

Abstract
Psychostimulant addiction is associated with dysfunctions in frontal cortex. Previous data demonstrated that repeated exposure to methamphetamine (METH) can alter prefrontal cortex (PFC) dependent functions. Here, we show that withdrawal from repetitive non-contingent METH administration (7 days, 1mg/kg) depressed voltage-dependent calcium currents (ICa) and increased IH amplitude and the paired-pulse ratio of evoked EPSCs in deep-layer pyramidal mPFC neurons. Most of these effects were blocked by systemic co-administration of the D1/D5 receptor antagonist SCH23390 (0.5 and 0.05 mg/kg). In vitro METH (i.e bath-applied to slices from naïve-treated animals) was able to emulate its systemic effects on ICa and evoked EPSCs paired-pulse ratio. We also provide evidence of altered mRNA expression of i) voltage-gated calcium channels P/Q-type Cacna1a (Cav2.1), N-type Cacna1b (Cav2.2), T-type Cav3.1 Cacna1g, Cav3.2 Cacna1h, Cav3.3 Cacna1i and the auxiliary subunit Cacna2d1 (α2δ1), ii) hyperpolarization-activated cyclic nucleotide-gated channels Hcn1 and Hcn2 and iii) glutamate receptors subunits AMPA-type Gria1, NMDA-type Grin1 and metabotropic Grm1 in the mouse mPFC after repeated METH treatment. Moreover, we show that some of these changes in mRNA expression were sensitive D1/5 receptor blockade.
Altogether these altered mechanisms affecting synaptic physiology and transcriptional regulation may underlie prefrontal cortex functional alterations that could lead to PFC impairments observed in METH-addicted individuals.
Keywords: dopamine receptors, glutamate, prefrontal cortex, methamphetamine, voltage-gated calcium channels
INTRODUCTION
The prefrontal cortex (PFC) has an essential role in cognition, emotion and reward (Goldman-Rakic et al., 2000). The PFC roles that are most pertinent to addiction include self-control, salience attribution and maintenance of motivational arousal (Berridge and Robinson, 1998; Schultz, 2007). Indeed, dysfunctions in the PFC synaptic circuits are thought to be responsible for the loss of control over drug taking that characterizes addicted individuals (Goldstein and Volkow, 2011). Repeated exposure to psychostimulants like methamphetamine (METH), which increase dopamine (DA) neurotransmission, causes abnormal changes in neurotransmitter activity and neuronal plasticity in different areas of the PFC that mediate cognition and motivation (Cadet et al., 2014; Kauer and Malenka, 2007). These neural deficits might be related to changes in the PFC glutamatergic transmission observed both in humans and animal models (Ernst and Chang, 2008; Parsegian and See, 2014). Glutamate NMDA, AMPA, and metabotropic receptors are key mediators of signal transduction events that convert environmental stimuli into genetic changes through regulation of gene transcription and epigenetic chromatin remodeling in neural cells (Cohen and Greenberg, 2008). The dynamic expression of glutamate receptors at synapses is a key determinant of synaptic plasticity, including neuroadaptations to drugs of abuse (Kalivas, 2007, Jayanthi et al., 2014).
Voltage-gated calcium channels (VGCC) control neurotransmitter release throughout their capacity to transiently increase intracellular calcium concentration ([Ca2+]i), influence neuronal excitability, regulate gene transcription and synaptic plasticity (Kawamoto et al., 2012). VGCC play a dual role in the CNS; they couple electrical activity to Ca2+ influx, thereby triggering myriad intracellular biochemical events, and they contribute to membrane properties that determine the precise nature of excitability in different cell types. Low T-type VGCCs have a hand in both of these roles that is readily distinguishable from their high P/Q-, N-, R- and L-types VGCCs counterparts, in part because they activate at potentials near the resting membrane potential (RMP). Hence, in addition to be involved in spike-induced Ca2+ entry, they allow Ca2+ influx at potentials below threshold. This influx can occur when cells are at rest or in response to subthreshold synaptic inputs (Talley et al., 1999; Kawamoto et al., 2012). The VGCC are heteromeric complexes comprised of a pore-forming α1 subunit with ancillary β, α2, and γ subunits. It was suggested that VGCC are implicated in drug-related plasticity and addictive behavior (Dolphin, 2012). Repeated non-contingent METH was found to increase α2δ1 subunit in frontal cortex (Kurokawa et al., 2010). This auxiliary α2δ1 subunit interacts with VGCC, including presynaptic release-regulating N-type and P/Q-type, as well as postsynaptic L-type channels, elevating the current amplitude and increasing VGCC trafficking (Dolphin, 2012).
Another conductance modulated by [Ca2+]i is the hyperpolarization-activated cation current (IH) (Lüthi and McCormick, 1998). IH is involved in the control of neuronal rhythmic activity, setting the RMP, as well as modulation of neurotransmission and synaptic plasticity. IH influx through hyperpolarization-activated cyclic nucleotide-gated channels (HCN) depolarizes the membrane potential, increasing the number of VGCC open and then inactivated at RMP. Decreases in Ca2+ influx, leading to the constraint of distal dendritic Ca2+ spikes may contribute to the sharpening of incoming signals that would be integrated at the soma (He et al., 2014).
Although it is reasonable to suggest that some of the PFC effects of METH might depend on the modulation of voltage-dependent channels that affect neuronal firing, direct evidence is lacking. Here we show that repeated METH treatment i) decreased Ca2+ influx, ii) increased IH amplitude, iii) decreased the frequency of mini excitatory post synaptic currents (EPSCs) and increased the paired-pulse ratio (PPR) of evoked EPSCs in mPFC deep-layer pyramidal neurons, through a D1/5 receptor-dependent mechanism. Interestingly, the same effects on Ca2+ influx and glutamate release probability were also observed when METH was bath-applied to naïve mPFC slices. We also provide evidence of altered mRNA expression of VGCC, HCN and glutamate receptors subunits in the mouse mPFC after repeated METH treatment. Moreover, we show that some of these changes in mRNA expression were sensitive D1/5 receptor blockade.
MATERIALS AND METHODS
Animals
C57BL/6 male mice (2-3 month-old) from the School of Exact and Natural Sciences of the University de Buenos Aires (UBA) were housed in a light- and temperature-controlled room. Principles of animal care were followed in accordance with ‘Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research’ (National Research Council, 2003) and approved by Universidad de Buenos Aires authorities (Protocol Number: A5801-01) using OLAW and ARENA directives (NIH, Bethesda, USA).
Drug treatments
The drugs used were (+)-methamphetamine hydrochloride (Sigma, St Louis, MO) and SCH23390 hydrochloride (TOCRIS bioscience, Ellisville, MO). All drugs were diluted with 0.9% saline. We have selected the METH dose based on previous studies showing that repeated non-contingent 7 days METH treatment (1 mg/kg) was able to induce cognitive impairments associated with deficits of the ERK1/2 pathway in the mPFC (González et al., 2014; also see Kamei et al., 2006). In the present study, METH was administered subcutaneously (s.c.) once a day for 7 days (1 mg/Kg, calculated as free base). To test D1/5 receptors effect, animals received intraperitoneal (i.p.) injections of the D1 receptor antagonist, SCH 23390 (0.5 and 0.05 mg/kg, for 7 days), 30 min before each injection of saline or METH. Vehicle groups received the same volume of sterile saline solution. Mice were killed 4 days after the last METH/saline injection.
