

Abstract
With the wider deployment of massively parallel, next generation sequencing, it is now possible to survey human genome data for research and clinical purposes. The reduced cost of producing short read sequencing has now shifted the burden to data analysis. Analysis of genome sequencing remains challenged by the complexity of the human genome, including redundancy and the repetitive nature of genome elements and the large amount of variation in individual genomes. Public databases of human genome sequence greatly facilitate interpretation of common and rare genetic variation, although linking database sequence information to detailed clinical information is limited by privacy and practical issues. Genetic variation is a rich source of knowledge for cardiovascular disease since many, if not all, cardiovascular disorders are highly heritable. The role of rare genetic variation in predicting risk and complications of cardiovascular diseases has been well established for hypertrophic and dilated cardiomyopathy, where the number of genes that are linked to these disorders is growing. Bolstered by family data, where genetic variants segregate with disease, rare variation can be linked to specific genetic variation that offers profound diagnostic information. Understanding genetic variation in cardiomyopathy is likely to help stratify forms of heart failure and guide therapy. Ultimately, genetic variation may be amenable to gene correction and gene editing strategies.
Keywords: cardiomyopathy, LQTS, exome, genome, ESP, 1000 genomes, variation, population, risk
INTRODUCTION
Advances in DNA sequencing technology have made it possible to define the variation between and among human genomes. For cardiovascular disease, human genetic variation is very powerful to identify those at risk for developing cardiomyopathy and heart failure, arrhythmias and vascular diseases. Massively parallel DNA sequencing produces millions of base pairs of DNA sequence simultaneously with the advantages of comparatively low cost and remarkably high output 1. The major disadvantage of this approach is that it generates short read sequencing, typically 75 to 150 bp in length. Typically, sequence is determined from small fragments of approximately 400–500 bp in length and both ends are sequenced (paired end sequencing). With short read data it is necessary to repetitively sequence regions to improve accuracy. Reads are then aligned against a reference sequence. With alignment, it is possible to identify differences between the reference genome and the genome under analysis. The computational needs for alignment and variant calling are substantial when the entire coding region or exome is determined. The burden for analysis rises exponentially when entire human genomes are sequenced because of the enormous number of reads and the redundant nature of noncoding regions. Furthermore, comparing variant call files from cohorts or populations of individuals is also challenging, given the large amount of variation present in individual human genomes.
Clinical genetic testing is now available for many inherited cardiovascular disorders including cardiomyopathy, arrhythmias like the Long QT and Brugada syndromes, and vascular disorders such as Marfan syndrome and other aortopathies. In many cases, rare mutations in multiple distinct genes lead to similar phenotypes. These phenocopies have led to gene panel testing where 10–100 genes are sequenced at the same time 2. Most clinical genetic testing employs massively parallel next generation sequencing that may be accompanied by confirmation using traditional Sanger sequencing. Interpretation of variants relies on frequency in the population and predicted effect of the variant on protein function. Genetic testing laboratories interpret findings using a grading scale. Generally, variants are classified as pathogenic, likely pathogenic, variant of unknown significance, likely benign, or benign. For inherited cardiovascular disorders, the biggest determinant of pathogenicity is the overall frequency of the genetic variant found in the population. Since these are considered rare disorders, frequencies above 0.1–1% are considered unlikely to cause disease and will be labeled as benign or likely benign. Some clinical laboratories will also consider whether functional, biological evidence supports the interpretation for pathogenicity and will often include information on whether the variant has been previously reported. For cardiomyopathy genetic testing, gene panels are growing in size and scope. However, the sensitivity for detecting a mutation may still only be around 50%. The large size of gene panels, particularly like those used to genetically diagnose cardiomyopathies, argues for broad-based sequencing approaches like those discussed below.
