

Abstract
Neuroinflammation occurs in AD. While AD genetic studies implicate inflammation-relevant genes and fibrillar amyloid β protein promotes inflammation, our understanding of AD neuroinflammation nevertheless remains incomplete. In this study we hypothesized damage-associated molecular pattern (DAMP) molecules arising from mitochondria, intracellular organelles that resemble bacteria, could contribute to AD neuroinflammation. To preliminarily test this possibility, we exposed neuronal and microglial cell lines to enriched mitochondrial lysates. BV2 microglial cells treated with mitochondrial lysates showed decreased TREM2 mRNA, increased TNFα mRNA, increased MMP-8 mRNA, increased IL-8 mRNA, redistribution of NFκB to the nucleus, and increased p38 MAPK phosphorylation. SH-SY5Y neuronal cells treated with mitochondrial lysates showed increased TNFα mRNA, increased NFκB protein, decreased IκBα protein, increased AβPP mRNA, and increased AβPP protein. Enriched mitochondrial lysates from SH-SY5Y cells lacking detectable mitochondrial DNA (ρ0 cells) failed to induce any of these changes, while mtDNA obtained directly from mitochondria (but not PCR-amplified mtDNA) increased BV2 cell TNFα mRNA. These results indicate at least one mitochondrial-derived DAMP molecule, mtDNA, can induce inflammatory changes in microglial and neuronal cell lines. Our data are consistent with the hypothesis that a mitochondrial-derived DAMP molecule or molecules could contribute to AD neuroinflammation.
Keywords: Alzheimer’s disease, amyloid β precursor protein, inflammation, mitochondria, mtDNA, TREM2
INTRODUCTION
Inflammation is observed in brain aging and Alzheimer’s disease (AD) [1-4]. Differences in peripheral cytokine levels may distinguish AD and control cohorts [3, 5], and recent genetic studies reveal inflammation-relevant genes influence AD risk [3]. Despite this, the causes and consequences of AD inflammatory changes remain incompletely understood.
Neuroinflammation in AD currently does not appear to reflect the presence of an externally acquired pathogen [6]. This implies an internally produced damage-associated molecular pattern (DAMP) molecule or molecules may contribute to AD neuroinflammation [7, 8]. Amyloid β protein (Aβ), which aggregates in AD, induces inflammation [9] but this does not exclude the possibility that other intrinsically generated molecules might also contribute.
To this point, mitochondrial dysfunction exists in AD brains [10] and in general mitochondrial dysfunction and inflammation changes co-localize [11, 12]. Mitochondria evolved from a symbiotic relationship with a prokaryote organism, and thus share characteristics with bacteria. Several molecules normally sequestered by mitochondria can induce inflammation, including mitochondrial DNA (mtDNA), mitochondrial transcription factor A (TFAM), cardiolipin, cytochrome c, formyl-peptides, high mobility group B protein 1 (HMGB1), and ATP [8, 13-23].
Mitochondrial-derived DAMPs activate inflammation in non-brain tissues. For example, mtDNA, when not sequestered within mitochondria, induces arthritis and drives inflammatory changes in heart failure models [24, 25]. To date, though, no one has specifically considered the potential contribution of mitochondria, through the release of mitochondrial-derived DAMPs, to chronic brain inflammation. To preliminarily assess the viability of this idea, we exposed microglial and neuronal cell lines to enriched mitochondrial lysates. This approach allowed us to explore ways in which mitochondrial-derived DAMP molecules may affect different central nervous system cell types. Our data suggest an mtDNA-containing DAMP component could indeed contribute to AD neuroinflammation, and also contribute to other AD-relevant molecular changes.
MATERIALS AND METHODS
Cell culture
Human neuronal SH-SY5Y cells and mouse microglial BV2 cells were cultured at 5% CO2 in high glucose DMEM supplemented with 10% fetal bovine serum (FBS) and 1% of a penicillin-streptomycin stock (catalogue number 30-001-CI, Fisher Scientific). mtDNA-depleted SH-SY5Y cells (ρ0 cells) were cultured at 5% CO2 in high glucose DMEM supplemented with 10% FBS, 1% penicillin-streptomycin stock, 100 μg/mL sodium pyruvate, and 50 μg/mL uridine. These ρ0 cells were generated as previously described [26]. ρ0 cell status was confirmed via PCR, and by demonstrating these cells cannot survive in the absence of uridine and pyruvate supplementation [27].
Mitochondrial lysates
Enriched mitochondrial lysates were generated from SH-SY5Y, SH-SY5Y ρ0, or BV2 cells by first suspending approximately 30 million cells from each line in ice-cold MSHE buffer (225 mM mannitol, 75 mM sucrose, 5 mM HEPES, 1 mM EGTA, pH 7.4). The suspended cells were disrupted via nitrogen cavitation, on ice and over 15 minutes, at a constant 900 PSI chamber pressure. Enriched mitochondrial pellets were subsequently generated by centrifugation (1000 g for 5 min at 4°C, followed by two 10 min spins at 20,000 g and 4°C). Mitochondria were re-suspended in sterile phosphate buffered saline (PBS), ruptured by three rapid freeze/thaw cycles, and protein concentrations were determined using a BCA Protein Assay kit (Bio-Rad). All procedures were completed using aseptic technique.