Whole-cell patch-clamp recordings
For slice recordings, 46 mice were included in this study. At 4 day of METH withdrawal, control or METH/SCH23390 treated mice were deeply anesthetized with tribromoethanol (250 mg/Kg; i.p.) followed by transcardial perfusion with ice-cold low sodium/antioxidants solution (composition in mM: 200 sucrose, 2.5 KCl, 3 MgSO4, 26 NaHCO3, 1.25 NaH2PO4, 20 d-glucose, 0.4 ascorbic acid, 2 pyruvic acid, 1 kynurenic acid, 1 CaCl2, and aerated with 95% O2/5% CO2, pH 7.4), and then decapitated as previously described (Urbano et al., 2009; Bisagno et al., 2010). Coronal brain slices, including mPFC (300 μm) were obtained gluing both hemispheres with the caudal part onto a vibrotome aluminum stage (Integraslicer 7550 PSDS, Campden Instruments, UK), submerged in a chamber containing chilled low-sodium/high-sucrose solution (composition in mM: 250 sucrose, 2.5 KCl, 3 MgSO4, 0.1 CaCl2, 1.25 NaH2PO4, 0.4 ascorbic acid, 3 myo-inositol, 2 pyruvic acid, 25 d-glucose, and 25 NaHCO3). Slices were cut sequentially and transferred to an incubation chamber at 37°C for 30 min containing a low Ca2+/high Mg2+ normal ACSF (composition in mM: 125 NaCl, 2.5 KCl, 3 MgSO4, 0.1 CaCl2, 1.25 NaH2PO4, 0.4 ascorbic acid, 3 myo-inositol, 2 pyruvic acid, 25 d-glucose, and 25 NaHCO3 and aerated with 95% O2/5% CO2, pH 7.4; Urbano et al., 2009, Bisagno et al., 2010). After incubation, slices were allowed to return to room temperature.
Whole-cell patch clamp recordings were made at room temperature (20-24°C) in normal ACSF with MgCl2 (1 mM) and CaCl2 (2 mM). Patch electrodes were made from borosilicate glass (2-3 MΩ) filled with a voltage-clamp high Cl—, high Cs+/QX314 solution (composition in mM: 110 CsCl, 40 HEPES, 10 TEA-Cl, 12 Na2phosphocreatine, 0.5 EGTA, 2 Mg-ATP, 0.5 Li-GTP, and 1 MgCl2. pH was adjusted to 7.3 with CsOH). To block Na+ currents and avoid postsynaptic action potentials, 10 mM N-(2,6-diethylphenylcarbamoylmethyl) triethylammonium chloride (QX-314) was added to the pipette solution (Urbano et al., 2009, Bisagno et al., 2010). Signals were recorded using a MultiClamp 700 amplifier commanded by pCLAMP 10.0 software (Molecular Devices, CA, USA). Data were filtered at 5 kHz, digitized and stored for off-line analysis. Capacitance and leak-currents were electronically subtracted using a standard pCLAMP P/N substraction protocol.
Pyramidal prelimbic cortex deep-V-VI layer neurons were used in this study (see Figure 1). At initial stages of this study, deep-layer V-VI pyramidal neurons were confirmed morphologically using calcium green intracellular containing (50 μM; Invitrogen) regular high Cs+/QX314 intracellular solution. Pyramids were visualized using Nomarski contrast on a upright BX-50WI microscope (40×, 0.8 numerical aperture [NA] water-immersion objective), illuminated with a monochromator (Polychrome V; Till Photonics) to excite calcium green at 488 nm, and fluorescence emission was recorded with a CCD camera (iXon+; Andor), controlled by Cell R software (Olympus).
Figure 1. Schematic figure showing morphological features of a recorded prelimbic cortex deep-layer V-VI pyramidal neuron.
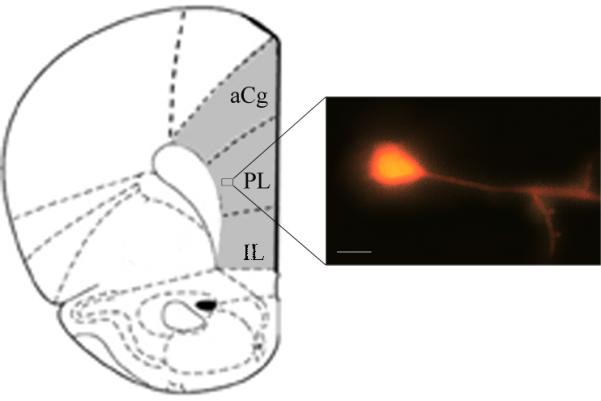
The gray area depicts the tissue zone isolated for RT-PCR assays. aCG: anterior cingulate cortex, PL: prelimbic cortex, IL: infralimbic cortex. Scale bar: 100 μm.
When recording AMPA receptor mediated miniature excitatory postsynaptic currents (mEPSC) tetrodotoxin (TTX, 3 μM), DL-2-amino-5-phosphonopentanoic acid (DL-AP5, 50μM) and bicuculline (20 μΜ) were included in the ACSF to block NMDA and GABA-A receptors, respectively. Holding potential was fixed at −70 mV. Spontaneous mIPSC were analyzed using Mini Analysis software (Version 6.0.7; Synaptosoft, Chapel Hill, NC, USA, www.synaptosoft.com). The threshold for mini detection was set at fivefold the RMS baseline noise. Cumulative amplitude and inter-event interval curves of mIPSCs/mEPSCs were obtained from each mPFC neuron recorded, and fitted to the exponential equation: f=y0+a*(1-exp(-Time(ms)/τ). Median amplitudes and mini intervals from each neuron recorded were statistically compared between experimental groups.
Evoked excitatory postsynaptic currents (EPSC) were evoked extracellularly (twice the threshold; 40–200 μs; 200–1000 μA) using a bipolar concentric electrode (FHC Inc, ME, USA) attached to a motorized micromanipulator MPC200/ROE200 (Sutter Instrument, CA, USA) and located on the deep-layers border of mPFC. Using the same high Cl− high Cs+/Qx-314intracellular solution described above and an extracellular ACSF solution containing DL-AP5 (50μM) and bicuculline (20 μΜ), sixteen stimuli of 10 Hz paired pulse protocol were delivered. EPSC paired pulse ratio calculated as the fraction of EPSC2 /EPSC1 amplitudes. Voltage dependent calcium currents (ICa) were obtained using a high-Cs+ pipette solution in the presence of excitatory (DL-AP5, 50μM; CNQX, 20μM), tetrodotoxin (TTX, 3μM) and inhibitory (Bicuculline, 20μM) synaptic receptors blockers. Square voltage pulses (200 msec, holding −70mV, pulses ranging from −55 to 30 mV, 5 mV steps) were used to elicit calcium currents.
IH recordings of mPFC deep-layer pyramidal neurons were implemented in the presence of TTX (3 μM), DL-AP5 (50 μM), CNQX (20 μM), and bicuculline (20 μM), with a slightly modified ACSF solution (in mM): 124 sodium chloride, 5 KCl , 10 dglucose, 26 NaHCO3, 2 CdCl2, 3 MgCl2, in combination with an intracellular high-K+ pipette solution (110 K-gluconate, 30 KCl, 10 HEPES, 10 Na-phosphocreatine, 0.2 EGTA, 2 Mg-ATP, 0.5 Li-GTP, and 1 MgCl2. pH was adjusted to 7.3 with KOH; Urbano et al., 2009). Holding potential was fixed at −40 mV throughout experiments, and 500 msec-long hyperpolarizing voltage holding pulses were transiently imposed from −60 to −140 mV, in 10 mV steps.
Real time PCR
Mice were killed 4 days after treatments and their brains were rapidly removed; mPFC tissues were dissected (Figure 1), placed on dry ice, and then stored at −70°C in RNAlater (Qiagen) until further assays. Total RNA was isolated using TRIZOL reagent (Invitrogen) according to the manufacturer's protocol. Five hundred nanograms of RNA were treated with DNAseI (Invitrogen) and reverse-transcribed in a 20 μL reaction using M-MLV reverse transcriptase (Promega) and random hexameres (Biodynamics). For quantitative real-time PCR (qPCR) primers sets were designed for the specific amplification of murine Cacna1a, Cacna1b, Cacna1c, Cacna1g, Cacna1h, Cacna1i, Cacna2d1, Hcn1, Hcn2, Gria1, Grin1, Grm1 and Gapdh as housekeeping control gene (sequences listed in Table 1). Each sample was assayed in duplicate using 4 pmol of each primer, 1X SYBR Green Master Mix (Applied Biosystems) and 2–20 ng of cDNA in a total volume of 13 μL. Amplification was carried out in an ABI PRISM 7500 Sequence Detection System (Applied Biosystems).