Whole Exome sequence (WES) and Whole Genome Sequencing (WGS)
Because of the complexity associated with cataloging and correlating human genome variation with clinical outcomes, most analysis has focused on the coding region of the genome known as whole exome sequencing (WES). Approximately 1% of the genome specifies proteins, and this 1% has less repetitive sequence than the noncoding regions. Thus, identifying and sequencing the coding region is computationally and analytically less intense. To analyze the coding region requires isolating the coding exons from the DNA sample to be sequenced, and this step is usually achieved using a capture method, where the exons are arrayed and available for hybridization. The exon arrays used for capture rely on proper informatic identification of exons, which is dependent on knowing the major transcript, which may be tissue specific and not adequately represented in transcript databases. The major transcript expressed in the heart may not be well cataloged in public databases, leading to a minor transcript being represented on the exome array. In this case, the major cardiac transcript may in fact not be assessed using conventional exon arrays.
An alternative sequencing approach relies on analyzing the entire human genome of a given DNA sample. Whole genome sequencing (WGS) does not rely on exon capture, but instead samples the majority of the human genome. Highly repetitive DNA regions may not be well sequenced using short read sequencing. While WGS is attractive from the standpoint of inclusiveness, it is far more challenging to analyze because alignment and variant calling includes the noncoding region in addition to the coding region. The repetitive nature and content of the noncoding regions of the genome are inherently more difficult to align reliably from short read sequencing. Nonetheless, there is a growing appreciation for the importance of noncoding regions in controlled expression of genes and modulation of transcript levels (reviewed in 3). These noncoding regions are particularly important for the heart and cardiovascular system.
Databases of Human Genetic Variation
An equally important advance in understanding and interpreting human genome variation is the emergence of large databases of human genetic variation. The first such database, the 1000 Genomes (1KG) project, represented an international effort to establish an ethnically diverse sample of human genomes 4. The 1KG database includes low coverage human genomes, with some whole exome data and targeted sequencing augmenting the findings. To reliably call genetic variation, particularly heterozygous single nucleotide variants and small insertion/deletions, requires a depth of sequence. In WES, it is common to sample each sequenced base 50–100 times (100 fold coverage). With WGS, 30 to 40 fold coverage is usually preferred for reliable alignment and variant calling. The 1KG genomes, as an early effort, averaged only 5 fold coverage. A major advantage of 1KG is its broad ethnic diversity including genomes from North America, Europe, Asia and Africa. While highly incomplete in its inclusion of all ethnicities, it represented a first view at the broad diversity across and between human populations.
A second large collection of human genome variation was supported by the National Heart Lung and Blood Institute and produced WES from more than 6500 individuals (see below) 5. This National Institutes of Health supported effort included two major US populations including those of European descent and those of African descent, offering the opportunity to compare variation in these two groups. The most recent consortium effort to catalog human genetic variation aggregated exomes from multiple sources (Exome Aggregation Consortium or ExAC, http://exac.broadinstitute.org) and, like the other databases, can be freely queried. For all of these large public databases of human genome/exome sequence, there are little to no phenotype correlates.
In 2012, Tennessen et al. described rare variation in the Exome Sequencing Project (ESP) 5. Exomes were captured and sequenced from 2440 individuals, including 1351 of European descent and 1088 of African descent. In total 15,585 genes were sampled using this approach, at an average of 100 fold depth, and over 500,000 single nucleotide variants (SNVs) were identified. Notably, 86% of SNVs had a minor allele frequency less than 0.5%, indicating that most SNVs are rarely seen in the population (Figure 1A). On average, each exome had more than 13,000 SNVs, and 95.7% of the SNVs that were predicted to change protein function were considered rare. This degree of protein variation was surprisingly large, and this diversity was thought to arise from rapid population growth and little selection pressure. These data are derived from two very different populations with respect to population diversity; those of European descent (EU) had less variation than African descent (AA).
Figure 1. Rare and potentially deleterious variants identified in the ESP.