Isolation and PCR amplification of mtDNA
Mitochondrial-enriched pellets (prepared as described above, only not exposed to the freeze/thaw steps) from BV2 or SH-SY5Y cells were suspended in 600 μl of lysis buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 0.1% SDS) and disrupted using 10 strokes of a Dounce homogenizer (clearance of 0.005-0.0025 inches). The homogenized mitochondria were transferred to microcentrifuge tubes, 60 μl of proteinase K (Qiagen) was added, and the tubes were vortexed and then incubated at 55°C for 2 h. During this 2 h incubation period we continued to vortex the samples every 30 min. Samples were next centrifuged at 8,000 g for 15 min, and the supernatants were transferred to new tubes. 600 μl of phenol:chloroform:isoamyl alcohol (Sigma) were added to the tubes, which were vortexed and centrifuged at 8,000 g for 15 min. Supernatants were transferred to new tubes, 500 μl of chloroform (Sigma) was added, and samples were vortexed and centrifuged at 8,000 g for 15 min. The supernatants were transferred to new tubes, and 40 μl of 3 M sodium acetate and 440 μl of 100% isopropanol were added. Samples were then incubated at −20°C overnight. The following day samples were centrifuged at 8,000 g for 15 min. The supernatants were discarded, and the pellets were washed with 750 μl of 70% ethanol and then centrifuged at 8,000 g for 8 min. The supernatants were discarded and the pellets were washed with 500 μl of 70% ethanol and centrifuged at 8,000 g for 8 min. After discarding the supernatants the pellets were gently air dried and re-suspended in 100 μl of nuclease-free water.
mtDNA was PCR-amplified using a Roche Expand Long Range dNTP pack. Primers used for human mtDNA amplification were previously published [28]. Mouse mtDNA primers were as follows: GCACTGAAAATGCTTAGATGG (forward), AGAGTTTTGGTTCACGGAAC (reverse). PCR amplification was performed using an Applied Biosystems 7300 PCR System. An initial denaturation step at 92°C for 2 min was followed by 10 cycles of 92°C for 15 sec (denaturation) followed by a 12 min 40 sec elongation time at 60°C. For cycles 11-30, for each successive individual cycle 20 sec was added to the elongation time from the previous cycle. An aliquot of the PCR mixture was resolved on a 1% Agarose gel (Bio-Rad) in TBE Buffer (Bio-Rad). PCR products were purified using a Qiagen PCR purification kit (Qiagen).
Treatment of cells with mitochondrial lysates and mtDNA
SH-SY5Y or BV2 cells were plated at approximately 80% confluence in 6-well dishes. The following day cells were treated with 50 μg of post-freeze/thaw mitochondrial lysate, 1 or 2 μg of purified mtDNA, 1 or 2 μg of PCR-amplified mtDNA, or vehicle (PBS) for approximately 24 h.
Immunoblotting
After the designated treatment period cells were lysed using MPER (Thermo-Pierce), or else nuclear and cytosolic fractions were generated using NE-PER (Thermo-Pierce) and differential centrifugation. Protein concentrations were determined using a BCA Protein Assay kit. Equal amounts of protein were resolved by SDS-PAGE using 4-20% Criterion TGX Tris-glycine polyacrylamide gels (Bio-Rad) and gel proteins were transferred to nitrocellulose membranes (Invitrogen). Membranes were blocked for 1 h at room temperature while rocking, and incubated in primary antibody at the indicated dilutions (see below) in blocking buffer overnight at 4°C while rocking. Membranes were washed 3 times with PBS-Tween 20 and placed in secondary antibody at 1:2000 (in blocking buffer) for 1 h at room temperature. Membranes were washed 3 times with PBS-Tween 20 and incubated with Supersignal West Femto chemiluminesence reagent (Thermo-Pierce). Images were captured using a Chemidoc imaging station (Bio-Rad). Primary antibodies used were to nuclear factor kappa B (NFκB) (Santa Cruz, 1:200), p38 mitogen activated protein kinase (p38 MAPK) (1:1000, Cell Signaling Technology), phospho-Thr180/Tyr182 p38 MAPK (1:2000, Cell Signaling Technology), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cell Signaling, 1:2000), actin (Cell Signaling, 1:1000), nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha (IκBα) (Santa Cruz, 1:200), amyloid precursor protein (AβPP) (Cell Signaling, 1:1000), cytochrome c oxidase subunit IV isoform 1 (COX4I1) (Cell Signaling, 1:1000), TATA binding protein (TATA BP) (Abcam, 1:500), Histone deacetylase 1 (HDAC1) (Cell Signaling; 1:1000).
Immunocytochemistry
After receiving a designated treatment, cells were fixed in 4% paraformaldehyde (in PBS, pH 7.4) for 1 h at room temperature. Cells were then incubated in blocking buffer (5% BSA, 0.2% Triton X-100 in PBS, pH 7.4) for 1 h at room temperature. Incubation at 4°C with a primary antibody to NFκB (Cell Signaling Technologies; 1:400) took place overnight in blocking buffer containing 10 μg/mL Hoechst 33258 (Invitrogen). Cells were washed 3 times with PBS followed by incubation with AlexaFlour 488 Donkey anti-Rabbit secondary antibody (Abcam 1:500) for 1 h at room temperature. Cells were washed 3 times in PBS, and placed in anti-fade reagent (Sigma). Images were captured using an Olympus IX71 fluorescent microscope. For each independent sample a minimum of 150 cells were counted.
Aβ enzyme-linked immunoabsorption assay (ELISA)
Conditioned media from cells exposed to different treatments was collected and 1 mM 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF) (Fisher), a serine protease inhibitor, was added. Media samples were analyzed using an Aβ1-40 ELISA kit (Invitrogen) at a dilution of 1:5 according to the manufacturer’s instructions.
Quantitative PCR
RNA was isolated using Tri Reagent (Ambion). Reverse transcription was performed on 1 μg of total RNA using an iScript RT qPCR master mix (BioRad). Amplifications were performed using an Applied Biosystems 7300 PCR System. Quantitative real-time PCR (qPCR) was performed using an iTaq universal probes supermix (Bio-Rad) and TaqMan Gene Expression Assays (Applied Biosystems) to quantify mRNA levels for tumor necrosis factor α (TNFα), matrix metalloproteinase-8 (MMP-8), interleukin-8 (IL-8), triggering receptor expressed on myeloid cells-2 (TREM2), C-C motif chemokine 11 (CCL11), and AβPP. GAPDH mRNA levels were used to normalize the expression levels of these genes.
We used Taqman primers to the nuclear 18S rRNA gene to determine the respective cycle threshold values for nuclear DNA derived from whole cells and from mitochondrial fractions. In qPCR, a 3.3 difference in cycle threshold values approximately reflects a 10-fold change in the amount of amplifiable template. The cycle threshold values obtained from whole cell DNA and enriched mitochondrial fraction DNA were further used to calculate the percentage of nuclear DNA that was eliminated during the preparation of the enriched mitochondrial fractions.
