

Abstract
Ultra-performance liquid chromatography (UPLC)-electrospray ionization (ESI)-tandem mass spectrometry (MS/MS) is typically employed for phosphoproteome analysis. Alternatively, capillary zone electrophoresis (CZE) - ESI-MS/MS has great potential for phosphoproteome analysis due to the significantly different migration times of phosphorylated and unphosphorylated forms of peptides. In this work, we systematically compared UPLC-MS/MS and CZE-MS/MS for phosphorylated peptide identifications (IDs) using an enriched phosphoproteome from the MCF-10A cell line. When the sample loading amount of UPLC was 10 times higher than that of CZE (2 μg vs. 200 ng), UPLC generated more phosphorylated peptide IDs than CZE (3,313 vs. 1,783). However, when the same sample loading amounts were used for CZE and UPLC (2–200 ng), CZE-MS/MS consistently and significantly outperformed UPLC-MS/MS in terms of phosphorylated peptide and total peptide IDs. This superior performance is most likely due to the higher peptide intensity generated by CZE-MS/MS. More importantly, compared with UPLC data from 2 μg sample, CZE-MS/MS can identify over 500 unique phosphorylated peptides from 200 ng sample, suggesting that CZE and UPLC are complementary for phosphorylated peptide IDs. With further improved loading capacity via a dynamic pH junction method, 2,313 phosphorylated peptides were identified with single-shot CZE-MS/MS in a 100 min analysis. This number of phosphorylated peptide IDs is over one order of magnitude higher than the number of phosphorylated peptide IDs previously reported by single-shot CZE-MS/MS.
Introduction
Capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry (CZE-ESI-MS/MS) is attracting greater attention for proteomics research in recent years.1–4 A number of innovations significantly improve the performance of CZE-MS/MS for proteomics, including more sensitive and robust CZE-MS interfaces,5–10 higher capacity CZE separation, 11,12 improved sample loading capacity 11,13,14 and fast and high-resolution mass spectrometers.15,16 For example, the Yates group coupled CZE to an Orbitrap Elite mass spectrometer via a sheathless CZE-MS interface for top-down proteomics of the Pyrococcus furiosus; 134 proteins and 291 proteoforms were identified in two hours of CZE-MS/MS analysis.4 Furthermore, our group collaborated with the Coon group at the University of Wisconsin to couple a high-capacity CZE system to an Orbitrap Fusion mass spectrometer (up to 20 HZ MS/MS acquisition) via a second-generation electro-kinetically pumped sheath flow interface.12 Over 10,000 peptide IDs from a HeLa cell proteome digest were produced with single-shot CZE-MS/MS in a ~100 min analysis, which significantly reduced the gap between CZE and ultra-performance liquid chromatography (UPLC) for shot-gun proteomics.12
Phosphorylation of serine, threonine, and tyrosine residues is a common biological event involved in cellular signaling processes including apoptosis, growth, and differentiation.17,18 UPLC-MS/MS based phosphoproteomics has allowed researchers to routinely monitor over ten thousand of phosphorylation events from tissues or cell lines.19–22 Because phosphorylated proteins usually have low abundance compared to the rest of the proteome, enrichment of proteins or/and peptides with titanium dioxide (TiO2)23 or immobilized metal affinity chromatography (IMAC)24,25 are typically used for global phosphoproteome analysis.
Although LC-MS/MS has become a very efficient method for phosphoproteome analysis, with 38,229 detectable phosphorylation events from a single human cancer cell line,21 the contributions of multidimensional LC separation to phosphoproteome analysis tend to be saturated. The estimated level of phosphorylation in the cell is well over 100,000 sites, and therefore alternative separation techniques are necessary for further improving the depth of the phosphoproteome.26
Phosphorylated peptides tend to be more hydrophilic than unmodified peptides, which decreases their retention on reversed-phase UPLC columns and leads to sample loss during loading. Several publications have demonstrated that CZE-MS/MS tends to identify more hydrophilic peptides compared to UPLC-MS/MS, and that CZE-MS/MS has higher sensitivity compared to typical nanoUPLC-MS/MS for shot-gun proteomics.13, 27–29 In addition, phosphorylated and unphosphorylated forms of peptides can be easily separated by CZE due to their significantly different size-to-charge ratios. Therefore, CZE-MS/MS has potential as an alternative platform to UPLC-MS/MS for phosphoproteome analysis.