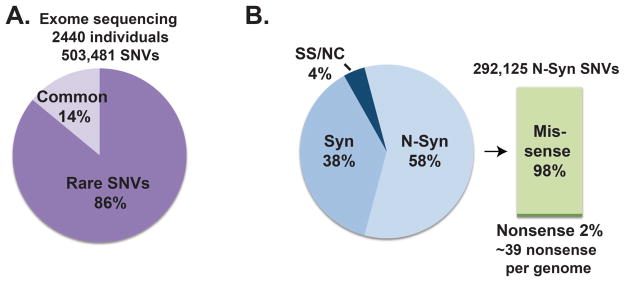
A). Exome sequence data from 2440 individuals identified 503,481 single nucleotide variants (SNVs) of which 432,993 (86%) had a minor allele frequency <0.5%, and were therefore considered rare. Therefore rare variation accounts for a larger percentage of genome variation. B.) 292,125 (58%) of the SNVs were nonsynonymous (N-Syn) changes including 285,960 missense and 6,165 nonsense variants. The remaining variants are synonymous (Syn) (38%) or splice site/noncoding (SS/NC) sequence (4%) 5.
Further analysis of ESP rare variants showed that 58% were nonsynonymous SNVs (Figure 1B). 57% of SNVs were observed in only one individual in the cohort. When evaluating variation across all genes sampled in the ESP, some genes displayed virtually no rare variation, consistent with very tight conservation across their coding regions. On the other hand, 110 genes had higher than expected variation. As anticipated, HLA-DQA2 had a high degree of rare variation. The AA population was observed to have more rare variation than the EU population. On average, each individual carried around 400 protein-altering variants that were predicted to be functional and ~39 nonsense variants. Overall, 318 individuals of the 2440 individual cohort were compound heterozygotes for nonsense variants in the same gene (13%). This percentage is on par with the presence of rare diseases in the population, which is estimated to be 10%. The sequenced population was drawn from 15 different research studies that included individuals with early myocardial infarction and stroke. Detailed clinical information was not available from individuals so it is not possible to link individual phenotypes to rare variants in these populations.
Fu et al. extended the ESP cohort by examining rare variation in >6500 genomes, composed of 4298 EU and 2217 AA individuals 6. The authors evaluated rare variants for predicted pathogenicity using several different algorithms that predict whether a given missense mutation is likely to be deleterious. These in silico protein prediction tools rely almost exclusively on conservation and may take into account structural domain information. Commonly used algorithms, and those used in evaluating rare variation in the ESP cohort, included Polyphen 2 (PP2), SIFT, Mutation Taster and GERP 7–10. The larger cohort examined by Fu and colleagues allowed better estimates of when variants arose in the population. Those variants shared between EU and AA cohorts are much older, with an average age of more than 100,000 years. Those variants found within the cohorts were much younger, EU average 5,400 years and AA average 15,300 years. This difference between old and new variation is explained by recent population growth, especially in the last 5,000 years. Variants that changed the protein including splice site, nonsense and missense changes, were 2–5 times younger than those variants that did not change the protein. When evaluating missense variants, those variants predicted to be deleterious were youngest. The authors suggest that this is consistent with new mutations arising in the population with little effect of selection pressure. Whether these data extrapolate to all populations is not known. These calculations assume a constant mutation rate over time and across populations. Genes with a higher than expected rate of deleterious variation after accelerated population growth differed between EA and AA individuals. AA individuals had 18 genes that showed excess deleterious variation while EA individuals had 3 genes that showed excess deleterious variation.
Fu et al. compared variation in genes linked to human diseases versus those not linked to human disease. The group of disease-associated genes included 829 genes linked to Mendelian diseases, 2663 genes linked to complex traits by GWAS, and another 1226 genes considered essential because based on mouse studies. The residual genes (n= 11711 genes) were considered non-disease associated. When comparing disease-associated genes to non-disease associated, the non-disease genes had a lower percentage of deleterious SNVs than disease-associated genes. However, this pattern was only observed in the EU population and not in the AA population. Fu et al. concluded that rare variation is likely to play an important role in heritability.
It has long been anticipated that common diseases like hypertension, myocardial infarction and heart failure would be caused by common genetic variation within the population. This common variant/common disease hypothesis formed the basis of many genomewide association studies (GWAS), and consistent with this, many genetic loci have been identified through GWAS studies for cardiovascular traits including hypertension, infarction, EKG intervals, heart failure and sudden death 11. The contribution of any given GWAS-identified locus to the phenotype in the population is relatively small. However, when aggregated, the sum of the contribution of loci for a given trait also fails to account for the known heritability. The missing heritability conundrum has lead geneticists to speculate that some traits may not be as heritable as predicted. Given the larger than expected degree of rare variation in the population, rare variation may contribute to the heritability of many common disorders 12, 13.
