

Abstract
Pseurotins comprise a family of structurally related Aspergillal natural products having interesting bioactivity. However, little is known about the biosynthetic steps involved in the formation of their complex chemical features. Here, we systematically deleted the pseurotin biosynthetic genes in A. fumigatus and performed in vivo and in vitro characterization of the tailoring enzymes to determine the biosynthetic intermediates and the gene products responsible for the formation of each intermediate. This allowed us to elucidate the main biosynthetic steps leading to the formation of pseurotin A from the predominant precursor, azaspirene. The study revealed the combinatorial nature of the biosynthesis of the pseurotin family of compounds and the intermediates. Most interestingly, we report the first identification of an epoxidase–C-methyltransferase bifunctional fusion protein PsoF that appears to methylate the nascent polyketide backbone carbon atom in trans.
Keywords: fungal natural products, pseurotin biosynthesis, redox enzyme, bifunctional enzyme, trans-acting enzyme
Pseurotins[1] make up a family of fungal secondary metabolites that exhibit wide-ranging biological activities of medicinal importance. For example, pseurotin A 8 was reported to inhibit monoamine oxidase[2] and induce cell differentiation in PC12 cells.[3] Previously, the gene cluster responsible for biosynthesizing 8 was predicted and identified by deletion and overexpression of the polyketide synthase–nonribosomal peptide synthetase (PKS–NRPS) hybrid enzyme gene, psoA, in Aspergillus fumigatus Af293.[4] PsoA was shown to be responsible for the biosynthesis of the core structure of 8, the1-oxa-7-azaspiro[4,4]non-2-ene-4,6-dione skeleton having five chiral centers. Recently, a detailed analysis of the pseurotin biosynthetic gene cluster describing its genetic organization was reported.[5] However, the mechanism of pseurotin biosynthesis, such as the formation of the spiro-ring core structure, still remains undefined (Scheme 1 and Figure 1A). Another interesting aspect of pseurotin biosynthesis is the formation of a large number of closely related bioactive compounds, such as azaspirene 2[6] and synerazol 7.[7] This diversity of pseurotin-type natural products is thought to be generated during the post-PKS–NRPS modification steps. However, it is often difficult to resolve the biosynthetic steps involving multiple enzymes and complex intermediates. Here, we carried out knockout of the pseurotin biosynthetic genes in the ΔpyrG/Δku70 strain of A. fumigatus A1159 named AfKW1 (See the Supporting Information) and conducted in vivo and in vitro characterization of four modification enzymes, PsoC, PsoD, PsoE and PsoF, to reveal the unique mechanism involved in the biosynthesis of pseurotin family of natural products.
Scheme 1.

Biosynthetic pathway for the transformation of 1 to pseurotins 8 and 9 involving methylation and multiple oxidation steps. Pathway enclosed within the broken-line box with thick arrows represents the proposed main pathway toward formation of 8 and 9.
Figure 1.

Modular organization of the uncharacterized iterative PKS-NRPS encoded by psoA (12.3 kb) and C-methylation activity of PsoF. Abbreviations: KS, ketosynthase; AT, acyltransferase; DH, dehydratase; MT, methyltransferase; MT*, partially active methyltransferase; KR, ketoreductase; ACP, acyl carrier protein; C, condensation; A, adenylation; T, thiolation; R, reductase; SAM, S-adenosyl-L-methionine. (A) Proposed biosynthetic pathway for the formations of 8 and 22. The iterative PKS–NRPS is shown with its predicted core domains. Pathways toward formation of 8 vs. 22 depending on the presence of PsoF are shown. (B) C-methylation of the polyketide backbone core by the MT domain of PsoF. Functional analysis was performed with a chemically synthesized NAC thioester 25, which was converted into a methylated product 26 by PsoF.
