

Abstract
Cadmium (Cd) is a toxic and carcinogenic metal naturally occurring in the earth’s crust. A common route of human exposure is via diet and cadmium accumulates in the liver. The effects of Cd exposure on gene expression in human hepatocellular carcinoma (HepG2) cells were examined in this study. HepG2 cells were acutely-treated with 0.1, 0.5, or 1.0 μM Cd for 24 hours; or chronically-treated with 0.01, 0.05, or 0.1 μM Cd for three weeks and gene expression analysis was performed using Affymetrix GeneChip® Human Gene 1.0 ST Arrays. Acute and chronic exposures significantly altered the expression of 333 and 181 genes, respectively. The genes most upregulated by acute exposure included several metallothioneins. Downregulated genes included the monooxygenase CYP3A7, involved in drug and lipid metabolism. In contrast, CYP3A7 was upregulated by chronic Cd exposure, as was DNAJB9, an anti-apoptotic J protein. Genes downregulated following chronic exposure included the transcriptional regulator early growth response protein 1. Ingenuity Pathway Analysis revealed that the top networks altered by acute exposure were lipid metabolism, small molecule biosynthesis, and cell morphology, organization, and development; while top networks altered by chronic exposure were organ morphology, cell cycle, cell signaling, and renal and urological diseases/cancer. Many of the dysregulated genes play important roles in cellular growth, proliferation, and apoptosis, and may be involved in carcinogenesis. In addition to gene expression changes, HepG2 cells treated with cadmium for 24 hours indicated a reduction in global levels of histone methylation and acetylation that persisted 72 hours post-treatment.
Introduction
Cadmium is a toxic and carcinogenic transition metal naturally occurring in the earth’s crust. It can also be introduced into the environment via anthropogenic sources. Cadmium is easily absorbed by plants from the soil, thus a common non-occupational route of human exposure is through diet [1]. Levels in the US food supply can range from 2-40 ppb [2]. Additionally, smokers are exposed to high levels of cadmium from tobacco, about 1.7 μg per cigarette [1]. Most cadmium accumulates in the liver and kidneys of individuals with low environmental exposures [3]. Occupational exposure to cadmium has been linked to cancers of the lung, prostate, kidney, liver, hematopoietic system, bladder, pancreas, testis, and stomach [4, 5], hence cadmium is classified as a class I human carcinogen by IARC [1]. The mechanisms of cadmium-induced carcinogenesis however, are not fully understood. While cadmium can be genotoxic at high doses, it is not highly mutagenic and does not form DNA adducts [6]. Cadmium is thought to induce oxidative stress by depleting glutathione and protein-bound sulfhydryl groups, leading to increased production of reactive oxygen species (ROS) and apoptosis [7, 8]. Cadmium is predominately non-genotoxic and therefore may induce carcinogenesis via epigenetic mechanisms by altering the expression levels of various critical genes [4] .
Carcinogenesis is a complex, multi-step process that involves many alterations to the cell. DNA damage, if left unrepaired, can lead to gene mutations. These mutations may be deleterious if they occur in genes controlling cell cycle, cell growth, or other critical processes. In addition, expression of various genes can be altered via epigenetic mechanisms. Epigenetic modifications alter chromatin structure, leading to changes in gene expression without affecting DNA structure or sequence. If these changes are inherited, then this process is considered an epigenetic effect. These epigenetic mechanisms include DNA methylation, micro RNAs, noncoding RNAs and post-translational modifications to histones [9, 10]. The N-terminal tails of histones can be altered post-translationally via methylation, acetylation, phosphorylation, SUMOylation, or other modifications [11]. These modifications can lead to relaxation or condensation of chromatin. Modifications can also recruit other histone modifying enzymes to initiate further alterations to the chromatin. These changes can dramatically alter gene expression, which can dysregulate normal cellular functions and lead to disease, transformation, and possibly carcinogenesis.
A common route of exposure to cadmium is via diet, and cadmium accumulates in the liver. Exposure has also been linked to liver disease and cancers of the liver [12, 13]. It is hypothesized that both acute and chronic exposures to cadmium induce gene expression changes in human hepatocarcinoma HepG2 cells. These differentially expressed genes will likely be involved in various carcinogenesis pathways. Previous gene expression studies on cadmium exposure in HepG2 studies have focused on higher, non-biologically relevant concentrations or very short exposure times (6 hours or less) [14, 15], and indicate an enrichment in carcinogenesis related genes and pathways. This study explored the changes in gene expression caused by acute and chronic treatment with low doses of CdCl2 in HepG2 cells and investigated the biological functions of differentially modified genes. HepG2 cells were treated with 0.1, 0.5, or 1.0 μM cadmium chloride for 24 hours, or 0.01, 0.05, or 0.1 μM cadmium chloride for three weeks and gene expression analysis was performed using Affymetrix GeneChip Human Gene 1.0 ST Arrays.
Gene expression changes are often accompanied by both gene-specific and global changes to histone modifications. Western blot analysis was used to investigate global changes to histone methylation and acetylation levels caused by acute treatment with 0, 0.5, 1.0, 2.5, and 5 μM cadmium chloride, as well as after a 72-hour recovery period to look for persistence of the changes. Cadmium has a half-life in the liver of up to 20 years, and levels can easily reach micromolar concentrations [16]. Additionally, uptake of insoluble cadmium particles can lead to high concentrations in the liver, as these particles are accumulated over time and not readily eliminated.
Materials and methods
Cell culture
Human hepatocarcinoma (HepG2) (ATCC) were cultured in Dulbecco’s modified minimum essential media (DMEM), low glucose (1 g/L) (Invitrogen), supplemented with 10% FBS and 100 U/ml penicillin and 100 mg/ml streptomycin (Invitrogen). For cadmium exposures, cells were seeded one day prior to treatment. Cadmium chloride hemipentahydrate (Acros Organics) was added to the media and evenly applied to the cultured cells. Cells were treated with 0.1, 0.5, or 1.0 μM CdCl2 for 24 hours (acute treatment), or 0.01, 0.05, or 0.1 μM CdCl2 for three weeks (chronic treatment). For chronic treatment, CdCl2 concentrations were maintained in the cell culture media. Both untreated control cells and treated cells were cultured in a 37 °C incubator with 5% CO2 until harvesting. For Western blots, cells were treated with 0.5, 1.0, 2.5, or 5 μM CdCl2 for 24 hours before cells were lysed for protein extraction. After 24 hour treatment with 5 μM CdCl2, cell culture media was replaced with fresh media containing no cadmium for washout samples. Cells were lysed for protein extraction after a 72 hour washout period.