Cardiomyopathy genetics: Individuals and Populations
Both dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM) are enriched in families. The primary mode of inheritance is autosomal dominant pattern with variable expressivity and penetrance 14. HCM is mainly linked to mutations in genes encoding sarcomere proteins, with two genes, MYH7 and MYBPC3, accounting for more than half of the known HCM-linked variants 15. If linkage or GWAS were conducted in an HCM population, signals would likely be observed at the MYH7 and MYBPC3 loci. Yet, it is individual and rare variation at these loci that accounts for the disease, rather than common variation.
For DCM, more than 50 different genes have been linked to DCM, and with current genetic testing, even large panels of more than 50 genes have only ~50% sensitivity to detect mutations 16–18. Despite this genetic heterogeneity, one gene has emerged as highly important for the genesis of cardiomyopathy. In 2012, Herman et al. demonstrated the truncating variants in TTN, the gene encoding the giant protein titin, are responsible for approximately 20% of DCM 19. In order to conduct this analysis, Herman and colleagues generated a TTN-specific capture array to capture all 363 exons in the TTN gene. The titin protein spans the length of half the sarcomere with a molecular mass of 3800 kDa; its carboxy-terminus resides in the M band while the amino-terminus is anchored in the Z disc 20. The TTN gene undergoes alternative splicing to produce several different forms of titin protein (Figure 2). The titin form referred to by UniProtKB entry Q8WZ42-1 (isoform 1; April 18, 2012; Version 4) has 34,350 amino acids. The N2B and N2BA isoforms of titin are enriched in the heart while the N2A form is mainly in skeletal muscle. There are other rare forms produced from the TTN gene, but these are though to be less abundant (Novex 1, 2, and 3). The Z band region of titin contains 9 immunoglobulin (Ig) domains, and the I band region contains additional Ig domains and the PEVK repeat units, so named for repeating amino acids. Herman et al. reported that many of the TTN truncating variants fell within the A band domain of titin 19.
Figure 2. TTN transcript variability in heart.

A.) Human titin (blue) spans half the sarcomere with the amino-terminus anchored at the Z disk and the carboxy-terminus located at the M line. B.) TTN produces a number of transcripts, including the cardiac specific N2BA and N2B that lack portions of the I-band region of titin. C.) Exon usage identified by Roberts et al. in LV from DCM hearts. Red areas indicate that all exons are transcribed in hearts, while lighter colors indicate less exon usage 22.
Norton and colleagues evaluated the contribution of TTN truncating variation in a cohort of 17 families with DCM 21. Using WES, 7 of 17 DCM families with DCM had TTN truncating variants. They also conducted a genomewide scan that provided statistical support for the TTN locus as a major contributing locus for DCM. Consistent with the idea that rare variation plays a major role in DCM, the individuals in this cohort had many different TTN truncating mutations. This analysis suggested a higher percentage of familial DCM attributed to TTN (7/17 or 41%). However, this sample was biased by the fact that these families were selected for inclusion in the study because conventional genetic testing did not show mutations in 16 genes previously associated with cardiomyopathy, and therefore this population was enriched for TTN mutations.
The TTN gene illustrates a limitation of using WES for analyzing cardiac genes. The TTN gene has 363 exons, and the splice forms that contribute to cardiac titin differ from that expressed in skeletal muscle. In order to fully assess TTN, a custom capture array was generated to access and evaluate all 360 exons of TTN 19. In contrast, the standard exome capture arrays used by Norton and colleagues captured only 315 exons of TTN and may miss exons expressed in the cardiac isoform of TTN 21. The design of exon capture arrays utilizes database information to identify the transcripts. Transcript selection and exon inclusion for any given gene may not be accurate for cardiac isoforms, since transcripts are often tissue specific, a point critically important for the heart. Importantly, transcripts may differ in disease states and exons used in failing hearts may differ from those in nonfailing hearts.
