

Abstract
Glass-supported lipid bilayers presenting freely diffusing proteins have served as a powerful tool for studying cell-cell interfaces, in particular, T cell–antigen presenting cell (APC) interactions, using optical microscopy. Here we expand upon existing protocols and describe the preparation of liposomes by an extrusion method, and describe how this system can be used to study immune synapse formation by Jurkat cells. We also present a method for forming such lipid bilayers on silica beads for the study of signaling responses by population methods, such as western blotting, flow cytometry, and gene-expression analysis. Finally, we describe how to design and prepare transmembrane-anchored protein-laden liposomes, following expression in suspension CHO (CHOs) cells, a mammalian expression system alternative to insect and bacterial cell lines, which do not produce mammalian glycosylation patterns. Such transmembrane-anchored proteins may have many novel applications in cell biology and immunology.
Keywords: supported lipid bilayers, synapse, signal transduction, diffusion, protein engineering, liposome extrusion, recombinant transmembrane proteins, suspension CHO cells, CHOs cells
Introduction
Cell-cell interactions govern diverse biological processes during development and in brain and immune function. Such cell-cell communication is dictated by interactions between membrane proteins expressed on the two cell surfaces. Our understanding of such membrane protein interactions is limited because tools to study such interactions are scarce. One useful tool that has emerged over the past two decades is the employment of glass-supported lipid bilayers as a surrogate for a second cell in the study of cell-cell interactions (Dustin, 2009). This system was first established by McConnell and colleagues (Watts et al., 1984, 1985, 1986; McConnell et al., 1986; Subramaniam et al., 1986) who, among many other applications, used it to stimulate T cells with peptide-loaded MHC molecules (pMHC). The methodology was adopted and further developed by Dustin and colleagues, who used it to study the immunological synapse, the two-dimensional interface formed between the T cell and an APC, presenting a cognate antigen (Dustin et al., 1996, 1998; Dustin, 1997; Grakoui et al., 1999). These studies highlighted the usefulness of this reductionist approach because: (a) one could have quantitative control over the density of molecules being incorporated into the bilayer, (b) the flat interface was ideal for optical imaging, and (c) the resulting lipid bilayers elicit immune function of the T cell. This has led to the use of bilayers beyond immunology, and similar reductionist approaches have become employed to study other systems in cell biology (Stroumpoulis et al., 2007; Thid et al., 2007; Yu et al., 2011, 2012, 2013; Luo et al., 2013).
Lipid bilayers are formed by the fusion of liposomes with an acid-etched glass surface. If the glass surface contains a metal coating, the metal coating prevents the formation of a bilayer on the underlying substrate. Researchers have taken advantage of this property by generating nano-patterned substrates that allow spatial patterning of the lipid bilayers, and thus the stimulus (Groves and Dustin, 2003; Mossman et al., 2005). This spatial patterning has been elegantly used to perform correlative light and electron microscopy experiments in the study of the immune synapse (Choudhuri et al., 2014). In this protocol, we present methods for making glass-supported lipid bilayers that complement several excellent previously published protocols (Dustin et al., 2007; Vardhana and Dustin, 2008; Luo et al., 2013; Zheng et al., 2015) based on what we have learned in the past few years. We hope that after reading this protocol, readers will be able to adapt this system to the study of any receptor-ligand interaction in any cell-cell context.
In adapting this protocol to one's specific research question, several experimental factors need to be considered before choosing the right mode of attachment of proteins to lipid bilayers. The easiest mode is based upon the interaction of soluble, polyhistidine (poly-His)–tagged proteins with a Ni-chelating NTA (Ni-NTA) headgroup containing lipids. Poly-His tags can be engineered onto one end of the extracellular domain of the protein of interest. The interaction of histidine with Ni-NTA is weak, and at least 10 histidine (His) tags are recommended (Nye and Groves, 2008). Other successful methods are to express the proteins as glycosyl-phosphatidyl-inositol (GPI)-anchored proteins (Dustin et al., 2007) or as transmembrane-anchored proteins (described in this unit). The potential pros and cons of each approach will be discussed. Alternatively, one can take advantage of biotinylated lipids and attach monobiotinylated proteins to the bilayers via streptavidin (Carrasco et al., 2004; Kaizuka et al., 2007). Monobiotinylation can be achieved either by reacting purified proteins with low concentrations of reactive biotin (Fleire and Batista, 2009) or by engineering a BirA tag and co-transfecting the biotin-ligase enzyme in the expression system of interest (Lesley and Groskreutz, 1997). Poly-His-tagged proteins can be either purchased commercially or purified following expression in diverse expression systems, such as E. coli, insect cells, or mammalian cells. One convenient mammalian expression system is the use of suspension CHO (CHOs) cells, which we have used to express GPI-anchored, poly-His-tagged, and transmembrane-anchored proteins.
Of interest to researchers studying the cell biology of immune cells, a convenient model is to stimulate mouse or human T cells or Jurkat cells with lipid bilayers containing intercellular adhesion molecule 1 (ICAM-1) and α-CD3 antibody, as this permits the stimulation of these cells regardless of their antigen specificity. Typically, in such experiments, α-CD3 is monobiotinylated and ICAM-1 is poly-His tagged (Kaizuka et al., 2007; Ilani et al., 2009; Hsu et al., 2012; Yi et al., 2012; Dillard et al., 2014; Furlan et al., 2014).
In this unit, we will first describe how to make liposomes using an extrusion method (Basic Protocol 1). The extrusion method presented here is quicker, as well as much less complex, and poses less risk of lipid oxidation during the process of liposome formation than conventional methods involving detergent dialysis. We then describe how to make glass-supported lipid bilayers in perfusion chambers (Basic Protocol 2). Basic Protocol 3 describes a procedure for studying the immune synapse formed between Jurkat cells and supported lipid bilayers containing α-CD3 antibodies and ICAM-1. Basic Protocol 4 describes how to form lipid bilayers on silica beads (also see Yokosuka et al., 2008) for use in the stimulation of cells for measuring signaling responses, by a variety of methods. Basic Protocol 5 is a procedure for stimulating T cells with artificial APCs prepared as glass-supported lipid bilayers on silica beads. Finally, we describe a new mode of attachment of proteins in lipid bilayers based on engineered transmembrane anchors (Basic Protocol 6).
Basic Protocol 1
Procedure for Making Liposomes of Ni-NTA or CAP-Biotin Lipids Using Extrusion
One of the published protocols for preparing liposomes involves dissolving lipids in detergents followed by dialysis (Dustin et al., 2007). Upon the removal of detergents by dialysis, lipids spontaneously form liposomes. One potentially severe drawback of this procedure is that it takes 3 to 4 days of dialysis to remove all detergent, increasing the likelihood of lipid oxidation. In addition, the success of the dialysis is heavily contingent on the quality of the dialysis membrane, and, under nonoptimal conditions, low concentrations of detergent may remain, which results in poor bilayer quality. When using an extrusion method (Lin et al., 2010), large volumes of liposomes can be prepared in a short amount of time (3 to 4 hr), potentially eliminating both of the previously mentioned complications associated with dialysis. As described in detail below, we perform our extrusion procedure using a commercial mini-extruder manufactured by Avanti Polar Lipids, Inc.
Materials
Tris-saline buffer (see recipe)
Nitrogen gas cylinder with regulator
High-purity chloroform
10 mg/ml Ni-NTA (1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiacetic acid)succinyl]) [nickel salt] lipid stocks (Avanti Polar Lipids, Inc.) in chloroform
10 mg/ml DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) lipid stocks (Avanti Polar Lipids) in chloroform
10 mg/ml CAP-biotin (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(CAP biotinyl)) lipid stocks (Avanti Polar Lipids) in chloroform
Argon gas cylinder with regulator
Gas dispersion tube with fritted end (Sigma, cat. no. CLS3953312C)
Screw-top glass vials
Lyophilizer
Mini-extruder kit, including retainer nut, extruder outer casing, Teflon bearing, internal membrane supports, O-rings, filter supports, air-tight 1-μl glass syringes, extruder stand, and extrusion membranes (0.1 μm in size) (Avanti Polar Lipids)
Autoclaved glass beaker
Bath sonicator
- The night before extrusion, prepare 500 ml of Tris-saline buffer as described in Reagents and Solutions. Purge the solution of oxygen by bubbling nitrogen gas through the solution using a gas dispersion tube for approximately 15 min. Store at 4°C until ready for use. Also prepare 10 to 20 screw-top glass vials that will be used to hold your liposome solutions. To clean the vials, rinse each vial at least three times with high-purity chloroform. After the last rinse, dry any remaining chloroform from each vial using a stream of nitrogen gas.Unused vials can be stored and kept in a clean container for use in future extrusions.
To prepare 2 ml of 10× (4 mM) Ni-NTA liposomes (6.25% Ni-NTA), pipet 52.6 μl of 10 mg/ml Ni-NTA lipids in chloroform and 589.6 μl of 10 mg/ml DOPC lipids in chloroform into one of the glass vials. To prepare 2 ml of 10× (4 mM) CAP-biotin liposomes (2% CAP-biotin), pipet 18 μl of 10 mg/ml CAP-biotin lipids in chloroform and 616.35 μl of 10 mg/ml DOPC lipids in chloroform into one of the glass vials.
Using a gentle stream of nitrogen, evaporate the chloroform from the vial in a fume hood until a white precipitate forms on the walls of the vial.
- Place the vial containing the precipitated lipids into a lyophilizer for 4 to 24 hr to remove virtually all of the remaining chloroform.The amount of time needed for complete removal of the chloroform will vary depending on the strength of the vacuum produced by a particular lyophilizer. We generally dry our lipids for 4 hr in a lyophilizer that produces a vacuum of 10 mTorr. If unsure, dry your precipitated lipids for 24 hr.
-
While the lipids are drying in the lyophilizer, assemble the mini-extruder according to the manufacturer's instructions.The full instructions, including illustrations, can be found at http://www.avantilipids.com, but we paraphrase them here and include tips that we have found to be useful in operating the extruder.
First, prepare an autoclaved glass beaker with approximately 10 ml of Tris-saline buffer and place (for each extrusion to be carried out) two filter supports, one polycarbonate membrane, and two rubber O-rings into the solution. Allow the supports and membranes to hydrate for approximately 5 min.
Remove the rubber O-rings from the solution and place them into the grooves on each of the internal membrane supports. Then, center one filter support within the circumference of each of the O-rings atop each of the internal membrane supports. (Fig. 24.5.1).
Place one of the internal membrane supports into the extruder outer casing. Into the retainer nut, first place the Teflon bearing and then the other internal membrane support.
- Remove the polycarbonate membrane from the beaker and place it atop the internal membrane support contained within the extruder outer casing.The membrane is slightly larger than the internal membrane support and should be centered such that it overlaps the rubber O-ring equally on all sides. The user must ensure that there are no air bubbles trapped between the filter support and the membrane.
Screw the retainer nut containing the second internal membrane support into the extruder outer casing until finger-tight.
Place the extruder assembly onto the extruder stand, making sure that the apex of the extruder outer casing's hex nut points downward.
- Finally, test the extruder for leaks before carrying out an extrusion by flowing Tris-saline buffer through the assembly. First, insert one of the two empty 1-μl glass syringes into the extruder with its plunger fully depressed. Close the latch over the syringe to secure it to the extruder stand. Fill the other glass syringe with 1 ml of the Tris-saline buffer from the beaker and load it into the other side of the extruder (Fig. 24.5.2). Secure it to the extruder stand using the second latch. Slowly depress the plunger of the full syringe, and liquid will be transferred through the extruder and into the second syringe.If the entire volume of liquid is transferred into the empty syringe, then the extruder has been properly assembled. If, however, the entire volume is not transferred, then the retainer nut likely needs to be tightened to make the seal water-tight.While pushing liquid through the extruder apparatus, it is helpful to use one finger of your free hand to apply gentle downward pressure on the syringe to stabilize it.