Table 1.
primer sequences
| Gene | Ac. Number | Primer forward | Primer reverse |
|---|---|---|---|
| Cacna1a | NM_007578 | CGGAGCACAATAACTTCCGG | CGAGCACAGGAAGATGAACG |
| Cacna1b | NM_001042528 | CTGTACAACCCCATCCCAGT | TCCGTGTCATCTAGTCGCTC |
| Cacna1c | NM_009781 | GCTGTACTGGATGCAAGACG | CCAGTCCAGGTAGCCTTTGA |
| Cacna1g | NM_009783 | TCTGCCCAATGACAGCTACA | TTGGGAAGCTGTAGGATGCA |
| Cacna1h | NM_021415 | TGGAGGAGAGCAACAAGGAG | CACAGATACTTTGCGCACGA |
| Cacna1i | NM 001044308 | TCTGTCATGGGCATCACACT | AGAGCCTGTACCACTGTGTC |
| Cacna2d1 | NM_001110843 | GCAGCCCAGATACCGAAAAG | TCGTTGCAGATCTGGGTTCT |
| Hcn1 | NM_010408 | GCTAACGCCGATCCCAATTT | CAGCAGGCATATCTCTCCGA |
| Hcn2 | NM_008226 | GATGGCTCCTATTTCGGGGA | TAGCCACAGTCTCAAAGGCA |
| Gria1 | NM_001113325 | CTGTGAATCAGAACGCCTCA | TCACTTGTCCTCCACTGCTG |
| Grin1 | NM_008169 | ACTCCCAACGACCACTTCAC | GTAGACGCGCATCATCTCAA |
| Grm1 | NM_016976 | CGAGAAGAGCTTTGATCGGC | TGTCTGCCCATCCATCACTT |
| Gapdh | NM_008084 | AGTGCCAGCCTCGTCCCGTAG | GTGCCGTTGAATTTGCCGTGAGTG |
Statistical analysis
Data are expressed as the mean ± SEM. Statistics were performed using either one-way (treatment) or two-way (voltage and treatment) ANOVA followed by Bonferroni post hoc tests. Data were transformed when required. For data that did not comply with parametric test assumptions Kruskal-wallis ANOVA on ranks was applied. Statistics were done with the software InfoStat 2010. A p value less than 0.05 was considered significant.
RESULTS
METH withdrawal reduced ICa and increased IH amplitude in mPFC pyramidal neurons
We analyzed ICa and IH current-density in deep-layer mPFC pyramidal neurons after 4 days withdrawal from repeated non-contingent 7 days METH administration (Fig. 2). We also evaluated the effect of D1/5 receptors by using D1/5 receptor antagonist SCH23390 with either a high dose of 0.5 mg/Kg (SCH 0.5) or a low dose of 0.05 mg/Kg (SCH 0.05). We found reduced ICa after METH compared with Control (p<0.001) (Fig. 2A, B). Repetitive SCH 0.5 mg/kg on its own also caused decreased ICa amplitude in deep-layer mPFC pyramidal neurons (p<0.001), but this effect was not observed with the lower SCH 0.05 dose (p>0.05). Importantly, both SCH 0.05 and 0.5 completely prevented METH effects on ICa. These results suggest that METH treatment induced a reduction in ICa through a D1/5-dependent mechanism.
Figure 2. METH withdrawal reduced ICa and increased IH amplitude in mPFC pyramidal neurons.

A, B. Voltage-gated calcium current-voltage (I-V) curves representing average current density (ICa in pA/pF) values recorded at holding potentials ranging from −55 mV to +30 mV in 5 mV steps. Representative ICa recordings corresponding to −40 mV, −30 mV, −20 mV, −10 mV and 0 mV holding potentials (left insets) were performed using a modified ACSF solution (see Materials and Methods) in the presence of TTX (3 μM), DL-AP5 (50 μM), CNQX (20 μM) and bicuculline (20 μM) to block voltage-gated sodium channels, NMDA receptors, AMPA/kainate receptors and GABA-A receptors, respectively. I-V curves were obtained during whole-cell patch clamp recordings of pyramidal mPFC neurons from two different groups of experiments using either SCH23390 0.5 mg/Kg (plot A, n=12 neurons) or SCH23390 0.05 mg/Kg (plot B, n=14 neurons). Also, control (n=18 neurons); METH withdrawal (n=27 neurons); SCH 0.5-METH (n=20 neurons) and SCH 0.05-METH (n=9 neurons) groups were displayed. Data was analyzed with Kruskal-Wallis two way ANOVA followed by post-hoc comparisons (plot A: H=52.5 p<0.0001, **: Control and SCH 0.5-METH different from METH and SCH 0.5 p<0.01; plot B: H=182.42, p<0.0001; **: METH different from Control, SCH 0.05 and SCH 0.05-METH p<0.01).
C, E. Comparison of average IH current densities versus voltage (I-V) relationships obtained by changing holding potential from −60 mV to −140 mV, in 10 mV, 500 msec. long hyperpolarizing square pulses, during whole-cell patch clamp recordings of pyramidal mPFC neurons. Representative IH recordings corresponding to −60 mV, −80 mV, −100 mV, −120 mV and −140 mV holding potentials (left insets) were obtained using a modified ACSF solution (see Materials and Methods) in the presence of TTX (3 μM), DL-AP5 (50 μM), CNQX (20 μM) and bicuculline (20 μM) from two different groups of experiments using either SCH23390 0.5 mg/Kg (plot C, n=9 neurons) or SCH23390 0.05 mg/Kg (plot E, n=8 neurons). Control (n=8 neurons); METH withdrawal (n=13 neurons); SCH 0.5-METH (n=14 neurons) and SCH 0.05-METH (n=9 neurons) groups were displayed. Data was analyzed with Kruskal-Wallis ANOVA followed by post-hoc comparisons (plot C: H=36.52 p<0.0001, plot E: H=66.05, p<0.0001, **: different from Control p<0.01).
d, f. Dotted line square insets from plots C and E representing in detail IH density values corresponding to the holding potentials −60, −70 and −80 mV for SCH 0.5 and SCH 0.05 groups. Data was analyzed with Kruskal-Wallis ANOVA followed by post-hoc comparisons (plot d: H=52 p<0.0001; plot f: H=65, p<0.0001, #: different from Control p<0.01; *: different from Control p<0.01; $: different from METH p<0.01).
Previous reports have shown that IH increases with DA stimulation in the PFC (Wu and Hablitz, 2005), and that D1/5 activation up-regulates IH in entorhinal pyramidal neurons (Rosenkranz and Johnston, 2006). Also, cocaine enhanced IH current density in thalamic neurons (Urbano et al., 2009). Therefore, we also investigated the effect of METH on HCN channels physiology. Figure 2 (C-F) shows METH induced increments on IH current density in deep-layer mPFC pyramidal neurons after METH compared with Control (p<0.001) (Fig. 2C, E). Repetitive treatment with the D1/5 antagonist (SCH 0.5 and 0.05 mg/kg) on its own caused increased IH current density (p<0.01), and neither of the concentrations of SCH23390 used prevented METH effects on IH. Moreover, SCH 0.05, SCH 0.5 and SCH 0.05–METH treatment groups drastically enhanced IH current density values at membrane potentials near mPFC pyramids resting membrane potentials (Fig. 2d,f; Vm values from −60 to −80 mV).
METH withdrawal increased paired-pulse ratio (PPR) and reduced spontaneous excitatory transmission in mPFC pyramidal neurons
To examine the effect of METH on evoked glutamate release probability we next analyzed the EPSCs PPR in deep-layer mPFC pyramidal neurons after 4-days withdrawal from repeated non-contingent 7 days METH administration (Fig. 3A, B). We also examined if these changes might be affected by pretreatment with D1/5 receptor antagonist.