Roberts et al. compared TTN exons between failed and nonfailed hearts and found that exon usage for TTN was comparable, indicating that splicing did not differ markedly 22. However, it has been shown that the N2BA form of titin is more highly expressed in failing hearts 23. The RNA-seq approach revealed a great deal of alternative splicing in TTN 22 (Figure 2). Using this as a backdrop for interpreting the location of TTN truncating variants, it was confirmed that DCM-associated TTN truncating variants fall preferentially within the A band region of TTN. In addition, the two major forms of TTN found in the heart N2B and N2BA were enriched for TTN truncating variants in DCM. In all these analyses, TTN truncating variations were identified as heterozygous.
One of the truly challenging areas is evaluating the degree of missense variation in TTN. In 5400 ESP cohort, the average number of TTN missense per individual was 23 (range 6 to 55) 21. One approach to determining what missense SNPs may be pathogenic is to filter variants based on frequency and to apply in silico tools. To estimate which TTN missense variants may be pathogenic in the cohort studied by Norton, they filtered based on frequency (<0.5%) and then applied protein prediction algorithms (either a PhastCons score >0.4 or a GERP score of >2). This reduced the number of TTN missense variants to 1.91 per individual in the DCM cohort. Whether TTN missense variants contribute to disease has been in debate. In some cases, the missense variants have been reported to segregate with disease in large families, providing statistical evidence to support causality. However, the observation that nearly all individuals have at least one missense TTN variant makes this conclusion difficult. More study is needed since TTN may serve as a modifier of outcome in cardiomyopathy.
Cardiomyopathy gene variants found in the general population
Databases of human genome variation are now available to estimate the genetic risk for cardiovascular disease, but this work is limited by the absence of linked clinical data for some of these cohorts. For example, Golbus et al. evaluated rare variation in the 1000 Genomes database and specifically queried rare variation associated with cardiomyopathy 24. Those variants previously reported as pathogenic and those predicted to be pathogenic using PP2 and SIFT were described. Twenty-one different rare missense variants were found in MYH7, including 7 that were previously reported as pathogenic. Twenty-three different missense variants were observed in MYBPC3 including 11 that were previously reported as pathogenic. Seventeen different TTN disruptive mutations were found in the cohort, including 6 that were in-frame variants. In some cases, the number of reported individuals indicated that these are not likely mutations as the frequency was too high. However, what is notable is that the frequency of reported pathogenic and predicted pathogenic variation far outstrips the presence of DCM and HCM in the population as a whole. This data suggests that 3–5% of the population may carry variants that could associate with cardiomyopathy.
Pan et al. examined variation in cardiomyopathy genes in the ESP cohort of exome sequences 25. In total 164 variants were seen in 5379 ESP in 8 cardiomyopathy genes ACTC1, TNNT2, TNNI3, MYL2, MYL3, TPM1, MYH7, and MYBPC3. Of these, 159 private missense variants were found (meaning found in only one individual in the cohort and not previously reported in other databases). Three private splice site and 2 private nonsense variants were found. In total, this could be interpreted to suggest that as much as 3% of the population carries variants that could place individuals at risk for cardiomyopathy. This is higher than anticipated based on the known population prevalence of HCM and DCM. The authors noted that nonsense mutations were far less likely to occur in cardiomyopathy genes compared to other Mendelian disease genes. Andreasen et al. conducted a similar analysis and concluded 1000X higher prevalence of mutations associated with cardiomyopathy than the population incidence of cardiomyopathy. Whether these are risk alleles or benign variants requires further study 26.
Long QT syndrome is linked to 15 genes, including many that encode ion channels important for normal cardiac action potential. Refsgaard et al. evaluated genetic variation in the 5400 individuals of the ESP specifically querying variants in LQTS genes including KCNQ1, KCNH2, SCN5A, ANK2, KcNE1, KCNE2, KCNJ2, CACNA1C, CAV3, SCN4B, AKAP9, SNTA1, and KCNJ5 27. In total, 33 variants were identified in 173 alleles. No rare variants were found in ANK2, KCNJ2, CACNA1C, SCN4B, or KCNJ5. Sixteen variants were found in SCN5A. This degree of variation is much higher than would be anticipated based on the known prevalence of LQTS in the population. Whether these alleles confer any increased risk is not known and will require tighter links between genotype and phenotype through careful clinical examination and evaluation of family members with both genetic and clinical assessment.