Remove the dried lipids from the lyophilizer and add 2 ml of Tris-saline buffer. After capping the vial, repeatedly alternate between vortexing for 30 sec and bath sonication for approximately 1 min until the dried lipid film forms a milky white suspension in solution. Repeat this procedure five to ten times to ensure that all of the dried lipids have entered into solution.
- As in step 5g, insert one empty glass syringe into the extruder assembly and secure. Fill the second syringe with the lipid suspension, insert into the other end of the extruder assembly, and secure. Gently depress the plunger of the syringe containing the lipid suspension until the whole volume has been transferred through the extruder and into the empty syringe. Likewise, pass the suspension back through the extruder to the first syringe. Repeat this process approximately 10 times.The resulting mixture will be 10× Ni-NTA or CAP-biotin liposomes.As the suspension is repeatedly passed through the extruder, the amount of force needed to push it through the apparatus will lessen and its color will gradually change from milky white to clear. The experimenter may also note that the solution has a slight violet color.
Eject the extruded 10× liposome solution into a clean sample vial.
To produce 1× liposome solutions, dilute the 10× liposome solution with Tris-saline buffer into the previously cleaned glass vials.
- Top the headspace of the vials containing the 1× liposome solutions with argon and cap. To take extra precautions against any oxygen entering the vials, seal each vial with Parafilm. Store at 4°C until use. After each subsequent opening of the vial, repeat the same procedure, filling the headspace with argon and resealing the vial with Parafilm.After each extrusion, be sure to follow the manufacturer's recommended cleaning instructions, which can be found on their Web site, mentioned above.
Figure 24.5.1.

Photograph of the various assembly parts of the mini-extruder from Avanti Polar Lipids, Inc., described in Basic Protocol 1.
Figure 24.5.2.

Image of the assembled extruder. The arrowhead shows the inlet where the syringe is inserted.
Basic Protocol 2
Procedure for Making Lipid Bilayers in Flow Cells
Lipid bilayers are prepared in flow or perfusion chambers for the study of the interactions between cell surface receptors on one cell and their respective ligands on the surfaces of other cells. In the past, this system has been used to study the interaction of the T cell receptor on the surface of T cells with peptide-loaded MHC molecules, costimulatory molecules, and adhesion molecules presented by fluid lipid bilayers, particularly using TIRF microscopy. This protocol is written expressly for use with the perfusion chambers manufactured by Bioptechs, Inc., but can be easily adapted to other commercially available flow cells.
Materials
HBS-BSA (see recipe)
1× Ni-NTA or CAP-biotin liposome solutions (Basic Protocol 1)
Poly-His-tagged or streptavidin-modified proteins of interest (the protein of interest would typically be a ligand of a receptor expressed on the cell type of interest)
Casein solution (see Support Protocol 3)
20-ml plastic syringes
Focht Chamber System (FCS2) (Bioptechs; http://www.bioptechs.com/Products/FCS2/fcs2.html)
Piranha solution–etched 40-mm-diamter cover glasses (Dustin et al., 2007)
Tygon tubing (R-3603; i.d. 1/16 in., o.d. 1/8 in.)
Two-way and three-way stopcocks (BioRad)
- Load a 20-ml plastic syringe with 20 ml of HBS-BSA. This solution is prone to frothing, and care must be taken to remove large bubbles from solution. This can be accomplished by holding the syringe with the plunger facing downward and tapping it on the edge of the bench. Bubbles will rise toward the syringe outlet and can be ejected into the sink. Lastly, attach the syringe to the inlet tubing for the flow cell and fully prime the line.It is crucial to remove as many bubbles as possible and avoid injecting them into the flow cell, as air will destroy the lipid bilayers.
Place the white retaining ring from the FCS2 system on the bench with the circular portion containing the perfusion tubes facing upward.
Place the circular rubber gasket with holes onto the perfusion tubes, ensuring that it is completely seated.
Place the microaqueduct slide atop the rubber gasket with the T-shaped flow channels facing upwards, again aligning the holes with the perfusion tubes and ensuring that it is completely seated.
Place the gasket (0.25 mm in thickness) with the rectangular cutout atop the microaqueduct slide, ensuring that the flow channels remain unobstructed.
Pipet 1 to 2 μl of 1× liposome solution (either Ni-NTA or CAP-biotin) into three or more distinct spots on the microaqueduct slide.
- Drop one Piranha solution–etched 40-mm-diameter cover glass onto the microaqueduct slide.Clean and etch the cover glass as described (Dustin et al., 2007). At this point, the lipid droplets will spread slightly. The distance between the droplets of liposome solution placed on the microaqueduct slide should be adjusted so that the spots do not merge when the cover glass is dropped onto the slide.
- Align the clamps of the stainless steel clamp base unit with the notches in the white retaining ring and lower it gently into place. Holding the clamp base and the white retaining ring together, flip the entire flow cell assembly over, and rotate the edge of the clamp base clockwise to firmly attach it to the microaqueduct slide.CAUTION: Over-tightening the clamp base can crack the cover glass. Tighten the assembly only enough to secure all the pieces of the flow cell firmly together.
With the clamps of the clamp base now facing upwards, mark the area where the droplets of liposome solution have spread onto the cover glass.
- Attach both the outlet tubing and the previously prepared syringe assembly containing HBS-BSA to the flow cell, and inject approximately 5 ml of HBS-BSA.Lipid bilayers form on the coverslip within at most 30 sec of dropping the liposome solution onto the glass coverslip. Care should be taken to assemble the flow cell and flow in the HBS-BSA as quickly as possible, as the bilayers can become immobile if allowed to sit for too long on the cover glass.
- Using the three-way valve in the inlet tubing, flow in 300 to 500 μl of solution containing His-tagged or streptavidin-modified proteins of interest at the concentrations required to achieve the required density of proteins on the bilayer. Incubate for 1 hr at room temperature.The procedure for calibrating the adsorbed amount of proteins has been described in great detail elsewhere (Dustin et al., 2007; Galush et al., 2008).
Rinse the flow cell with 3 to 5 ml of HBS-BSA.
Inject 1 ml of 37°C casein solution into the flow cell and incubate for 30 min to 1 hr.
- Finally, flush the casein out of the flow cell with 5 ml of HBS-BSA.At this point, cells can be injected into the flow cell and imaging of the cells' interaction with the bilayers can be performed.
Basic Protocol 3
Procedure for Studying the Immune Synapse Formed Between Jurkat Cells and Supported Lipid Bilayers Containing α-CD3 Antibodies and ICAM-1
Several laboratories have taken advantage of Jurkat cells to address basic cell biological questions surrounding the formation of the immunological synapse. This procedure becomes particularly useful for labs that do not maintain a mouse colony and do not have access to T cells from TCR transgenic mice. Jurkat cells will form an immunological synapse on lipid bilayers containing lipid-anchored α-CD3 and ICAM-1. LFA-1 is not very active on Jurkat cells and does not undergo a significant affinity change due to inside-out signaling and, for this reason, the amount of ICAM-1 incorporated in the bilayer needs to be less than when studying primary T cells. To incorporate α-CD3 antibodies in the supported bilayer, they are first mono-biotinylated. The antibody is attached to bilayers containing biotin lipids via streptavidin. Since streptavidin is tetravalent, having more than one biotin per antibody can create multimeric antibody-streptavidin complexes. We will describe in this protocol how to avoid attaching more than one antibody per streptavidin molecule. Antibodies are monobiotinylated following the procedure described by Fleire and Batista (2009) and will not be presented here. Following mono-biotinylation, the antibody can be conjugated to a fluorescent dye. Jurkat cells are of human origin, and since human LFA-1 can bind to mouse ICAM-1 (Johnston et al., 1990), ICAM-1 from either species can be used in these experiments. On the contrary, mouse LFA-1 cannot bind to human ICAM-1 (Johnston et al., 1990), and therefore mouse ICAM-1 needs to be used for experiments involving mouse T cells. Here, fluorescently-labeled ICAM-1 is incorporated in lipid bilayers via poly-His tags. T cells purified from peripheral blood mononuclear cells can also be used in this model.
Materials
1× Ni-NTA and 1× CAP-biotin liposome solutions (Basic Protocol 1)
Mono-biotinylated α-CD3 (Fleire and Batista, 2009)
Streptavidin
Poly-His-tagged ICAM-1 (Sino Biological, cat. no. 50440-M08H-20)
Casein solution (See Support Protocol 3)
HBS-BSA (see recipe)
Additional reagents and equipment for preparing glass-supported lipid bilayers in flow cells (Basic Protocol 2)
Prepare glass-supported lipid bilayers using a 1:1 mixture of 1× Ni-NTA liposomes (6.25%) and 1× CAP-biotin liposomes (2%) as described in Basic Protocol 2.
- Prepare 1:1 complexes of monobiotinylated α-CD3 and streptavidin by mixing 250 μl of 2 μg/ml (13.3 nM) of monobiotinylated α-CD3 with 250 μl of a two-fold molar excess of streptavidin (27 nM, 1.43 μg/ml) and incubate the mixture for 10 min at room temperature.It is important to prepare the dilute solutions in two separate tubes and then mix them rapidly, as the biotin-streptavidin affinity is very high. We also recommend a titration of antibody concentrations (0.1 to 10 μg/ml).
- To this mixture, add poly-His-tagged ICAM-1 such that the final site density on the bilayer is 10 molecules/μm2. Incubate this mixture containing the antibody-streptavidin complexes and ICAM-1 with the bilayer for 1 hr at room temperature.The protein concentration required to achieve a specific site density on the lipid bilayer can be determined by following a procedure described previously (Dustin et al., 2007).
Rinse the flow cell with 3 to 5 ml of HBS-BSA and block with a solution of casein as described in Basic Protocol 2.
- Rinse the flow cell with 3 to 4 ml of HBS-BSA and heat it at 37°C using the provided flow chamber heater.At this point, Jurkat cells or human T cells can be injected into the flow cell and immune synapse formation can be visualized.
Basic Protocol 4
Procedure for Preparing Lipid Bilayers on Silica Beads
Similar to the formation of fluid lipid bilayers on glass coverslips for studying the dynamics of receptor-ligand interactions using TIRF microscopy, fluid lipid bilayers may also be formed on silica beads for use in stimulating cells and studying the downstream signaling that results. In this protocol, we describe the process of forming fluid lipid bilayers on silica beads for this purpose, and follow with the protocol (see Basic Protocol 5) for stimulating cells with the beads and performing western blot analysis of the resulting signaling. We term these beads coated with functionalized fluid lipid bilayers artificial antigen-presenting cells (artificial APCs).
Materials
1× Ni-NTA and 1× CAP-biotin liposome solutions (Basic Protocol 1)
5-μm silica bead suspension (supplied as 10% solid; Bangs Laboratories, cat. no. SS05N)
HBS-BSA (see recipe)
Proteins of interest
96-well V-bottom microtiter plate or 1.5-ml microcentrifuge tubes
Plate-sealing tape
Benchtop centrifuge with microtiter plate adapter
Orbital microtiter plate shaker
Place 5 μl of 1× liposome solution (either Ni-NTA or CAP-biotin prepared as in Basic Protocol 1) in the desired numbers of wells of a 96-well V-bottom microtiter plate.
- To each well add 2.5 μl of the 5-μm silica bead suspension and cover the plate with sealing tape.To prepare silica-coated bilayers on a bead suspension of more than 20 μl, the beads can be spun down, the supernatant removed, and the beads resuspended in liposome solution at twice the volume of the bead suspension in 1.5-ml microcentrifuge tubes.
- Pulse the plate on a vortexer three times, each time for approximately 10 sec.Ensure that the plate-sealing tape is securely adhered to the surface of the plate so as to avoid mixing of samples from adjacent wells. If not using all 96 wells in the plate, use of only every other well in the plate is advisable to avoid spillover between wells during vortexing.