Sequence analysis (See the Supporting Information) indicated that PsoF is a single polypeptide comprised of an unusual combination of two domains, one homologous to a methyltransferase (MT) and another to an FAD-containing monooxygenase (FMO). Deletion of psoF abolished production of 8, 9, 13 and 14, indicating the essential role of PsoF in pseurotin biosynthesis (Figure 2 i vs. ii). Surprisingly, the deletion also resulted in the formation of 22 (Supporting Information, Table S15 and Figures S45–S48), a C3-desmethylated analog of 2 (Figure 2 i vs. ii). A mixture of a geometric isomer 23 and a reduced product 24, both lacking the C3 methyl group, (Table S16 and Figures S49–S52) were also isolated from ΔpsoF/AfKW1 (Figure S5). Close analysis revealed that the MT domain of PsoA actually exhibited a residual activity as judged by a marginal formation of 4 in the ΔpsoF/AfKW1 strain (Figure S5 ii and viii). These results allowed us to propose a mechanism of how the C3 methyl group is introduced into the pseurotin scaffold (Figure 1A). For the FMO domain of PsoF, lack of formation of pseurotin-type compounds 8, 9, 13 and 14 suggested its role in the formation of the 10,11-epoxide.
Figure 2.
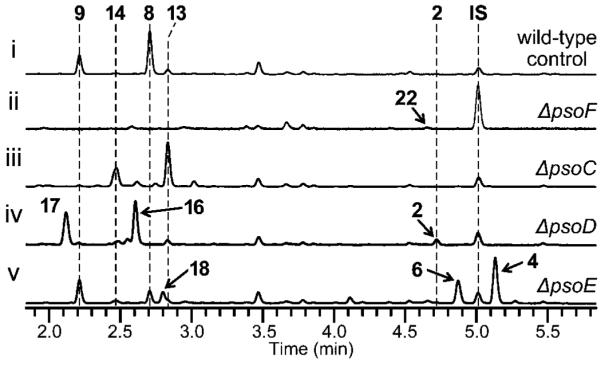
HPLC traces of metabolic extracts from the cultures of various A. fumigatus A1159-based deletion strains to identify the genes responsible for the biosynthetic steps involved in the formation of 8 during the pseurotin biosynthesis. All deletion strains were prepared in AfKW1 by replacing the target gene with pyrG via homologous recombination. All cultures were grown in MYG medium. All HPLC traces were monitored at 280 nm. Extract of the culture of (i) AfKW1 harboring pKW20088 for the expression of pyrG as a wild-type control, (ii) ΔpsoF strain, (iii) ΔpsoC strain, (iv) ΔpsoD strain and (v) ΔpsoE strain. IS, internal standard N-Boc-L-tryptophan methyl ester (Boc-Trp-OMe).
To characterize in detail the biochemical functions of C-MT and FMO domains of PsoF, we cloned psoF from mRNA and expressed it heterologously in Escherichia coli (See the Supporting Information). The C-methylation step was examined in vitro using an N-acetylcysteamine (NAC) thioester[8] that mimics the natural substrate, a phosphopanteteine-linked 25 (See the Supporting Information). The purified PsoF was able to catalyze methylation of 25 to form 26 (Figure 1B and Figure 3). Interestingly, we did not observe the formation of epoxidated products of 25 or 26, suggesting those early intermediates are not recognized as substrates by the FMO domain of PsoF. For functional analysis of the FMO domain, initially 2 was subjected to an in vitro assay with the recombinant PsoF. The assay revealed that PsoF was completely unreactive to 2. Since the presence of an epoxide moiety in synerazol 7 (Table S7 and Figure S22) hinted that 7 could be formed by the activity of an FMO, we subjected the mixture of geometric isomers 4 and 6 to PsoF as substrates in an in vitro