Statistics
Data were summarized by means and standard errors. To compare means between three groups we used one-way analysis of variance (ANOVA) followed by Fisher’s least significant difference (LSD) post-hoc testing. p-values less than 0.05 were considered statistically significant.
RESULTS
Mitochondrial lysate characterization
The mitochondrial lysates used for these experiments were assessed for mitochondrial purity and enrichment (Fig. 1). Relative to what was observed in SH-SY5Y and BV2 whole cell lysates, SH-SY5Y and BV2 mitochondrial lysates showed a robust reduction in GAPDH, a primarily non-mitochondrial cytosolic protein, and TATA BP, a nuclear protein. For DNA analyzed from SH-SY5Y whole cells and SH-SY5Y mitochondrial lysates, subtracting the whole cell cycle threshold from the mitochondrial lysate cycle threshold yielded a cycle threshold difference of 2.1. This indicates a 6.4-fold reduction in nuclear DNA occurred during the mitochondrial enrichment process, or that 84.4%, of the nuclear DNA was eliminated. For DNA prepared from BV2 whole cells and BV2 mitochondrial lysates, subtracting the whole cell cycle threshold from the mitochondrial lysate cycle threshold yielded a cycle threshold difference of 2.5. This indicates a 7.6-fold reduction in nuclear DNA occurred during the mitochondrial enrichment process, or that 86.8% of the nuclear DNA was eliminated. For DNA prepared from SH-SY5Y ρ0 whole cells and ρ0 mitochondrial lysates, subtracting the whole cell cycle threshold from the mitochondrial lysate cycle threshold yielded a cycle threshold difference of 2.7. This indicates an 8.2-fold reduction in nuclear DNA occurred during the mitochondrial enrichment process, or that 87.8% of the nuclear DNA was eliminated.
Fig. 1.
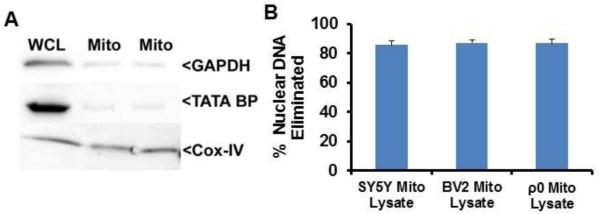
Characterization of mitochondrial lysates. (A) 50 μg of whole cell lysate (WCL) or mitochondrial lysate were resolved by electrophoresis and transferred to a nitrocellulose membrane. Representative immunochemistry data for COX-4I1 (mitochondrial marker), TATA BP (nuclear marker), and GAPDH (cytosolic marker) protein are shown. (B) The cycle threshold difference between DNA from whole cells and DNA from enriched mitochondria was used to calculate the amount of nuclear DNA that was eliminated during the mitochondrial enrichment process.
Effect of mitochondrial lysates on BV2 cell inflammatory gene expression
We measured levels of DAMP response-associated genes in BV2 mouse microglial cells treated with mitochondrial lysates from either BV2 cells (containing mtDNA) or SH-SY5Y human ρ0 cells lacking mtDNA. We used this strategy because a BV2 ρ0 cell line does not currently exist, and because BV2 mitochondrial lysates should overall appear less foreign to BV2 cells than SH-SY5Y mitochondrial lysates. Specific changes observed in BV2 cells treated with BV2-derived mitochondrial lysates but not SH-SY5Y ρ0 mitochondrial lysates, therefore, could indicate mtDNA, an mtDNA-containing complex, mtRNA, or mtDNA-encoded proteins act as DAMP molecules.
TNFα mRNA levels showed a 4.3 fold increase following treatment with mitochondrial lysates containing mtDNA. TNFα mRNA did not significantly increase when mitochondrial lysates from mtDNA-depleted cells were used (Fig. 2A). mRNA levels of TREM2, a gene whose protein product suppresses the production of cytokines and pushes immune cells towards a more phagocytic phenotype, decreased by 48% in cells treated with mitochondrial lysates that contained mtDNA. No change in TREM2 mRNA was observed in cells treated with mitochondrial lysates prepared from mtDNA-depleted cells (Fig 2B).
Fig. 2.

Effect of mitochondrial lysates on microglial cell inflammatory gene expression. BV2 cells were treated overnight with either PBS vehicle, 50 μg of mitochondrial lysate from BV2 cells, or 50 μg of mitochondrial lysate from SH-SY5Y ρ0 cells. Nine independent samples were analysed for each condition. (A) TNFα mRNA level. (B) TREM2 mRNA level. (C) IL-8 mRNA level. (D) MMP-8 mRNA level. Values shown are relative group means ± SEM, with the control group set to 1. *indicates p<0.05; ** indicates p<0.005.
Prior studies have implicated IL-8 and MMP-8 in mitochondria-induced DAMP responses [13]. We observed an approximate 7.6 fold increase in IL-8 mRNA and an approximate 7.0 fold increase in MMP-8 mRNA in cells treated with mitochondrial lysates from BV2 cells (Fig. 2C,D). IL-8 and MMP-8 mRNA levels were unchanged in cells treated with SH-SY5Y ρ0 mitochondrial lysates (Fig. 2C,D). When BV2 cells were treated with mitochondrial lysates from standard SH-SY5Y cells (which contain mtDNA), the results were similar to those obtained when BV2 cells were treated with mitochondrial lystates from BV2 cells (data not shown). Therefore, mtDNA overall appeared to play either a direct or indirect role in generating these responses.
Effect of mitochondrial lysates on BV2 cell NFκB expression and compartmentalization
The NFκB transcription factor helps regulate TNFα production and other inflammation parameters [29]. We therefore examined total cell protein levels of NFκB and its repressor, IκBα, and quantified nuclear and cytoplasmic NFκB protein levels. Cells treated with BV2 cell mitochondrial lysates displayed a 33% increase in total NFκB protein (Fig. 3A,C). Total NFκB protein levels did not change in cells treated with mitochondrial lysates prepared from SH-SY5Y ρ0 cells that lacked detectable mtDNA (Fig. 3A,C). IκBα, which sequesters NFκB in the cytoplasm and prevents NFκB-mediated transcription did not change, although a non-significant trend towards a lower total level was perhaps observed in cells exposed to BV2 cell mitochondrial lysates (p=0.085) (Fig. 3B,C). Despite this, the NFκB nucleus:cytoplasm ratio increased by 220% in cells treated with BV2 mitochondrial lysates (Fig 3D-G). The NFκB nucleus:cytoplasm ratio did not change in cells treated with SH-SY5Y ρ0 mitochondrial lysates.