Ng et al. examined 870 exomes from individuals not known to have arrhythmia, cardiomyopathy or sudden death 28. The exome data focused on 41 cardiomyopathy genes and 22 arrhythmia-associated genes. From this cohort, 1367 variants in the cardiomyopathy genes were identified, and 360 variants were found in the arrhythmia-associated genes. These variants were further stratified based on frequency and predicted effect with focus on those most likely to be damaging or previously reported as mutations in the Human Gene Mutation Database (HGMD). Six subjects were identified as having disease-causing mutations. The authors extended their findings by clinically examining the individuals linked to these mutations, and found 1 individual with DCM, 2 with HCM, 1 with LVNC and 2 with LQTS. The clinical link between genetic mutation and the presence of disease indicates that genetic screening can be used to identify those at risk for disease. Notably, these findings indicated a higher than expected frequency of DCM and HCM (estimated ~1:500) and LQTS (1:3000) than what was expected based on population-based estimates.
A recent report demonstrated that rare variation in two genes, APOA5 and LDLR, was associated with risk for myocardial infarction 29. LDLR is linked to familial hypercholesterolemia, and the finding that rare variation across the population accounts for risk for MI validates that Mendelian genes can play an important role in population based genetic assessment of cardiovascular risk. This study was conducted by examining the genomes of 1088 individuals with early MI (<50 year of age for males, <60 for females) and comparing findings to 978 individuals who did not have early MI. A number of distinct rare variants were found that created risk alleles for adult onset common disease.
Actionable variants from WES/WGS
The explosion of WES/WGS sequencing had led to the concern for incidental findings. Incidental findings are those variants found to predict disease unrelated to the original clinical query. The problem of incidental findings led the American College of Medical Genetics to identify a list of 56 genes from which results are considered actionable 30. Nearly half the genes in this list of 56 genes are those that cause inherited cardiovascular disease, and of these many are linked to sudden death risk. The cardiovascular genes on this list include those that cause DCM, HCM, arrhythmogenic right ventricular cardiomyopathy, LQTS and Marfan Syndrome as well as genes linked to other aortopathies. The remaining non-cardiovascular genes on the list are those related to cancer syndromes. These disorders are considered actionable since they would change medical screening and management. Not all have embraced the practice of returning results on “actionable” genes since many of the variants may be of uncertain significance.
Dorschner et al. examined actionable variants in 1000 randomly selected individuals from the ESP cohort with focus on 114 genes that included the 56 ACMG actionable genes 31. 3.4% of the EU population and 1.2% of the AA population were found to have variants in the 114 genes. The decision whether to consider a variant actionable relied heavily on whether the variant had previously been reported as pathogenic and specifically its presence in HGMD. 239 variants were reported in HGMD, and of these 72 were too highly represented in ESP to be considered mutations. 123 variants were seen only once but many exceeded the known population frequency of disease. Because this yielded a high number of potentially actionable variants, more stringent criteria for considering a mutation were used. It was required that the variant be reported in three unrelated individuals with disease, and/or segregate with disease in a large family. By applying these more stringent criteria, only 17 actionable variants were identified. Amendola et al. extended this same approach to 6500 exomes in ESP using 112 genes. They identified 0.7% of the EU population as having a pathogenic mutation with 0.1% having disruptive mutations 32. An additional 1.2% was found to have likely pathogenic mutations. With the increasing size of population databases, reclassification of variants may occur 33.