Add 150 μl of HBS-BSA to each of the wells and mix by pipetting up and down several times. Spin the plate in a centrifuge for 2 min at 600 × g, room temperature. Aspirate the supernatant from each well, paying careful attention not to disturb the pelleted beads and leaving behind a few microliters of supernatant. Completely removing the supernatant will destroy the bilayers that have formed on the beads. To remove excess lipid left in the wells, repeat the wash two or more times.
To attach proteins of interest to the bilayers, pipet 150 μl of protein-containing HBS-BSA at the concentration necessary to reach the desired protein density into each well. Pipet up and down several times to ensure that the beads are well mixed with the protein-containing solution.
Again, seal the plate with plate-sealing tape and place the plate on an orbital microtiter plate shaker for 30 min.
After shaking the plate, repeat step 4 to remove excess protein from each of the wells.
Resuspend the beads in each well with 150 μl of HBS-BSA until ready to use for cell-stimulation experiments (e.g., Basic Protocol 5).
Basic Protocol 5
Stimulating T Cells with Artificial APCs
When glass-supported lipid bilayers are prepared in flow chambers, the geometry is ideal for microscopic observation of signaling processes. One is however limited to a few sets of conditions, as it becomes prohibitively time consuming to image more than 10 flow chambers in a single day. Under such circumstances, artificial APCs (see Basic Protocol 4) are more useful, as one can process multiple stimulation conditions for monitoring a large number of signaling parameters either by flow cytometry or western blot analysis. We have typically used silica beads with diameters between 5 and 6 μm. For stimulating T cells, we recommend a ratio of beads to T cells between 2 and 5 to maximize the likelihood of interaction between the cells and the beads. The beads are supplied as a 10% solid suspension in deionized water. A quick calculation reveals that there are approximately 2 million beads in 1 μl of this suspension. Cells can be stimulated in 96- to 12-well plates, depending on the number of cells that need to be stimulated. We do not recommend carrying out cell stimulations in 6-well plates or plates with larger wells, but rather duplicating the conditions in 12-well plates if more cells need to be stimulated. See Table 24.5.1 for choice of the optimum stimulation geometry for a given number of cells.
Table 24.5.1. Recommended Tissue Culture Plate and Number of Lipid-Bilayer-Coated Silica Beads for Stimulating a Given Number of T Cells.
| Plate size | Well area (cm2) | Stimulation volume (μl) | Number of T cells | Number of beads |
|---|---|---|---|---|
| 96-well flat bottom | 0.32 | 200 | 100,000-200,000 | 500,000 |
| 48-well | 0.95 | 500 | 500,000 | 1,000,000 |
| 24-well | 1.9 | 1000 | 1,000,000 | 4,000,000 |
| 12-well | 3.8 | 2000 | 2,000,000-5,000,000 | 20,000,000 |
Materials
T cells, or other cells used for stimulation (see appropriate units of Coligan et al., 2015)
Artificial APCs (Basic Protocol 4)
FACS buffer (see recipe), chilled
Primary antibodies against the extracellular portion of the proteins of interest or against intracellular proteins or phosphorylated proteins of interest
Fluorescently labeled secondary antibodies against primary antibody isotype, if necessary
FACS buffer (see recipe) containing 0.05% (w/v) saponin
2% (w/v) paraformaldehyde in PBS (see appendix 2a for PBS)
Phosphate-buffered saline (PBS; appendix 2a)
Lysis buffer (see recipe)
6× SDS loading dye (unit 6.1; Gallagher, 2007)
Molecular weight markers for SDS-PAGE (also see unit 6.1; Gallagher, 2007)
5% (w/v) nonfat dry milk in PBS (see appendix 2a for PBS)
Flat-bottom microtiter plate, number of wells determined by Table 24.5.1
Benchtop centrifuge with microtiter plate adapters
37°C water bath or 37°C, 5% CO2 humidified incubator
2-ml U-bottom centrifuge tubes
Boiling water bath
Nitrocellulose membranes (see unit 6.1; Gallagher, 2007)
Additional reagents and equipment for flow cytometry (Robinson et al., 2015), SDS-PAGE (unit 6.1; Gallagher, 2007), and western blotting (immunoblotting; unit 6.2; Gallagher et al., 2011)
Stimulate cells
- Determine the number of T cells to be stimulated and spin them for 5 min at 300 × g,room temperature. Resuspend the cells in HBS-BSA at room temperature.The volume of buffer to be used for resuspension is based on the plate geometry, as given in Table 24.5.1.
- Spin the artificial APCs, as prepared in Basic Protocol 4, to which pMHC complexes, adhesion and/or costimulatory molecules have been attached, for 2 min at 600 × g, room temperature. Aspirate the supernatant and resuspend the artificial APCs in the solution containing the T cells (from step 1) and dispense them into the wells of the appropriate plate in the appropriate stimulation volume.Determine the plate size, stimulation volume, number of T cells, and the number of beads required for a given stimulation condition from Table 24.5.1.
Spin the plate 30 to 60 sec at 60 × g, room temperature, so that the cells and beads sediment to the base of the well.
- Incubate the plate at 37°C to allow signaling to occur, either in a water bath for short stimulation times (2 to 15 min) or in a humidified 37°C, 5% CO2 incubator for longer times.Cells can be stimulated in HBS-BSA for a maximum period of 6 hr, after which they may become unhealthy due to lack of nutrients found in complete culture medium.
After the end of the incubation, add an equal volume of chilled FACS buffer to each well. Gently transfer the cells into 2-ml U-bottom centrifuge tubes, which have been chilled on ice. Spin the tubes 3 min at 600 × g, 4°C.
Stain cells or lyse for western blot analysis
At this stage, the cells can be used for one of three different kinds of assays: (a) they can be stained for cell surface markers in cold FACS buffer; (b) they can be stained with antibodies against intracellular phosphorylated proteins to analyze signaling responses, or (c) they can be lysed for western blot analysis. The steps for each of these processes are described below.
6a. Perform cell surface staining
- Prepare 100 μl of the appropriate dilution of primary antibody against cell surface proteins of interest in FACS buffer for each of the stimulation conditions.Concentration of antibody at which saturation binding is observed should be determined independently. Typically, the saturation concentration ranges between 1 and 10 μg/ml.If all conditions are identical, a master dilution of antibody consisting of (number of conditions × 100 μl) can be prepared. Do not forget to prepare one aliquot of unstained cells for use as a blank control condition.
Resuspend the stimulated cells from step 5 in 100 μl of diluted antibody from step 1 in a 1.5-ml microcentrifuge tube and incubate for 1 hr on ice.
Wash the cells once in 1 ml of 4°C FACS buffer by spinning for 3 min at 600 × g, 4°C.
- If it is necessary to stain the cells with fluorescently labeled secondary antibodies, prepare 100 μl of the appropriate dilution of secondary antibody for each of the stimulation conditions.As in step 6a, substep i, if all the conditions are identical, then a master dilution consisting of (number of conditions × 100 μl) can be prepared.
Resuspend the primary antibody labeled cells from step 3 and resuspend them in 100 μl of diluted secondary antibody from step 4 in a 1.5-ml microcentrifuge tube and incubate for 1 hr on ice.
Wash the cells once in 1 ml of 4°C FACS buffer by spinning for 3 min them at 600 × g, 4°C. Resuspend the cells in 100 μl of FACS buffer and analyze by flow cytometry (Robinson et al., 2015).
6b. Perform stain for intracellular proteins
- For permeabilization, prepare 100 μl of the appropriate dilution of primary antibody against intracellular proteins or against phosphorylated proteins of interest in cold FACS buffer containing 0.05% saponin for each of the stimulation conditions.The concentration of antibody at which saturation binding is observed should be determined independently. Typically, the saturation concentration ranges between 1 and 10 μg/ml.If all conditions are identical, then a master dilution of antibody consisting of (number of conditions × 100 μl) can be prepared. Do not forget to prepare one aliquot of unstained cells for use as a blank control condition.
Fix the stimulated cells from step 5 in 500 μl of 2% paraformaldehyde prepared in PBS in a 1.5-ml microcentrifuge tube for 10 min at room temperature.
Wash the cells once with 1 ml FACS buffer by spinning them 3 min at 600 × g, room temperature.
Resuspend the cells in 100 μl of diluted antibody from substep i in a 1.5-ml microcentrifuge tube and incubate for 1 hr at room temperature.
Wash the cells once with 1 ml FACS buffer by spinning them 3 min at 600 × g, room temperature.
- If it is necessary to stain the cells with fluorescently labeled secondary antibodies, prepare 100 μl of the appropriate dilution of secondary antibody for each of the stimulation conditions.As in step 6a, substep i, if all the conditions are identical, then a master dilution consisting of (number of conditions × 100 μl) can be prepared.
Resuspend the primary antibody–labeled cells from substep v in 100 μl of diluted secondary antibody from substep vi in a 1.5-ml microcentrifuge tube and incubate for 1 hr at room temperature.
Wash the cells once in 1 ml of FACS buffer by spinning them 3 min at 600 × g, room temperature. Resuspend the cells in 100 μl of FACS buffer and analyze them by flow cytometry.
6c. Perform western blot analysis
Resuspend 3 to 5 million stimulated cells from step 5 in 100 μl lysis buffer on ice for 30 min.
Microcentrifuge the lysate at 13,000 rpm for 10 min at 4°C.
Transfer 90 μl of the supernatant, taking care not to disturb the pelleted cellular debris, to a 1.5-ml microcentrifuge tube. Add 18 μl of 6× SDS loading dye and heat the sample in a boiling water bath for 5 min.
- Load 10 to 20 μl of sample lysate from substep iii into each well of a linear or gradient polyacrylamide gel and perform SDS-PAGE (see unit 6.1; Gallagher, 2007).The choice of gel will depend on the molecular weight of the protein. Do not forget to load a lane with molecular weight markers.
Following electrophoresis, transfer the protein onto a nitrocellulose membrane using commercially available transfer apparatus (unit 6.2; Gallagher et al., 2011).
Block the nitrocellulose membrane with 5% nonfat dry milk suspension prepared in PBS for 1 hr at room temperature.
- Process the membrane with primary and secondary antibodies according to standard protocols for western blot analysis (unit 6.2; Gallagher et al., 2011).We recommend imaging the membranes using CCD camera–based phosphorimagers for quantitative analysis.
Basic Protocol 6
Design, Construction, and Purification of Tailless Transmembrane-Anchored Proteins for Incorporation in Lipid Bilayers
In the earliest demonstration of this system, single-pass transmembrane-anchored proteins were purified from detergent-solubilized cells and incorporated in liposomes; however, when glass-supported bilayers were made from such liposomes, the proteins did not diffuse because the cytoplasmic region of the protein became adsorbed to the underlying glass substrate (McConnell et al., 1986). For this reason, subsequently, proteins were expressed as GPI-anchored proteins to permit lateral diffusion in supported lipid bilayers (Grakoui et al., 1999). For some research questions, however, it might be advantageous to incorporate a transmembrane-anchored protein in supported bilayers such that it undergoes free diffusion. The transmembrane or membrane proximal regions of the protein may possess unique properties that researchers may wish to study.