RNA extraction and microarray hybridization
At the end of the treatment, cells were collected in Trizol (Invitrogen) and RNA was isolated and purified using RNeasy PlusMicro Kit (Qiagen, Valencia, CA). 100 ng of total RNA was used to synthesize double-stranded cDNA (dsDNA). cRNA was synthesized from dsDNA template, and subsequently used to produce sense single-stranded cDNA (ssDNA) with incorporated deoxyuridine triphosphate. The ssDNAs were fragmented, end-labeled, and hybridized to Affymetrix Human Gene 1.0 ST Array (Affymetrix). Arrays were hybridized and scanned using a standard protocol.
Microarray data analysis
Microarray data analysis was performed using GeneSpring v12.0 (Agilent Technologies). Differentially expressed genes were identified using one-way ANOVA (p<0.05). Hierarchical cluster analysis using Euclidean distance was performed to cluster genes and samples to generate a heat map. Gene network and pathway analysis was performed using Ingenuity Pathway Analysis (http://www.ingenuity.com).
Real time PCR
250 ng of total RNA extracted from control and cadmium-treated cells was converted to single stranded cDNA using Superscript® III (Invitrogen). Quantitative real-time PCR analysis was performed on 20 ng of each cDNA using SYBR green (Applied Biosystems) on ABI prism 7900HT (Applied Biosystems). Relative gene expression levels were normalized to β-actin expression. All PCR reactions were performed in triplicate. Primers were obtained from Invitrogen. Results were presented as fold change relative to the level expressed in untreated control cells using the ΔΔCT method. The primer sequences are listed in table 1.
Table 1.
Primer sequences used for qPCR validation of microarray results
| MT1G-F: | GCCAGCTCCTGCAAGTGCAA | MT1G-R: | TCTCCGATGCCCTTTGCAG |
| MT1F-F: | TCCTGCAAGTGCAAAGAGTG | MT1F-R: | TCCTGCAAGTGCAAAGAGTG |
| MT1M-F: | TCCGGGTGGGCCTAGCAGTCG | MT1M-R: | AATGCAGCAAATGGCTCAGTATCGTATTG |
| CYP3A7-F: | GATCTCATCCCAAACTTGGCCG | CYP3A7-R: | CATAGGCTGTTGACAGTCATAAATA |
| NT5E-F: | GGGCGGAAGGTTCCTGTAG | NT5E-R: | GAGGAGCCATCCAGATAGACA |
| SPINK1-F: | TGTCTGTGGGACTGATGGAA | SPINK1-R: | TCAACAATAAGGCCAGTCAGG |
| NPNT-F: | GCTGGTATCCTCGCTCTACC | NPNT-R: | TCCTCCCACCATAACGACAT |
| DNAJB9-F: | GCCATGAAGTACCACCCTGACA | DNAJB9-R: | TCGTCTATTAGCATCTGAGAGTGT |
| EGR1-F: | CTTCAACCCTCAGGC GGACA | EGR1-R: | GGAAAAGCGGCCAGTATAGGT |
| ADH4-F: | GAAACCATGAAAGCAGCCCT | ADH4-R: | CCAACCACCAAAGAATGTTCC |
| ID1-F: | CCAACGCGCCTCGCCGGATC | ID1-R: | CTCCTCGCCAGTGCCTCAG |
| β actin-F: | CACCATTGGCAATGAGCGGTTC | β actin-R: | AGGTCTTTGCGGATGTCCACGT |
Whole cell lysis and protein isolation
HepG2 cells were lysed using boiling buffer (1% SDS, 1 mM Na3VO4, 10 mM Tris-Cl, pH 7.4). 0.5 mL of 100 °C preheated boiling buffer was added to 10 cm cell culture dishes at a sub-confluent density after two washes with 1X PBS. The lysate was denatured at 100 °C for 5 minutes then sonicated using a Diagenode Bioruptor at a maximum setting for 10 minutes. Samples were then centrifuged at 4 °C for 15 minutes at 21,000 rpm. Protein was quantified using a Nanodrop 2000 Spectrophotometer.
Western Blot
Mini-PROTEAN TGX precast gels (Bio-Rad) were loaded with protein in Laemmli sample buffer (Bio-Rad) containing 5% (v/v) 2-mercaptoethanol (Sigma). The Precision Plus Protein Kaleidoscope standard (Bio-Rad) was used to determine protein size. Electrophoresis took place in 1X Tris/glycine/SDS buffer (Bio-Rad) at 100 volts at room temperature. The protein was transferred onto Immuno-Blot PVDF Membrane (Bio-Rad) in 1X Tris/glycine buffer (Bio-Rad) at 22 volts overnight at 4 °C. The membranes were blocked for 30 minutes with 5% (w/v) Blotting-Grade Blocker (Bio-Rad) in TBST at room temperature. Membranes were incubated with primary antibodies for one hour at room temperature or overnight at 4 °C. Membranes were incubated with secondary antibodies (goat anti-mouse IgG-HRP [sc-2005, Santa Cruz Biotechnology, Dallas, TX] or goat anti-rabbit IgG-HRP [sc-2004, Santa Cruz Biotechnology]) for one hour at room temperature. Protein bands were detected using the Pierce ECL Western Blotting Substrate (Thermo). Relative band intensities were determined using ImageJ software (NIH).
Results
A distinct set of genes were up- and downregulated due to acute and chronic treatment with cadmium
Heatmaps of differentially expressed genes are shown in figure 1 for all treatment groups (left, acute treatment; right, chronic treatment). Differential genes are shown either in red (upregulation) or blue (downregulation) (p>0.05). The intensity of the color indicates the level up/down regulation. The samples were grouped by treatment dose and showed an overall global change in expression for all genes. Hierarchical clustering analysis indicated a detectable difference in gene expression patterns between sample groups. The fifteen top up- and down-regulated genes are listed for the 0.5 μM acute treatment (table 2) and 0.1 μM chronic treatment (table 3), as these were the treatment groups with the highest number of genes with altered expression for each exposure duration. Expression levels for those top genes are also given for the other treatment doses. Expression of 333 genes was significantly changed by acute exposure in HepG2 cells, while 181 genes were altered by chronic exposure. Of the 333 genes significantly altered by acute cadmium exposure, 171 genes were upregulated and 162 were downregulated. Of the 181 genes significantly altered by chronic exposure, 88 genes were upregulated while 93 genes were downregulated. There were four genes significantly upregulated by both acute and chronic exposures and no genes were commonly downregulated by the treatments.
Figure 1.