Though CZE-MS/MS has been established for shotgun proteomics, there are few reports regarding its ability for phosphoproteome analysis.7,30,31 Sarg et al. analyzed the post-translational modifications of histones with CZE-MS/MS and UPLC-MS/MS.30 A total of 55 phosphopeptides were identified from the IMAC enriched histone digest with CZE and UPLC; 22 of these peptides were unique to CZE-MS, and 19 were unique to LC-ESI-MS, suggesting the complementarity of the two techniques. Very recently, Faserl et al. performed quantitative proteomic analysis of yeast with a sheathless interface based CZE-MS/MS system, and 28,538 peptides that correspond to 3,272 proteins were quantified with CZE-MS/MS from 182 UPLC fractions of yeast digest in close to 200 hours of instrument time.31 About 1,500 phosphorylated peptides were also identified from those UPLC fractions. Recently, our group also demonstrated the identification of 170 phosphorylated peptides with single-shot CZE-MS/MS from Xenopus laevis egg proteome digest in ~100 min separation without phosphopeptide enrichment.7
In this work, we systematically compared the UPLC-MS/MS and CZE-MS/MS for phosphorylated peptide identifications on the proteome scale with an enriched phosphoproteome sample from the MCF-10A cell line. CZE-MS/MS consistently and significantly outperformed UPLC-MS/MS in terms of phosphorylated peptide and total peptide IDs for 2–200 ng sample loading amounts. Over 2,300 phosphorylated peptides were identified with single-shot CZE-MS/MS in a 100 min analysis duration.
Experimental Methods
Sample Preparation and Enrichment
MCF-10A human mammary cells were grown to 70% confluence and lysed in 8 M urea with 75 mM NaCl, 50 mM Tris-HCl (pH 8.2), 10 mM sodium pyrophosphate, 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF, 1 mM β-glycerophosphate, and 1 EDTA-free protease inhibitor cocktail. 3 mg of protein was denatured by at 37 °C for 1 hour, reduced with 5 mM DTT at 37 °C for 1 hour, and alkylated with 14 mM iodoacetamide for 30 minutes at room temperature. The alkylation reaction was quenched with addition of 5 mM DTT at RT for 25 minutes. Proteins were diluted with 25 mM Tris-HCl (pH 8.2) and digested with trypsin in the presence of 1 mM CaCl2 overnight at 37 °C. The resulting peptides were desalted and enriched for phosphorylated peptides using TiO2 beads (GL Sciences, Tokyo, Japan) in a 4:1 bead to peptide ratio. Peptides were then desalted and split into two samples for subsequent UPLC-MS/MS and CZE-MS/MS analysis. An additional 2 mg of protein was digested and enriched for further analysis by only CZE-MS/MS.
UPLC-MS/MS and CZE-MS/MS Analysis
For comparison of UPLC-MS/MS and CZE-MS/MS, enriched samples containing 2 ng, 20 ng, and 200 ng of phosphorylated peptides were analyzed by each technique. An additional 2,000 ng run was performed using UPLC-MS/MS. For UPLC a nanoACQUITY UltraPerformance LC system (Waters, Milford, MA) was used. Peptides were separated on a commercial C18 reversed-phase column (Waters, 100μm × 100 mm, 1.7 μm particle, BEH130C18) at 1 μL/min with a 3-step gradient separation totaling 100 minutes. The eluted peptides were analyzed by a Q-Exactive mass spectrometer (Thermo Fisher Scientific) with Top 20 data-dependent acquisition.
CZE analysis used a linear polyacrylamide (LPA) coated capillary32 (50/360 μm i.d./o.d., 85 and 90 cm length; the exterior of the distal tip of the capillary was etched to ~80 μm outer diameter. The background electrolyte was 5% acetic acid (v/v). Enriched peptides were dissolved in 30% (v/v) ACN containing 0.07% (v/v) FA. Samples were injected with 250 mbar pressure and separated over 100 minutes. 200 mbar of pressure was applied for the last 15 minutes of the separation. A third generation electro-kinetically pumped sheath flow interface7 was used to couple the CZE system to a Q-Exactive mass spectrometer (Thermo Fisher Scientific) operating with identical parameters as the UPLC analysis. For single-shot CZE analysis with the dynamic pH junction, peptides were dissolved in 20 μL of 10 mM ammonium acetate (pH ~6.0) and separated with an 85 cm LPA coated capillary. Sample injection was performed using 250 mbar pressure for 0.8 minutes. All other parameters were identical to those described above.