Fig. 3.

Effect of mitochondrial lysates on microglial cell NFκB expression and protein compartmentalization. BV2 cells were treated overnight with PBS vehicle, 50 μg of mitochondrial lysate from BV2 cells, or 50 μg of mitochondrial lysate from SH-SY5Y ρ0 cells. In (A) through (G), seven to ten independent samples were analysed for each condition. (A) Total cell NFκB protein. (B) Total cell IκBα protein. (C) Representative whole cell immunochemistry data. (D) Cytosolic NFκB protein. (E) Nuclear NFκB protein. (F) Nucleus:cytosol NFκB protein ratio. (G) Representative nucleus and cytosol immunochemistry data. (H) Immunocytochemistry with a primary antibody to NFκB and Hoechst staining was performed on BV2 cells treated with PBS vehicle or 50 μg of BV2 cell mitochondrial lysate. Cells were visualized using confocal microscopy. The NFκB staining is green and the nuclear Hoechst staining is blue. Arrowheads indicate cells with intense nuclear NFκB staining. (I) The percentage of BV2 cells in each group with intense NFκB nuclear staining is indicated. Six independent samples were prepared and analysed for each condition. Values shown in (A) through (F) are relative group means ± SEM, with the control group set to 1. * indicates p<0.05. In (H), ** indicates p<0.005 by Student’s T-test.
To validate our Western blot-based protein compartmentalization approach, we additionally performed immunocytochemistry using a primary antibody to NFκB. With this technique we observed that BV2 cells exposed to mtDNA-containing mitochondrial lysates had a higher percentage of cells that showed intense nuclear NFκB staining (Fig. 3H-I). Immunocytochemistry data, therefore, are essentially consistent with Western blot data that indicate mitochondrial lysates from mtDNA-containing cells induce a relative redistribution of NFκB to cell nuclei.
Effect of mitochondrial homogenates on BV2 cell p38 protein and phosphorylation levels
NFκB expression and activation is mediated by the p38 MAPK pathway [30, 31]. No change in total p38 protein was observed in cells treated with mitochondria containing or lacking mtDNA (Fig. 4A,D). Since p38 is activated through distinct phosphorylation events, we assessed p38 Thr180/Tyr182 phosphorylation. In cells treated with mitochondrial lysates that contained mtDNA, phosphorylated p38 levels were significantly increased when normalized to GAPDH (an approximate 4 fold increase) or total p38 protein (an approximate 3 fold increase) (Fig. 4B-D). Cells treated with mitochondrial lysates from ρ0 cells did not show significant changes in p38 Thr180/Tyr182 phosphorylation when normalized to either GAPDH or total p38 protein.
Fig. 4.

Effect of mitochondrial lysates on microglial cell p38 protein and phosphorylation levels. BV2 cells were treated overnight with PBS vehicle, 50 μg of mitochondrial lysate from BV2 cells, or 50 μg of mitochondrial lysate from SH-SY5Y ρ0 cells. Cells were lysed using MPER, equal protein concentrations were resolved by electrophoresis, gel proteins were transferred to nitrocellulose membranes, and the membranes were immunoblotted for p38, GAPDH, and phosphorylated p38 protein. (A) Total p38 normalized to GAPDH. (B) Phosphorylated p38 normalized to GAPDH. (C) Phosphorylated p38 normalized to total p38. (D) Representative immunochemistry data. Values shown are relative group means ± SEM, with the control group set to 1. ** indicates p<0.005.
Effect of mitochondrial lysates on SH-SY5Y cell inflammatory genes
We next assessed cytokine production in SH-SY5Y cells treated with mitochondrial lysates prepared from SH-SY5Y and SH-SY5Y ρ0 cells. Cells treated with mitochondrial lysates from mtDNA-containing cells showed an approximate 3.3 fold increase in their TNFα mRNA level (Fig. 5A). TNFα mRNA levels did not change in SH-SY5Y cells treated with ρ0 cell mitochondrial lysates (Fig. 5A). When SH-SY5Y cells were treated with SH-SY5Y cell mitochondrial lysates, CCL11 mRNA levels were unchanged across the groups (ANOVA p=0.10), although on post-hoc analysis a trend towards increased CCL11 mRNA was observed (2.3 fold increase, p=0.04 on LSD test) (Fig. 5B). Cells treated with SH-SH5Y ρ0 mitochondrial lysates showed no evidence of CCL11 mRNA upregulation (Fig. 5B). IL-8 and MMP-8 mRNA levels were undetectable in SH-SY5Y cells either preceding or following exposure to mitochondrial lysates (data not shown).
Fig. 5.
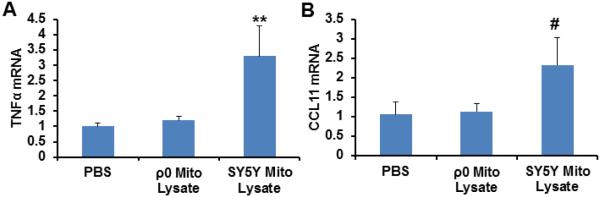
Effect of mitochondrial lysates on neuronal cell line inflammatory gene expression. SH-SY5Y cells were treated overnight with PBS vehicle, 50 μg of mitochondrial lysate from SH-SY5Y cells, or 50 μg of mitochondrial lysate from SH-SY5Y ρ0 cells. Twelve independent samples were prepared and analysed for each condition. (A) TNFα mRNA level. (B) CCL11 mRNA level. Values shown are relative group means ± SEM, with the control group set to 1. ** indicates p<0.005; # indicates a non-significant ANOVA, but with p<0.05 between the control group and the indicated group on LSD post hoc testing.