Heatmaps of differentially expressed genes due to acute (left) and chronic (right) cadmium treatment in HepG2 cells (over 1.25-fold change, p>0.05). Red indicates upregulation while blue indicates downregulation of gene expression relative to control, untreated cells.
Table 2.
Top fifteen up- and downregulated genes (p<0.05) by 24 hour treatment with 0.5 μM CdCl2 in HepG2 cells. Fold changes for the other treatments also listed. # fold change >1.25
| Gene Symbol | Gene Description | Fold Change (0.5 μM) |
Fold Change (0.1 μM) |
Fold Change (1.0 μM) |
Regulation |
|---|---|---|---|---|---|
| MT1G | metallothionein 1G | 2.215775 | 1.5027326 | 2.6238236 | up |
| MT1F | metallothionein 1F | 1.9121428 | 1.405878 | 2.2424114 | up |
| MT1M | metallothionein 1M | 1.8444134 | # | 6.988504 | up |
| MT1B | metallothionein 1B | 1.7858652 | # | 5.00817 | up |
| MT1X | metallothionein 1X | 1.6122478 | 1.4785146 | 1.8759271 | up |
| MT1P3 | metallothionein 1 pseudogene 3 | 1.5198572 | 1.307409 | 1.7234105 | up |
| CSH2 | chorionic somatomammotropin hormone 2 | 1.5099572 | # | 1.3089397 | up |
| KRTAP10-2 | keratin associated protein 10-2 | 1.5006515 | # | 1.2620568 | up |
| MT1JP|MT1M | metallothionein 1J (pseudogene) | metallothionein 1M |
1.4997663 | 1.2292569 | 1.8385758 | up |
| MT1P2 | metallothionein 1 pseudogene 2 | 1.4846494 | 1.2776792 | # | up |
| MT1H | metallothionein 1H | 1.4614742 | # | 2.0551221 | up |
| SCXA|SCXB | scleraxis homolog A | scleraxis homolog B | 1.4582701 | # | 1.2557648 | up |
| SCXA|SCXB | scleraxis homolog A | scleraxis homolog B | 1.4572202 | # | 1.25512 | up |
| TMEM61 | transmembrane protein 61 | 1.4298121 | # | # | up |
| MT1DP | metallothionein 1D (pseudogene) | 1.4165779 | 1.2316265 | # | up |
| GBP2 | guanylate binding protein 2, interferon inducible |
-1.5112702 | -1.4250351 | -1.4131097 | down |
| MIR21 | microRNA 21 | -1.5275458 | -1.2404983 | -1.6964701 | down |
| ZNF224 | zinc finger protein 224 | -1.572604 | -1.2397038 | -1.3674877 | down |
| SAA4 | serum amyloid A4, constitutive | -1.573077 | -1.3437153 | -1.5190501 | down |
| ACTR3C | ARP3 actin-related protein 3 homolog C (yeast) |
-1.5773983 | -1.2445956 | # | down |
| SNORD50B | small nucleolar RNA, C/D box 50B | -1.5776544 | # | -1.527359 | down |
| TRA2A | transformer 2 alpha homolog (Drosophila) | -1.6224118 | -1.2303989 | -1.5386964 | down |
| CD109 | -1.659105 | # | -1.5179392 | down | |
| CYP3A7 | cytochrome P450, family 3 subfamily A, polypeptide 7 |
-1.664616 | -1.3983974 | -1.483224 | down |
| NT5E | 5ʹ-nucleotidase, ecto (CD73) | -1.691666 | -1.5449091 | -1.5966983 | down |
| SPINK1 | serine peptidase inhibitor, Kazal type 1 | -1.700486 | -1.39644 | -1.4552168 | down |
| SNORD14E | small nucleolar RNA, C/D box 14E | -1.7864679 | # | -1.8290889 | down |
| SNORD75 | small nucleolar RNA, C/D box 75 | -1.8150924 | # | -1.5868542 | down |
| UCA1 | urothelial cancer associated 1 (non-protein coding) |
-1.8260639 | -1.5522093 | -1.6103948 | down |
| MIR15A | microRNA 15a | -1.9946613 | -1.3650888 | -1.779063 | down |
Table 3.
Top fifteen up- and downregulated genes (p<0.05) by three week treatment with 0.1 μM CdCl2 in HepG2 cells. Fold changes for other treatments are also listed. ## fold change <1.25
| Gene Symbol | Gene Description | Fold Change (0.1 μM) |
Fold Change (0.05 μM) |
Fold Change (0.01 μM) |
Regulation |
|---|---|---|---|---|---|
| GSTTP1| GSTTP2| |
glutathione S-transferase theta pseudogene 1 |
1.72073 | 1.4901183 | 1.379154 | up |
| SNRPN | small nuclear ribonucleoprotein polypeptide N |
1.6563919 | 1.6350169 | ## | up |
| CYP3A7 | cytochrome P450, family 3, subfamily A, polypeptide 7 |
1.5384295 | 1.3135703 | ## | up |
| NPNT | NPNT nephronectin | 1.5199842 | 1.4056195 | ## | up |
| CAB39L | calcium binding protein 39-like | 1.5002468 | 1.240774 | ## | up |
| ZBTB38 | zinc finger and BTB domain containing 38 | 1.4971563 | 1.3198949 | ## | up |
| SNORD116-13 | Small Nucleolar RNA, C/D Box 116-13 | 1.4758297 | 1.2465166 | 1.2062092 | up |
| FBLN5 | fibulin 5 | 1.4431939 | 1.4760754 | 1.3243747 | up |
| DNAJB9 | DnaJ (Hsp40) homolog, subfamily B, member 9 |
1.4407821 | 1.4589071 | 1.2065107 | up |
| C6orf138 | chromosome 6 open reading frame 138 | 1.4336088 | 1.3983722 | ## | up |
| C5orf41 | CREB3 regulatory factor | 1.422349 | 1.3452742 | ## | up |
| MGAM | maltase-glucoamylase (alpha-glucosidase) | 1.4202621 | 1.3677275 | ## | up |
| SNORD109A| SNORD109B |
small nucleolar RNA, C/D box 109A/109B | 1.4162974 | 1.3215218 | 1.3739192 | up |
| SNORD109A| SNORD109B |
small nucleolar RNA, C/D box 109A/109B | 1.4149537 | 1.3213891 | 1.3719118 | up |
| SCARNA9L | small Cajal body-specific RNA 9-like | 1.4086239 | 1.220644 | 1.3410316 | up |
| RRM2 | ribonucleotide reductase M2 | −1.4714146 | −1.3555955 | ## | down |
| BTBD11 | BTB (POZ) domain containing 11 | −1.4720253 | −1.4257193 | ## | down |
| CTGF | connective tissue growth factor | −1.4752284 | −1.3320917 | ## | down |
| HIST1H3B | histone cluster 1, H3b | −1.485579 | −1.3974302 | ## | down |
| EGR1 | early growth response 1 | −1.517825 | −1.4408088 | ## | down |
| TGM2 | transglutaminase 2 | −1.5214871 | −1.4627025 | ## | down |
| ANKRD1 | ankyrin repeat domain 1 | −1.5234282 | −1.4508984 | ## | down |
| HIST1H2AK| HIST1H2BN |
histone cluster 1, H2ak/H2bn | −1.537931 | ## | ## | down |
| MIXL1 | Mix paired-like homeobox | −1.5498296 | −1.3671521 | ## | down |
| SLC16A6 | solute carrier family 16, member 6 | −1.5749238 | −1.6012409 | −1.322911 | down |