Data Analysis
All raw mass spectrometer files were analyzed in Proteome Discoverer 1.4 (Thermo Fischer Scientific) with MASCOT version 2.2.4 against the UniProt human database (89,601 sequences). Reverse sequence databases were searched to determine the false discovery rate (FDR). Percolator (version 2.04) software was used to evaluate search results, and reported peptides corresponded to an FDR level below 1%. The peptide identification lists from CZE and UPLC are listed in supporting material I. Raw files were also searched using MaxQuant33 version 1.3.0.5. with the Andromeda search engine34. FDR was set to 0.01 for peptide and protein identifications. Extended descriptions of all materials and methods can be found in supporting material II.
Results and Discussion
Sarg et al. demonstrated that CZE-MS/MS is complementary to UPLC-MS/MS for histone post-translational modifications analysis, especially phosphorylation, and about 50 phosphorylated peptides were identified by CZE and UPLC.30 It is valuable to further evaluate the complementarity of CZE and UPLC for phosphorylated peptide analysis with a proteome scale dataset. To do this, we enriched phosphorylated peptides from 3 mg of MCF-10A cell proteome digest with TiO2 beads and divided the enriched peptides into two equal aliquots for CZE and UPLC. It is important to note that we assumed a 5% yield during TiO2 enrichment process in order to estimate the sample loading amounts.
CZE-MS/MS produced a wider separation window than UPLC, as seen in Figure S-1 in supporting material II. For CZE, the separation window is approximately 80 min (from 20 min to 100 min) for 200 ng loading amount; for UPLC, the separation window is limited to 70 min (from ~10–80 min) for 200 ng and 2,000 ng loading amounts due to sample loading, column flushing, and column equilibration time. Peaks seen in the chromatograms of S-Figure 1 after 80 minutes are not likely related to peptide signals. CZE-MS/MS consistently produced higher base peak intensities than UPLC-MS/MS for 2–200 ng loading amounts (S-Figure 1) demonstrating the higher sensitivity of CZE-MS, which agrees with previous reports.13,27–30 When 2 μg of sample was loaded for UPLC, the base peak intensity from UPLC is about two times higher than that from CZE with the 200 ng loading amount.
We further compared the peptide and protein identifications with CZE-MS/MS and UPLC-MS/MS from enriched phosphorylated peptide samples (Table 1). CZE-MS/MS significantly outperforms UPLC-MS/MS in terms of peptide, phosphorylated peptide, and protein IDs when the same sample amounts (2–200 ng) were loaded for analysis, which is most likely due to the reduced potential sample loss and higher sensitivity of CZE. As expected, the phosphorylated peptide IDs from 2 μg of sample with UPLC-MS/MS are higher than that from 200 ng of sample with CZE-MS/MS (3,313 vs. 1,783).
Table 1.
Summary of phosphoproteome results from single-shot CZE-MS/MS and UPLC-MS/MS.*
| Single-shot CZE-MS/MS | Single-shot UPLC-MS/MS | |||||
|---|---|---|---|---|---|---|
| Loading amounts (ng) | Total Peptide IDs | Phosphopeptide IDs | Protein IDs | Total Peptide IDs | Phosphopeptide IDs | Protein IDs |
| 2000 | 5486 | 3313 | 1931 | |||
| 200 | 2725 | 1783 | 1159 | 1179 | 743 | 549 |
| 20 | 1286 | 926 | 577 | 82 | 44 | 58 |
| 2 | 175 | 119 | 100 | 49 | 6 | 28 |
The data listed in the table is from Proteome Discoverer 1.4. The number of peptide and phosphopeptide IDs listed in the table are the number of unique peptide sequences.
We next determined the overlap of phosphorylated peptides identified by CZE-MS/MS and UPLC-MS/MS (Figure 1). Although single-shot CZE-MS/MS generated significantly higher phosphorylated peptide IDs than single-shot UPLC-MS/MS when 200 ng of sample was loaded (1,783 vs. 743), only 66% of the phosphorylated peptides identified by UPLC are covered by CZE data, while 247 phosphorylated peptides are unique to UPLC (Figure 1A). When data from duplicate UPLC runs were compared (Figure 1B), the overlap between CZE and UPLC is similar to Figure 1A. Single-shot UPLC-MS/MS yields a greater number of phosphorylated peptide IDs than single-shot CZE-MS/MS (3,313 vs. 1,783) when 2,000 ng and 200 ng of materials were loaded, respectively. Only 70% of the phosphorylated peptides identified by CZE are covered by UPLC data, while 518 phosphorylated peptides are unique to CZE-MS/MS (Figure 1C). About 4,000 phosphorylated peptides were identified by UPLC in duplicate runs (2,000 ng/run), which is over two times higher than the single-shot CZE-MS/MS data (200 ng). However, 22% of the phosphorylated peptides identified by CZE-MS/MS were still not detected by UPLC (Figure 1D). These results clearly demonstrate that CZE and UPLC are complementary for phosphorylated peptide identifications, which agrees with previous findings.30
Figure 1.