Effect of mitochondrial lysates on SH-SY5Y cell NFκB protein
SH-SY5Y cells treated with mitochondrial lysates from mtDNA-containing cells displayed a 2.9 fold increase in NFκB total protein (Fig. 6A,C). No significant change was observed in NFκB protein levels in SH-SY5Y cells treated with mitochondrial lysates from cells that lacked detectable mtDNA (Fig. 6A,C). The IκBα protein level fell by 38% in cells treated with mitochondrial lysates that contained mtDNA, but no reduction was observed in cells treated with mitochondrial lysates that lacked mtDNA (Fig. 6B,C). While NFκB nucleus:cytoplasm ratios were comparable by ANOVA when SH-SY5Y cells were treated with mitochondrial lysates that contained or lacked mtDNA (p=0.11), the post hoc analysis showed perhaps a trend towards an increase in cells exposed to the mtDNA-containing mitochondrial lysate (p=0.05 on LSD test) (Fig. 6D-G).
Fig. 6.

Effect of mitochondrial lysates on neuronal cell NFκB protein levels and compartmentalization. SH-SY5Y cells were treated overnight with PBS vehicle, 50 μg of mitochondrial lysate from SH-SY5Y cells, or 50 μg of mitochondrial lysate from SH-SY5Y ρ0 cells. Ten to twelve independent samples were prepared and analysed for each condition. (A) Total cell NFκB protein. (B) Total cell IκBα protein. (C) Representative whole cell immunochemistry data. (D) Cytosolic NFκB protein. (E) Nuclear NFκB protein. (F) Nucleus:cytosol NFκB protein ratio. (G) Representative nucleus and cytosol immunochemistry data. Values shown are relative group means ± SEM, with the control group set to 1. * indicates p<0.05; # indicates a non-significant ANOVA, but with p<0.05 between the control group and the indicated group on LSD post hoc testing.
Effect of mitochondrial lysates on SH-SY5Y cell AβPP mRNA, AβPP protein, and secreted Aβ
AβPP transcription is activated by NFκB [32, 33]. Given the results presented above, as well as reported links between AβPP and AD, we assessed AβPP levels in SH-SY5Y cells treated with SH-SY5Y and SH-SY5Y ρ0 cell mitochondrial lysates. AβPP mRNA and protein levels increased when cells were exposed to mitochondrial lysates from cells that contained mtDNA (61% and 160% increases, respectively), but did not change following exposure to mitochondrial lysates from cells that lacked mtDNA (Fig. 7A-C). No changes, though, in secreted Aβ protein were observed. The Aβ protein concentration in the lysate-untreated control cells was 29.6 ± 0.5 pg/ml, for the cells treated with 50 μg of ρ0 cell mitochondrial lysates it was 29.3 ± 0.2 pg/ml, and for cells treated with 50 μg of SH-SY5Y cell mitochondrial lysates it was 31.5 ± 1.1 pg/ml.
Fig. 7.

Effect of mitochondrial lysates on neuronal cell AβPP mRNA and protein levels. SH-SY5Y cells were treated overnight with PBS vehicle, 50 μg of mitochondrial lysate from SH-SY5Y cells, or 50 μg of mitochondrial lysate from SH-SY5Y ρ0 cells. Ten to twelve independent samples were prepared and analysed for each condition. (A) AβPP mRNA levels. (B) AβPP protein levels. (C) Representative immunochemistry data. Values shown are relative group means ± SEM, with the control group set to 1. * indicates p<0.05; ** indicates p<0.005.
Direct effect of mtDNA on SH-SY5Y and BV2 cell TNFα expression
We next tested the ability of mtDNA to specifically induce TNFα expression. We utilized enriched mtDNA obtained directly from BV2 cells, as well as through a long PCR amplification of BV2 mtDNA (Fig. 8A). When BV2 cells were treated with either 1 μg of enriched native mtDNA or 1 μg of PCR mtDNA, TNFα mRNA levels did not change although there was perhaps an upward trend in cells exposed to the enriched native mtDNA (p=0.085 on post hoc LSD test) (Fig. 8B). The BV2 TNFα mRNA level did increase when cells were treated with 2 μg of enriched native mtDNA prepared directly from BV2 cells, but did not change when BV2 cells were treated with an equivalent amount of PCR-generated mtDNA (Fig. 8C). SH-SY5Y cell TNFα mRNA levels did not change following treatment with either 1 μg or 2 μg of enriched mitochondrial lysate mtDNA or PCR-amplified mtDNA (data not shown).
Fig. 8.

Effect of mtDNA on microglial cell TNFα expression. (A) Long PCR of mtDNA. The band shown is approximately 15kb in size. Neg=negative control PCR. (B) BV2 cells were treated overnight with either 1 μg of native enriched mtDNA from BV2 cells or 1 μg of PCR-amplified mtDNA. (C) BV2 cells were treated overnight with either 2 μg of native enriched mtDNA from BV2 cells or 2 μg of PCR-amplified mtDNA. In (B) and (C), twelve independent samples were prepared and analysed for each condition. Values shown are relative group means ± SEM, with the control group set to 1. * indicates p<0.05.
DISCUSSION
This study tested the ability of mitochondrial lysates to induce inflammation in microglial and neuronal cell lines, as well as the ability of mitochondrial lysates to influence specific AD-associated genes including TREM2 and AβPP. We found that BV2 microglial cells exposed to mitochondrial lysates from cells containing mtDNA, but not mitochondrial lysates prepared from mtDNA-depleted cells, increased mRNA levels of several inflammation-relevant genes (TNFα, IL-8, and MMP-8), decreased TREM2 mRNA, increased total NFκB protein, shifted NFκB to the nucleus, and increased p38 MAPK phosphorylation. We also found that in SH-SY5Y neuronal cells mitochondrial lysates from cells containing mtDNA, but not mitochondrial lysates prepared from mtDNA-depleted cells, increased TNFα mRNA, perhaps increased CCL11 mRNA, increased total NFκB protein, increased AβPP mRNA, and increased AβPP protein. Direct treatment with enriched mtDNA, but not PCR-amplified mtDNA, also increased BV2 cell TNFα expression.
