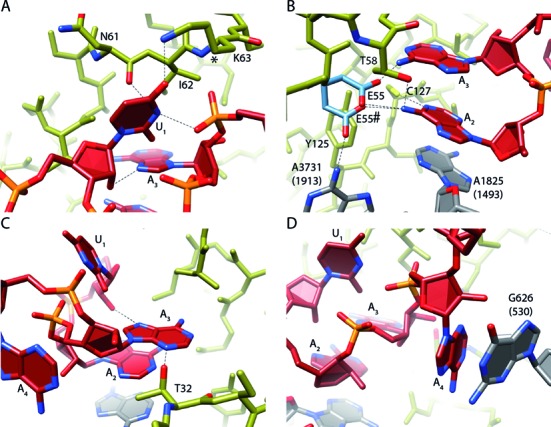
Interaction of eRF1 with the UAA(A) stop codon. (A) The Uracil 1 (U1) nucleotide of the stop codon interacts with N7 of adenosine 3 (A3) and the A3 backbone phosphate to adopt U-turn geometry of the stop codon. Further, U1 interacts with N61 and K63 of the eRF1 TAS-NIKS motif. The hydroxylation site of K63 is located at C4 (*). (B) Adenosine 2 (A2) interacts with Hs A1825 (h44) and C127 (YxCxxxF motif). Possible rotamer conformations of E55 (E55 and E55#) for the interaction with A2 are depicted in light blue. Y125 (YxCxxxF motif) stabilizes the shifted conformation of Hs A3731 (H69). (C) A3 mainly interacts with T32 (GTS motif) and the 2′OH of U1. (D) Adenosine 4 (A4) stacks on the rRNA base Hs G626 (Ec G530). Interactions are indicated by dotted lines.
The second position in a UNR-turn can in principle be occupied by any base, however, in our structure A2 is stacked between A3 of the stop codon and Hs A1825 of h44 (Figure 5B). Such stacking interaction is more stable for purines than for pyrimidines (55). Further, A2 interacts with the YxCxxxF motif, likely forming a direct contact with Cys127 that would be possible only with the purines A and G, thus further discriminating against pyrimidines in position 2. Although we do not see any density for the nearby Glu55 of eRF1, it is likely that it adopts a canonical rotamer conformation to interact with the amino groups of A2 and/or A3 (Figure 5B). This interaction would not be possible with guanosine which may explain why eRF1 does not decode UGG. A role of Glu55 in discriminating against two Gs in position 2 and 3 would be in agreement with previous findings suggesting the participation of Glu55 in recognizing the identity of the bases in these positions (56,57).
The third position in a UNR-turn is always a purine, therefore, meeting the requirement in this position to exclude pyrimidines and allowing only adenosine or guanine. In addition to the hydrogen bonding with the ribose in position 1, A3 is stabilized by Thr32 of the eRF1 GTS motif, an interaction that would also be possible for a G (Figure 5C). Directly after the stop codon the A in position 4 stacks onto Hs G626 (Ec G530) and thereby provides the frame for the stable compacted conformation of the stop codon itself (Figure 5D). A stronger stacking interaction of purines may explain why eukaryotic stop codons have a significant bias in the fourth position, particularly for adenosine (58).
Taken together, we find that eRF1 together with the ribosome recognizes the UAA(A) stop codon by a combination of probing the base identities with monitoring the capacity to adopt a stable U-turn-like geometry, most likely via an induced-fit mechanism. In contrast, stop codon recognition in bacteria by two specific release factors relies mostly on the recognition of the individual bases (6–9). Divergent evolution towards U-turn geometry recognition in eukaryotes would rationalize the unique capacity of eRF1 to recognize three distinct codons with different base identities in two positions.
ACCESSION NUMBERS
The cryo-electron microscopy map for the human termination complex has been deposited in the EMDataBank with accession code EMD-3099. The respective coordinates for electron-microscopy-based model of the ribosome-bound eRF1 are deposited in the ProteinDataBank (5a8l).