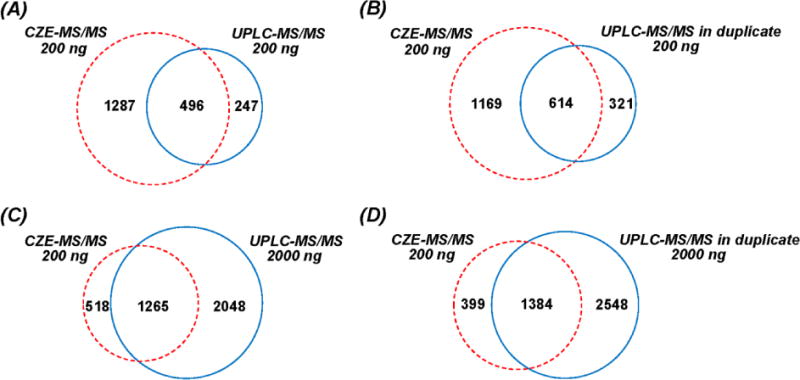
Overlap of identified phosphorylated peptides with CZE-MS/MS and UPLC-MS/MS. (A) single-shot CZE (200 ng) vs. single-shot UPLC (200 ng); (B) single-shot CZE (200 ng) vs. UPLC in duplicate runs (200 ng/run); (C) single-shot CZE (200 ng) vs. single-shot UPLC (2 000 ng); (D) single-shot CZE (200 ng) vs. UPLC in duplicate runs (2 000 ng/run). Data from Proteome Discoverer 1.4 were used.
To gain further information from this data, we used MaxQuant33 to obtain the peptide intensity information for CZE and UPLC comparisons. We first compared the peptide intensity from CZE and UPLC for 300 ng of the MCF-10A cell proteome digest without TiO2 enrichment (Figure 2A). We summed the peptide intensity from both CZE and UPLC data for peptides identified in both datasets (overlapping peptides) based on MS/MS spectra and used the summed peptide intensity to represent the peptide abundance in the sample. On average, CZE produces peptide spectra with 2.6 times higher intensity than UPLC, which agrees with our recent report.7 For the enriched phosphopeptide sample from MCF-10A cell proteome digest, CZE generates spectra with an average of 6.7 times higher peptides intensity than UPLC (Figure 2B). The difference between Figure 2A and 2B may be due to the lower pH of the separation buffer of CZE (5% acetic acid, pH ~2.4) than for UPLC (0.1% formic acid, pH ~2.7), which benefits the electrospray ionization of the phosphorylated peptides. Figure 2C shows the correlation of sample loading amounts and summed intensity of all the peptides identified in each experiment from CZE and UPLC. CZE yields significantly higher total peptide intensity than UPLC for 2–200 ng loading amounts, and UPLC generates reasonably higher total peptide intensity than CZE when 10 times higher loading amount was used for UPLC (2,000 ng for UPLC vs. 200 ng for CZE). For both CZE and UPLC, the logarithm of peptide intensity increases in a linear fashion with the logarithm of sample loading amount. The slope of the log-log plot for the CZE data is very close to 1, which demonstrates a linear dependence of signal on the loading amount. In contrast, the slope of the log-log plot for UPLC has a slope of 1.33, which implies that the signal for UPLC increases with peptide concentration, raised to the 1.33 power. This nonlinear behavior is likely due to peptide loss for small sample loadings in UPLC.
Figure 2.