TNFα, an inflammatory cytokine, is activated by inflammatory molecules and can also amplify inflammatory signaling cascades [34, 35]. TREM2 suppresses cytokine production and genetic ablation of TREM2 reduces microglial phagocytosis [36]. IL-8 is a macrophage-generated and secreted inflammatory chemokine that binds Toll-like receptors and plays a role in innate immune system responses [37, 38]. The matrix metalloproteinase MMP-8, when aberrantly produced by microglial cells, facilitates neuroinflammatory responses through its effects on TNFα [39]. Demonstrating effects on the expression of these genes could prove relevant to AD, a disorder that features neuroinflammation, elevated TNFα levels, reduced TREM2 levels [36, 40], and an increased risk for individuals that carry loss-of-function TREM2 sequence variants [2]. To our knowledge, IL-8 and MMP-8 levels have not been reported in AD brains, although our data are consistent with prior studies that indicate mtDNA induces IL-8 and MMP-8 expression [13].
NFκB, a transcription factor, plays a role in inflammatory cascades. NFκB signaling is induced by TNFα, and in turn NFκB can induce TNFα expression [34]. In our experiments mtDNA-containing mitochondrial lysates increased NFκB expression, decreased IκBα levels in SH-SY5Y cells, and caused either absolute or relative (to cytosolic levels) increases in nuclear NFκB. These findings may prove relevant to AD, a disorder that exhibits NFκB activation [41].
The canonical inflammatory p38 MAPK pathway also modulates NFκB activation [42]. Mitochondrial lysates prepared from mtDNA-containing cells, but not mitochondrial lysates prepared from mtDNA-depleted cells, induced p38 phosphorylation. The presence of increased p38 phosphorylation is consistent with a state of increased NFκB activation. p38 MAPK signaling is increased in human AD subject brains [43].
AβPP expression increases in AD subjects [44, 45]. Therefore, our finding that mitochondrial lysates increase both AβPP mRNA and protein levels could be AD-relevant. Existing data indicate the AβPP gene promoter contains putative NFκB binding sites [32, 33]. Although we did not mechanistically address this possibility, it seems reasonable to consider that AβPP upregulation in our experiments could reflect increased NFκB activity. Of further interest to this point, AP1 and SP1, transcription factors that are also linked to inflammatory signaling, may additionally induce AβPP transcription [46].
The observed mitochondrial lysate-induced increase in AβPP protein, though, did not cause a detectable increase in soluble extracellular Aβ protein. The fate of the AβPP protein in our experiments, therefore, remains unclear. While an increase in AβPP mRNA suggests the increase in AβPP protein reflects increased production, as opposed to decreased degradation, increased AβPP protein production would not necessarily require AβPP degradation rates to also increase. Alternatively, AβPP degradation could be proceeding at an increased rate through the alpha secretase-mediated non-amyloidogenic pathway, or it could be proceeding at an increased rate through an intracellular beta secretase-mediated event. More detailed studies to resolve this question are indicated.
Mitochondria, which resemble bacteria in many ways, contain a pool of known DAMP molecules. mtDNA, TFAM, cardiolipin, cytochrome c, formyl-peptides, HMGB1, and ATP have previously been shown to induce a DAMP response in various models [8, 13-23]. Our data argue mtDNA, at least in its native form, may play a particularly important role. We cannot, however, completely rule out the possibility that other lysate components acted as DAMP molecules and contributed to DAMP responses in our experiments. Specifically, due to their lack of mtDNA ρ0 cells cannot synthesize mitochondrial RNA or mtDNA-encoded proteins and this, in turn, should eliminate formyl-peptide synthesis. It therefore remains possible that mitochondrial RNA or formyl-peptides may have partly driven changes in our inflammation-associated endpoints. TFAM protein levels may also change in ρ0 cells.
The mitochondrial fractions used throughout this study were robustly enriched for mitochondrial content but were not completely pure. We thus need to consider the possibility that potential non-mitochondrial contaminants, such as nuclear DNA, may have contributed to our observed DAMP responses. While the enrichment procedure we used eliminated approximately 85% of the nuclear DNA initially present in our cell cultures, substantial amounts of nuclear DNA were no doubt present in our mitochondrial lysate and mtDNA preparations. However, equivalently enriched mitochondrial lysates obtained from ρ0 cells, which also contained nuclear DNA contamination but lacked mtDNA, consistently failed to elicit an inflammatory response. We therefore conclude, as have others, that even if nuclear DNA can act as a DAMP molecule in this respect it is less functionally potent than mtDNA [25]. We further conclude that in our experiments, either mtDNA or an mtDNA-containing complex functioned as a DAMP molecule. We again wish to emphasize, though, that this does not rule out the possibility that other mitochondrial components contributed to the DAMP responses observed in these experiments.
Several factors could account for the finding that PCR-amplified mtDNA did not increase TNFα mRNA levels despite the fact that native enriched mtDNA did. In general, certain DNA features are not perpetuated during PCR amplification. These include DNA tertiary structure, epigenetic modifications, oxidative changes, and bound proteins (although one would predict proteinase K treatment should remove most or all mtDNA-bound proteins).
A recent study reported that retinal ganglion cells release mitochondria from axons, and that these mitochondria are then engulfed and degraded by surrounding glial cells [47]. Therefore, neurons have the ability to shed mitochondria, and if this process becomes compromised inflammation could ensue. Given this context, our findings are consistent with the possibility that bioenergetic dysfunction and failure of axonal mitochondrial degradation could give rise to chronic brain inflammation through the release of mitochondrial components. Further studies are warranted to determine if mitochondrial components induce chronic brain inflammation in vivo.
ACKNOWLEDGEMENTS
This work was supported by the University of Kansas Alzheimer’s Disease Center (P30AG035982), the Frank and Evangeline Thompson Alzheimer’s Treatment Program Fund, the Hugh and Betty Libby Foundation, the Greater Kansas City Automobile Dealers Association, the Gene and Marge Sweeney Chair, the University of Kansas Alzheimer’s Disease Center Pilot Program, the University of Kansas Landon Center on Aging, the University of Kansas Frontiers Heartland Institute for Clinical and Translational Research, and the University of Kansas Medical Center’s Biomedical Research Training Program.