Supplementary Material
Acknowledgments
We thank Andrea Gilmozzi and Susi Rieder for technical assistance, Daniel N. Wilson, Bertrand Beckert and Daniel Sohmen for critical discussions. Further, we thank Christian M.T. Spahn for providing the coordinates of the human 80S ribosome model.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft (DFG) [FOR 1805 and SFB646 to R.B.]; R.B. acknowledges support by the Center for Integrated Protein Science Munich (CiPS-M), the Graduate School of Quantitative Biosciences Munich (QBM) and the European Research Council (Advanced Grant CRYOTRANSLATION). Funding for open access charge: European Research Council (ERC, CRYOTRANSLATION).
Conflict of interest statement. None declared.
REFERENCES
- 1.Ogle J.M., Carter A.P., Ramakrishnan V. Insights into the decoding mechanism from recent ribosome structures. Trends Biochem. Sci. 2003;28:259–266. doi: 10.1016/S0968-0004(03)00066-5. [DOI] [PubMed] [Google Scholar]
- 2.Demeshkina N., Jenner L., Westhof E., Yusupov M., Yusupova G. A new understanding of the decoding principle on the ribosome. Nature. 2012;484:256–259. doi: 10.1038/nature10913. [DOI] [PubMed] [Google Scholar]
- 3.Zhou J., Korostelev A., Lancaster L., Noller H.F. Crystal structures of 70S ribosomes bound to release factors RF1, RF2 and RF3. Curr. Opin. Struct. Biol. 2012;22:733–742. doi: 10.1016/j.sbi.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korostelev A.A. Structural aspects of translation termination on the ribosome. RNA. 2011;17:1409–1421. doi: 10.1261/rna.2733411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodnina M.V. The ribosome as a versatile catalyst: reactions at the peptidyl transferase center. Curr. Opin. Struct. Biol. 2013;23:595–602. doi: 10.1016/j.sbi.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 6.Korostelev A., Asahara H., Lancaster L., Laurberg M., Hirschi A., Zhu J., Trakhanov S., Scott W.G., Noller H.F. Crystal structure of a translation termination complex formed with release factor RF2. Proc. Natl. Acad. Sci. U.S.A. 2008;105:19684–19689. doi: 10.1073/pnas.0810953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laurberg M., Asahara H., Korostelev A., Zhu J., Trakhanov S., Noller H.F. Structural basis for translation termination on the 70S ribosome. Nature. 2008;454:852–857. doi: 10.1038/nature07115. [DOI] [PubMed] [Google Scholar]
- 8.Weixlbaumer A., Jin H., Neubauer C., Voorhees R.M., Petry S., Kelley A.C., Ramakrishnan V. Insights into Translational Termination from the Structure of RF2 Bound to the Ribosome. Science (80-.). 2008;322:953–956. doi: 10.1126/science.1164840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin H., Kelley A.C., Loakes D., Ramakrishnan V. Structure of the 70S ribosome bound to release factor 2 and a substrate analog provides insights into catalysis of peptide release. Proc. Natl. Acad. Sci. U.S.A. 2010;107:8593–8598. doi: 10.1073/pnas.1003995107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertram G., Bell H.A., Ritchie D.W., Fullerton G., Stansfield I. Terminating eukaryote translation: domain 1 of release factor eRF1 functions in stop codon recognition. RNA. 2000;6:1236–1247. doi: 10.1017/s1355838200000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song H., Mugnier P., Das A.K., Webb H.M., Evans D.R., Tuite M.F., Hemmings B.A., Barford D. The crystal structure of human eukaryotic release factor eRF1–mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell. 2000;100:311–321. doi: 10.1016/s0092-8674(00)80667-4. [DOI] [PubMed] [Google Scholar]