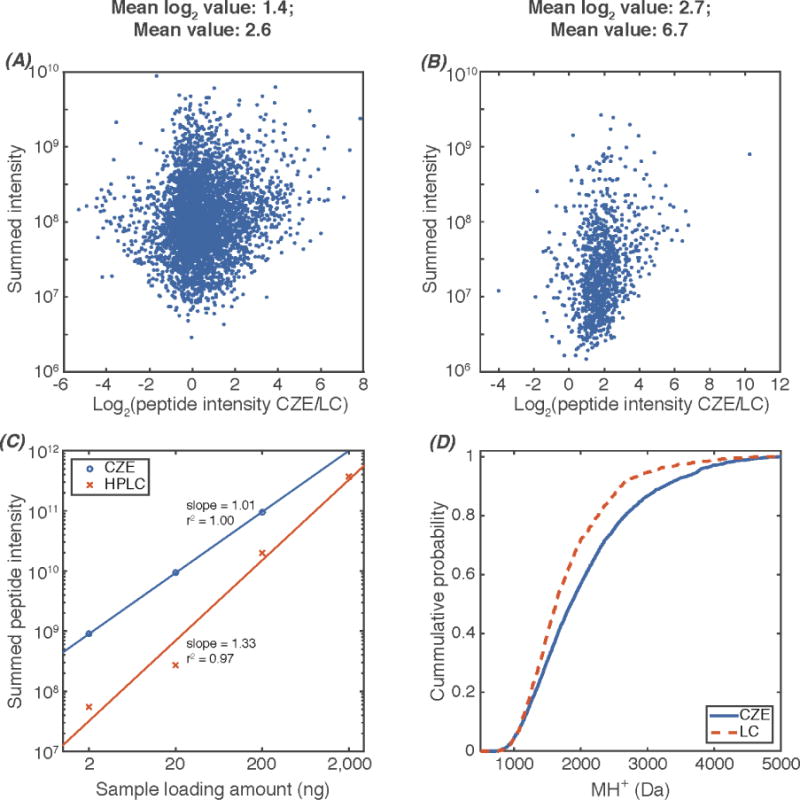
Comparisons of CZE-MS/MS and UPLC-MS/MS. (A) Log2 (peptide intensity CZE/LC) vs. summed peptide intensity, data from MCF-10A cell proteome digest without TiO2 enrichment. 300 ng proteome digest was loaded for CZE and UPLC; (B) Log2 (peptide intensity CZE/LC) vs. summed peptide intensity, data from enriched phosphorylated peptides from MCF-10A cell proteome digest with TiO2. 200 ng sample was loaded for CZE and UPLC; (C) sample loading amount vs. summed peptide intensity; (D) Molecular weight distributions of identified peptides with CZE and UPLC from 200 ng samples. Data from MaxQuant were used.
We further determined the molecular weight distribution of identified peptides with CZE and UPLC from 200 ng samples (Figure 2D). CZE tends to identify larger peptides compared with UPLC, which agrees with our previous reports7,12,13
For the CZE experiments discussed above, 30% (v/v) ACN containing 0.07% (v/v) FA was used as the sample buffer. The sample buffer’s conductivity is much lower than the background electrolyte (5% (v/v) acetic acid), which results in significant sample stacking at the beginning of the separation, improving the loading capacity of CZE. However, in our previous report, we demonstrated that the dynamic pH junction method produced better enrichment performance than the typical stacking method when the sample loading volume was very large.14 To test the identification capacity of single-shot CZE-MS/MS for complex phosphoproteome samples, we performed phosphopeptide enrichment with TiO2 from 2 mg of MCF-10A cell proteome digest, and dissolved the sample in 20 μL of 10 mM ammonium acetate (pH ~6.0), followed by dynamic pH junction based single-shot CZE-MS/MS analysis with about 200 nL loading volume (Figure 3). This loading amount is approximately 1 μg. After 100-min single-shot CZE-MS/MS analysis, 2,313 phosphorylated peptides were identified, which represents the largest number of phosphorylated peptide IDs from single-shot CZE-MS/MS. These results are over one order of magnitude higher than the phosphorylated peptide IDs previously reported in the literature with single-shot CZE-MS/MS.
Figure 3.