The simplest approach for designing a transmembrane-anchored protein that would diffuse freely in the bilayer is to truncate the protein after the last hydrophobic residue of the transmembrane domain. We, however, found that some proteins would get expressed as a GPI-anchored protein if the cytoplasmic region were removed, as the transmembrane region serves as a GPI-anchoring signal sequence. There are other examples in the literature where such a phenomenon has been reported (Mitchell et al., 1991; Bell et al., 1994; Jacobs et al., 1996). Fortunately, the characteristics of a GPI-anchoring signal sequence are well understood, and there exist bioinformatics programs that can predict with confidence whether a protein would become GPI-anchored when expressed (Eisenhaber et al., 1998; Eisenhaber et al., 1999). Here, we describe the construction and purification of a transmembrane-anchored CD80 construct that contains fluorescent protein between the extracellular domain and the transmembrane domain. We made glass-supported lipid bilayers with liposomes containing this purified protein, and showed that the protein undergoes free diffusion in supported lipid bilayers. The protein was also functional, as T cells could cluster this protein in the immunological synapse. Researchers can take advantage of the principles described here to engineer proteins of their interest to be incorporated in supported lipid bilayers as transmembrane-anchored proteins.
Using bioinformatics tools to predict GPI-anchoring signal sequences within transmembrane regions
-
Download the amino acid sequence of the transmembrane protein of interest,in our case CD80, from either PubMed (http://www.pubmed.gov) or Uniprot (http://uniprot.org).
>sp|Q00609|CD80_MOUSE T-lymphocyte activation
antigen CD80 OS=Mus musculus GN=Cd80 PE=1 SV=1
MACNCQLMQDTPLLKFPCPRLILLFVLLIRLSQVSSDVDEQLSKSVKDKVLLPCRYNSPHEDESEDRIYWQKHDKVVLSVIAGKLKVWPEYKNRTLYDNTTYSLIILGLVLSDRGTYSCVVQKKERGTYEVKHLALVKLSIKADFSTPNITESGNPSADTKRITCFASGGFPKPRFSWLENGRELPGINTTISQDPESELYTISSQLDFNTTRNHTIKCLIKYGDAHVSEDFTWEKPPEDPPDSKNTLVLFGAGFGAVITVVVIVVIIKCFCKHRSCFRRNEASRETNNSLTFGPEEALAEQTVFL
-
Determine the transmembrane domain using one of several transmembrane-region predicting tools found on http://web.expasy.org. We have used TMHMM Server v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM-2.0/).As shown in Figure 24.5.3, the output of this program shows that amino acid residues from 249-271 (in bold and underlined) comprise the transmembrane region and 272-306 the cytoplasmic region.
>sp|Q00609|CD80_MOUSE T-lymphocyte activation
antigen CD80 OS=Mus musculus GN=Cd80 PE=1 SV=1
MACNCQLMQDTPLLKFPCPRLILLFVLLIRLSQVSSDVDEQLSKSVKDKVLLPCRYNSPHEDESEDRIYWQKHDKVVLSVIAGKLKVWPEYKNRTLYDNTTYSLIILGLVLSDRGTYSCVVQKKERGTYEVKHLALVKLSIKADFSTPNITESGNPSADTKRITCFASGGFPKPRFSWLENGRELPGINTTISQDPESELYTISSQLDFNTTRNHTIKCLIKYGDAHVSEDFTWEKPPEDPPDSKNTLVLFGAGFGAVITVVVIVVIIKCFCKHRSCFRRNEASRETNNSLTFGPEEALAEQTVFL -
As can be seen in the previous step, amino acid 271 of CD80 (phenylalanine) is the last hydrophobic residue of the transmembrane region and, hence, the protein should be truncated after this residue for incorporation in supported lipid bilayers. Take the following truncated amino acid sequence and test for GPI-linkage using BIG-PI tool (http://mendel.imp.ac.at/sat/gpi/gpi_server.html).
>sp|Q00609|CD80_MOUSE T-lymphocyte activation
antigen CD80 OS=Mus musculus GN=Cd80 PE=1 SV=1
MACNCQLMQDTPLLKFPCPRLILLFVLLIRLSQVSSDVDEQLSKSVKDKVLLPCRYNSPHEDESEDRIYWQKHDKVVLSVIAGKLKVWPEYKNRTLYDNTTYSLIILGLVLSDRGTYSCVVQKKERGTYEVKHLALVKLSIKADFSTPNITESGNPSADTKRITCFASGGFPKPRFSWLENGRELPGINTTISQDPESELYTISSQLDFNTTRNHTIKCLIKYGDAHVSEDFTWEKPPEDPPDSKNTLVLFGAGFGAVITVVVIVVIIKCFAs shown in Figure 24.5.4, a potential GPI-modification site was found. The score of this site was close to zero and hence was assigned the quality “S” (scores between −4.86 and 4.66), interpreted as needing experimental validation. A prediction with a score greater than 4.66 is assigned a quality “P” or predicted.
Users can also take advantage of an alternative prediction program called GPI-SOM (http://gpi.unibe.ch/). The results from Big-PI and GPI-SOM are similar and complementary.
-
For our purposes, we next screened many cell surface proteins to find a transmembrane domain that would not have a GPI-modification site (see Table 24.5.2). Based on these results, we decided to choose the transmembrane domain of CD28 to replace the transmembrane region of CD80 to circumvent GPI-anchorage. We assembled the following sequence:
MACNCQLMQDTPLLKFPCPRLILLFVLLIRLSQVSSDVDEQLSKSVKDKVLLPCRYNSPHEDESEDRIYWQKHDKVVLSVIAGKLKVWPEYKNRTLYDNTTYSLIILGLVLSDRGTYSCVVQKKERGTYEVKHLALVKLSIKADFSTPNITESGNPSADTKRITCFASGGFPKPRFSWLENGRELPGINTTISQDPESELYTISSQLDFNTTRNHTIKCLIKYGDAHVSEDFTWEKPPEDPPDSKNSGEFASMVSKGEELIKENMHMKLYMEGTVNNHHFKCTSEGEGKPYEGTQTMRIKVVEGGPLPFAFDILATSFMYGSRTFINHTQGIPDFFKQSFPEGFTWERVTTYEDGGVLTATQDTSLQDGCLIYNVKIRGVNFPSNGPVMQKKTLGWEANTEMLYPADGGLEGRTDMALKLVGGGHLICNFKTTYRSKKPAKNLKMPGVYYVDHRLERIKEADKETYVEQHEVAVARYCDLPSKLGHKLNSGAAAAGFWALVVVAGVLFCYGLLVTVALCVIWTThe sequence in italics is the extracellular domain of CD80, the region in bold is the sequence for the monomeric red fluorescent protein (RFP), and the underlined sequence is the transmembrane domain of CD28. RFP was incorporated into the protein so that the behavior of the protein once incorporated into lipid bilayers could be monitored. As shown in Figure 24.5.5, no potential GPI-modification site was found for this sequence. In Support Protocol 2, we describe how to experimentally verify the presence of GPI linkages.Standard molecular biology techniques can be used to generate plasmids that contain cDNA encoding the truncated CD80 constructs described in the previous section. The cDNA is cloned into a mammalian expression vector such as pcDNA3.1. The transmembrane domain was added using synthesized oligos using a method known as annealed oligo cloning (see for example, https://www.addgene.org/plasmid-protocols/annealedoligo-cloning/).
Figure 24.5.3.

Output of the transmembrane domain prediction program showing the amino acids that comprise the extracellular domain, the transmembrane domain, and the intracellular region or cytoplasmic tail.
Figure 24.5.4.

Output of the Big-PI predictor tool for the tailless CD80 protein as input sequence. Potential GPI-modification site in the tailless CD80 protein is found, and the amino acid is highlighted in red. The quality and score of the site are also shown.
Table 24.5.2. Transmembrane Domains that Would Not Have a GPI-Modification Sitea.
| Protein | Result | Domain length | Big-PI score |
|---|---|---|---|
| H-2Kb alpha chain | GPI | 23 | 2.92 |
| I-Ek beta chain | GPI | 22 | 6.42 |
| ICAM-1 | GPI | 24 | 3.4 |
| CD11c | GPI | 20 | 0.69 |
| MHCI related protein | GPI | 20 | 3.36 |
| B7H | GPI | 21 | 4.4 |
| CD4 | TM | 23 | −5.13 |
| LDL R. | GPI | 22 | 6.38 |
| MICA | GPI | 21 | 0.5 |
| MICB | GPI | 21 | 0.5 |
| CD45 | GPI | 21 | 6.16 |
| EPO R | GPI | 23 | 6.92 |
| Cadherin | GPI | 24 | 9.42 |
| CD19 | GPI | 24 | 4.78 |
| Insulin R | GPI | 21 | 2.94 |
| EGFR | GPI | 20 | 7.20 |
| NOTCH-1 | GPI | 21 | 4.25 |
| LFA-1 | GPI | 21 | 7.79 |
| NACR alpha4 TM4 (ACH receptor) | TM | 21 | −8.56 |
| CD2 | GPI | 26 | 4.53 |
| ICOS | GPI | 21 | 4.54 |
| Hematopoietic cell transducer | TM | 21 | −11.94 |
| CD28 | TM | 27 | −11.12 |
| CD8 | GPI | 21 | 3.31 |
| CD80 | GPI | 22 | −4.51 |
| Nicastrin | GPI | 21 | −0.33 |
Sequence comprising the extracellular domain of CD80, red fluorescent protein, and transmembrane domains from the proteins below were assembled as in step 4 of Basic Protocol 6 and were analyzed using the Big-PI predictor tool. We found transmembrane domains of some proteins that would not cause the proteins to become GPI-anchored when expressed. We chose the transmembrane domain of CD28 for use in generating the transmembrane protein expression constructs described herein.
Figure 24.5.5.

The output of the Big-PI predictor tool for the input protein sequence consisting of the extracellular domain of CD80, red fluorescent protein, and transmembrane domain of CD28. No potential GPI-modification site was found in this sequence.
Support Protocol 1
Culturing and Transfection of CHOs Cells for the Expression of Poly-His Tagged, GPI-Anchored, and Transmembrane-Anchored Proteins
Since the introduction of the lipid-bilayer system for the study of cell-cell interactions, many different cell expression systems have been employed for the expression of the recombinant proteins required, including S2 insect cell lines, adherent BHK cells, and adherent CHO cells. We have found that suspension CHO cells (CHOs), available from Life Technologies, are highly suitable for the expression and purification of poly-His-tagged, GPI-anchored, and transmembrane-anchored proteins. In this section, we detail the cell-culture procedure that we have used to maintain such CHOs cell cultures, transfect them via Amaxa Nucleofection Technology, and generate stable cell lines expressing high amounts of transfected protein. In this support protocol, we focus on the expression of poly-His-tagged and transmembrane-anchored proteins. In the case of poly-His-tagged proteins, plasmid constructs are generated so that the protein is expressed in a vector containing an internal ribosomal entry sequence and EGFP, in such a way that the protein and EGFP are transcribed independently by the cellular machinery during expression. EGFP is then used to sort for cells that are highly expressing the poly-His-tagged protein. In the case of transmembrane-anchored proteins, the proteins are engineered to contain a fluorescent protein that may be monitored directly (see Support Protocol 2). The expression of GPI-anchored proteins may be monitored by FACS analysis using an antibody against the extracellular portion of the expressed protein.