| SLC2A3 | solute carrier family 2 member 3 | −1.5907145 | −1.4265127 | ## | down |
| ADH4 | alcohol dehydrogenase 4 (class II) | −1.6070219 | −1.3255012 | −1.2636199 | down |
| LOC1720 | dihydrofolate reductase pseudogene | −1.632394 | −1.3695079 | −1.207613 | down |
| ID1 | inhibitor of DNA binding 1 | −1.6802144 | −1.5311089 | ## | down |
| C8orf4 | chromosome 8 open reading frame 4 | −1.7252662 | −1.5806782 | ## | down |
Gene Expression Validation
Microarray results for both acute and chronic exposures were validated for three top up- and down-regulated genes by quantitative real-time PCR (qRT-PCR) of RNA from treated and untreated HepG2 cells. Results are shown in table 4. qRT-PCR results shown represent means of triplicates from two independent experiments, represented as fold change to level of expression in control HepG2 cells. qPCR is a more robust and quantitative measure of mRNA levels, thus fold changes are not identical to those measured in the microarrays. The direction of the change, however, was consistent between microarray and qPCR for all of the genes studied.
Table 4.
qRT-PCR validation of microarray results. qRT-PCR data represents means of triplicates from two independent experiments, represented as fold change to level of expression in control HepG2 cells.
| 0.5 µM Acute Treatment | ||
|---|---|---|
| Gene | Microarray Fold Change | qRT-PCR Fold Change |
| MT1G | 2.216 | 2.074 |
| MT1F | 1.912 | 3.510 |
| MT1M | 1.844 | 7.162 |
| CYP3A7 | −1.665 | −2.098 |
| NT5E | −1.692 | −2.306 |
| SPINK1 | −1.701 | −1.523 |
| 0.1 µM Chronic Treatment | ||
| Gene | Microarray Fold Change | qRT-PCR Fold Change |
| CYP3A7 | 1.538 | 1.871 |
| NPNT | 1.520 | 3.680 |
| DNAJB9 | 1.441 | 1.776 |
| EGR1 | −1.518 | −2.413 |
| ADH4 | −1.607 | −1.269 |
| ID1 | −1.680 | −1.258 |
Genes altered by 24 hour treatment with cadmium
Several metallothionein (MT) genes were among the genes most upregulated with acute treatment with cadmium, including MT1G, MT1F, MT1M, MT1B, MT1X and MT1H (table 2). Not surprisingly, ten of the top 15 upregulated genes due to acute treatment were MT genes or pseudogenes. Downregulated genes included the cytochrome p450 monooxygenase CYP3A7, 5'-nucleotidase ecto (NT5E/CD73), serine peptidase inhibitor, Kazal type 1 (SPINK1), and the transcription factor zinc finger protein 224 (ZZF224).
Genes altered by chronic treatment with cadmium
Genes most upregulated by three week chronic treatment with cadmium included CYP3A7, the extracellular matrix protein nephronectin (NPNT), the J family member of 70 kD heat shock protein (Hsp70) chaperones DNAJB9, and transcriptional activator zinc finger and BTB domain containing 38 ZBTB38 (table 3). Downregulated genes included transcriptional regulator early growth response regulator EGR1, alcohol dehydrogenase ADH4, helix-loop-helix protein inhibitor of DNA binding 1 (ID1), transporters SLC16A6 and SLC2A3, and connective tissue growth factor CTGF.
Gene expression and pathway analysis
Heatmaps of differentially expressed genes are shown in figure 1. Genes highlighted in red indicate upregulation versus untreated control cells, while blue indicates downregulation in the treated cells. Hierarchical clustering analysis shows a detectable difference in gene expression patterns between sample groups. Genes whose expression was altered at least 1.25-fold were uploaded into Ingenuity Pathway Analysis for further investigation. Top networks altered by acute exposure to 0.5 μM cadmium, as determined by IPA, were lipid metabolism, small molecule biochemistry; vitamin and mineral metabolism; and cell morphology, assembly, organization, and development (figure 2). Top molecular and cellular functions altered by acute exposure included cell death and survival, and cellular assembly and organization. Top diseases and disorders that were enriched were cancer, gastrointestinal diseases, and renal and urological disease. Top canonical pathways were retinol biosynthesis, nicotine degradation, and lipopolysaccharide (LPS)/interleukin-1 (IL-1) mediated inhibition of retinoid X receptor (RXR) function. Top toxicological functions were liver hyperplasia/hyperproliferation, hepatocellular carcinoma, liver proliferation, degeneration, and cirrhosis.
Figure 2.

Top networks altered by acute exposure to 0.5μM cadmium in HepG2 cells were lipid metabolism, small molecule biochemistry, and vitamin and mineral metabolism (left), and cellular morphology, assembly, organization, and development (right). Red – upregulation; green - downregulation.
Top networks altered by chronic exposure were organ morphology, cell cycle, and cancer; developmental disorder, renal and urological diseases/cancer; and cellular growth and proliferation, and cancer (figure 3). Top molecular and cellular functions altered by chronic exposure included cell-to-cell signaling and interaction, cell death and survival, and cellular growth and proliferation. Top diseases and disorders were cancer, reproductive system disease, and endocrine system disorders. Top canonical pathways were tyrosine degradation I, pregnane X receptor/retinoid X receptor (PXR/RXR) activation, and pyrimidine deoxyribonucleotides de novo biosynthesis I. Top upstream regulators were IL-6, hypoxia inducible factor 1α (HIF1α), platelet-derived growth factor (PDGF) BB, tumor necrosis factor (TNF), and transcription factor E2F4. Top toxicological functions were liver hyperplasia/hyperproliferation, hepatocellular carcinoma, liver regeneration, liver necrosis, and liver proliferation.