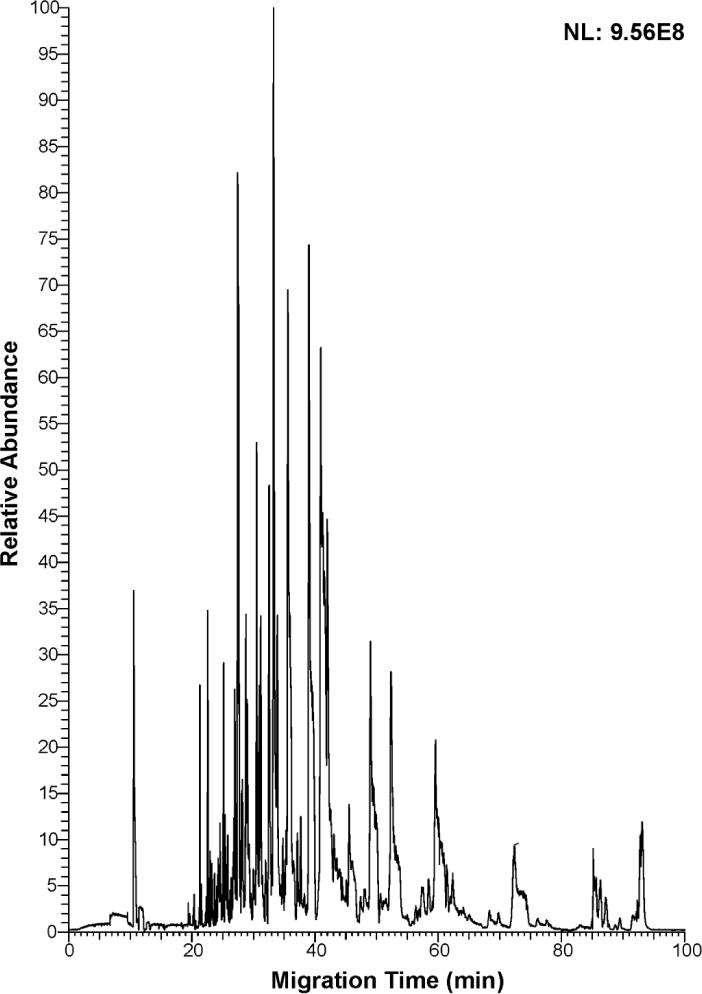
Electropherogram of enriched phosphopeptides from MCF-10A cell proteome digest analyzed by single-shot CZE-MS/MS. 2 mg of MCF-10A cell proteome digest was used for phosphopeptide enrichment, and the enriched sample was dissolved in 20 μL of 10 mM ammonium acetate (pH ~6.0), followed by dynamic pH junction based single-shot CZE-MS/MS analysis with about 200 nL loading volume (~1 µg loading amount).
We then evaluated the overlap of phosphorylated peptides identified by the CZE-MS/MS discussed above and the UPLC-MS/MS run with 2 μg sample loading amount. 1,430 phosphorylated peptides were identified by both CZE and UPLC, representing 62% of the total phosphorylated peptides from CZE (2,313 phosphorylated peptides in total). Combination of CZE and UPLC produces 27% more phosphorylated peptide IDs than UPLC alone (4,196 vs. 3,313), which clearly indicates the value of CZE-MS/MS for phosphoproteomics.
We further analyzed the migration time/retention time distributions of the identified phosphorylated and unphosphorylated peptides from the CZE-MS/MS run mentioned above and the UPLC-MS/MS run with 2 μg sample loading amount (Figure 4). For CZE-MS/MS, the phosphorylated peptides tend to move through the capillary more slowly than unphosphorylated peptides. This is likely due to the reduction of positive charge of phosphorylated peptides because of the negatively charged phosphate group, which yields larger size-to-charge ratio than unphosphorylated peptides. For UPLC-MS/MS, the retention of phosphorylated peptides on UPLC column tends to be slightly weaker than that of unphosphorylated peptides, which is likely due to their increased hydrophilicity from the phosphate group. The results here suggest that CZE cis superior to UPLC for separating phosphorylated and unphosphorylated peptide pools from complex proteome digests, which benefits phosphorylated peptide identifications by decreasing interferences from unphosphorylated peptides. This feature makes CZE-MS/MS a promising tool for large-scale phosphoproteomics.
Figure 4.