Materials
CD CHO medium (Life Technologies)
HT supplement (Life Technologies)
100× penicillin-streptomycin (e.g., Life Technologies)
l-glutamine (e.g., Life Technologies)
Phenol red (e.g., Life Technologies; optional)
Suspension CHO (CHOs) cells (Life Technologies)
Amaxa transfection Kit V, containing transfection solution V, transfection cuvettes, and plastic droppers (Lonza)
Plasmid DNA construct for protein to be expressed, including G418 (or similar) resistance cassette
G418 sulfate (e.g., Life Technologies)
Clumping reagent (Life Technologies; optional)
Hanks' balanced salt solution (HBSS; see recipe in appendix 2a or use commercial version) containing 5% bovine serum albumin (BSA) and 1× penicillin-streptomycin
6-well flat bottom cell culture plate
37°C humidified 5% CO2 incubator with orbital shaker
Conical centrifuge tubes (e.g., Corning Falcon)
125-ml (Corning, cat. no. 431143) and 250-ml (Corning, cat. no. 431144) conical culture flasks
Amaxa Nucleofector™ 2b device (Lonza)
50-μm nylon mesh filter
FACS tubes
Fluorescence-activated cell sorter (FACS)
Additional reagents and equipment for basic cell culture techniques including determining cell viability by trypan blue exclusion (unit 1.1; Phelan and May, 2015), and fluorescence-activated cell sorting (Robinson et al., 2015)
- Prepare CHOs medium by supplementing CD CHO medium with 10 ml/liter of HT supplement (100×), 10 ml/liter of penicillin-streptomycin (100×), and l-glutamine at a final concentration of 8 mM. Add phenol red at a concentration of 11 mg/liter, if desired, as discussed below. Propagate the initial CHOs cell culture supplied by the manufacturer in a 37°C, 5% CO2 incubator capable of orbital shaking, rotating the culture at 125 rpm.As CHOs cells propagate extremely rapidly, addition of phenol red to the culture allows for quick visual analysis of the degree of growth from day to day based on the level of acidification as indicated by the color of the phenol red. Especially when cells are to be used for transfection, it is optimal to use cells that have not proliferated past a density 1 × 106 cells/ml; thus, when a transfection from a particular culture is intended, it is important to manually determine the density of the culture daily using standard procedures.
- At least 1 hr before transfection, place 2 ml of fully supplemented CHOs cell medium from step 1 in each well of a 6-well flat-bottom cell culture plate and place into a 37°C, 5% CO2 incubator without shaking.The number of wells necessary will be determined depending upon the number of CHOs cells used for transfection. Approximately 2 × 106 cells/ml will be used for each transfection reaction. It has been our experience that it is optimal to resuspend cells at a density of 0.5 × 106 cells/ml in 25 ml of medium in a 125-ml culture flask post transfection; therefore, six transfection reactions are usually suitable for each intended culture.
At the time of transfection, aliquot the desired numbers of CHOs cell aliquots into conical centrifuge tubes, each containing 2 × 106 cells. Spin the cells for 5 min at 90 × g, room temperature.
- Gently resuspend each aliquot of 2 × 106 cells into 100 μl of Amaxa Transfection Solution V, containing 5 μg of plasmid DNA, transfer to a supplied transfection cuvette, and transfect the cells using Program U-024. Add 0.5 ml of 37°C culture medium from one well of the 6-well plate to the transfection cuvette, and, using the supplied plastic dropper, transfer the transfected cells drop-by-drop into the well of the prepared 6-well plate from step 2. Rest the cells in the 37°C, 5% CO2 incubator, without shaking, overnight.It is best to resuspend and transfect each cell aliquot one by one, rather than resuspending all of aliquots at one time so that each aliquot is in contact with the transfection solution for the shortest amount of time possible.
- The next morning:
- Pre-equilibrate 25 ml of fully supplemented CHOs medium (see step 1) in a 125-ml conical flask in a 37°C, 5% CO2 incubator for 1 hr.
- During this time, pool the cells from all wells and determine their viability using standard methods of choice [e.g., trypan blue exclusion; see unit 1.1(Phelan and May, 2015); the viability should be >90%]. Transfer the cells into the flask and place into the shaking incubator.
- After 24 hr, spin down the cells and resuspend in fresh CHOs medium supplemented with 1 mg/ml G418 sulfate.
The selection antibiotic will depend on the expression vector. A killing curve is recommended to determine the optimum dose of the appropriate antibiotic. The transfection efficiency may also be determined at this time by using flow cytometry to check for fluorescent protein expression, in the case of poly-His-tagged and transmembrane-anchored protein expression. In all cases, the transfection efficiency should easily range between 80% to 95%.At this point, the expansion rate of the culture in G418 sulfate will depend heavily on both the health of the cells and the expression system used. Determine the cell density every 24 hr and adjust the volume of medium such that the density of live cells remains at 0.5 × 106 cells/ml. Split the cells into separate flasks if the volume grows to greater than 50 ml, with a minimum volume of 25 ml per flask. When 100 ml of volume has been achieved, transfer the culture to a 250-ml shaking flask.unit 1.1 (Phelan and May, 2015) includes protocols for basic cell culture. - After passaging the cells into the 250-ml flask, allow the culture to expand to adensity of 2–3 × 106 cells/ml, while changing the medium every 2 to 3 days.If phenol red has been included into the culture, the medium will turn from red to a yellow/orange color after this period of time, as the cells are rapidly expanding. It is not unusual for these cells to form large clumps as they expand, which will fall to the bottom of the flask. During each passage, filter the cells through a 50-μm nylon mesh to remove the clumps. Since adoption of this cell line in our lab, the manufacturer has developed a clumping reagent to prevent this behavior in the cells. Although we do not know what effect this reagent will have on the described cell culture outcomes, it could be added to CD CHO medium, if desired.
- Further allow the culture to expand to a maximum volume of 150 ml. Every 48 hr, determine the viable number of cells (unit 1.1; Phelan and May, 2015), allowing the culture to expand to 2–3 × 106 cells/ml.During each passage, filter the cells through a 50-μm nylon mesh to remove the clumps.
- To prepare for sorting for high-expressing cells, passage the cells into multiple 250-ml flasks at a density of 1 × 106 cells/ml and allow to expand to 2–3 × 106 cells/ml, as before.In our experience, it usually necessary to sort 100 × 106 cells to obtain a truly high-expressing cell population in one sort.
- On the day of the sort, filter the cells through 50-μm nylon mesh and resuspend the cells to be sorted into FACS tubes in sterile HBSS containing 5% BSA and 1× penicillin-streptomycin at a density of 5 × 106 cells/ml. Also, prepare 50 ml of fully supplemented CD CHO medium (without G418; see step 1) in which the cells will be captured post-sort.Be sure to check with your sorting facility as to any restrictions they may have regarding cell density, sorting medium compositions, etc.
- For sorting (see Robinson et al., 2015), perform two sorts, preferably on the same day. Perform the first sort for the top 10% of cells expressing your protein. After this sort, perform another identical sort on the previously sorted population for the top 10% of cells expressing your protein, to rid the culture of virtually of all low-expressing/nonexpressing cells.The results of a typical sort are shown in Figure 24.5.6.
Immediately after sorting, return your cells to the shaking incubator in a 125-ml flask in 25 ml of fully supplemented CD CHO medium without G418. Allow the cells to recover overnight.
The following day, check the cells for viability and resuspend them in CD CHO medium containing G418. Follow the culturing procedure described above until the cells reach a density of 2–3 × 106 cells/ml, and freeze down several vials of your highly-expressing cell line for future use according to the manufacturer's instructions.
- To harvest cell supernatant or cell pellets, passage the cells into 250-ml flasks as described above and expand them to a density of approximately 5 × 106 cells/ml. At this point, collect the supernatant for purification of poly-His tagged proteins, or the cells for purification of GPI-anchored or transmembrane-anchored proteins.Although the medium will become quite yellow, and the culture will appear very dense, CHOs cells are very robust and should survive nicely under these conditions. To be certain, check the viability of the cells regularly during expansion. If viability becomes a problem before the desired density is reached, passage the cells and collect cells and supernatants at a lower cell density. It is also a good idea at this point to freeze down several aliquots of these cells that have been adapted to grow at high density for future use.
- Finally, purify GPI- and transmembrane-anchored proteins using affinity chromatography as described (Dustin et al., 2007); purify poly-His-tagged soluble proteins from supernatant using a Ni-column, according to the manufacturer'sinstructions (for example, see, https://www.qiagen.com/us/resources/resourcedetail?id=6b440493-e246-412a-b903-c465e0a9ae8d&lang=en).If purification is not to be carried out immediately after harvesting, supernatants and cell pellets should be stored in a −80°C freezer until use.Unfortunately, it is difficult to predict, for any given protein, the volume of supernatant or number of cells needed to yield a significant amount of protein. In our lab, we have purified 1 mg of poly-His-tagged CD80 from 100 ml of culture supernatant and purified GPI-anchored CD80 from 10 g of CHOs cell pellet, and obtained 2 ml of liposomes. The typical site density of GPI-CD80 on glass-supported lipid bilayers from such liposomes was 600 to 900 molecules/μm2.
Figure 24.5.6.

In each panel, the red curve indicates CHOs cells that have been transfected with CD80-IRES-EGFP, and the blue curve indicates untransfected cells. (A) Transfected CHOs cells before sorting, (B) Transfected CHOs cells after a first sort for the top 10% of EGFP-expressing cells, and (C) Transfected CHOs cells after a second sequential sort for the top 10% of EGFP- expressing cells.
Support Protocol 2
Phosphatidylinositol-Specific Phospholipase C Treatment for Test of GPI Linkage
As described in Basic Protocol 6, a transmembrane-anchored protein may become GPI-anchored if its cytoplasmic tail is truncated. A bioinformatics program predicted that CD80 may be expressed as a GPI-anchored protein if its cytoplasmic region is removed; on the other hand, if the transmembrane (TM) domain of CD28 is substituted for this region, it is not predicted to be GPI-linked. We briefly describe a procedure for experimentally testing the presence of a GPI-anchor.
Materials
CHOs cells or other mammalian cell line
Amaxa transfection Kit V, containing transfection solution V, transfection cuvettes, and plastic droppers (Lonza)
Phosphatidylinositol-specific phospholipase C (PIPLC; from Bacillus cereus; Sigma, cat. no. P5542)
10 mM Tris·Cl, pH 7.4 (appendix 2a), containing 144 mM NaCl and 0.05% (w/v) BSA
Plasmid DNA encoding CD80-TM and CD80-CD28-TM [cDNA encoding CD80-TM and CD80-CD28-TM were generated according to the steps outlined in Basic Protocol 6; these constructs are available as a courtesy from our lab; please contact Rajat Varma (rajat.varma@nih.gov)]
HBS-BSA (see recipe)
FACS buffer (see recipe), cold
Amaxa Nucleofector™ 2b device (Lonza)
24-well plates
Refrigerated centrifuge with microtiter plate adapter
Additional reagents and equipment for flow cytometry (Robinson et al., 2015)
To test for GPI-linkage, express CD80-TM and CD80-CD28-TM transiently in suspension CHO cells or any other mammalian cells using AMAXA-based electroporation (Support Protocol 1, steps 2 to 4) or any other transient transfection procedure for 4 to 24 hr.
Reconstitute PIPLC from Bacillus cereus at 1000 U/ml in 10 mM Tris·Cl, pH 7.4, containing 144 mM NaCl and 0.05% (w/v) BSA.
Wash 0.5–1 × 106 cells, transfected with CD80-TM and CD80-CD28-TM, twice in 5 ml of HBS-BSA by spinning for 5 min at 300 × g, room temperature.
Resuspend the cells in 1 ml of fresh HBS-BSA in a 24-well plate. Add 100 μl of PIPLC stock solution to the wells to achieve a concentration of 100 U/ml. At the same time, prepare a parallel condition in which the cells are not treated with PIPLC. Incubate the cells at 37°C for 1 hr.
Following 1 hr of PIPLC treatment, wash the cells with 5 ml of cold FACS buffer by spinning the cells 5 min at 300 × g, 4°C.
Stain the cells with 2.5 μg/ml of fluorescently labeled antibody against the extracellular domain of CD80 (Alexa647 conjugated 1610A1 Biolegend) in 0.5 ml for 1 hr on ice, as described in Basic Protocol 5.
- Wash the cells once with 5 ml FACS buffer by spinning the cells for 5 min at 300 × g, 4°C, and analyze by flow cytometry (Robinson et al., 2015).As shown in Figure 24.5.7, the CD80-TM protein is sensitive to PIPLC digestion but not CD80-CD28-TM, demonstrating that the former became GPI-anchored.
Figure 24.5.7.