REFERENCES
- [1].Cameron B, Landreth GE. Inflammation, microglia, and Alzheimer's disease. Neurobiol Dis. 2010;37:503–509. doi: 10.1016/j.nbd.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jiang T, Yu JT, Zhu XC, Tan L. TREM2 in Alzheimer's disease. Mol Neurobiol. 2013;48:180–185. doi: 10.1007/s12035-013-8424-8. [DOI] [PubMed] [Google Scholar]
- [3].Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin CF, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau MT, Choi SH, Reitz C, Pasquier F, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Letenneur L, Moron FJ, Rubinsztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fievet N, Huentelman MW, Gill M, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuiness B, Larson EB, Green R, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossu P, Spalletta G, Proitsi P, Collinge J, Sorbi S, Sanchez-Garcia F, Fox NC, Hardy J, Deniz Naranjo MC, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Matthews F, European Alzheimer's Disease I, Genetic, Environmental Risk in Alzheimer's D, Alzheimer's Disease Genetic C, Cohorts for H, Aging Research in Genomic E. Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Lannefelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alvarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson KL, Gu W, Razquin C, Pastor P, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O'Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley TH, Jr., Bennett DA, Harris TB, Fratiglioni L, Holmes C, de Bruijn RF, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie K, Lunetta KL, Kauwe JS, Boerwinkle E, Riemenschneider M, Boada M, Hiltuenen M, Martin ER, Schmidt R, Rujescu D, Wang LS, Dartigues JF, Mayeux R, Tzourio C, Hofman A, Nothen MM, Graff C, Psaty BM, Jones L, Haines JL, Holmans PA, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, van Duijn CM, Van Broeckhoven C, Moskvina V, Seshadri S, Williams J, Schellenberg GD, Amouyel P. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schott JM, Revesz T. Inflammation in Alzheimer's disease: insights from immunotherapy. Brain. 2013;136:2654–2656. doi: 10.1093/brain/awt231. [DOI] [PubMed] [Google Scholar]
- [5].Mrak RE, Griffin WS. Potential inflammatory biomarkers in Alzheimer's disease. J Alzheimers Dis. 2005;8:369–375. doi: 10.3233/jad-2005-8406. [DOI] [PubMed] [Google Scholar]
- [6].Hensley K. Neuroinflammation in Alzheimer's disease: mechanisms, pathologic consequences, and potential for therapeutic manipulation. J Alzheimers Dis. 2010;21:1–14. doi: 10.3233/JAD-2010-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, Vandenabeele P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011;32:157–164. doi: 10.1016/j.it.2011.01.005. [DOI] [PubMed] [Google Scholar]
- [9].Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14:463–477. doi: 10.1038/nri3705. [DOI] [PubMed] [Google Scholar]
- [10].Swerdlow RH. Mitochondria and cell bioenergetics: increasingly recognized components and a possible etiologic cause of Alzheimer's disease. Antioxid Redox Signal. 2012;16:1434–1455. doi: 10.1089/ars.2011.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hagberg H, Mallard C, Rousset CI, Thornton C. Mitochondria: hub of injury responses in the developing brain. Lancet Neurol. 2014;13:217–232. doi: 10.1016/S1474-4422(13)70261-8. [DOI] [PubMed] [Google Scholar]
- [12].Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- [13].Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cauwels A, Rogge E, Vandendriessche B, Shiva S, Brouckaert P. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis. 2014;5:e1102. doi: 10.1038/cddis.2014.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Riteau N, Gasse P, Fauconnier L, Gombault A, Couegnat M, Fick L, Kanellopoulos J, Quesniaux VF, Marchand-Adam S, Crestani B, Ryffel B, Couillin I. Extracellular ATP is a danger signal activating P2X7 receptor in lung inflammation and fibrosis. Am J Respir Crit Care Med. 2010;182:774–783. doi: 10.1164/rccm.201003-0359OC. [DOI] [PubMed] [Google Scholar]
- [16].Flohe SB, Bruggemann J, Lendemans S, Nikulina M, Meierhoff G, Flohe S, Kolb H. Human Heat Shock Protein 60 Induces Maturation of Dendritic Cells Versus a Th1-Promoting Phenotype. The Journal of Immunology. 2003;170:2340–2348. doi: 10.4049/jimmunol.170.5.2340. [DOI] [PubMed] [Google Scholar]
- [17].Panjwani NN, Popova L, Srivastava PK. Heat Shock Proteins gp96 and hsp70 Activate the Release of Nitric Oxide by APCs. The Journal of Immunology. 2002;168:2997–3003. doi: 10.4049/jimmunol.168.6.2997. [DOI] [PubMed] [Google Scholar]
- [18].Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107–15112. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- [19].Wan M, Hua X, Su J, Thiagarajan D, Frostegard AG, Haeggstrom JZ, Frostegard J. Oxidized but not native cardiolipin has pro-inflammatory effects, which are inhibited by Annexin A5. Atherosclerosis. 2014;235:592–598. doi: 10.1016/j.atherosclerosis.2014.05.913. [DOI] [PubMed] [Google Scholar]
- [20].Pullerits R, Bokarewa M, Jonsson IM, Verdrengh M, Tarkowski A. Extracellular cytochrome c, a mitochondrial apoptosis-related protein, induces arthritis. Rheumatology (Oxford) 2005;44:32–39. doi: 10.1093/rheumatology/keh406. [DOI] [PubMed] [Google Scholar]
- [21].Gao X, Hu X, Qian L, Yang S, Zhang W, Zhang D, Wu X, Fraser A, Wilson B, Flood PM, Block M, Hong JS. Formyl-methionyl-leucyl-phenylalanine-induced dopaminergic neurotoxicity via microglial activation: a mediator between peripheral infection and neurodegeneration? Environ Health Perspect. 2008;116:593–598. doi: 10.1289/ehp.11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Little JP, Simtchouk S, Schindler SM, Villanueva EB, Gill NE, Walker DG, Wolthers KR, Klegeris A. Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Mol Cell Neurosci. 2014;60:88–96. doi: 10.1016/j.mcn.2014.04.003. [DOI] [PubMed] [Google Scholar]