Figure 3. METH withdrawal increased paired-pulse ratio (PPR) and reduced spontaneous excitatory transmission in mPFC pyramidal neurons.

A. Representative excitatory postsynaptic currents (EPSC) recorded using whole-cell patch clamp of mPFC deep-layer pyramidal neurons, in the presence of bicuculline (20 μM) and DL-2-amino-5-phosphonopentanoic acid (DL-AP5, 50μM), during 10-Hz paired-pulse stimuli for the six different drug treatments (Control, METH withdrawal, SCH 0.5, SCH 0.05, SCH 0.5-METH and SCH 0.05-METH groups). Each trace was obtained averaging 16 stimuli. Note how METH withdrawal treatment induced a clear facilitation (i.e., EPSC amplitude of second paired pulse stimulus was bigger than first EPSC amplitude). B. Mean EPSC paired pulse ratio (i.e., fraction of EPSC2 /EPSC1 amplitudes). Individual ratio paired-pulse values are shown for each treatment (see dots on each bar). ANOVA-Bonferroni, **: p<0.001 different from Control, SCH 0.5, SCH 0.05, SCH 0.5-METH and SCH 0.05-METH groups. C. Representative miniature excitatory postsynaptic currents (mEPSC) recorded using whole-cell patch clamp of mPFC deep-layer pyramidal neurons after tetrodotoxin (TTX, 3 μM), DL-2-amino-5-phosphonopentanoic acid (DL-AP5, 50μM) and bicuculline (20 μM) were included in the ACSF to block voltage-gated sodium channels, NMDA and GABA-A receptors, respectively. Each trace was obtained from different mPFC neurons. Note how mEPSC frequency was higher when recording from SCH0.5-treated mPFC neurons compared to control, METH. D, E. Average mEPSC median amplitude (D) and inter-events intervals (E) plots for control, SCH and METH groups. ANOVA-followed by Tukey's post hoc comparisons: * p<0.01 different from Control. # p<0.01 different from Control. & p<0.001 different from SCH 0.5.
The PPR, which is the ratio of the amplitude of a second post-synaptic response to that of the first, is commonly used to detect changes in the probability of neurotransmitter release: PPR goes down when the probability of release increases, and it goes up when the probability of release decreases (Zucker and Regehr, 2002), although it needs to be mentioned that in some synapses the direct relation between PPR and probability of release has not been fully elucidated (Branco and Staras, 2009). In this study PPR was evaluated during 10 Hz stimulation of excitatory synaptic transmission in the presence of DL-AP5 (50μM) and bicuculline (20μM) (to block NMDA and GABA-A-mediated transmission) (Fig. 3A, B). We found that METH treatment significantly increased evoked EPSCs PPR in deep-layer mPFC pyramidal neurons, which was completely prevented by both SCH 0.5 and 0.05 mg/kg pre-treatment (p<0.01) (Fig. 3B). Evoked EPSCs PPR increases after METH suggest a presynaptic reduction in the probability of glutamate release. These presynaptic effects might be due to the strong reduction in ICa through a D1/5-dependent mechanism described in this study. Repetitive SCH treatment had no effect on its own at any dose on evoked EPSCs PPR (Fig. 3B).
We also analyzed if spontaneous glutamate-mediated miniature EPSCs were affected by SCH and METH treatments (Fig. 3C-E). We found that METH did not affect EPSCs minis median amplitude (Fig. 3D) but significantly increased the inter-event intervals compared to Control (p<0.01), which was fully prevented by both SCH 0.05 and 0.5 mg/kg administration (Fig. 3E), suggesting D1/5-dependent mechanisms. Repetitive SCH 0.05 mg/kg treatment had no effect on its own, but SCH 0.5 mg/kg caused decreased mEPSCs inter-event intervals compared to Control (p<0.05). Such increment in the probability of spontaneous glutamate release (i.e., increasing frequency of mEPSCs) by SCH 0.5 mg/kg would compensate for the reduction in ICa current-density values, thus leaving unaffected mean PPR values. We also evaluated spontaneous GABA-A-mediated miniature amplitudes and inter-event intervals in mPFC pyramids from METH group compared to Control and found no differences (data not shown).
METH withdrawal alters VGCC, HCN and glutamate receptors subunits mRNA expression in mPFC
METH can cause substantial changes in gene expression in many brain regions including the cortex, the striatum and midbrain (Cadet el al., 2009; Martin et al., 2012; Thomas et al., 2004). Thus, we sought to examine the mRNA expression of VGCC, HCN and glutamate receptors subunits in the mPFC after treatments (Fig. 4). For VGCC, we found elevated expression of P/Q-type Cav2.1 Cacna1a (p<0.01), N-type Cav2.2 Cacna1b (p<0.001), T-type Cav3.1 Cacna1g (p<0.001), Cav3.2 Cacna1h (p<0.001) and Cav3.3 Cacna1i (p<0.001), as well as the auxiliary subunit α2δ1 Cacna2d1 (p<0.05) at METH withdrawal compared to control mice. For Cacna1a, Cacna1h, Cacna1i and Cacna2d1 expression, SCH 0.5 mg/kg had no effect on its own and was able to prevent the METH-induced elevated expression, whereas for Cacna1g SCH 0.5mg/kg pre-treatment was not able to block the METH-induced elevated expression. For Cacna1b the SCH 0.5 and SCH 0.5-METH groups also showed elevated mRNA expression compared to controls (p<0.01). No changes were detected for the postsynaptic L-type Cav1.2 Cacna1c after any treatment. For HCN, we found elevated Hcn2 at METH withdrawal compared to control mice (p<0.01); we also found elevated expression in SCH 0.5 and SCH 0.5-METH groups compared to controls (p<0.01 and p<0.05, respectively). Interestingly, Hcn2 expression might be related to the increased IH in mPFC pyramids found in METH, SCH and SCH-METH groups found in this study. In contrast, we found diminished Hcn1 expression at METH withdrawal that was not modulated by SCH 0.5 mg/kg pre-treatment (p<0.001). For glutamate receptors subunits we found elevated AMPA GluA1, Gria1 (p<0.001), NMDA GluN1 Grin1 (p<0.05) and metabotropic mGluR1 Grm1 (p<0.01) after METH and SCH 0.5 treatments compared to controls. SCH 0.5 mg/kg treatment before METH had no effect on Gria1, which remained elevated compared to control (p<0.001), but was able to normalize Grin1 and Grm1 expression. In addition, we also tested SCH 23390 at 0.05 mg/kg (co-administered in vivo with METH) showed effects on its own for those markers that the antagonist at a high dose (i.e., 0.5 mg/kg): Cacna1b, Hcn2, Gria1, Grin1, Grm1. We found that the lower dose of the antagonist, 0.05 mg/kg, also showed increased mRNA expression compared to control values in all five markers and only blocked METH increases in Grm1 and Hcn2 (data non shown).
Figure 4. METH and SCH23390 affected VGCC, HCN and glutamate receptors subunits mRNA expression in the mPFC.

The mRNA expression of VGCC subunits Cacna1a (P/Q Cav2.1), Cacna1b (N Cav2.2), Cacna1c (L Cav1.2), Cacna1g (T-type Cav3.1), Cacna1h (T-type Cav3.2) Cacna1g (T-type Cav3.3), as well as the auxiliary subunit α2δ1 Cacna2d1, HCN subunits Hcn1 and Hcn2 and glutamate receptors subunits Gria1 (AMPA GluA1), Grin1 (NMDA GluN1) and Grm1 (metabotropic mGluR1) was evaluated by RT-PCR. ANOVA-Bonferroni (N=5), * p<0.05, ** p<0.01, *** p<0.001 different from Control; $: p<0.05, $$: p<0.01, $$$: p<0.001 different from METH.
These results suggest compensatory mechanisms at the nuclear level on METH-induced DA and Ca2+ alterations observed at the membrane level.