A caveat of this approach was the reliance exclusively on genetic data, literature and HGMD reports in the absence of clinical findings. In general, interpreting genetic variation in the absence of clinical data is challenging, and does not reflect clinical practice. In clinical practice, the identification of a potential mutation linked to cardiomyopathy or LQTS, for example, would likely trigger examination using cardiac imaging and testing. Furthermore for cardiovascular genetics, correlating genotype and phenotype requires a detailed knowledge of both genetics and cardiovascular testing. Thus a close working relationship between the cardiologist and geneticist is required. Alternatively, “cardiogenetics” may be an emerging field requiring training in both cardiology and genetics in order to best interpret variation in the context of clinical findings. In clinical practice, genetic testing may produce variants of unknown significance, and the management of this genetic information in the context of families and individuals requires clinical correlation. In population-based research studies, it may be reasonable to dismiss variants of unknown significance. But in the clinical setting, variants of unknown significance require further testing. Evaluating additional family members with both genetic testing and careful clinical testing may help establish a link between genotype and phenotype.
Conclusions
The data above highlight the complexity in interpreting rare variation as being causal for cardiomyopathy. Not all of these variants cause cardiomyopathy, and for those that may increase risk for cardiomyopathy, their likely effect is variable. Mutations exert their effect based on effect on protein structure, and in silico tools are not sufficiently reliable for this prediction. However, mutations also exert their effect in the context of other genetic variation as well as environmental effects. Given that risk for heart failure and arrhythmias can be mitigated through medical and occasionally device management, this calls for better interpretation of variants in the context of clinical phenotype by those knowledgeable about the given phenotype including more subtle findings. Where possible, robust clinical phenotyping should be carried out and extended to family members to help establish a link between genotype and phenotype.
The increased knowledge of genetic mutations for cardiomyopathy is expected to lead to better classification that may inform the clinician and researcher about progression rates and complications. For example, LMNA gene mutations, the second most common genetic cause of inherited DCM, are associated with a high risk for arrhythmias. A multicenter cohort of 269 subjects with LMNA mutations identified that those with frameshifting mutations and reduced left ventricular function were more prone to ventricular tachycardia 34. This information can be used to better stratify who may benefit most from device implantation.
Genetic classification of cardiomyopathies may also lead to better treatment. High throughput screens have been conducted to identify compounds that target sarcomeric myosin as treatments for heart failure 35. The identification of PCSK9 mutations in hyperlipidemia has led to compounds that target this pathway 36. Moreover, genetic correction strategies are being pursued using anti-sense oligonucleotides to reduce or correct the mutant alleles. The capacity to generate patient-specific induced pluripotent stem cells offers a ready model in which to test therapies for molecular correction. The capacity for gene specific corrections with precision using CRISPR/Cas 9 gene editing heralds a new era in personalized medicine and hope for correcting genetic diseases.
References
- 1.Punetha J, Hoffman EP. Short read (next-generation) sequencing: a tutorial with cardiomyopathy diagnostics as an exemplar. Circ Cardiovasc Genet. 2013;6:427–434. doi: 10.1161/CIRCGENETICS.113.000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Puckelwartz MJ, McNally EM. Genetic profiling for risk reduction in human cardiovascular disease. Genes (Basel) 2014;5:214–234. doi: 10.3390/genes5010214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pai AA, Pritchard JK, Gilad Y. The genetic and mechanistic basis for variation in gene regulation. [accessed May 15, 2015];PLoS Genet. 2015 11:e1004857. doi: 10.1371/journal.pgen.1004857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Genomes Project C. Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu W, O’Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 9.Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 10.Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ [accessed May 15, 2015];PLoS Comput Biol. 2010 6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith JG, Newton-Cheh C. Genome-wide association studies of late-onset cardiovascular disease. J Mol Cell Cardiol. 