- [23].Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Collins LV, Hajizadeh S, Holme E, Jonsson IM, Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol. 2004;75:995–1000. doi: 10.1189/jlb.0703328. [DOI] [PubMed] [Google Scholar]
- [26].Miller SW, Trimmer PA, Parker WD, Jr., Davis RE. Creation and characterization of mitochondrial DNA-depleted cell lines with "neuronal-like" properties. J Neurochem. 1996;67:1897–1907. doi: 10.1046/j.1471-4159.1996.67051897.x. [DOI] [PubMed] [Google Scholar]
- [27].Swerdlow RH, Redpath GT, Binder DR, Davis JN, 2nd, VandenBerg SR. Mitochondrial DNA depletion analysis by pseudogene ratioing. J Neurosci Methods. 2006;150:265–271. doi: 10.1016/j.jneumeth.2005.06.023. [DOI] [PubMed] [Google Scholar]
- [28].Binder DR, Dunn WH, Jr., Swerdlow RH. Molecular characterization of mtDNA depleted and repleted NT2 cell lines. Mitochondrion. 2005;5:255–265. doi: 10.1016/j.mito.2005.04.003. [DOI] [PubMed] [Google Scholar]
- [29].Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Baeza-Raja B, Munoz-Canoves P. p38 MAPK-induced nuclear factor-kappaB activity is required for skeletal muscle differentiation: role of interleukin-6. Mol Biol Cell. 2004;15:2013–2026. doi: 10.1091/mbc.E03-08-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- [32].Grilli M, Goffi F, Memo M, Spano P. Interleukin-1beta and glutamate activate the NF-kappaB/Rel binding site from the regulatory region of the amyloid precursor protein gene in primary neuronal cultures. J Biol Chem. 1996;271:15002–15007. doi: 10.1074/jbc.271.25.15002. [DOI] [PubMed] [Google Scholar]
- [33].Grilli M, Ribola M, Alberici A, Valerio A, Memo M, Spano P. Identification and characterization of a kappa B/Rel binding site in the regulatory region of the amyloid precursor protein gene. J Biol Chem. 1995;270:26774–26777. doi: 10.1074/jbc.270.45.26774. [DOI] [PubMed] [Google Scholar]
- [34].Barnes PJ. Nuclear factor-kappa B. Int J Biochem Cell Biol. 1997;29:867–870. doi: 10.1016/s1357-2725(96)00159-8. [DOI] [PubMed] [Google Scholar]
- [35].Bradley JR. TNF-mediated inflammatory disease. J Pathol. 2008;214:149–160. doi: 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- [36].Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleo A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sanchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014;6:243ra286. doi: 10.1126/scitranslmed.3009093. [DOI] [PubMed] [Google Scholar]
- [37].Semple BD, Kossmann T, Morganti-Kossmann MC. Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab. 2010;30:459–473. doi: 10.1038/jcbfm.2009.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Park C, Lee S, Cho IH, Lee HK, Kim D, Choi SY, Oh SB, Park K, Kim JS, Lee SJ. TLR3-mediated signal induces proinflammatory cytokine and chemokine gene expression in astrocytes: differential signaling mechanisms of TLR3-induced IP-10 and IL-8 gene expression. Glia. 2006;53:248–256. doi: 10.1002/glia.20278. [DOI] [PubMed] [Google Scholar]
- [39].Lee EJ, Han JE, Woo MS, Shin JA, Park EM, Kang JL, Moon PG, Baek MC, Son WS, Ko YT, Choi JW, Kim HS. Matrix metalloproteinase-8 plays a pivotal role in neuroinflammation by modulating TNF-alpha activation. J Immunol. 2014;193:2384–2393. doi: 10.4049/jimmunol.1303240. [DOI] [PubMed] [Google Scholar]
- [40].Alvarez A, Cacabelos R, Sanpedro C, Garcia-Fantini M, Aleixandre M. Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol Aging. 2007;28:533–536. doi: 10.1016/j.neurobiolaging.2006.02.012. [DOI] [PubMed] [Google Scholar]
- [41].Kaltschmidt B, Uherek M, Volk B, Baeuerle PA, Kaltschmidt C. Transcription factor NF-kappaB is activated in primary neurons by amyloid beta peptides and in neurons surrounding early plaques from patients with Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:2642–2647. doi: 10.1073/pnas.94.6.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005;15:11–18. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- [43].Sun A, Liu M, Nguyen XV, Bing G. P38 MAP kinase is activated at early stages in Alzheimer's disease brain. Exp Neurol. 2003;183:394–405. doi: 10.1016/s0014-4886(03)00180-8. [DOI] [PubMed] [Google Scholar]
- [44].Vignini A, Morganti S, Salvolini E, Sartini D, Luzzi S, Fiorini R, Provinciali L, Di Primio R, Mazzanti L, Emanuelli M. Amyloid precursor protein expression is enhanced in human platelets from subjects with Alzheimer's disease and frontotemporal lobar degeneration: a real-time PCR study. Exp Gerontol. 2013;48:1505–1508. [PubMed] [Google Scholar]
- [45].Matsui T, Ingelsson M, Fukumoto H, Ramasamy K, Kowa H, Frosch MP, Irizarry MC, Hyman BT. Expression of APP pathway mRNAs and proteins in Alzheimer's disease. Brain Res. 2007;1161:116–123. doi: 10.1016/j.brainres.2007.05.050. [DOI] [PubMed] [Google Scholar]
- [46].Theuns J, Van Broeckhoven C. Transcriptional regulation of Alzheimer's disease genes: implications for susceptibility. Hum Mol Genet. 2000;9:2383–2394. doi: 10.1093/hmg/9.16.2383. [DOI] [PubMed] [Google Scholar]
- [47].Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, Kinebuchi M, Phan S, Zhou Y, Bihlmeyer NA, Nguyen JV, Jin Y, Ellisman MH, Marsh-Armstrong N. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A. 2014;111:9633–9638. doi: 10.1073/pnas.1404651111. [DOI] [PMC free article] [PubMed] [Google Scholar]