Effects of bath-applied METH, SCH23390 and SKF38383 in mPFC
Given the substantial effects on mPFC Ca2+ currents and evoked EPSCs PPR induced by METH in vivo administration we next investigated whether there were similar effects of this compound in vitro (i.e. METH bath-applied in slices containing mPFC from naïve mice, Fig. 5 and 6). For that purpose we used a solution exchange system at a 1.8 ml/min speed of perfusion. Bath-applied drugs reached the recording chamber after 1 minute, and maximum effects were observed after 15 minutes. Figure 5A shows the time course of ICa reduction by bath-applied METH (1 μM), reaching its maximum effect after 12 minutes. Figure 5B shows how bath-applied SCH23390 (10 μM) was also able to reduce ICa, followed by a further reduction in ICa when METH (1 μM) was co-applied in the presence of SCH23390 (10 μM). Importantly, both METH and SCH23390 reduced ICa without altering its voltage-dependence (i.e., showing similar current-voltage, I-V plots) as shown in Figure 5C and D, respectively. Moreover, Figure 5D and E illustrate dose-dependent reduction in ICa by the D1/5 antagonist SCH23390 as well as the D1/5 agonist SKF38383. We quantified the percentage of ICa reduction of bath-applied METH 1 μM, SKF38383 1 and 10 μM and SCH23390 1 and 10 μM, as well as METH 1 μM co-incubated with SCH23390 1 or 10 μM (Fig. 5F). We found that METH-induced ICa reduction was similar to that of D1/5 agonist SKF38383 10 μM (56.4 ± 5.2 and 63.8 ± 8.5, respectively), and higher than SKF38383 1 μM (26.6 ± 3.4) (p<0.01). Significant differences were also found when comparing METH 1 μM and SKF38383 10 μM percentage of ICa reduction with that of D1/5 antagonist SCH23390 1 μM (20.0 ± 5.4) and SCH23390 10 μM (40.0 ± 3.9) (p<0.01). Additionally, co-incubation of SCH 1 μM and METH 1 μM partially reduced the METH blocking effect on ICa (p<0.05), whereas co-incubation of SCH 10 μM +METH 1 μM was no different than METH 1 μM alone.
Figure 5. Effects of bath-applied METH, SCH23390 and SKF38383 in mPFC.

A. Time course of ICa block during bath-application of METH (1 μM) from a mPFC deep-layer pyramidal neuron. METH application is indicated by the solid gray bar. ICa were recorded in the presence of TTX (3 μM), DL-AP5 (50 μM), CNQX (20 μM) and bicuculline (20 μM) to block voltage-gated sodium channels, NMDA receptors, AMPA/kainate receptors and GABA-A receptors, respectively. Inset: Representative currents corresponding to the time points noted in parenthesis.
B. Time course of ICa block during bath-application of SCH23390 (10 μM) and SCH23390 (10 μM) + METH (1 μM) from a mPFC deep-layer pyramidal neuron. SCH/SCH+METH applications are indicated by the solid black and gray bars, respectively. Inset: Representative currents corresponding to the time points noted in parenthesis.
C. Calcium current-voltage (I-V) curves representing acute, bath-applied METH blocking effect on ICa (1 μM, >15 minutes application). D. I-V curves representing acute, bath-applied SCH23390 blocking effect on ICa (1 μM and 10 μM; >15 minutes application). E. I-V curves representing acute, bath-applied SKF38383 blocking effect on ICa (1 μM and 10 μM; >15 minutes application).
F. Bar plot showing the mean ICa percentage reduction of peak amplitude after bath-application of METH (1 μM, diagonal lines filled bar), SKF38383 (10 μM and 1 μM, solid black bars), SCH23390 (1 μM and 10 μM, solid grey bars), SCH23390 (1 μM) + METH (1 μM) (vertical lines filled bar) and SCH23390 (10 μM) + METH (1 μM) (horizontal lines filled bar), Number of mPFC recorded are included in parenthesis in each bar. ANOVA-Tukey **: p<0.01. Student's t-test, METH (1 μM) vs. SCH23390 (1 μM) + METH (1 μM), #: p<0.05.
Figure 6. Effect of acute, bath-applied, METH, SCH23390 and SCH23390+METH on paired pulse EPSC ratio in mPFC pyramidal neurons.

A. Excitatory postsynaptic currents (EPSC) recorded in the presence of DL-AP5 (50μM) and bicuculline (20 μM), using whole cell patch clamp recordings of mPFC deep-layer pyramidal neurons from a control mouse during 10-Hz paired-pulse stimuli before (black trace; paired pulse ratio=1.0) and after 15 minutes bath-applied METH (1 μM; grey trace; paired-pulse ratio=1.3). Note how EPSC amplitudes during both paired-pulse stimuli were drastically reduced while paired pulse ratio increased in the presence of METH. B. Box plot representing mean change in paired-pulse ratio (PPR, in % of control) after >15 minutes acute, bath-applied, METH (1 μM), SCH23390 (1 μM) and SCH23390 (1 μM) + METH (1 μM). White lines in boxes representing the median value of PPR change. ANOVA-Bonferroni: **: p<0.01.
Figure 6 shows the effect of in vitro bath-application of METH and SCH23390 (1μM) on evoked EPSCs PPR. Figure 6A shows representative traces ilustrating the effect of bath-applied METH 1 μM (15 minutes, grey line) during patch-clamp recording of a mPFC pyramid of a naïve slice at 10 Hz paired-pulse stimulation. Note how METH 1 μM increased PPR, while reducing EPSCs current amplitude. Mean amplitude of the first EPSC during paired pulse stimulation was reduced by a 54±10% (n=6), 30±3% (n=4) and 10±10% (n=6) after bath-application of METH 1 μM, SCH 1 μM and the co-incubation of SCH 1 μM and METH 1 μM in naïve slices, respectively. Mean reduction of the first EPSCs amplitude observed after METH showed increased values compared to the combination of SCH 1 μM and METH 1 μM (p<0.05). However, no significant differences were observed comparing SCH 1 μM with METH, SCH and METH application (p>0.05).
We then quantified the percentage of PPR change after bath-applied METH 1 μM, SCH 1 μM and the co-incubation of SCH 1 μM and METH 1 μM in naïve slices (>15 minutes; Fig. 6B). We found that METH 1 μM application increased evoked EPSCs PPR, and the co-incubation with SCH 1 μM was able to block the effect (Fig. 6B, p<0.01). SCH 1 μM alone had no effect.
Our in vitro results regarding ICa and evoked EPSCs PPR effects induced by METH mimic the effects observed in animals treated with METH and suggest a possible mechanism mediated by the activation of D1/5 receptors since METH effects were blocked by the co-incubation with the D1/5 receptor antagonist SCH23390. However, it needs to be noted that, although of a reduced magnitude compared to METH effects, SCH23390 by itself show decreased Ca2+ currents similar to the lower concentration of the D1/5 agonist SKF38393 1 μM suggesting that, under some conditions, this compound can exert agonist-like actions (Wachtel and White 1995).
DISCUSSION
This is the first study describing reduced voltage-dependent Ca2+ currents, reduced spontaneous (mEPSCs) and evoked (EPSCs) glutamate release and increased IH amplitude in deep-layer pyramidal neurons as part of METH-induced detrimental effects on PFC function. These METH-induced effects on ICa and mEPSCs/evoked EPSCs were dependent on D1/5 receptor activation, since pre-treatment with the D1/5 antagonist SCH23390 was able to fully prevent these effects. DA signaling in the mPFC circuitry is central for cognitive and reward processing (Berridge and Robinson, 1998). Thus, METH-induced supraphysiological increases in DA release (Cadet et al., 2014) likely promote maladaptive changes that may be behind the alterations in glutamate, ICa and IH homeostasis observed in the present study. It needs to be noted that METH effects on Ca2+-dependent mechanisms were seen both at early withdrawal from non-contingent METH and also when METH was acutely applied in the recording bath, strongly suggesting that METH-induced Ca2+ and glutamate alterations may not be dependent on the behavioral contingency of the drug administration. Our results are consistent with similar findings observed by Mair and Kauer (2007) with the related molecule amphetamine, where bath-applied 10 μM amphetamine, acting through D1/5 receptors, significantly depressed glutamate release probability and excitatory field potentials (EPSPs) in mPFC.