- 12.Cheng Z., Saito K., Pisarev A. V, Wada M., Pisareva V.P., Pestova T. V, Gajda M., Round A., Kong C., Lim M., et al. Structural insights into eRF3 and stop codon recognition by eRF1. Genes Dev. 2009;23:1106–1118. doi: 10.1101/gad.1770109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Preis A., Heuer A., Barrio-Garcia C., Hauser A., Eyler D.E., Berninghausen O., Green R., Becker T., Beckmann R. Cryoelectron microscopic structures of eukaryotic translation termination complexes containing eRF1-eRF3 or eRF1-ABCE1. Cell Rep. 2014;8:59–65. doi: 10.1016/j.celrep.2014.04.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pisarev A. V, Skabkin M.A., Pisareva V.P., Skabkina O. V, Rakotondrafara A.M., Hentze M.W., Hellen C.U.T., Pestova T. V. The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol. Cell. 2010;37:196–210. doi: 10.1016/j.molcel.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shoemaker C.J., Green R. Kinetic analysis reveals the ordered coupling of translation termination and ribosome recycling in yeast. Proc. Natl. Acad. Sci. U.S.A. 2011;108:E1392–E1398. doi: 10.1073/pnas.1113956108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alkalaeva E.Z., Pisarev A.V., Frolova L.Y., Kisselev L.L., Pestova T.V. In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3. Cell. 2006;125:1125–1136. doi: 10.1016/j.cell.2006.04.035. [DOI] [PubMed] [Google Scholar]
- 17.Frolova L., Le Goff X., Zhouravleva G., Davydova E., Philippe M., Kisselev L. Eukaryotic polypeptide chain release factor eRF3 is an eRF1- and ribosome-dependent guanosine triphosphatase. RNA. 1996;2:334–341. [PMC free article] [PubMed] [Google Scholar]
- 18.Muhs M., Hilal T., Mielke T., Skabkin M.A., Sanbonmatsu K.Y., Pestova T.V., Spahn C.M.T. Cryo-EM of ribosomal 80S complexes with termination factors reveals the translocated cricket paralysis virus IRES. Mol. Cell. 2015;57:422–432. doi: 10.1016/j.molcel.2014.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Behrmann E., Loerke J., Budkevich T.V., Yamamoto K., Schmidt A., Penczek P.A., Vos M.R., Bürger J., Mielke T., Scheerer P., et al. Structural Snapshots of Actively Translating Human Ribosomes. Cell. 2015;161:845–857. doi: 10.1016/j.cell.2015.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barthelme D., Dinkelaker S., Albers S.-V., Londei P., Ermler U., Tampé R. Ribosome recycling depends on a mechanistic link between the FeS cluster domain and a conformational switch of the twin-ATPase ABCE1. Proc. Natl. Acad. Sci. U.S.A. 2011;108:3228–3233. doi: 10.1073/pnas.1015953108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Becker T., Franckenberg S., Wickles S., Shoemaker C.J., Anger A.M., Armache J.-P., Sieber H., Ungewickell C., Berninghausen O., Daberkow I., et al. Structural basis of highly conserved ribosome recycling in eukaryotes and archaea. Nature. 2012;482:501–506. doi: 10.1038/nature10829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Des Georges A., Hashem Y., Unbehaun A., Grassucci R.A., Taylor D., Hellen C.U.T., Pestova T.V., Frank J. Structure of the mammalian ribosomal pre-termination complex associated with eRF1.eRF3.GDPNP. Nucleic Acids Res. 2014;42:3409–3418. doi: 10.1093/nar/gkt1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frolova L., Seit-Nebi A., Kisselev L. Highly conserved NIKS tetrapeptide is functionally essential in eukaryotic translation termination factor eRF1. RNA. 2002;8:129–136. doi: 10.1017/s1355838202013262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong L.E., Li Y., Pillay S., Frolova L., Pervushin K. Selectivity of stop codon recognition in translation termination is modulated by multiple conformations of GTS loop in eRF1. Nucleic Acids Res. 2012;40:5751–5765. doi: 10.1093/nar/gks192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conard S.E., Buckley J., Dang M., Bedwell G.J., Carter R.L., Khass M., Bedwell D.M. Identification of eRF1 residues that play critical and complementary roles in stop codon recognition. RNA. 2012;18:1210–1221. doi: 10.1261/rna.031997.