Figure 3.

Top networks altered by chronic exposure to 0.1μM cadmium in HepG2 cells were organ morphology, cell cycle, and cancer (left), and developmental disorder, renal and urological diseases and cancer (right). Red – upregulation; green – downregulation
Global changes in histone methylation and acetylation marks
The concentrations used for Western blot analysis were determined based on preliminary experiments in which doses below 0.5 μM cadmium chloride did not yield detectable changes to histone modifications (data not shown). Due to the slow growth rate of HepG2 cells, a washout (WO) period of 72 hours, in which the cadmium chloride was completely removed from the cell culture media, was used to ascertain the persistence of the epigenetic changes for at least three generations post-treatment.
Both histone H3 and H4 pan acetylation levels decreased with cadmium levels in HepG2 cells, however neither decrease was observed at the 0.5 μM dose. After a 72 hour washout period, the acetylation levels remained diminished, but not to the levels seen directly following treatment. H4K16ac levels decreased with increasing cadmium dose and were nearly depleted with both 2.5 and 5 μM cadmium. A reduction in protein expression of nearly 20% was observed at 0.5 μM. Diminished acetylation levels did not recover 72 hours post-treatment. Global H3K4me3 levels decreased with cadmium dose in HepG2 cells, however this effect was not entirely dose dependent, as levels were higher after treatment with 1.0 versus 0.5 μM cadmium. Levels remained slightly reduced 72 hours post-treatment. Reduced global H3K9me2 levels were observed after treatment with 2.5 and 5 μM cadmium. This decrease was sustained even 72 hours after the metal was removed from the media (figure 4).
Figure 4.
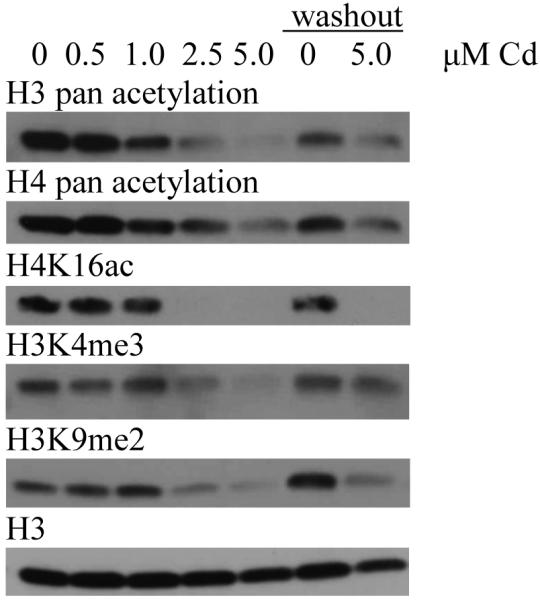

Western blot images (A) and graphical representations (B) of relative intensities calculated by ImageJ for cadmium treatment in HepG2 cells. Cells were treated for 24 hours with varying doses of cadmium and cells were lysed post-treatment. Cells treated with 5 μM cadmium were allowed to recover for 72 hours after treatment before lysis (washout). Blots and graphs are representative of two independent experiments. WO; washout
Discussion
The genes most upregulated by acute exposure to cadmium chloride included several metallothioneins (MTs), including MT1G, MT1F, MT1M, MT1B, MT1X and MT1H. MT genes are usually expressed at low levels, but their expression is often induced in the presence of metal ions, as their encoded cysteine-rich proteins are involved in metal storage, transport, and detoxification. MTs are also involved in regulation of homeostasis of essential trace metals [17]. Dysregulated MT expression has been observed in many cancers [10, 17], including hepatocellular carcinoma [18] as well as liver disease [19]. Upregulated MT expression indicates a cellular response to the cadmium treatment and mechanism to ameliorate the toxic effects of the metal.
Downregulated genes included the cytochrome p450 monooxygenase CYP3A7, which is involved in drug metabolism and lipid synthesis [20]. NT5E [5'-nucleotidase, ecto (CD73)] was also downregulated following to acute exposure. NT5E encodes a plasma membrane protein that catalyzes the conversion of extracellular nucleosides to membrane permeable nucleosides. NT5E functions as the rate-limiting enzyme in the conversion of pro-inflammatory ATP to immunosuppressive adenosine [21]. Its downregulation may contribute to a pro-inflammatory state. NT5E appears to play key regulatory roles in cancer cell proliferation, migration, and invasion and in tumor angiogenesis. Its upregulation has been reported in some carcinomas including gallbladder [22], prostate [23], and those of the gastrointestinal tract [24]. It has recently been identified as a novel prognostic indicator of colorectal cancer [21]. Its expression can be regulated by cytokines or by hypoxia [25], perhaps via a potential HIF-1 binding site [26]. The role of NT5E in human liver cancers outside of metastases has not been widely reported.
Serine peptidase inhibitor, Kazal type 1 (SPINK1), also downregulated following acute exposure, is secreted by pancreas cells to inhibit activation of zymogens. SPINK1 is aberrantly expressed in a number of human cancers and was found to promote epithelial to mesenchymal transition in prostate cancer [27]. SPINK1 was also found to increase breast cancer invasiveness and chemoresistance [28]. It has recently been identified as a possible marker for hepatocellular carcinoma (HCC) [29]. While downregulation of SPINK1 as it relates to HCC has not been reported, loss of SPINK1 was found to correlate with aggressive urothelial carcinomas of the bladder [30].
Transformer 2 alpha homolog (TRA2A) was also downregulated with acute cadmium treatment. This nuclear protein with several RNA recognition motif domains plays a role in the regulation of pre-mRNA splicing. TRA2A, as well as other proteins in the spliceosome pathway, was found to be dysregulated in a meta-analysis of human HCC [31]. Thus, cadmium may affect proper mRNA splicing, which could lead to aberrant gene expression.
While CYP3A7 expression was significantly downregulated following acute exposure, this monooxygenase gene was found to be one of the most upregulated due to chronic exposure. CYP3A7 has been observed to be upregulated in non-alcoholic steatohepatitis (fatty liver disease) and cirrhosis, as well after exposure to hepatotoxic compounds. The upregulation of CYP3A7 appears to be a response of stressed hepatocytes both in vitro and in vivo [20].
Nephronectin (NPNT), also upregulated due to chronic exposure, is an extracellular matrix protein that regulates cell adhesion, spreading, and survival in the kidney. While the role of NPNT in the liver is not known, its expression is upregulated in hepatitis mouse models and in liver samples from patients exhibiting hepatitis, fatty liver, and cirrhosis [20]. NPNT may play a role in inflammation, as it localizes to inflammatory foci [32]. Inflammation, especially as induced by hepatitis B infection, is a known precursor to HCC, thus the upregulation of NPNT during inflammatory injury to the liver as well as HCC warrants further study.