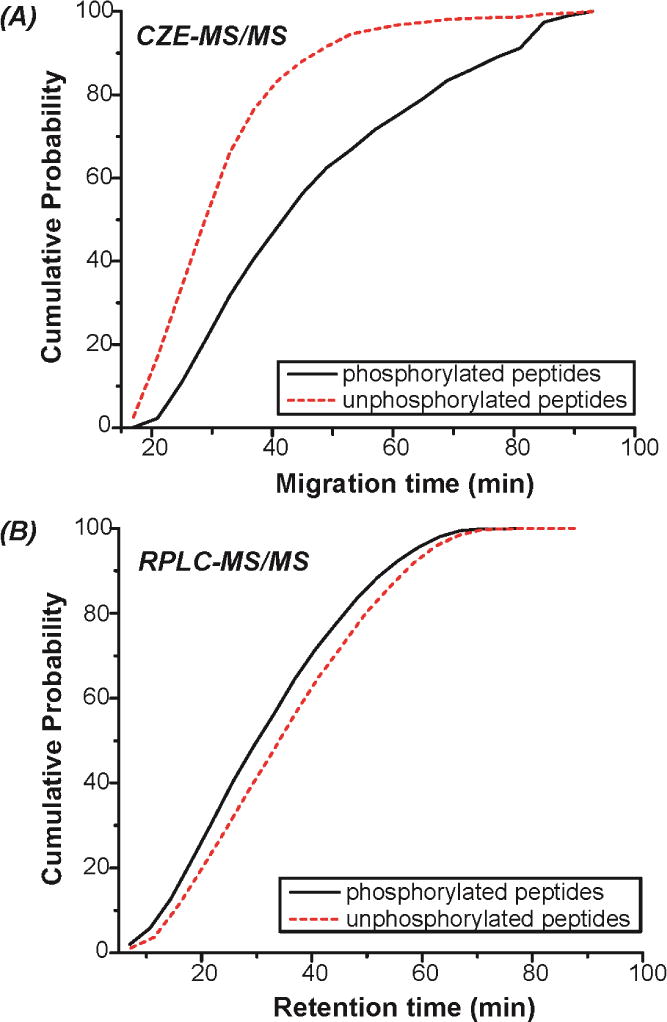
(A) Migration time distributions of phosphorylated and unphosphorylated peptides identified by single-shot CZE-MS/MS. The experimental conditions are the same as Figure 3. (B) Retention time distributions of phosphorylated and unphosphorylated peptides identified by single-shot UPLC-MS/MS with 2 μg sample loading amount.
Conclusions
CZE-ESI-MS/MS can produce better peptide sensitivity than UPLC-MS/MS, and CZE outperforms UPLC for 2–200 ng sample loading amounts in terms of phosphorylated peptide IDs. CZE and UPLC are complementary separations for phosphopeptide identifications, and the combination of both techniques can improve the coverage of phosphoproteomes. Over 2 300 phosphorylated peptides can be identified with single-shot CZE-MS/MS in 100 min analysis duration, which clearly suggests the great potential of CZE-MS/MS for large-scale phosphoproteome analysis.
We will employ UPLC based phosphopeptide prefractionation and both CZE-MS/MS and UPLC-MS/MS for large-scale qualitative and quantitative phosphoproteome analysis in our future work. CZE-MS/MS and UPLC-MS/MS data will be combined to improve the phosphoproteome coverage.
Supplementary Material
Acknowledgments
We thank Dr. William Boggess in the Notre Dame Mass Spectrometry and Proteomics Facility for his help with this project. This work was funded by the National Institutes of Health (R01GM096767-NJD), (R01GM110406-ABH), and the National Science Foundation (CAREER Award, CHE-1351595 to ABH). The Walther Cancer Foundation provided salary support for ABH.
Footnotes
Supporting Information
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Sun L, Zhu G, Yan X, Dovichi NJ. Curr Opin Chem Biol. 2013;17:795–800. doi: 10.1016/j.cbpa.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun L, Zhu G, Yan X, Champion MM, Dovichi NJ. Proteomics. 2014;14:622–628. doi: 10.1002/pmic.201300295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heemskerk AA, Deelder AM, Mayboroda OA. Mass Spectrom Rev. 2014 doi: 10.1002/mas.21432. [DOI] [PubMed] [Google Scholar]
- 4.Han X, Wang Y, Aslanian A, Bern M, Lavallée-Adam M, Yates JR., 3rd Anal Chem. 2014;86:11006–11012. doi: 10.1021/ac503439n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wojcik R, Dada OO, Sadilek M, Dovichi NJ. Rapid Commun Mass Spectrom. 2010;17:2554–60. doi: 10.1002/rcm.4672. [DOI] [PubMed] [Google Scholar]
- 6.Sun L, Zhu G, Zhao Y, Yan X, Mou S, Dovichi NJ. Angew Chem, Int Ed Engl. 2013;51:13661–4. doi: 10.1002/anie.201308139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun L, Zhu G, Zhang Z, Mou S, Dovichi NJ. J Proteome Res. 2015;14:2312–21. doi: 10.1021/acs.jproteome.5b00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moini M. Anal Chem. 2007;79:4241–4246. doi: 10.1021/ac0704560. [DOI] [PubMed] [Google Scholar]
- 9.Maxwell EJ, Zhong X, Zhang H, van Zeijl N, Chen DD. Electrophoresis. 2010;31:1130–1137. doi: 10.1002/elps.200900517. [DOI] [PubMed] [Google Scholar]
- 10.Wang C, Lee CS, Smith RD, Tang K. Anal Chem. 2013;85:7308–7315. doi: 10.1021/ac401202c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Busnel JM, Schoenmaker B, Ramautar R, Carrasco-Pancorbo A, Ratnayake C, Feitelson JS, Chapman JD, Deelder AM, Mayboroda OA. Anal Chem. 2010;82:9476–9483. doi: 10.1021/ac102159d. [DOI] [PubMed] [Google Scholar]