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seit-Nebi A. Conversion of omnipotent translation termination factor eRF1 into ciliate-like UGA-only unipotent eRF1. EMBO Rep. 2002;3:881–886. doi: 10.1093/embo-reports/kvf178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyazaki K. MEGAWHOP cloning: a method of creating random mutagenesis libraries via megaprimer PCR of whole plasmids. Methods Enzymol. 2011;498:399–406. doi: 10.1016/B978-0-12-385120-8.00017-6. [DOI] [PubMed] [Google Scholar]
- 28.Feng T., Yamamoto A., Wilkins S.E., Sokolova E., Yates L.A., Münzel M., Singh P., Hopkinson R.J., Fischer R., Cockman M.E., et al. Optimal Translational Termination Requires C4 Lysyl Hydroxylation of eRF1. Mol. Cell. 2014;53:645–654. doi: 10.1016/j.molcel.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mikami S., Kobayashi T., Imataka H. Cell-free protein synthesis systems with extracts from cultured human cells. Methods Mol. Biol. 2010;607:43–52. doi: 10.1007/978-1-60327-331-2_5. [DOI] [PubMed] [Google Scholar]
- 30.Beckmann R., Spahn C.M., Eswar N., Helmers J., Penczek P.A., Sali A., Frank J., Blobel G. Architecture of the protein-conducting channel associated with the translating 80S ribosome. Cell. 2001;107:361–372. doi: 10.1016/s0092-8674(01)00541-4. [DOI] [PubMed] [Google Scholar]
- 31.Halic M., Becker T., Pool M.R., Spahn C.M., Grassucci R.A., Frank J., Beckmann R. Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature. 2004;427:808–814. doi: 10.1038/nature02342. [DOI] [PubMed] [Google Scholar]
- 32.Li X., Mooney P., Zheng S., Booth C.R., Braunfeld M.B., Gubbens S., Agard D.A., Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen J.Z., Grigorieff N. SIGNATURE: a single-particle selection system for molecular electron microscopy. J. Struct. Biol. 2007;157:168–173. doi: 10.1016/j.jsb.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 34.Frank J., Radermacher M., Penczek P., Zhu J., Li Y., Ladjadj M., Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 35.Loerke J., Giesebrecht J., Spahn C.M.T. Multiparticle cryo-EM of ribosomes. Methods Enzymol. 2010;483:161–177. doi: 10.1016/S0076-6879(10)83008-3. [DOI] [PubMed] [Google Scholar]
- 36.Kucukelbir A., Sigworth F.J., Tagare H.D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scheres S.H.W., Chen S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods. 2012;9:853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernández J.J., Luque D., Castón J.R., Carrascosa J.L. Sharpening high resolution information in single particle electron cryomicroscopy. J. Struct. Biol. 2008;164:170–175. doi: 10.1016/j.jsb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 39.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 40.Emsley P., Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 41.Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhushan S., Meyer H., Starosta A.L., Becker T., Mielke T., Berninghausen O., Sattler M., Wilson D.N., Beckmann R. Structural basis for translational stalling by human cytomegalovirus and fungal arginine attenuator peptide. Mol. Cell. 2010;40:138–146. doi: 10.1016/j.molcel.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 43.Degnin C.R., Schleiss M.R., Cao J., Geballe A.P. Translational inhibition mediated by a short upstream open reading frame in the human cytomegalovirus gpUL4 (gp48) transcript. J. Virol. 1993;67:5514–5521. doi: 10.1128/jvi.67.9.5514-5521.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Janzen D.M., Frolova L., Geballe A.P. Inhibition of translation termination mediated by an interaction of eukaryotic release factor 1 with a nascent peptidyl-tRNA. Mol. Cell. Biol. 2002;22:8562–8570. doi: 10.1128/MCB.22.24.8562-8570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson J.E., Pestova T. V, Hellen C.U., Sarnow P. Initiation of protein synthesis from the A site of the ribosome. Cell. 2000;102:511–520. doi: 10.1016/s0092-8674(00)00055-6. [DOI] [PubMed] [Google Scholar]