DNAJB9, a J protein family member, was also upregulated due to chronic treatment. J proteins function as part of the heat shock 70 kD protein (Hsp70) machinery that guides proper protein folding, drives protein transport, and mediates protein-protein interactions [33]. DNAJB9 is induced by endoplasmic reticulum (ER) stress and helps protect stressed cells from apoptosis. DNAJB9 was found to be induced by p53 under genotoxic conditions and to inhibit the pro-apoptotic function of p53 [34]. The upregulation of DNAJB9 may inhibit p53-induced apoptosis of cells under stress and allow for transformation to occur after exposure to cadmium.
CREB3 Regulatory Factor (CREBRF/C5orf41) was also upregulated by chronic exposure to cadmium. This protein, induced by ER stress, acts as a negative regulator of the ER stress response and unfolded protein response by repressing the transcriptional activity of CREB3 transcription factor [35]. CREB3 is involved in cell proliferation and migration, tumor suppression, and the inflammatory response. It promotes cell survival against ER stress-induced apoptotic cell death during the unfolded protein response. Upregulation of CREB3RF can lead to repression of CREB3, which could cause aberrant cell proliferation or dysregulate the unfolded protein response, leading to apoptosis.
Genes downregulated due to chronic exposure included the transcriptional regulator Early Growth Response 1 (EGR1). EGR1 encodes an ERK-induced transcriptional regulator whose target genes have various roles in differentiation and mitogenesis. EGR1 was found to arrest cell mobility, inhibit migration and induce apoptosis in human non-small-cell lung carcinoma cells [36]. It has previously been observed to be downregulated in HCC [20]. It has been suggested that EGR1 is a tumor suppressor gene, thus its downregulation due to cadmium exposure requires further investigation.
ADH4 was also downregulated by chronic exposure. ADH4, an alcohol dehydrogenase, metabolizes a wide array of substrates including ethanol, retinol, hydroxysteroids, and lipid peroxidation products. Diseases associated with ADH4 include alcoholic liver cirrhosis and alcohol dependence. Reduced expression of ADH4 has been suggested as a potential prognostic indicator of HCC [37].
Inhibitor of DNA binding 1 (ID1), also downregulated due to chronic cadmium treatment, is a helix-loop-helix protein that interacts with other HLH transcription factors. ID1 inhibits DNA binding and transcriptional activation activity of its interacting proteins. ID1 plays a role in cell growth, differentiation, and senescence. It has been observed to be downregulated in cirrhosis, HCC, and other carcinomas [20]. Reduced expression of ID1 can inhibit transcriptional activation of downstream genes, leading to irregular cell growth or senescence.
C8orf4, also known as human thyroid cancer protein 1 (TC-1), was also downregulated with chronic treatment. TC-1 protein functions as a positive regulator of the Wnt/β-catenin signaling pathway that regulates cell migration, polarity, and fate determination during embryogenesis. Mutations or epigenetic inactivation of elements of this pathway can lead to birth defects or cancer [38]. TC-1 is thought to competitively block interactions between β-catenin and a repressor protein, leading to upregulation of β-catenin target genes [39]. TC-1 may also play a role in NF-κB and ERK signaling pathways [40]. Downregulation TC-1 can thus lead to aberrant signaling, causing disrupted embryonic development or carcinogenesis.
A distinct set of genes was altered due to acute and chronic treatment in HepG2 cells. Acute exposure to cadmium altered the expression of genes encoding proteins involved in detoxification of metals and xenobiotics and metabolism of drugs, lipids, nucleosides, and other small molecules. Thus, acute exposure may disrupt metabolism and upregulate genes involved in detoxification and sequestration of the cadmium. Alternatively, chronic treatment caused dysregulation of genes involved in various cell signaling pathways, including immune, NFAT, and insulin signaling. Genes involved in the inflammatory response, unfolded protein response, and stress response were also dysregulated, indicating increased cellular stress and the initiation of an inflammatory state. Disruption of signaling pathways, stress response, proliferation, and cell cycle, as well as increased cellular stress and inflammation are all potential mechanisms to induce carcinogenesis in HepG2 cells chronically exposed to cadmium.
IPA revealed that top networks altered by acute exposure were lipid metabolism, small molecule biochemistry; vitamin and mineral metabolism; and cell morphology, assembly, organization, and development. Top molecular and cellular functions altered by acute exposure included cell death and survival, and cellular assembly and organization. Top networks altered by chronic exposure were organ morphology, cell cycle, and cancer; developmental disorder, renal and urological diseases/cancer; and cellular growth and proliferation, and cancer. Top molecular and cellular functions altered by chronic exposure included cell-to-cell signaling and interaction, cell death and survival, and cellular growth and proliferation.
Gene expression can be modulated epigenetically via DNA methylation, microRNAs or posttranslational modifications to histones. While cadmium has been found to alter global methylation levels in various human cell lines [6, 41, 42], changes to histone modifications have not been reported in HepG2 cells. Both methylation and acetylation are important histone posttranslational modifications that can have dramatic effects on gene expression. Methylation of histones may activate or suppress gene expression, depending on the particular residue modified and the level of methylation. Trimethylated H3 lysine 4 (H3K4me3) is found in promoter regions of transcriptionally active genes, [43] while dimethylated H3K9 (H3K9me2) is localized to regulatory regions of transcriptionally silent genes [44]. Altered global levels of H3K4me3 and H3K9me2 are associated with aberrant gene expression and have been linked with several human cancers [45, 46]. Cadmium treatment increased global levels of both histone marks in human bronchial epithelial cells [47]. Effects of cadmium on global H3K4me3 and H3K9me2 levels have not been previously reported in HepG2 cells.
Histone acetylation is typically associated with transcriptional activation. Addition of an acetyl group to a lysine reside on a histone N-terminal tail neutralizes its positive charge. This weakens contact between the lysine and DNA backbone, opening up the chromatin and facilitating loading of the transcription machinery onto DNA [48]. Both hyperacetylation and deacetylation of histones have been observed in human cancers and depend on the target gene or genes affected [49]. Acetylated histone H4K16 (H4K16ac) regulates chromatin remodeling by inhibiting the formation of condensed 30 nm fiber structures [50]. Loss of H4K16 is commonly seen in human cancers [51]. Effects of cadmium on H4K16ac levels have not been reported.