- 12.Sun L, Hebert AS, Yan X, Zhao Y, Westphall MS, Rush MJ, Zhu G, Champion MM, Coon JJ, Dovichi NJ. Angew Chem, Int Ed Engl. 2014;53:13931–3. doi: 10.1002/anie.201409075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu G, Sun L, Yan X, Dovichi NJ. Anal Chem. 2013;85:2569–73. doi: 10.1021/ac303750g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu G, Sun L, Yan X, Dovichi NJ. Anal Chem. 2014;86:6331–6. doi: 10.1021/ac5004486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelstrup CD, Young C, Lavallee R, Nielsen ML, Olsen JV. J Proteome Res. 2012;11:3487–97. doi: 10.1021/pr3000249. [DOI] [PubMed] [Google Scholar]
- 16.Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS, Coon JJ. Mol Cell Proteomics. 2014;13:339–47. doi: 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harsha HC, Pandey A. Mol Oncol. 2010;4:482–95. doi: 10.1016/j.molonc.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iliuk A, Liu XS, Xue L, Liu X, Tao WA. Mol Cell Proteomics. 2012;11:629–39. doi: 10.1074/mcp.O112.018010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yue XS, Hummon AB. J Proteome Res. 2013;12:4176–86. doi: 10.1021/pr4005234. [DOI] [PubMed] [Google Scholar]
- 20.Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJ, Mohammed S. J Proteome Res. 2013;12:260–71. doi: 10.1021/pr300630k. [DOI] [PubMed] [Google Scholar]
- 21.Sharma K, D’Souza RC, Tyanova S, Schaab C, Wiśniewski JR, Cox J, Mann M. Cell Rep. 2014;8:1583–94. doi: 10.1016/j.celrep.2014.07.036. [DOI] [PubMed] [Google Scholar]
- 22.Wang F, Song C, Cheng K, Jiang X, Ye M, Zou H. Anal Chem. 2011;83:8078–85. doi: 10.1021/ac201833j. [DOI] [PubMed] [Google Scholar]
- 23.Thingholm TE, Jørgensen TJ, Jensen ON, Larsen MR. Nat Protoc. 2006;1:1929–35. doi: 10.1038/nprot.2006.185. [DOI] [PubMed] [Google Scholar]
- 24.Villén J, Gygi SP. Nat Protoc. 2008;3:1630–8. doi: 10.1038/nprot.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou H, Ye M, Dong J, Corradini E, Cristobal A, Heck AJ, Zou H, Mohammed S. Nat Protoc. 2013;8:461–80. doi: 10.1038/nprot.2013.010. [DOI] [PubMed] [Google Scholar]
- 26.Lemeer S, Heck AJ. Curr Opin Chem Biol. 2009;13:414–20. doi: 10.1016/j.cbpa.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Champion MM, Sun L, Champion PA, Wojcik R, Dovichi NJ. Anal Chem. 2012;84:1617–22. doi: 10.1021/ac202899p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Fonslow BR, Wong CC, Nakorchevsky A, Yates JR., 3rd Anal Chem. 2012;84:8505–13. doi: 10.1021/ac301091m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faserl K, Sarg B, Kremser L, Lindner HH. Anal Chem. 2011;83:7297–305. doi: 10.1021/ac2010372. [DOI] [PubMed] [Google Scholar]
- 30.Sarg B, Faserl K, Kremser L, Halfinger B, Sebastiano R, Lindner HH. Mol Cell Proteomics. 2013;12:2640–56. doi: 10.1074/mcp.M112.024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Faserl K, Kremser L, Müller M, Teis D, Lindner HH. Anal Chem. 2015;87:4633–40. doi: 10.1021/acs.analchem.5b00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu G, Sun L, Dovichi NJ. Talanta. doi: 10.1016/j.talanta.2015.06.003. [DOI] [Google Scholar]
- 33.Cox J, Mann M. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 34.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. J Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.