Curves in each panel represent staining of cells with Alexa647 conjugated antibody against CD80. In each panel, the red curve represents staining of mock-transfected CHO cells, the blue curve represents staining of CD80-TM or CD80-CD28-TM transfected cells, and the green curve represents staining of CD80-TM or CD80-CD28-TM transfected cells treated with PIPLC. (A) PIPLC sensitivity of CD80-CFP-CD80TM-ΔT. (B) PIPLC sensitivity of CD80-YFP-CD80TM-ΔWAT. (C) PIPLC sensitivity of CD80-CFP-CD28TM-ΔT and (D) PIPLC sensitivity of CD80-YFP-CD28TM-ΔWΔT. Based on these results, we concluded that the CD80-TM domain was causing the protein to become GPI-anchored, while the CD28-TM domain was not.
Purification and reconstitution of TM-anchored proteins in liposomes
Dustin and colleagues have described a detailed protocol for purification of GPI-anchored proteins and the subsequent incorporation of the purified protein in DOPC-containing liposomes (Dustin et al., 2007). The same protocol is used to purify TM-anchored proteins expressed in suspension CHO cells (See Support Protocol 1). As with any liposome preparation, it is important to store it under argon to prevent oxidation.
Characterization of lipid bilayers containing TM-anchored proteins
Our goal was to reconstitute TM-anchored proteins in supported lipid bilayers such that they undergo free diffusion. We constructed three different versions of CD80-XFP-CD28TM: (1) CD80-RFP-CD28TM, (2) CD80-CFP-CD28TM-ΔT, where the last amino acid of the TM domain is removed, and (3) CD80-YFP-CD28TM-ΔWΔT, where the last two amino acids of the TM domain are removed. When we designed these TM-anchored proteins, we anticipated that the last amino acid of the TM domain might still have some interaction with the underlying glass substrate, thus preventing its free diffusion. We therefore designed two other versions of the protein, in which terminal amino acids were sequentially removed. We tested the fluidity of the TM-anchored proteins in supported bilayers by performing fluorescence recovery after photobleaching (FRAP) experiments. We found that the percentage recovery for all three proteins was comparable and high, demonstrating that all three proteins were free to diffuse in the lipid bilayer (Fig. 24.5.8). To further determine if these TM-anchored proteins engaged their receptors on the surface of T cells, we imaged the interaction of mouse T cells with lipid bilayers containing CD80-YFP-CD28TM-ΔTΔW, pMHC, and ICAM-1. As shown in Figure 24.5.9, YFP fluorescence localizes to punctate structures reminiscent of TCR microclusters, showing that the protein was functional and engaging its receptors (CD28 and/or CTLA-4) on the T cell surface.
Figure 24.5.8.

Fluorescence recovery after photobleaching (FRAP) experiment was performed on supported lipid bilayers containing either 100 molecules/mm2 of CD80-YFP-CD28TM-ΔWΔT (A) or CD80-CFP-CD28TM-ΔT (B). The laser beam was focused to a diffraction-limited spot in the bilayer plane by filling the back aperture of the objective and was used to perform photobleaching in a small area. Images of the bilayers were collected before, immediately at, and after indicated times after photobleaching. Percentage recovery was calculated from background-subtracted images using the formula %recovery = [(intensity(t) – intensitypostbleach)/(intensityprebleach)] × 100, where the intensities are average fluorescence intensities from an area bounding the bleach spot. Data are average and standard deviation from six independent bleach spots.
Figure 24.5.9.
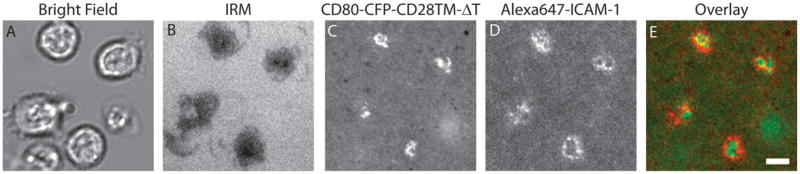
Glass-supported lipid bilayers were prepared consisting of 5 molecules/μm2 of MCC loaded poly-His-tagged I-Ek, 100 molecules/μm2 of Alexa647 poly-His-tagged ICAM-1, and 100 molecules/μm2 of CD80-CFP-CD28TM-ΔT. T cells derived from AND TCR transgenic mice were incubated on these bilayers at 37°C and wide-field microscopy was performed to collect bright-field (A) and interference reflection microscopy (B) images. Panel (C) represents CFP fluorescence associated with CD80-CFP-CD28TM-ΔT, and panel (D) represents Alexa647 fluorescence associated with ICAM-1. Panel E represents the overlay of panel C (green) and panel D (red). CD80-CFP-CD28TM-ΔT localizes to the boundary of cSMAC and pSMAC as reportedly previously. Scale bar, 4 μm.
Support Protocol 3
Preparation of Casein Reagent
Casein reagent is used as a blocking agent when conducting imaging experiments in perfusion chambers. After lipid bilayer spots have been formed on acid-etched coverslips, casein is injected into the perfusion chamber and adsorbs to bare glass where no lipid bilayer is present.
Materials
Casein
10 N NaOH
10× phosphate-buffered saline (PBS; see recipe for 1× PBS in appendix 2a)
500-ml plastic bottle with screw cap
Magnetic stir plate and stir bar
pH meter
Ultracentrifuge with Beckman 45 Ti rotor and appropriate ultracentrifuge tubes
Ring stand with clamps
250-ml 0.22-μm Millipore Stericup filter
FACS tubes
Add 18 g of casein to 250 ml of water in a 500-ml plastic bottle with a screw top, containing a stir bar. Stir slowly on a magnetic stirrer.
Measure the pH of the resulting solution, which should be close to 6.0.
Add 100 μl of 10 N NaOH and allow the pH to stabilize.
- Repeat step 3, adding 100 μl aliquots of 10 N NaOH, while stirring slowly, until the pH reaches 8.0.This step may take 2 to 3 hr to complete.
Move the bottle and magnetic stirrer into a 4°C cold room and allow to stir overnight.
The next day, add 36 ml of 10× PBS and adjust the final volume to 360 ml with water.
Dispense the solution into ultracentrifuge tubes and ultracentrifuge for 2 hr at 100,000 × g (40,000 rpm. in a Beckman 45 Ti rotor) at 4°C.
- Remove the tubes from the ultracentrifuge and carefully place them on ice.At this point, the solution will have separated into three layers: a particulate layer on the bottom of the tube, a clear middle layer (the casein solution), and an upper opaque layer.
Being careful not to disturb the layers, clamp each tube to a ring stand. Aspirate the opaque upper layer using the laboratory vacuum, being sure to catch it in a waste flask.
Using a Pipetman with a 1- or 5-ml tip, collect the clear middle layer from each tube and collect in a suitable container, being careful not to disturb the particulate matter at the bottom of the tube.
Filter the collected casein reagent using a 250-ml, 0.22 μm Millipore Stericup filter system.
Aliquot the filtered casein reagent into 1.5 ml aliquots in FACS tubes.
Store the aliquots at −20°C until use.
Reagents and Solutions
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see appendix 2a.
FACS buffer (1×)
HBS-BSA (see recipe)
0.02% (w/v) sodium azide
Store up to 3 months at 4°C
HBS-BSA
20 mM HEPES, pH 7.2
137 mM NaCl
5 mM KCl
0.7 mM Na2HPO4
6 mM d-glucose
2 mM MgCl2
1 mM CaCl2
1% (w/v) BSA
Filter through 0.22-μm filter
Store up to 1 month at 4°C
Lysis buffer
1% (v/v) Triton X-100
20 mM Tris-Cl, pH 7.5 (appendix 2a)
150 mM NaCl
2 mM EDTA
0.1% (v/v) SDS
1 × protease and phosphatase inhibitor cocktails (Roche Applied Sciences)
Store in aliquots up to 1 year at −20°C (once thawed do not refreeze; use for experiment on the same day)
Tris-saline buffer (1×)
25 mM Tris-Cl, pH 8.0 (appendix 2a)
0.15 M NaCl
Filter through 0.22-μm filter
Store up to 1 year at 4°C
On the day liposomes are being prepared, purge in sterile hood with nitrogen for 15 min and top bottle with argon.
Commentary
Background Information
Over the past two decades, the use of supported lipid bilayers has proved to be an effective model system to understand the spatio-temporal regulation of receptor-ligand binding and signal transduction in a variety of cell-cell interfaces (Campi et al., 2005; Yokosuka et al., 2005; Fleire et al., 2006; Tolar et al., 2009; Huppa et al., 2010; Beemiller et al., 2012; Guy et al., 2013; Le Floc'h et al., 2013; Mattila et al., 2013; Natkanski et al., 2013; O'Donoghue et al., 2013; Suzuki et al., 2014). Many laboratories have contributed to the development of this technology and have published excellent protocols that describe the preparation of liposomes and formation of lipid bilayers in different kinds of perfusion chambers. We have published a protocol for efficient transfection of primary T cells using Amaxa Nucleofection Technology for transient expression of GFP-tagged signaling molecules (Crites et al., 2012). Many groups have used such transfected T cells to study signal transduction by incubating them with supported lipid bilayers. In this protocol, we have, in addition, shown how to adapt this protocol to the study of the Jurkat cell immune synapse, provided a method for stimulating cells with silica bead–supported bilayers, and introduced a new method for incorporating transmembrane-anchored proteins in such supported lipid bilayers. The method of stimulating cells with lipid bilayer–coated silica beads is very useful, as large number of cells can be simultaneously stimulated and analyzed for phosphorylated proteins by western blots or mass spectrometry. Additionally, RNA can be extracted for gene expression analysis. Flow cytometry can also be used to analyze cells stimulated with silica bead–supported bilayers for phosphorylated proteins using fluorescently tagged phospho-specific antibodies and gene expression using GFP-based transcriptional reporters, albeit on a much smaller scale.
One of the important features of the supported lipid bilayer system is that proteins incorporated in it undergo unrestricted diffusion. This, however, is not true for endogenously expressed proteins on the cell surface because the actin cytoskeleton restricts diffusion of most membrane proteins (Kusumi et al., 2005). The hope behind designing transmembrane-anchored proteins was that some interaction between the TM domain and the underlying glass substrate would provide a more restricted diffusion, mimicking the diffusion of proteins in cell membranes. We did not observe this in the proteins we designed; however, our efforts were not exhaustive, and other researchers might take advantage of our experience and experiment with other terminal amino acids in the transmembrane domain.
While there can be many other uses for transmembrane-anchored proteins, we cite one example here where transmembrane-anchored proteins could be especially useful—in the design of MHC class II molecules. MHC class II molecules are expressed on the cell surface as a heterodimer of the alpha chain and the beta chain in complex with a peptide. Currently, MHC class II molecules are incorporated in lipid bilayers following purification as soluble proteins bearing poly-His tags or biotin tags, or as GPI-anchored proteins (Dustin et al., 2007). There is an advantage in purifying MHC molecules in a peptide-free form so that they can be loaded with diverse antigenic peptides, but, due to the complex nature of the interaction between the alpha chain and the beta chain (Loset et al., 2014), in the absence of the transmembrane domain, only some alleles of MHC class II can be purified in the peptide free form (Wettstein et al., 1991; Wallny et al., 1995). To stabilize the alpha and beta chain heterodimer in solution, researchers have engineered dimerization domains from Fos and Jun proteins on the alpha and beta chains to hold the chains together (Scott et al., 1996). For some alleles, even this approach is insufficient in generating stable dimeric forms in solution. Under such circumstances, a covalent peptide needs to be engineered on the beta chain (Kozono et al., 1994; Rees et al., 1999), limiting the number of antigens that can be studied. The transmembrane domains of various alleles of MHC class II alpha and beta chains are fairly conserved, and, using the bioinformatics approach presented here, we have determined that none of these proteins would become GPI-anchored if their cytoplasmic tails are truncated. Due to the presence of the native transmembrane domain of alpha and beta chain, they are also likely to maintain a stable dimeric state without the peptide. This method has the potential to generate any allele of recombinant, peptide-free MHC class II molecules that can be incorporated in supported lipid bilayers as freely diffusing stable dimers.