In the current study, cadmium treatment caused a decrease in histone H3 and H4 acetylation levels. After a 72 hour recovery period, the acetylation levels remained slightly diminished. H4K16ac levels decreased with increasing cadmium dose and were nearly depleted at the higher concentrations. Acetylation levels did not recover 72 hours post-treatment. Global H3K4me3 and H3K9me2 levels decreased with increased cadmium concentrations. H3K4me3 levels remained slightly reduced 72 hours post-treatment. Reduced H3K9me2 persisted 72 hours post treatment. These results indicate that while acute cadmium treatment can cause significant changes to the global levels of methylation and acetylation of histones, these changes do not always persist after the cadmium is removed. Changes to H4K16ac and H3K9me2 did appear persistent; at least 72 hours post treatment. These results suggest a change to the epigenetic landscape induced by acute exposure to cadmium.
Conclusion
Treatment with low concentrations of cadmium, both acute and chronic, caused significant changes to expression levels of hundreds of different genes. A different set of genes was altered depending on the length of the treatment. Dysregulated genes were involved in many important pathways and processes including metabolism, cell death and survival, and cell-to-cell signaling. Changes to global histone modifications were also observed with acute treatment with various doses of cadmium. Global acetylation and methylation levels both decreased after exposure. Some of these changes persisted 72 hours after treatment. Histone methylation and acetylation can dramatically alter gene expression via the unwinding or condensing of chromatin. It is clear that cadmium can induce significant changes to both gene expression and epigenetic marks. Further studies into whether these changes occur in the promoter or regulatory regions of dysregulated genes will provide further insight into the epigenetic mechanisms by which cadmium can affect gene expression.
Highlights.
A common route of human exposure to the carcinogenic metal cadmium is via diet
HepG2 cells were treated acutely or chronically with varying doses of cadmium
Gene expression analysis was performed using Affymetrix Human Gene 1.0 Arrays
Acute and chronic exposures altered the expression of 333 and 181 genes, respectively
Acute cadmium exposure altered global levels of histone methylation and acetylation
Acknowledgments
This work was supported by National Institutes of Health, National Institute of Environmental Health Sciences Grants R01ES023174 P30ES000260 and R01ES022935.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors declare that they have no conflicts of interest with the contents of this article.
References
- 1. Cadmium and Cadmium Compounds. 13 ed. Report on Carcinogens. 2014: National Toxicology Program, Department of Health and Human Services.
- 2.Satarug S, Haswell-Elkins MR, Moore MR. Safe levels of cadmium intake to prevent renal toxicity in human subjects. Br J Nutr. 2000;84(6):791–802. [PubMed] [Google Scholar]
- 3.Bernard A. Cadmium & its adverse effects on human health. Indian J Med Res. 2008;128(4):557–64. [PubMed] [Google Scholar]
- 4.Wang B, et al. Cadmium and its epigenetic effects. Curr Med Chem. 2012;19(16):2611–20. doi: 10.2174/092986712800492913. [DOI] [PubMed] [Google Scholar]
- 5.Jin YH, et al. Cadmium is a mutagen that acts by inhibiting mismatch repair. Nat Genet. 2003;34(3):326–9. doi: 10.1038/ng1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brocato J, Costa M. Basic mechanics of DNA methylation and the unique landscape of the DNA methylome in metal-induced carcinogenesis. Crit Rev Toxicol. 2013;43(6):493–514. doi: 10.3109/10408444.2013.794769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stohs SJ, et al. Oxidative mechanisms in the toxicity of chromium and cadmium ions. J Environ Pathol Toxicol Oncol. 2001;20(2):77–88. [PubMed] [Google Scholar]
- 8.Oh SH, Lim SC. A rapid and transient ROS generation by cadmium triggers apoptosis via caspase-dependent pathway in HepG2 cells and this is inhibited through N-acetylcysteine-mediated catalase upregulation. Toxicol Appl Pharmacol. 2006;212(3):212–23. doi: 10.1016/j.taap.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 9.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luevano J, Damodaran C. A review of molecular events of cadmium-induced carcinogenesis. J Environ Pathol Toxicol Oncol. 2014;33(3):183–94. doi: 10.1615/jenvironpatholtoxicoloncol.2014011075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21(3):381–95. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hyder O, et al. Cadmium exposure and liver disease among US adults. J Gastrointest Surg. 2013;17(7):1265–73. doi: 10.1007/s11605-013-2210-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waalkes MP. Cadmium carcinogenesis. Mutat Res. 2003;533(1-2):107–20. doi: 10.1016/j.mrfmmm.2003.07.011. [DOI] [PubMed] [Google Scholar]
- 14.Kawata K, et al. Classification of heavy-metal toxicity by human DNA microarray analysis. Environ Sci Technol. 2007;41(10):3769–74. doi: 10.1021/es062717d. [DOI] [PubMed] [Google Scholar]
- 15.Fabbri M, et al. Whole genome analysis and microRNAs regulation in HepG2 cells exposed to cadmium. ALTEX. 2012;29(2):173–82. doi: 10.14573/altex.2012.2.173. [DOI] [PubMed] [Google Scholar]
- 16.Faroon O, et al. Toxicological Profile for Cadmium. Atlanta (GA): 2012. [PubMed] [Google Scholar]
- 17.Gumulec J, et al. Metallothionein - immunohistochemical cancer biomarker: a meta-analysis. PLoS One. 2014;9(1):e85346. doi: 10.1371/journal.pone.0085346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanda M, et al. Detection of metallothionein 1G as a methylated tumor suppressor gene in human hepatocellular carcinoma using a novel method of double combination array analysis. Int J Oncol. 2009;35(3):477–83. doi: 10.3892/ijo_00000359. [DOI] [PubMed] [Google Scholar]
- 19.Nagamine T, Nakajima K. Significance of metallothionein expression in liver disease. Curr Pharm Biotechnol. 2013;14(4):420–6. doi: 10.2174/1389201011314040006. [DOI] [PubMed] [Google Scholar]
- 20.Grinberg M, et al. Toxicogenomics directory of chemically exposed human hepatocytes. Arch Toxicol. 2014;88(12):2261–87. doi: 10.1007/s00204-014-1400-x. [DOI] [PubMed] [Google Scholar]