Deep-layer pyramidal neurons integrate afferent innervations from dopaminergic cells in the ventral tegmental area, GABAergic inputs from local interneurons as well as glutamatergic inputs from the thalamus, hippocampus, amygdala and neighboring PFC pyramidal cells. The mesocortical DA input to PFC is crucial for processing working memory, and an optimal level of D1/5 receptor activation in this circuit is required to optimal processing of short-term-memory-related activities (Goldman-Rakic et al., 2000). There is a normal range of DA signaling through D1/5 receptor in PFC that is classically described as an “inverted-U” relationship between DA transmission and the integrity of working memory. Both over-stimulation and under-stimulation of D1/5 receptors impairs performance on PFC-sensitive behavioral tasks (Rinaldi et al., 2006). In this work we have found that METH and SCH23390 can elicit similar responses on some parameters of ICa (both in vivo and in vitro) and IH (in vivo) physiology, which are consistent with DA U-shaped mechanisms through post-synaptic D1/5 signaling. However, another possible explanation for these observations is that SCH23390 could, under certain conditions, have D1-agonist like effects. This was observed previously for this compound in the rat nucleus accumbens, where SCH23390 administration mimicked DA actions eliciting quinpirole responses that require prior D1 activation (Wachtel and White, 1995). Other reports showed dose-dependency in agonist-like effects, where low doses of SCH23390 mimicked SKF38393-induced excessive grooming in rodents (Starr and Starr, 1986; Wachtel et al., 1992) and acetylcholine release in the striatum in freely moving conditions (Ajima et al., 1990).
We found that both repeated METH and SCH23390 (at a high dose of 0.5 mg/kg) reduced ICa in mPFC pyramids. Our results showing a D1/5 receptor-mediated ICa reduction after METH is consistent with previous findings of DA actions in the PFC. It was shown that DA, acting through D1 receptor, suppresses Ca2+ spikes transients amplitude (Young and Yang, 2004; Kisilevsky et al., 2008). Another study stimulating and recording from paired deep-layer pyramidal cells reported that DA application depressed EPSPs, an effect blocked by SCH23390 and mimicked by the D1/5 agonist SKF33989 (Gao et al., 2001). Also, it was shown that endogenous DA activates presynaptic D1/5 to inhibit glutamate release (Gao et al., 2001, Seamans et al., 2001; Tritsch and Sabatini, 2012), suggesting that the impairment of PFC-sensitive tasks associated with D1/5 supra-activation could involve activation of this presynaptic D1/5 mechanism (Mair and Kauer, 2007). Moreover, it was recently shown that METH self-administration reduced glutamate basal levels in the mPFC (Parsegian and See, 2014). Interestingly, the evoked EPSCs paired-pulse ratio (PPR) was enhanced only in mice treated with METH, but not with SCH23390. An enhancement in PPR is consistent with a presynaptic reduction in the probability of glutamate release after the strong reduction in ICa through a D1/5 over-activation mechanism described in this work. Altogether, these studies provide solid evidence that D1/5 receptor activation inhibits Ca2+ influx and glutamate release in the mPFC consistent with results presented here.
Of a related interest to this discussion, we have also found increased IH and HCN mRNA alterations after METH and SCH23390. The HCN channels are highly expressed in all mesocorticolimbic areas including the PFC and they participate in the modulation of DA neurotransmission (Chu and Zhen, 2010); still the IH role in psychostimulant addiction is almost unexplored. Among the HCN subunits, the HCN2 is the most abundant in the mesocorticolimbic structures and it was found elevated after cocaine (Santos-Vera et al., 2013). However, HCN1 is highly distributed in distal apical-dendritic compartments of mPFC pyramids (Santoro et al., 2000; Lörincz et al., 2002), whose activation actually prevents initiation of distal calcium spikes (Berger et al., 2003). We found direct Hcn2 mRNA expression modulation by D1/5 which can explain increased IH amplitude after repeated SCH23390 and METH treatment. We also found decreased Hcn1 mRNA expression after METH that was not responsive to SCH23390 pre-treatment, which could be related to other DA receptors mechanism. Our results are consistent with other reports showing that IH increases with DA stimulation in the PFC (Wu and Hablitz, 2005), and that D1/5 activation upregulates IH in entorhinal pyramidal neurons (Rosenkranz and Johnston, 2006). Also, cocaine enhanced IH current density (previously described to be mediated by HCN2 channels, Santoro et al., 2000) in thalamic neurons (Urbano et al., 2009). Thus, sustained activation of D1/5 receptors in mPFC could be the mechanism behind METH-induced IH increments and mEPSC frequency reduction observed here that, together with reduced ICa indicate functional alterations of mPFC pyramidal neurons that could reflect PFC impairment. It is important to mention that increased HCN signaling was also associated with other conditions presenting with elevated DA tone and impaired PFC functionality, like stress and schizophrenia (Arnsten, 2011). We and others have previously demonstrated that the repeated METH administration used in this study induces cognitive impairments (González et al., 2014; Kamei et al., 2006). As IH elevation seems to be detrimental for cortical integrity and cognition, IH inhibition in PFC by HCN channel blockade or HCN1 channel knockdown improves cognitive performance (Nolan et al., 2004; Wang et al., 2007). IH reduction would result in significant enhancement of local recurrent network activity in cortical networks, presumably through enhanced effectiveness of dendritic synaptic potentials to initiate action potential activity (Magee, 1999; Nolan et al., 2004). Interestingly, recent studies have shown that psychostimulant exposure in animal models of self-administration induces hypoactivity of mPFC similar to what is seen in human addicts (Chen et al., 2013; Lu et al., 2014). Further research needs to be conducted in order to clarify the IH and HCN subunits contribution to METH abuse and dependence.
In the PFC, D1/5 activation effects are linked to Ca2+ influx modulation through VGCC regulation (Young and Yang, 2004; Kisilevsky et al., 2008; Tritsch and Sabatini, 2012). Previous studies have found increased auxiliary subunit α2δ1 mRNA and protein expression after repeated METH in frontal cortex that was dependent on DA signaling (Kurokawa et al., 2010). In this work, we found that repeated METH also increased mRNA expression of Cacna2d1 at early withdrawal, together with increased high VGCC subunits Cacna1a and Cacna1b and low VGCC Cacna1g, Cacna1h and Cacna1i. These results suggest the activation of compensatory mechanisms in the nucleus in response to the METH-induced DA and [Ca2+]i dynamics alterations observed at the membrane level. The fact that we observed elevated VGCC subunits expression but diminished Ca2+ influx could be explained by multiple G protein-dependent modulatory mechanisms affecting these channels. In this sense, it was previously shown in the striatum that D1 activation can decrease Ca2+ influx by G protein-dependent mechanisms (Surmeier et al., 1995). D1/5 activation and diminished Ca2+ influx could be related to direct N-type VGCC inhibition. This relationship between D1 and N-type VGCC was shown in mPFC neurons, where D1 receptor was found to form a signaling complex with N-type VGCC, that in basal conditions enhanced its membrane expression and after D1 agonist administration potently inhibited the channel activity and removed it from the membrane (Kisilevsky et al., 2008). This strong association between N-type VGCCs and D1 receptors was evident in our study at the mRNA level. We found that not only METH but also SCH23390 alone or before METH also caused increased Cacna1b expression. Cacna1a, Cacna1g, Cacna1h, Cacna1i and Cacna2d1 mRNA expression behaved differently than Cacna1b since they were responsive to repeated METH-induced D1/5 stimulation but were irresponsive to repeated SCH23390 alone treatment. The Cacna1a and Cacna2d1 expression could be more related to the pre-synaptic D1/5 mechanism of glutamate release inhibition, which could explain SCH23390-alone lack of effect. Also, this could be related to the differential distribution of N- and P/Q-type VGCC in the PFC, where P/Q-type are located closer to release sites and are less affected by G-protein-mediated inhibition than are N-type VGCC (Colecraft et al., 2000). Less is known about the synaptic localization and function of T-type VGCC in the PFC, although the three of them were found in the neocortex by in situ hybridization (Talley et al., 1999). Interestingly, Cacna1h was found at very high levels in a subset of layer V pyramidal neurons (Talley et al., 1999), and in the ethorinal cortex this isoform was found to form a pre-synaptic complex with HCN1, where its activation would inhibit glutamate release by deactivating the low threshold Cav3.2 (Huang et al., 2011).