reaction. In this reaction, PsoF converted the substrates into four products: the epoxide-containing 7 and spontaneously hydrolyzed epoxide products, 8, 9 and 18 (Figure 4). The chemical structures of all compounds, 4 (Table S5 and Figures S14–S17), 6 (Table S6 and Figures S18–S21), 7, 8 (Table S8 and Figure S23), 9 (Table S9 and Figures S24–S27) and the new compound 18 (Table S13 and Figures S40–S43), were elucidated by HRESIMS, 1H NMR and 13C NMR (See the Supporting Information). In vitro transformation of 4 into 18 by PsoF indicated that the FMO domain is capable of accepting 13E-containing compound as its substrate. However, a complete lack of formation of any 13E-containing products in the wild-type strain (Figure 2 i) suggests that 13Z-configured compounds are likely the true substrate of PsoF. Furthermore, formation of epoxidated or hydroxylated products from the 11Z-containing compound 6 was not observed in the reaction. Thus, the preferred substrate of PsoF is considered to be 5, an 11E,13Z-configured isomer. Based on the chemical structures of the products 8 and 9, we propose that those products are formed from 5 via epoxidation catalyzed by the FMO domain of PsoF. After epoxidation of 5 by PsoF to form 7, 7 can be attacked by a water molecule nonenzymatically at two different positions to yield two diol products 8 and 9 via SN2 and SN2’ reaction, respectively (Scheme 2). For the formation of 8, the water molecule attacks 7 at C11 from the opposite face of the epoxide in an anti-periplanar manner to yield the 10S,11S-diol of 8. On the other hand, 9 is formed by the water molecule attacking 7 at C13. Subsequent allyl migration and epoxide opening yields the diol product. Taking these results together, PsoF was shown to be a unique bifunctional enzyme catalyzing two different types of reactions at completely separate steps of the pseurotin biosynthetic pathway.
Figure 3.
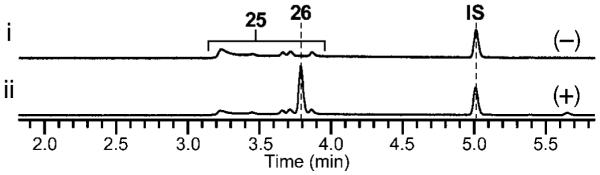
HPLC analysis of 26 biosynthesis using purified PsoF. HPLC traces of (i) control reaction without PsoF and (ii) in vitro reaction with PsoF, which afforded the methylated product 26 after 1 h at 30 °C with 0.2 mM of substrate, which was a keto–enol equilibrated mixture of 25 and its geometric isomers, 1 mM SAM and 0.1 M NaCl in 0.1 M Tris-HCl (pH 7.4). All HPLC traces were monitored at 280 nm. IS, internal standard Boc-Trp-OMe.
Figure 4.
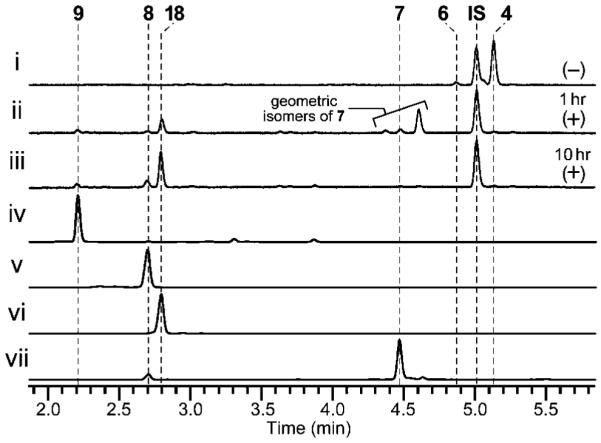
In vitro characterization of PsoF for the formation of 7, 8, 9 and 18. All HPLC traces were monitored at 280 nm. Analysis of in vitro reaction of PsoF with a mixture of geometric isomers 4 and 6 as a substrate. (i) Negative control using heat-inactivated PsoF. (ii) Epoxide intermediates were formed from 4 and 6 by PsoF after 1 h at 30 °C. (iii) Three products 8, 9 and 18 were formed from the substrates by PsoF at 30 °C after 10 h. Authentic references of (iv) 9, (v) 8, (vi) 18 and (vii) 7 are also shown. IS, internal standard Boc-Trp-OMe.
Scheme 2.