There are a couple of minor differences between the procedures described here and those described by Dustin et al. (2007). We recommend preparing 6.25% Ni-NTA liposomes as opposed to 25%. We found that the fluidity of the bilayer was highly variable if 25% Ni-NTA liposomes were prepared, depending on the batch of the lipids from the manufacturer. When preparing lipid bilayers using 6.25% Ni-NTA, the maximum density of poly-histidine tagged protein achievable is 500 molecules/μm2. The second difference relates to the amount of time liposomes are made to interact with etched glass surface for bilayer formation to take place. We found that it was not necessary to incubate the liposomes with the glass coverslip for 20 min, as bilayer formation occurred within 30 sec to 1 min.
Critical Parameters
As has been mentioned in other protocols, one of the most critical parameter relates to the storage of liposomes. It is very critical to store liposome suspensions under argon at 4°C with the tubes sealed with Parafilm. Liposomes containing GPI- or transmembrane- anchored proteins should be handled in a sterile environment, preferably a biosafety cabinet. It is important to characterize the site density of incorporated proteins no matter the mode of attachment. This becomes a critical parameter for studying signaling responses. Ideally a kinetic analysis of signaling in response to different doses of stimulus should be performed.
We recommend that the stimulation of cells with silica bead–coated bilayers be performed in HBS-BSA for a maximum period of 6 hr. After 6 hr, the cells may become unhealthy because of lack of nutrients. If long stimulation times are desired, then it is recommended that complete medium supplemented with serum be used. In this case, we also recommend that poly-His-tagged proteins not be used to attach proteins to bilayers. An unknown combination of components in complete medium causes poly-His proteins to dissociate from Ni-NTA bilayers. This can be avoided if the complete medium is passed over a Ni-NTA agarose column; however, this may also deplete the medium of nutrients that are necessary to maintain cell health during longer incubation times.
Troubleshooting
The quality of the bilayer is reflected in the fluidity of the proteins incorporated. This can be tested most simply by using FRAP assays or, in a more complex manner, by single-particle tracking or fluorescence correlation spectroscopy. If proteins do not exhibit at least 70% mobility, it is likely that the liposomes have deteriorated in quality via oxidation of lipids or that the glass surface is not being etched properly by Piranha solution. One crude test for a properly etched glass surface is that it becomes hydrophilic. If the glass is dipped in water and removed, the etched surface will be uniformly wet. If droplets of water form on the surface, the glass is not sufficiently hydrophilic. Only certified ACS sulfuric acid and hydrogen peroxide should be purchased. Sulfuric acid bottles should be stored with their caps tightened, as sulfuric acid is very hygroscopic and in humid months can absorb a lot of moisture, reducing its molarity/normality. When stimulating cells with silica bead–coated bilayers, it is advisable to be very gentle with the cells. p38 MAP kinase, for example, is a stress-activated kinase, and we found that shear stress imposed by repeated pipetting of the cells could cause activation of the protein in the absence of TCR stimulation. To solve this problem, we truncated the ends of Pipetman tips to reduce the shear stress on the cells, which resulted in a large reduction in the background levels of p38 activation.
Anticipated Results
Using the methods described here, we have recently published a manuscript describing the relationship between ligand binding and TCR microcluster formation (Crites et al., 2014). In that manuscript, we prepared liposomes using the method of extrusion described here, prepared glass supported lipid bilayers in flow chambers and on silica beads, and performed imaging-based and biochemical experiments. Several groups have also published studies concerning immune synapse formation by Jurkat cells interacting with α-CD3 containing bilayers (Kaizuka et al., 2007; Ilani et al., 2009; Hsu et al., 2012; Yi et al., 2012; Dillard et al., 2014; Furlan et al., 2014).
Time Considerations
The most time-consuming aspect of this system is the generation of proteins for incorporation into the lipid bilayer. If the user is fortunate, the protein of interest might be commercially available as a recombinant poly-His-tagged soluble protein. Alternatively, it might be available in sufficient quantity such that it can be mono-biotinylated. When using commercially available recombinant proteins, users should make their own evaluation of the purity of the proteins. It is also important to consider whether the protein was expressed in bacteria, insect cells, or mammalian cells, because glycosylation can be important for some protein functions. Shown below is the typical timeline for expression and purification of recombinant proteins in CHOs cells.
Generation of expression construct using molecular biology techniques, 2 to 3 weeks
Generation of stable cell CHOs line expressing a high amount of recombinant protein through sequential sorting, 2 to 3 months
Scale-up of cells for purification, 2 weeks
Purification, incorporation in liposomes (for GPI- or transmembrane- anchored proteins) and characterization, 3 weeks.
The production and purification of antibodies for use in affinity chromatography can start when the generation of expression construct is initiated, and will be complete by the time pellets of high-expressing cells are ready. It typically takes 1 week to prepare primary T cell cultures and about the same amount of time to have a culture of Jurkat cells from a frozen vial. Western blot analysis can take 2 to 3 days, and all other procedures described in this protocol can be completed within a day.
Acknowledgments
Animals used in this study were maintained in a specific-pathogen-free environment, and the National Institutes of Allergy and Infection Diseases Animal Care and Use Committee approved all experiments. We would like to acknowledge the Flow Cytometry Section of the Research Technologies Branch of NIAID, NIH, for help with sorting cells. This work has been funded by the Intramural Research Program of NIAID, NIH.
Footnotes
Key References: Dustin et al., 2007. See above.
Vardana and Dustin, 2008. See above
These are key supporting references for this unit. The first reference provides a detailed step-by-step procedure for purifying GPI-anchored proteins and their subsequent incorporation in liposomes. We have followed the same protocol for the purification of tailless transmembrane-anchored proteins described in Basic Protocol 6 and their incorporation in liposomes following purification. The second reference provides a video protocol for formation of bilayers in FCS2 perfusion chambers described here in Basic Protocol 2. Both references contain important steps on how to clean cover glasses with Piranha solution for formation of lipid bilayers.
Literature Cited
- Beemiller P, Jacobelli J, Krummel MF. Integration of the movement of signaling microclusters with cellular motility in immunological synapses. Nat Immunol. 2012;13:787–795. doi: 10.1038/ni.2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell LM, Solomon KR, Gold JP, Tan KN. Cytoplasmic tail deletion of T cell receptor (TCR) beta-chain results in its surface expression as glycosylphosphatidylinositol-anchored polypeptide on mature T cells in the absence of TCR-alpha. J Biol Chem. 1994;269:22758–22763. [PubMed] [Google Scholar]
- Campi G, Varma R, Dustin ML. Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J Exp Med. 2005;202:1031–1036. doi: 10.1084/jem.20051182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco YR, Fleire SJ, Cameron T, Dustin ML, Batista FD. LFA-1/ICAM-1 interaction lowers the threshold of B cell activation by facilitating B cell adhesion and synapse formation. Immunity. 2004;20:589–599. doi: 10.1016/s1074-7613(04)00105-0. [DOI] [PubMed] [Google Scholar]
- Choudhuri K, Llodra J, Roth EW, Tsai J, Gordo S, Wucherpfennig KW, Kam LC, Stokes DL, Dustin ML. Polarized release of T-cell-receptor-enriched mi-crovesicles at the immunological synapse. Nature. 2014;507:118–123. doi: 10.1038/nature12951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coligan JE, Bierer B, Marguiles DH, Shevach EM, Strober W, editors. Current Protocols in Immunology. John Wiley & Sons; Hoboken, N.J: 2015. [Google Scholar]
- Crites TJ, Chen L, Varma R. A TIRF microscopy technique for real-time, simultaneous imaging of the TCR and its associated signaling proteins. J Vis Exp. 2012;3892 doi: 10.3791/3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crites TJ, Padhan K, Muller J, Krogsgaard M, Gudla PR, Lockett SJ, Varma R. TCR Microclusters pre-exist and contain molecules necessary for TCR signal transduction. J Immunol. 2014;193:56–67. doi: 10.4049/jimmunol.1400315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillard P, Varma R, Sengupta K, Limozin L. Ligand-mediated friction determines morphodynamics of spreading T Cells. Biophys J. 2014;107:2629–2638. doi: 10.1016/j.bpj.2014.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML. Adhesive bond dynamics in contacts between T lymphocytes and glass-supported planar bilayers reconstituted with the immunoglobulin-related adhesion molecule CD58. J Biol Chem. 1997;272:15782–15788. doi: 10.1074/jbc.272.25.15782. [DOI] [PubMed] [Google Scholar]
- Dustin ML. Supported bilayers at the vanguard of immune cell activation studies. J Struct Biol. 2009;168:152–160. doi: 10.1016/j.jsb.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dustin ML, Miller JM, Ranganath S, Vignali DA, Viner NJ, Nelson CA, Unanue ER. TCR-mediated adhesion of T cell hybridomas to planar bilayers containing purified MHC class II/peptide complexes and receptor shedding during detachment. Eur J Immunol. 1996;157:2014–2021. [PubMed] [Google Scholar]
- Dustin ML, Olszowy MW, Holdorf AD, Li J, Bromley S, Desai N, Widder P, Rosenberger F, van der Merwe PA, Allen PM, Shaw AS. A novel adaptor protein orchestrates receptor patterning and cytoskeletal polarity in T-cell contacts. Cell. 1998;94:667–677. doi: 10.1016/s0092-8674(00)81608-6. [DOI] [PubMed] [Google Scholar]
- Dustin ML, Starr T, Varma R, Thomas VK. Supported planar bilayers for study of the immunological synapse. Curr Protoc Immunol. 2007;76:18.13.1–18.13.35. doi: 10.1002/0471142735.im1813s76. [DOI] [PubMed] [Google Scholar]
- Eisenhaber B, Bork P, Eisenhaber F. Sequence properties of GPI-anchored proteins near the omega-site: Constraints for the polypeptide binding site of the putative transamidase. Protein Eng. 1998;11:1155–1161. doi: 10.1093/protein/11.12.1155. [DOI] [PubMed] [Google Scholar]
- Eisenhaber B, Bork P, Eisenhaber F. Prediction of potential GPI-modification sites in proprotein sequences. J Mol Biol. 1999;292:741–758. doi: 10.1006/jmbi.1999.3069. [DOI] [PubMed] [Google Scholar]
- Fleire SJ, Batista FD. Studying cell-to-cell interactions: An easy method of tethering ligands on artificial membranes. Methods Mol Biol. 2009;462:145–154. doi: 10.1007/978-1-60327-115-8_9. [DOI] [PubMed] [Google Scholar]
- Fleire SJ, Goldman JP, Carrasco YR, Weber M, Bray D, Batista FD. B cell ligand discrimination through a spreading and contraction response. Science. 2006;312:738–741. doi: 10.1126/science.1123940. [DOI] [PubMed] [Google Scholar]