In this study, we applied a non-contingent repeated METH treatment capable of inducing cognitive impairments associated with deficits of the ERK1/2 pathway in the mPFC (González et al., 2014; also see Kamei et al., 2006). Synchronization between DA and glutamate transmission is central for ERK1/2 signaling to occur in the PFC and other mesocorticolimbic areas, and this mechanism has been shown to play important roles in addiction (Girault et al., 2007; Cahill et al., 2014). It was previously shown that D1 activation facilitates the strengthening of excitatory synapses by increasing AMPA and NMDA subunits trafficking to the membrane of mPFC neurons (Sun et al., 2005; Gao and Wolf, 2008). We have found elevated expression of AMPA, NMDA and metabotropic glutamate receptor subunits Gria1, Grin1 and Grm1 after repeated METH and also SCH23390 treatments. These results highlight a D1/5 dependency of glutamate receptors subunits mRNA expression. An abnormal engagement of this D1-dependent mechanism controlling glutamate receptor subunits expression during unregulated DA release may account for maladaptive plasticity after repeated exposure to psychostimulants (Sun et al., 2005). Interestingly, Grin1 and Grm1 effects were returned to control values in the group that received both SCH23390 and METH.
In conclusion, our study provides novel evidence of the detrimental mechanisms activated by METH in the PFC. METH was able to blunt [Ca2+] dynamics and excitatory neurotransmission in mPFC pyramidal neurons. PFC is critically involved in addictive behaviors and represents a potential target for therapeutic interventions (Goldstein and Volkow, 2011). Our results suggest that behavioral or pharmacological approaches to treat psychostimulant addiction should be oriented to increase PFC functionality aimed to return the addicted brain to homeostasis. Furthermore, alterations in IH physiology described here are very novel and offer new targets for pharmacological approaches to treat PFC abnormalities seen in psychostimulant addicts.
Acknowledgements
Dr. Bisagno has been authorized to study drug abuse substances in animal models by A.N.M.A.T. (National Board of Medicine Food and Medical Technology, Ministerio de Salud, Argentina). Authors would like to thank Dr. Jose Luis Rozas for his suggestions to improve this manuscript and Dr. M. Gustavo Murer for kindly provide the SCH23390 used in this study. This work is supported by grants PIP 11420100100072, FONCYT-Agencia Nacional de Promoción Científica y Tecnológica; BID 1728 OC.AR. T 2012-0924 Argentina (to Dr. Bisagno) and FONCYT-Agencia Nacional de Promoción Científica y Tecnológica; BID 1728 OC.AR. PICT-2012-1769, Argentina and UBACYT 2014-2017 #20120130101305BA (to Dr. Urbano). In addition, this work was supported by NIH award P20 GM103425 to the Center for Translational Neuroscience, UAMS, USA.
Footnotes
Authors contributions: VB, FJU and BG were responsible for the study design. BG, FJU, CRE and JAM performed the experiments and analyzed data. VB, FJU, BG, JLC, EGR provided critical revision of the manuscript for important intellectual content. All authors have critically reviewed content and approved final version submitted for publication.
REFERENCES
- Ajima A, Yamaguchi T, Kato T. Modulation of acetylcholine release by D1, D2 dopamine receptors in rat striatum under freely moving conditions. Brain Res. 1990;518(1-2):193–198. doi: 10.1016/0006-8993(90)90972-e. [DOI] [PubMed] [Google Scholar]
- Berger T, Senn W, Lüscher HR. Hyperpolarization-activated current Ih disconnects somatic and dendritic spike initiation zones in layer V pyramidal neurons. J Neurophysiol. 2003;90(4):2428–2437. doi: 10.1152/jn.00377.2003. [DOI] [PubMed] [Google Scholar]
- Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev. 1998;28:309–369. doi: 10.1016/s0165-0173(98)00019-8. [DOI] [PubMed] [Google Scholar]
- Bisagno V, Raineri M, Peskin V, Wikinski SI, Uchitel OD, Llinás RR, Urbano FJ. Effects of T-type calcium channel blockers on cocaine-induced hyperlocomotion and thalamocortical GABAergic abnormalities in mice. Psychopharmacology (Berl) 2010;212:205–214. doi: 10.1007/s00213-010-1947-z. [DOI] [PubMed] [Google Scholar]
- Branco T, Staras K. The probability of neurotransmitter release: variability and feedback control at single synapses. Nat Rev Neurosci. 2009;10(5):373–383. doi: 10.1038/nrn2634. [DOI] [PubMed] [Google Scholar]
- Cadet JL, Bisagno V, Milroy CM. Neuropathology of substance use disorders. Acta Neuropatho. 2014;l127:91–107. doi: 10.1007/s00401-013-1221-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet JL, Krasnova IN, Ladenheim B, Cai NS, McCoy MT, Atianjoh FE. Methamphetamine preconditioning: differential protective effects on monoaminergic systems in the rat brain. Neurotox Res. 2009;15:252–259. doi: 10.1007/s12640-009-9026-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill E, Salery M, Vanhoutte P, Caboche J. Convergence of dopamine and glutamate signaling onto striatal ERK activation in response to drugs of abuse. Front Pharmacol. 2014;4:172. doi: 10.3389/fphar.2013.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BT, Yau HJ, Hatch C, Kusumoto-Yoshida I, Cho SL, Hopf FW, Bonci A. Rescuing cocaine-induced prefrontal cortex hypoactivity prevents compulsive cocaine seeking. Nature. 2013;496:359–362. doi: 10.1038/nature12024. [DOI] [PubMed] [Google Scholar]
- Chu HY, Zhen X. Hyperpolarization-activated, cyclic nucleotide-gated (HCN) channels in the regulation of midbrain dopamine systems. Acta Pharmacol Sin. 2010;31(9):1036–1043. doi: 10.1038/aps.2010.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Greenberg ME. Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu Rev Cell Dev Biol. 2008;24:183–209. doi: 10.1146/annurev.cellbio.24.110707.175235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colecraft HM, Patil PG, Yue DT. Differential occurrence of reluctant openings in G-protein-inhibited N- and P/Q-type calcium channels. J Gen Physiol. 2000;115:175–192. doi: 10.1085/jgp.115.2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. Calcium channel auxiliary α2δ and β subunits: trafficking and one step beyond. Nat Rev Neurosci. 2012;13:542–555. doi: 10.1038/nrn3311. [DOI] [PubMed] [Google Scholar]
- Ernst T, Chang L. Adaptation of brain glutamate plus glutamine during abstinence from chronic methamphetamine use. J Neuroimmune Pharmacol. 2008;3:165–172. doi: 10.1007/s11481-008-9108-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao C, Wolf ME. Dopamine receptors regulate NMDA receptor surface expression in prefrontal cortex neurons. J Neurochem. 2008;106(6):2489–2501. doi: 10.1111/j.1471-4159.2008.05597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WJ, Krimer LS, Goldman-Rakic PS. Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proc Natl Acad Sci U S A. 2001;98:295–300. doi: 10.1073/pnas.011524298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girault JA, Valjent E, Caboche J, Hervé D. ERK2: a logical AND gate critical for drug-induced plasticity? Curr Opin Pharmacol. 2007;7:77–85. doi: 10.1016/j.coph.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Muly EC, 3rd, Williams GV D(1) receptors in prefrontal cells and circuits. Brain Res Brain Res Rev. 2000;31:295–301. doi: 10.1016/s0165-0173(99)00045-4. [DOI] [PubMed] [Google Scholar]