2015 doi: 10.1016/j.yjmcc.2015.04.004. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 13.Moutsianas L, Morris AP. Methodology for the analysis of rare genetic variation in genome-wide association and re-sequencing studies of complex human traits. Brief Funct Genomics. 2014;13:362–370. doi: 10.1093/bfgp/elu012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123:19–26. doi: 10.1172/JCI62862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho CY. Genetic considerations in hypertrophic cardiomyopathy. Prog Cardiovasc Dis. 2012;54:456–460. doi: 10.1016/j.pcad.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ho CY, Charron P, Richard P, Girolami F, Van Spaendonck-Zwarts KY, Pinto Y. Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc Res. 2015;105:397–408. doi: 10.1093/cvr/cvv025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mestroni L, Brun F, Spezzacatene A, Sinagra G, Taylor MR. Genetic Causes of Dilated Cardiomyopathy. Prog Pediatr Cardiol. 2014;37:13–18. doi: 10.1016/j.ppedcard.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547. doi: 10.1038/nrcardio.2013.105. [DOI] [PubMed] [Google Scholar]
- 19.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.LeWinter MM, Granzier HL. Titin is a major human disease gene. Circulation. 2013;127:938–944. doi: 10.1161/CIRCULATIONAHA.112.139717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Norton N, Li D, Rampersaud E, Morales A, Martin ER, Zuchner S, et al. Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ Cardiovasc Genet. 2013;6:144–153. doi: 10.1161/CIRCGENETICS.111.000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7:270ra276. doi: 10.1126/scitranslmed.3010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, et al. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation. 2004;110:155–162. doi: 10.1161/01.CIR.0000135591.37759.AF. [DOI] [PubMed] [Google Scholar]
- 24.Golbus JR, Puckelwartz MJ, Fahrenbach JP, Dellefave-Castillo LM, Wolfgeher D, McNally EM. Population-based variation in cardiomyopathy genes. Circ Cardiovasc Genet. 2012;5:391–399. doi: 10.1161/CIRCGENETICS.112.962928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan S, Caleshu CA, Dunn KE, Foti MJ, Moran MK, Soyinka O, et al. Cardiac structural and sarcomere genes associated with cardiomyopathy exhibit marked intolerance of genetic variation. Circ Cardiovasc Genet. 2012;5:602–610. doi: 10.1161/CIRCGENETICS.112.963421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andreasen C, Refsgaard L, Nielsen JB, Sajadieh A, Winkel BG, Tfelt-Hansen J, et al. Mutations in genes encoding cardiac ion channels previously associated with sudden infant death syndrome (SIDS) are present with high frequency in new exome data. Can J Cardiol. 2013;29:1104–1109. doi: 10.1016/j.cjca.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 27.Refsgaard L, Holst AG, Sadjadieh G, Haunso S, Nielsen JB, Olesen MS. High prevalence of genetic variants previously associated with LQT syndrome in new exome data. Eur J Hum Genet. 2012;20:905–908. doi: 10.1038/ejhg.2012.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng D, Johnston JJ, Teer JK, Singh LN, Peller LC, Wynter JS, et al. Interpreting secondary cardiac disease variants in an exome cohort. Circ Cardiovasc Genet. 2013;6:337–346. doi: 10.1161/CIRCGENETICS.113.000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Do R, Stitziel NO, Won HH, Jorgensen AB, Duga S, Angelica Merlini P, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518:102–106. doi: 10.1038/nature13917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, Gallego CJ, et al. Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am J Hum Genet. 2013;93:631–640. doi: 10.1016/j.ajhg.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amendola LM, Dorschner MO, Robertson PD, Salama JS, Hart R, Shirts BH, et al. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res. 2015;25:305–315. doi: 10.1101/gr.183483.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Das KJ, Ingles J, Bagnall RD, Semsarian C. Determining pathogenicity of genetic variants in hypertrophic cardiomyopathy: importance of periodic reassessment. Genet Med. 2014;16:286–293. doi: 10.1038/gim.2013.138. [DOI] [PubMed] [Google Scholar]
- 34.van Rijsingen IA, Nannenberg EA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur J Heart Fail. 2013;15:376–384. doi: 10.1093/eurjhf/hfs191. [DOI] [PubMed] [Google Scholar]
- 35.Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, et al. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. 2011;331:1439–1443. doi: 10.1126/science.1200113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Desai NR, Sabatine MS. PCSK9 inhibition in patients with hypercholesterolemia. Trends Cardiovasc Med. 2015 doi: 10.1016/j.tcm.2015.01.009. in press. [DOI] [PubMed] [Google Scholar]