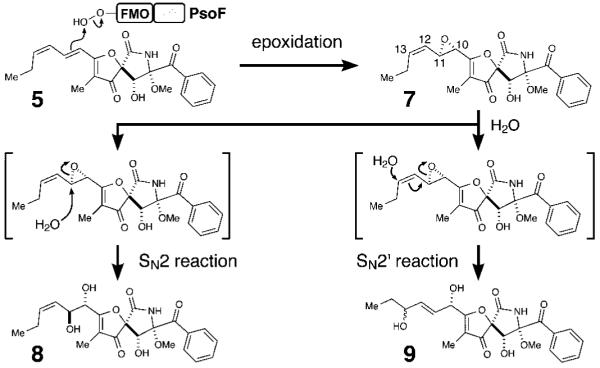
Proposed reaction mechanism of diol formation via SN2 or SN2’ reaction from the epoxide substrate 7 in 8 and 9 by PsoF.
Next, we examined psoC and psoD, which are predicted to code for a MT and a cytochrome P450, respectively. To understand the function of PsoC, we analyzed the mutant strain, ΔpsoC/AfKW1. The UV traces from HPLC analysis of the ΔpsoC/AfKW1 culture extract showed accumulation of O8-unmethylated products 13 (Table S10 and Figures S28–S31) and 14, with loss of 8 and 9 (Figure 2 i vs. iii). This result indicated that PsoC likely performs O-methylation of the hydroxyl group of C8 in 3 to form 4 (Scheme 1). To characterize the O-methylation activity of PsoC in detail, we examined recombinant PsoC in vitro using 13 as a substrate. Interestingly, PsoC did not convert 13 to 8. This result indicates that PsoC does not tolerate modifications to the diene side chain of the azaspirene skeleton as its substrate. To investigate the role of PsoD, we prepared another mutant strain, ΔpsoD/AfKW1. HPLC analysis of the extract showed an accumulation of 16 (Table S11 and Figures S32–S35) and 17 (Table S12 and Figures S36–S39) and a loss of 8 and 9 (Figure 2 i vs. iv), indicating that this cytochrome P450 is responsible for installing the ketone group at C17 (Scheme 1). To confirm the activity of PsoD, in vivo conversion of 2 fed exogenously to the culture of Saccharomyces cerevisiae expressing the psoD gene (Figure 5), as well as in vitro assay for the same conversion (Figure S12), were conducted. The observed conversion of 2 into 3 confirmed the idea that psoD codes for an oxidoreductase that is responsible for the oxidation of C17 in 2 and, by extension, 10. Lack of formation of O8-methylated products in the ΔpsoD/AfKW1 mutant indicates that PsoC requires the presence of the C17 ketone group in its substrate for activity. This places the PsoD-catalyzed step upstream of the PsoC-catalyzed step in the overall pseurotin biosynthetic pathway. In addition, inactivity of PsoD against 16 and 17 (Figure S57) indicates that, like PsoC, PsoD is also sensitive to modifications to the diene side chain. Lastly, based on the proposed activities of PsoC and PsoD, we predict that 22 isolated from the ΔpsoF strain is derived from 21, the analog of 2 that is lacking the C3 methyl group due to the absence of PsoF.
Figure 5.
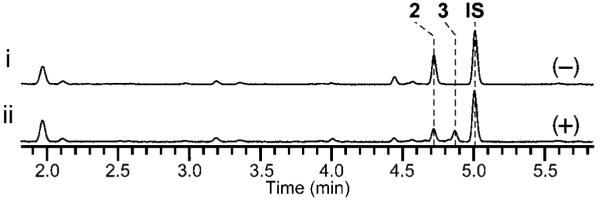
In vivo characterization of the cytochrome P450 PsoD for the formation of 3. UV trace at 280 nm from HPLC analysis of the conversion of the substrate 2 to a product 3 by PsoD. (i) Saccharomyces cerevisiae BY4705 without psoD as a negative control. (ii) S. cerevisiae BY4705 harboring the expression vector carrying the psoD gene. Cultures were incubated at 30 °C for 3 h after feeding 2 to the strains. IS, internal standard Boc-Trp-OMe.