- Goldstein RZ, Volkow ND. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci. 2011;12:652–669. doi: 10.1038/nrn3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González B, Raineri M, Cadet JL, García-Rill E, Urbano FJ, Bisagno V. Modafinil improves methamphetamine-induced object recognition deficits and restores prefrontal cortex ERK signaling in mice. Neuropharmacology. 2014;87:188–197. doi: 10.1016/j.neuropharm.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Chen F, Li B, Hu Z. Neurophysiology of HCN channels: from cellular functions to multiple regulations. Prog Neurobiol. 2014;112:1–23. doi: 10.1016/j.pneurobio.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Huang Z, Lujan R, Kadurin I, Uebele VN, Renger JJ, Dolphin AC, Shah MM. Presynaptic HCN1 channels regulate Cav3.2 activity and neurotransmission at select cortical synapses. Nat Neurosci. 2011;14(4):478–486. doi: 10.1038/nn.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayanthi S, McCoy MT, Chen B, Britt JP, Kourrich S, Yau HJ, Ladenheim B, Krasnova IN, Bonci A, Cadet JL. Methamphetamine downregulates striatal glutamate receptors via diverse epigenetic mechanisms. Biol Psychiatry. 2014;76(1):47–56. doi: 10.1016/j.biopsych.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. Cocaine and amphetamine-like psychostimulants: neurocircuitry and glutamate neuroplasticity. Dialogues Clin Neurosci. 2007;9:389–397. doi: 10.31887/DCNS.2007.9.4/pkalivas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei H, Nagai T, Nakano H, Togan Y, Takayanagi M, Takahashi K, Kobayashi K, Yoshida S, Maeda K, Takuma K, Nabeshima T, Yamada K. Repeated methamphetamine treatment impairs recognition memory through a failure of novelty-induced ERK1/2 activation in the prefrontal cortex of mice. Biol Psychiatry. 2006;59:75–84. doi: 10.1016/j.biopsych.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kawamoto EM, Vivar C, Camandola S. Physiology and pathology of calcium signaling in the brain. Front Pharmacol. 2012;3:61. doi: 10.3389/fphar.2012.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisilevsky AE, Mulligan SJ, Altier C, Iftinca MC, Varela D, Tai C, Chen L, Hameed S, Hamid J, Macvicar BA, Zamponi GW. D1 receptors physically interact with N-type calcium channels to regulate channel distribution and dendritic calcium entry. Neuron. 2008;58:557–570. doi: 10.1016/j.neuron.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Kurokawa K, Shibasaki M, Ohkuma S. Methamphetamine-induced up-regulation of α2/δ subunit of voltage-gated calcium channels is regulated by DA receptors. Synapse. 2010;64:822–828. doi: 10.1002/syn.20797. [DOI] [PubMed] [Google Scholar]
- Lörincz A, Notomi T, Tamás G, Shigemoto R, Nusser Z. Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci. 2002;5(11):1185–1193. doi: 10.1038/nn962. [DOI] [PubMed] [Google Scholar]
- Lu H, Zou Q, Chefer S, Ross TJ, Vaupel DB, Guillem K, Rea W, Peoples LL, Stein EA. Abstinence from Cocaine and Sucrose Self-administration Reveals Altered Mesocorticolimbic Circuit Connectivity. Brain Connect. 2014;4:499–510. doi: 10.1089/brain.2014.0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüthi A, McCormick DA. Periodicity of thalamic synchronized oscillations: the role of Ca2+-mediated upregulation of Ih. Neuron. 1998;20:553–563. doi: 10.1016/s0896-6273(00)80994-0. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic lh normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2(6):508–514. doi: 10.1038/9158. [DOI] [PubMed] [Google Scholar]
- Mair RD, Kauer JA. Amphetamine depresses excitatory synaptic transmission at prefrontal cortical layer V synapses. Neuropharmacology. 2007;52:193–199. doi: 10.1016/j.neuropharm.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Martin TA, Jayanthi S, McCoy MT, Brannock C, Ladenheim B, Garrett T, Lehrmann E, Becker KG, Cadet JL. Methamphetamine causes differential alterations in gene expression and patterns of histone acetylation/hypoacetylation in the rat nucleus accumbens. PLoS One. 2012;7(3):e34236. doi: 10.1371/journal.pone.0034236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsáki G, Siegelbaum SA, Kandel ER, Morozov A. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119(5):719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Parsegian A, See RE. Dysregulation of dopamine and glutamate release in the prefrontal cortex and nucleus accumbens following methamphetamine self-administration and during reinstatement in rats. Neuropsychopharmacology. 2014;39:811–22. doi: 10.1038/npp.2013.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz JA, Johnston D. Dopaminergic regulation of neuronal excitability through modulation of Ih in layer V entorhinal cortex. J Neurosci. 2006;26:3229–3244. doi: 10.1523/JNEUROSCI.4333-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci. 2000;20(14):5264–5275. doi: 10.1523/JNEUROSCI.20-14-05264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Vera B, Vázquez-Torres R, Marrero HG, Acevedo JM, Arencibia-Albite F, Vélez-Hernández ME, Miranda JD, Jiménez-Rivera CA. Cocaine sensitization increases I h current channel subunit 2 (HCN2) protein expression in structures of the mesocorticolimbic system. J Mol Neurosci. 2013;50:234–245. doi: 10.1007/s12031-012-9920-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowski TJ. Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layer V prefrontal cortex neurons. Proc Natl Acad Sci U S A. 2001;98:301–306. doi: 10.1073/pnas.011518798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr BS, Starr MS. Differential effects of dopamine D1 and D2 agonists and antagonists on velocity of movement, rearing and grooming in the mouse. Implications for the roles of D1 and D2 receptors. Neuropharmacology. 1986;25(5):455–463. doi: 10.1016/0028-3908(86)90168-1. [DOI] [PubMed] [Google Scholar]
- Sun X, Zhao Y, Wolf ME. Dopamine receptor stimulation modulates AMPA receptor synaptic insertion in prefrontal cortex neurons. J Neurosci. 2005;25:7342–7351. doi: 10.1523/JNEUROSCI.4603-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Bargas J, Hemmings HC, Jr, Nairn AC, Greengard P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. 1995;14:385–397. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- Tritsch NX, Sabatini BL. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron. 2012;76:33–50. doi: 10.1016/j.neuron.2012.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbano FJ, Bisagno V, Wikinski SI, Uchitel OD, Llinás RR. Cocaine acute “binge” administration results in altered thalamocortical interactions in mice. Biol Psychiatry. 2009;66:769–776. doi: 10.1016/j.biopsych.2009.04.026. [DOI] [PubMed] [Google Scholar]
- Wachtel SR, Brooderson RJ, White FJ. Parametric and pharmacological analyses of the enhanced grooming response elicited by the D1 dopamine receptor agonist SKF 38393 in the rat. Psychopharmacology (Berl) 1992;109(1-2):41–48. doi: 10.1007/BF02245478. [DOI] [PubMed] [Google Scholar]
- Wachtel SR, White FJ. The dopamine D1 receptor antagonist SCH 23390 can exert D1 agonist-like effects on rat nucleus accumbens neurons. Neurosci Lett. 1995;199(1):13–16. doi: 10.1016/0304-3940(95)12025-y. [DOI] [PubMed] [Google Scholar]
- Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, Mazer JA, McCormick DA, Arnsten AF. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129(2):397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Wu J, Hablitz JJ. Cooperative activation of D1 and D2 dopamine receptors enhances a hyperpolarization-activated inward current in layer I interneurons. J Neurosci. 2005;25(27):6322–6328. doi: 10.1523/JNEUROSCI.1405-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young CE, Yang CR. Dopamine D1/D5 receptor modulates state-dependent switching of soma-dendritic Ca2+ potentials via differential protein kinase A and C activation in rat prefrontal cortical neurons. J Neurosci. 2004;24:8–23. doi: 10.1523/JNEUROSCI.1650-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]