- 46.Anger A.M., Armache J.-P., Berninghausen O., Habeck M., Subklewe M., Wilson D.N., Beckmann R. Structures of the human and Drosophila 80S ribosome. Nature. 2013;497:80–85. doi: 10.1038/nature12104. [DOI] [PubMed] [Google Scholar]
- 47.Khatter H., Myasnikov A.G., Natchiar S.K., Klaholz B.P. Structure of the human 80S ribosome. Nature. 2015;520:640–645. doi: 10.1038/nature14427. [DOI] [PubMed] [Google Scholar]
- 48.Alderete J.P., Jarrahian S., Geballe A.P. Translational Effects of Mutations and Polymorphisms in a Repressive Upstream Open Reading Frame of the Human Cytomegalovirus UL4 Gene. 1999;73:8330–8337. doi: 10.1128/jvi.73.10.8330-8337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Youngman E.M., Brunelle J.L., Kochaniak A.B., Green R. The active site of the ribosome is composed of two layers of conserved nucleotides with distinct roles in peptide bond formation and peptide release. Cell. 2004;117:589–599. doi: 10.1016/s0092-8674(04)00411-8. [DOI] [PubMed] [Google Scholar]
- 50.Arenz S., Meydan S., Starosta A.L., Berninghausen O., Beckmann R., Vázquez-Laslop N., Wilson D.N. Drug Sensing by the Ribosome Induces Translational Arrest via Active Site Perturbation. Mol. Cell. 2014;56:446–452. doi: 10.1016/j.molcel.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gutell R.R., Cannone J.J., Konings D., Gautheret D. Predicting U-turns in ribosomal RNA with comparative sequence analysis. J. Mol. Biol. 2000;300:791–803. doi: 10.1006/jmbi.2000.3900. [DOI] [PubMed] [Google Scholar]
- 52.Blanchet S., Rowe M., Haar T., Fabret C., Demais S., Howard M.J., Namy O. New insights into stop codon recognition by eRF1. Nucleic Acids Res. 2015;43:3298–3308. doi: 10.1093/nar/gkv154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saito K., Ito K. Genetic analysis of L123 of the tRNA-mimicking eukaryote release factor eRF1, an amino acid residue critical for discrimination of stop codons. Nucleic Acids Res. 2015;43:4591–4601. doi: 10.1093/nar/gkv376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chavatte L., Seit-Nebi A., Dubovaya V., Favre A. The invariant uridine of stop codons contacts the conserved NIKSR loop of human eRF1 in the ribosome. EMBO J. 2002;21:5302–5311. doi: 10.1093/emboj/cdf484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Friedman R.A., Honig B. A free energy analysis of nucleic acid base stacking in aqueous solution. Biophys. J. 1995;69:1528–1535. doi: 10.1016/S0006-3495(95)80023-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Conard S.E., Buckley J., Dang M., Bedwell G.J., Carter R.L., Khass M., Bedwell D.M. Identification of eRF1 residues that play critical and complementary roles in stop codon recognition. RNA. 2012;18:1210–1221. doi: 10.1261/rna.031997.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kryuchkova P., Grishin A., Eliseev B., Karyagina A., Frolova L., Alkalaeva E. Two-step model of stop codon recognition by eukaryotic release factor eRF1. Nucleic Acids Res. 2013;41:4573–4586. doi: 10.1093/nar/gkt113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tate W.P., Mannering S.A. Three, four or more: the translational stop signal at length. Mol. Microbiol. 1996;21:213–219. doi: 10.1046/j.1365-2958.1996.6391352.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.