Characterization of the disruption mutants and heterologously produced enzymes also identified multiple analogs arising from cis/trans isomerization of the diene portion of the azaspirene core. Disruption of psoE, a predicted glutathione S-transferase (GST) gene, resulted in a dramatic loss of 8, new formation of 18, and a significant accumulation of 4 and 6 as compared to the wild-type control (Figure 2 v vs. i). Compounds 4 and 6 are both a 13E isomer, and they are predicted to be formed immediately before the PsoF-catalyzed epoxidation step. As discussed earlier, the preferred substrate for PsoF is predicted to be 13Z-configured compound. Thus, accumulation of 4 and 6 in the ΔpsoE strain and previous reports of isomerase activity of certain GSTs[9] suggest that PsoE may be involved in the trans-to-cis isomerization of the C13 olefin. As shown in the in vitro assay of PsoF (Figure 4 ii and iii), 18 is formed via C10,C11 epoxidation by PsoF at low efficiency as 4 accumulates in the ΔpsoE cell. On the other hand, formation of 8 in this mutant strain can be explained by the low level of isomerization of the C13 olefin that converted 4 into 5. This isomerization was also observed in the in vitro assay of PsoF, where a small amount of 8 was formed in the absence of 13Z-containing substrate (Figure 4 iii). As to the stereoisomerism at C11, concomitant accumulation of 11E- and 11Z-configured geometric isomers 22 and 23 in the ΔpsoF/AfKW1 strain and similar accumulation of 4 and 6 upon disruption of another gene, psoE, suggest that cis/trans isomerization of C11 olefin is non-enzymatic. We also observed the formation of 24 in the ΔpsoF strain and cephalimysin A 19[10] in the ΔpsoB strain (Figure S6, Table S14 and Figure S44). Those compounds appear to be formed via reduction by a non-specific oxidoreductase, since no other oxidoreductase gene is found in or near the gene cluster.
In this study, we combined targeted gene deletion, heterologous in vivo bioconversion and in vitro assay to identify the main pathway leading to the formation of pseurotins through the formation of azaspirene and synerazol (Scheme 1). Through the study, we identified eight of the intermediates formed and the enzymes responsible for the oxidation and methylation steps taken during the transformation of 2 to 8. PsoF is unique in that its gene is encoded separately from the pseurotin biosynthetic gene cluster and embedded within the unrelated fumagillin biosynthetic gene cluster.[5] However, even more unexpected was that PsoF predominantly performed an apparent trans C-methylation of the backbone of the growing polyketide chain, with the MT domain of the PKS–NRPS enzyme PsoA maintaining residual methyltransferase activity. Furthermore, PsoF turned out to be a bifunctional enzyme, where its FMO domain catalyzes the stereospecific epoxidation of the C11,12 olefin. Presumed spontaneous conversion of the epoxide to diol by SN2 or SN2’ reaction leads to the formation of pseurotin A 8 and D 9, as well as 13, 14, 16 and 17. The in vitro assay of PsoF with the NAC thioester of 25 showed that PsoF catalyzed the C-methylation to form 26 (Figure 3). However, 26 was converted to neither epoxide nor diol product. This result suggests that PsoF performs the C3 methylation prior to epoxidation, and the FMO domain requires extension of the polyketide chain with a phenylalanine residue and possibly spiro-ring formation for activity. While CTB3 from Cercospora nicotianae involved in the cercosporin biosynthesis was also reported to be a potential bifunctional enzyme predicted to have an N-terminal O-MT and C-terminal FMO-like domain,[11] PsoF is the first enzyme of the kind whose activity has been characterized in detail. While bifunctional enzymes often catalyze two consecutive steps of a metabolic pathway,[12] PsoF represents an example of a highly unusual enzyme that is experimentally shown to catalyze two completely unrelated steps within the same biosynthetic pathway. Our current study also identified that the enzymes responsible for the post-NRPS–PKS assembly modifications, such as PsoC, PsoD and PsoF, are capable of accepting more than one compound as their substrate. Moreover, possible combination of enzymatic and non-enzymatic isomerization of the conjugated side chain further multiplies the number of compounds generated. These findings reiterate the notion[13] that redox enzymes and MTs possess considerable substrate tolerance and play a vital role in forming various intermediates while introducing complexity into the chemical structures of those compounds.