- 21.Pancione M, et al. Immune escape mechanisms in colorectal cancer pathogenesis and liver metastasis. J Immunol Res. 2014;2014:686879. doi: 10.1155/2014/686879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiong L, et al. NT5E and FcGBP as key regulators of TGF-1-induced epithelial-mesenchymal transition (EMT) are associated with tumor progression and survival of patients with gallbladder cancer. Cell Tissue Res. 2014;355(2):365–74. doi: 10.1007/s00441-013-1752-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Q, Du J, Zu L. Overexpression of CD73 in prostate cancer is associated with lymph node metastasis. Pathol Oncol Res. 2013;19(4):811–4. doi: 10.1007/s12253-013-9648-7. [DOI] [PubMed] [Google Scholar]
- 24.Lu XX, et al. Expression and clinical significance of CD73 and hypoxia-inducible factor-1alpha in gastric carcinoma. World J Gastroenterol. 2013;19(12):1912–8. doi: 10.3748/wjg.v19.i12.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Longhi MS, et al. Biological functions of ecto-enzymes in regulating extracellular adenosine levels in neoplastic and inflammatory disease states. J Mol Med (Berl) 2013;91(2):165–72. doi: 10.1007/s00109-012-0991-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Synnestvedt K, et al. Ecto-5'-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110(7):993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang C, et al. Serine protease inhibitor Kazal type 1 promotes epithelial-mesenchymal transition through EGFR signaling pathway in prostate cancer. Prostate. 2014;74(7):689–701. doi: 10.1002/pros.22787. [DOI] [PubMed] [Google Scholar]
- 28.Soon WW, et al. Combined genomic and phenotype screening reveals secretory factor SPINK1 as an invasion and survival factor associated with patient prognosis in breast cancer. EMBO Mol Med. 2011;3(8):451–64. doi: 10.1002/emmm.201100150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marshall A, et al. Global gene expression profiling reveals SPINK1 as a potential hepatocellular carcinoma marker. PLoS One. 2013;8(3):e59459. doi: 10.1371/journal.pone.0059459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rink M, et al. Loss of SPINK1 expression is associated with unfavorable outcomes in urothelial carcinoma of the bladder after radical cystectomy. Urol Oncol. 2013;31(8):1716–24. doi: 10.1016/j.urolonc.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 31.Xu W, et al. Meta-analysis of gene expression profiles indicates genes in spliceosome pathway are up-regulated in hepatocellular carcinoma (HCC) Med Oncol. 201532(4):425. doi: 10.1007/s12032-014-0425-6. [DOI] [PubMed] [Google Scholar]
- 32.Inagaki FF, et al. Nephronectin is upregulated in acute and chronic hepatitis and aggravates liver injury by recruiting CD4 positive cells. Biochem Biophys Res Commun. 2013;430(2):751–6. doi: 10.1016/j.bbrc.2012.11.076. [DOI] [PubMed] [Google Scholar]
- 33.Kampinga HH, Craig EA. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol. 2010;11(8):579–92. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee HJ, et al. Genotoxic stress/p53-induced DNAJB9 inhibits the pro-apoptotic function of p53. Cell Death Differ. 2015;22(1):86–95. doi: 10.1038/cdd.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Audas TE, et al. A novel protein, Luman/CREB3 recruitment factor, inhibits Luman activation of the unfolded protein response. Mol Cell Biol. 2008;28(12):3952–66. doi: 10.1128/MCB.01439-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, et al. EGR1 decreases the malignancy of human non-small cell lung carcinoma by regulating KRT18 expression. Sci Rep. 2014;4:5416. doi: 10.1038/srep05416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei RR, et al. Identification of ADH4 as a novel and potential prognostic marker in hepatocellular carcinoma. Med Oncol. 2012;29(4):2737–43. doi: 10.1007/s12032-011-0126-3. [DOI] [PubMed] [Google Scholar]
- 38.Tian J, He H, Lei G. Wnt/beta-catenin pathway in bone cancers. Tumour Biol. 2014;35(10):9439–45. doi: 10.1007/s13277-014-2433-8. [DOI] [PubMed] [Google Scholar]
- 39.Su K, et al. TC-1 (c8orf4) enhances aggressive biologic behavior in lung cancer through the Wnt/beta-catenin pathway. J Surg Res. 2013;185(1):255–63. doi: 10.1016/j.jss.2013.05.075. [DOI] [PubMed] [Google Scholar]
- 40.Wang YD, et al. TC1 (C8orf4) is involved in ERK1/2 pathway-regulated G(1)- to S-phase transition. BMB Rep. 2008;41(10):733–8. doi: 10.5483/bmbrep.2008.41.10.733. [DOI] [PubMed] [Google Scholar]
- 41.Yuan D, et al. Long-term cadmium exposure leads to the enhancement of lymphocyte proliferation via down-regulating p16 by DNA hypermethylation. Mutat Res. 2013;757(2):125–31. doi: 10.1016/j.mrgentox.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 42.Zhou ZH, Lei YX, Wang CX. Analysis of aberrant methylation in DNA repair genes during malignant transformation of human bronchial epithelial cells induced by cadmium. Toxicol Sci. 2012;125(2):412–7. doi: 10.1093/toxsci/kfr320. [DOI] [PubMed] [Google Scholar]
- 43.Santos-Rosa H, et al. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419(6905):407–11. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 44.Rice JC, et al. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell. 2003;12(6):1591–8. doi: 10.1016/s1097-2765(03)00479-9. [DOI] [PubMed] [Google Scholar]
- 45.Baylin SB. Resistance, epigenetics and the cancer ecosystem. Nat Med. 2011;17(3):288–9. doi: 10.1038/nm0311-288. [DOI] [PubMed] [Google Scholar]
- 46.Seligson DB, et al. Global levels of histone modifications predict prognosis in different cancers. Am J Pathol. 2009;174(5):1619–28. doi: 10.2353/ajpath.2009.080874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao C, et al. Cadmium induces histone H3 lysine methylation by inhibiting histone demethylase activity. Toxicol Sci. 2015 doi: 10.1093/toxsci/kfv019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Verdone L, Caserta M, Di Mauro E. Role of histone acetylation in the control of gene expression. Biochem Cell Biol. 2005;83(3):344–53. doi: 10.1139/o05-041. [DOI] [PubMed] [Google Scholar]
- 49.Archer SY, Hodin RA. Histone acetylation and cancer. Curr Opin Genet Dev. 1999;9(2):171–4. doi: 10.1016/s0959-437x(99)80026-4. [DOI] [PubMed] [Google Scholar]
- 50.Shogren-Knaak M, et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science. 2006;311(5762):844–7. doi: 10.1126/science.1124000. [DOI] [PubMed] [Google Scholar]
- 51.Fraga MF, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37(4):391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 52.Jo WJ, et al. Acetylated H4K16 by MYST1 protects UROtsa cells from arsenic toxicity and is decreased following chronic arsenic exposure. Toxicol Appl Pharmacol. 2009;241(3):294–302. doi: 10.1016/j.taap.2009.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]