- Furlan G, Minowa T, Hanagata N, Kataoka-Hamai C, Kaizuka Y. Phosphatase CD45 both positively and negatively regulates T cell receptor phosphorylation in reconstituted membrane protein clusters. J Biol Chem. 2014;289:28514–28525. doi: 10.1074/jbc.M114.574319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher SR. One-dimensional SDS gel electrophoresis of proteins. Curr Protoc Cell Biol. 2007;37:6.1.1–6.1.38. doi: 10.1002/0471143030.cb0601s37. [DOI] [PubMed] [Google Scholar]
- Gallagher S, Winston SE, Fuller SA, Hurrell JG. Immunoblotting and immunodetection. Curr Protoc Cell Biol. 2011;52:6.2.1–6.2.28. doi: 10.1002/0471143030.cb0602s00. [DOI] [PubMed] [Google Scholar]
- Galush WJ, Nye JA, Groves JT. Quantitative fluorescence microscopy using supported lipid bilayer standards. Biophys J. 2008;95:2512–2519. doi: 10.1529/biophysj.108.131540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse: A molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- Groves JT, Dustin ML. Supported planar bilayers in studies on immune cell adhesion and communication. J Immunol Methods. 2003;278:19–32. doi: 10.1016/s0022-1759(03)00193-5. [DOI] [PubMed] [Google Scholar]
- Guy CS, Vignali KM, Temirov J, Bettini ML, Overacre AE, Smeltzer M, Zhang H, Huppa JB, Tsai YH, Lobry C, Xie J, Dempsey PJ, Crawford HC, Aifantis I, Davis MM, Vignali DA. Distinct TCR signaling pathways drive proliferation and cytokine production in T cells. Nat Immunol. 2013;14:262–270. doi: 10.1038/ni.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CJ, Hsieh WT, Waldman A, Clarke F, Huseby ES, Burkhardt JK, Baumgart T. Ligand mobility modulates immunological synapse formation and T cell activation. PloS One. 2012;7:e32398. doi: 10.1371/journal.pone.0032398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schutz GJ, Davis MM. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilani T, Vasiliver-Shamis G, Vardhana S, Bretscher A, Dustin ML. T cell antigen receptor signaling and immunological synapse stability require myosin IIA. Nat Immunol. 2009;10:531–539. doi: 10.1038/ni.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs H, Iacomini J, van de Ven M, Tonegawa S, Berns A. Domains of the TCR beta-chain required for early thymocyte development. J Exp Med. 1996;184:1833–1843. doi: 10.1084/jem.184.5.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SC, Dustin ML, Hibbs ML, Springer TA. On the species specificity of the interaction of LFA-1 with intercellular adhesion molecules. Eur J Immunol. 1990;145:1181–1187. [PubMed] [Google Scholar]
- Kaizuka Y, Douglass AD, Varma R, Dustin ML, Vale RD. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc Natl Acad Sci U S A. 2007;104:20296–20301. doi: 10.1073/pnas.0710258105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozono H, White J, Clements J, Marrack P, Kappler J. Production of soluble MHC class II proteins with covalently bound single peptides. Nature. 1994;369:151–154. doi: 10.1038/369151a0. [DOI] [PubMed] [Google Scholar]
- Kusumi A, Nakada C, Ritchie K, Murase K, Suzuki K, Murakoshi H, Kasai RS, Kondo J, Fujiwara T. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: High-speed single-molecule tracking of membrane molecules. Annu Rev Biophys Biomol Struct. 2005;34:351–378. doi: 10.1146/annurev.biophys.34.040204.144637. [DOI] [PubMed] [Google Scholar]
- Le Floc'h A, Tanaka Y, Bantilan NS, Voisinne G, Altan-Bonnet G, Fukui Y, Huse M. Annular PIP3 accumulation controls actin architecture and modulates cytotoxicity at the immunological synapse. J Exp Med. 2013;210:2721–2737. doi: 10.1084/jem.20131324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesley SA, Groskreutz DJ. Simple affinity purification of antibodies using in vivo biotinylation of a fusion protein. J Immunol Methods. 1997;207:147–155. doi: 10.1016/s0022-1759(97)00113-0. [DOI] [PubMed] [Google Scholar]
- Lin WC, Yu CH, Triffo S, Groves JT. Supported membrane formation, characterization, functionalization, and patterning for application in biological science and technology. Curr Protoc Chem Biol. 2010;2:235–269. doi: 10.1002/9780470559277.ch100131. [DOI] [PubMed] [Google Scholar]
- Loset GA, Frigstad T, Sandlie I, Bogen B. Disulphide bond-stabilized functional soluble MHC class II heterodimers. Patent application number 20140349315 2014
- Luo W, Yu CH, Lieu ZZ, Allard J, Mogilner A, Sheetz MP, Bershadsky AD. Analysis of the local organization and dynamics of cellular actin networks. J Cell Biol. 2013;202:1057–1073. doi: 10.1083/jcb.201210123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila PK, Feest C, Depoil D, Treanor B, Montaner B, Otipoby KL, Carter R, Justement LB, Bruckbauer A, Batista FD. The actin and tetraspanin networks organize receptor nanoclusters to regulate B cell receptor-mediated signaling. Immunity. 2013;38:461–474. doi: 10.1016/j.immuni.2012.11.019. [DOI] [PubMed] [Google Scholar]
- McConnell HM, Watts TH, Weis RM, Brian AA. Supported planar membranes in studies of cell-cell recognition in the immune system. Biochim Biophys Acta. 1986;864:95–106. doi: 10.1016/0304-4157(86)90016-x. [DOI] [PubMed] [Google Scholar]
- Mitchell RN, Shaw AC, Weaver YK, Leder P, Abbas AK. Cytoplasmic tail deletion converts membrane immunoglobulin to a phosphatidylinositol-linked form lacking signaling and efficient antigen internalization functions. J Biol Chem. 1991;266:8856–8860. [PubMed] [Google Scholar]
- Mossman KD, Campi G, Groves JT, Dustin ML. Altered TCR signaling from geometrically repatterned immunological synapses. Science. 2005;310:1191–1193. doi: 10.1126/science.1119238. [DOI] [PubMed] [Google Scholar]
- Natkanski E, Lee WY, Mistry B, Casal A, Molloy JE, Tolar P. B cells use mechanical energy to discriminate antigen affinities. Science. 2013;340:1587–1590. doi: 10.1126/science.1237572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nye JA, Groves JT. Kinetic control of histidine-tagged protein surface density on supported lipid bilayers. Langmuir. 2008;24:4145–4149. doi: 10.1021/la703788h. [DOI] [PubMed] [Google Scholar]
- O'Donoghue GP, Pielak RM, Smoligovets AA, Lin JJ, Groves JT. Direct single molecule measurement of TCR triggering by agonist pMHC in living primary T cells. eLife. 2013;2:e00778. doi: 10.7554/eLife.00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan K, May KM. Basic techniques in mammalian cell tissue culture. Curr Protoc Cell Biol. 2015;66:1.1.1–1.1.22. doi: 10.1002/0471143030.cb0101s66. [DOI] [PubMed] [Google Scholar]
- Rees W, Bender J, Teague TK, Kedl RM, Crawford F, Marrack P, Kappler J. An inverse relationship between T cell receptor affinity and antigen dose during CD4+ T cell responses in vivo and in vitro. Proc Natl Acad Sci. 1999;96:9781–9786. doi: 10.1073/pnas.96.17.9781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JP, Darzynkiewicz Z, Dean PN, Orfao A, Rabinovitch PS, Stewart CC, Tanke HJ, Wheeless LL, editors. Current Protocols in Cytometry. John Wiley & Sons; Hoboken, N.J: 2015. [Google Scholar]
- Scott CA, Garcia KC, Carbone FR, Wilson IA, Teyton L. Role of chain pairing for the production of functional soluble IA major histocompatibility complex class II molecules. J Exp Med. 1996;183:2087–2095. [Google Scholar]
- Stroumpoulis D, Zhang H, Rubalcava L, Gliem J, Tirrell M. Cell adhesion and growth to Peptide-patterned supported lipid membranes. Langmuir. 2007;23:3849–3856. doi: 10.1021/la062375p. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Seul M, McConnell HM. Lateral diffusion of specific antibodies bound to lipid monolayers on alkylated substrates. Proc Natl Acad Sci U S A. 1986;83:1169–1173. doi: 10.1073/pnas.83.5.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, Leach S, Liu W, Ralston E, Scheffel J, Zhang W, Lowell CA, Rivera J. Molecular editing of cellular responses by the high-affinity receptor for IgE. Science. 2014;343:1021–1025. doi: 10.1126/science.1246976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thid D, Bally M, Holm K, Chessari S, Tosatti S, Textor M, Gold J. Issues of ligand accessibility and mobility in initial cell attachment. Langmuir. 2007;23:11693–11704. doi: 10.1021/la701159u. [DOI] [PubMed] [Google Scholar]
- Tolar P, Hanna J, Krueger PD, Pierce SK. The constant region of the membrane immunoglobulin mediates B cell-receptor clustering and signaling in response to membrane antigens. Immunity. 2009;30:44–55. doi: 10.1016/j.immuni.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardhana S, Dustin M. Supported planar bilayers for the formation of study of immunological synapses and kinapse. J Vis Exp. 2008 doi: 10.3791/947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallny HJ, Sollami G, Karjalainen K. Soluble mouse major histocompatibility complex class II molecules produced in Drosophila cells. Eur J Immunol. 1995;25:1262–1266. doi: 10.1002/eji.1830250520. [DOI] [PubMed] [Google Scholar]
- Watts TH, Brian AA, Kappler JW, Marrack P, McConnell HM. Antigen presentation by supported planar membranes containing affinity-purified I-Ad. Proc Natl Acad Sci U S A. 1984;81:7564–7568. doi: 10.1073/pnas.81.23.7564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts TH, Gariepy J, Schoolnik GK, Mc-Connell HM. T-cell activation by peptide antigen: Effect of peptide sequence and method of antigen presentation. Proc Natl Acad Sci U S A. 1985;82:5480–5484. doi: 10.1073/pnas.82.16.5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts TH, Gaub HE, McConnell HM. T-cell-mediated association of peptide antigen and major histocompatibility complex protein detected by energy transfer in an evanescent wave-field. Nature. 1986;320:179–181. doi: 10.1038/320179a0. [DOI] [PubMed] [Google Scholar]
- Wettstein DA, Boniface JJ, Reay PA, Schild H, Davis MM. Expression of a class II major histocompatibility complex (MHC)heterodimer in a lipid-linked form with enhanced peptide/soluble MHC complex formation at low pH. J Exp Med. 1991;174:219–228. doi: 10.1084/jem.174.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Wu XS, Crites T, Hammer JA. Actin retrograde flow and actomyosin II arc contraction drive receptor cluster dynamics at the immunological synapse in Jurkat T cells. Mol Biol Cell. 2012;23:834–852. doi: 10.1091/mbc.E11-08-0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, Dustin ML, Saito T. Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat Immunol. 2005;6:1253–1262. doi: 10.1038/ni1272. [DOI] [PubMed] [Google Scholar]
- Yokosuka T, Kobayashi W, Sakata-Sogawa K, Takamatsu M, Hashimoto-Tane A, Dustin ML, Tokunaga M, Saito T. Spatiotemporal regulation of T cell costimulation by TCR-CD28 microclusters and protein kinase C theta translocation. Immunity. 2008;29:589–601. doi: 10.1016/j.immuni.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CH, Law JB, Suryana M, Low HY, Sheetz MP. Early integrin binding to Arg-Gly-Asp peptide activates actin polymerization and contractile movement that stimulates outward translocation. Proc Natl Acad Sci U S A. 2011;108:20585–20590. doi: 10.1073/pnas.1109485108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CH, Luo W, Sheetz MP. Spatialtemporal reorganization of activated integrins. Cell Adh Migr. 2012;6:280–284. doi: 10.4161/cam.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CH, Rafiq NB, Krishnasamy A, Hartman KL, Jones GE, Bershadsky AD, Sheetz MP. Integrin-matrix clusters form podosome-like adhesions in the absence of traction forces. Cell Rep. 2013;5:1456–1468. doi: 10.1016/j.celrep.2013.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P, Bertolet G, Chen Y, Huang S, Liu D. Super-resolution imaging of the natural killer cell immunological synapse on a glass-supported planar lipid bilayer. J Vis Exp. 2015;96 doi: 10.3791/52502. [DOI] [PMC free article] [PubMed] [Google Scholar]