Supplementary Material
Footnotes
This work was supported by Japan Society for the Promotion of Science (JSPS) through the “Funding Program for Next Generation World-Leading Researchers,” initiated by the Council for Science and Technology Policy (No. LS103) (K.W.) and by Industrial Technology Research Grant Program in 2009 (No. 09C46001a) from New Energy and Industrial Technology Development Organization (NEDO) of Japan (K.W.). These works were also supported in part by The Uehara Memorial Foundation (K.W.), by Mochida Memorial Foundation for Medical and Pharmaceutical Research (K.W.), by The Naito Foundation Japan (K.W.), by Nagase Science and Technology Foundation Japan (K.W.) and by JSPS Fellowship for Research In Japan (K.H.).
References
- [1].Breitenstein W, Chexal KK, Mohr P, Tamm C. Helv. Chim. Acta. 1981;64:379–388. [Google Scholar]
- [2].Maebayashi Y, Horie Y, Satoh Y, Yamazaki M. Mycotoxins. 1985;1985:33–34. [Google Scholar]
- [3].Komagata D, Fujita S, Yamashita N, Saito S, Morino T. J. Antibiot. (Tokyo) 1996;49:958–959. doi: 10.7164/antibiotics.49.958. [DOI] [PubMed] [Google Scholar]
- [4].Maiya S, Grundmann A, Li X, Li SM, Turner G. Chembiochem. 2007;8:1736–1743. doi: 10.1002/cbic.200700202. [DOI] [PubMed] [Google Scholar]
- [5].Wiemann P, Guo CJ, Palmer JM, Sekonyela R, Wang CC, Keller NP. Proc. Natl. Acad. Sci. USA. 2013;110:17065–17070. doi: 10.1073/pnas.1313258110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Asami Y, Kakeya H, Onose R, Yoshida A, Matsuzaki H, Osada H. Org. Lett. 2002;4:2845–2848. doi: 10.1021/ol020104+. [DOI] [PubMed] [Google Scholar]
- [7].Igarashi Y, Yabuta Y, Furumai T. J. Antibiot. (Tokyo) 2004;57:537–540. doi: 10.7164/antibiotics.57.537. [DOI] [PubMed] [Google Scholar]
- [8].Yue S, Duncan JS, Yamamoto Y, Hutchinson CR. J. Am. Chem. Soc. 1987;109:1253–1255. [Google Scholar]
- [9].Wu B, Dong D. Trends in pharmacological sciences. 2012;33:656–668. doi: 10.1016/j.tips.2012.09.007. [DOI] [PubMed] [Google Scholar]
- [10].Yamada T, Imai E, Nakatuji K, Numata A, Tanaka R. Tetrahedron Lett. 2007;48:6294–6296. [Google Scholar]
- [11].Dekkers KL, You BJ, Gowda VS, Liao HL, Lee MH, Bau HJ, Ueng PP, Chung KR. Fungal. Genet. Biol. 2007;44:444–454. doi: 10.1016/j.fgb.2006.08.005. [DOI] [PubMed] [Google Scholar]
- [12].Kim J, Almo SC. BMC Struct. Biol. 2013;13:5. doi: 10.1186/1472-6807-13-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Wang P, Gao X, Tang Y. Curr. Opin. Chem. Biol. 2012;16:362–369. doi: 10.1016/j.cbpa.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ishiuchi K, Nakazawa T, Yagishita F, Mino T, Noguchi H, Hotta K, Watanabe K. J. Am. Chem. Soc. 2013;135:7371–7377. doi: 10.1021/ja402828w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.