

Abstract
Purpose
To investigate whether prenatal exposure to indoor fine particulate matter (PM2.5) and environmental tobacco smoke (ETS) affects susceptibility to respiratory tract infections (RTIs) in infancy, to compare their effects between prenatal and postnatal exposure, and to determine whether genetic factors modify these environmental effects.
Methods
The study population consisted of 307 birth cohort infants. A diagnosis of RTIs was based on parental report of a physician's diagnosis. Indoor PM2.5 and ETS levels were measured during pregnancy and infancy. TaqMan was used for genotyping of nuclear factor erythroid 2-related factor (Nrf2) (rs6726395), glutathione-S-transferase-pi (GSTP) 1 (rs1695), and glutathione-S-transferase-mu (GSTM) 1. Microarrays were used for genome-wide methylation analysis.
Results
Prenatal exposure to indoor PM2.5 increased the susceptibility of lower RTIs (LRTIs) in infancy (adjusted odds ratio [aOR]=2.11). In terms of combined exposure to both indoor PM2.5 and ETS, prenatal exposure to both pollutants increased susceptibility to LRTIs (aOR=6.56); however, this association was not found for postnatal exposure. The Nrf2 GG (aOR=23.69), GSTM1 null (aOR=8.18), and GSTP1 AG or GG (aOR=7.37) genotypes increased the combined LRTIs-promoting effects of prenatal exposure to the 2 indoor pollutants. Such effects of prenatal indoor PM2.5 and ETS exposure were not found for upper RTIs.
Conclusions
Prenatal exposure to both indoor PM2.5 and ETS may increase susceptibility to LRTIs. This effect can be modified by polymorphisms in reactive oxygen species-related genes.
Keywords: Prenatal exposure, particulate matter, tobacco smoke, respiratory tract infections, polymorphism, methylation
INTRODUCTION
Environmental tobacco smoke (ETS), which consists of a mixture of gaseous and particulate pollutants, is major indoor air pollution. Indoor particulate matter (PM) is also emitted from cooking, cleaning, and other human activities as well as from smoking.1,2 Indoor PM and ETS are major indoor air pollutants and may modify the effects of each other.3,4 Therefore, it is important to understand the combined effect of indoor PM and ETS on health and to elucidate the mechanisms involved. Although there is some epidemiologic evidence on the combined effect of ETS and ambient air pollutants on childhood respiratory outcomes,5,6,7,8,9 results on combined exposure to indoor PM and ETS are limited, particularly prenatal exposure.
The prenatal period is critical in terms of the later development of respiratory disorders in childhood because prenatal air pollutant exposure is associated with adverse effects on fetal growth10 and immune responses in early life.11,12,13,14 Prenatal and postnatal exposure involve different routes: prenatal air pollutant exposure occurs via transplacental absorption, whereas postnatal exposure occurs via the respiratory route. Therefore, prenatal air pollutant exposure may affect health via a different mechanism from postnatal exposure. Based on these results, we hypothesized that prenatal indoor PM and ETS exposure compared to postnatal exposure would more severely affect the lower respiratory tract than the upper respiratory tract. Since early-childhood respiratory disorders, especially lower-respiratory tract infections (LRTIs), can develop into chronic respiratory impairment later in life,15,16,17 it is important to identify modifiable early life determinants of adverse respiratory outcomes, especially those operating in the prenatal period. However, the impact of prenatal indoor air pollutant exposure, especially the interaction between indoor PM and ETS, on the susceptibility to LRTIs remains poorly understood.
A mechanism through which PM and ETS may lead to respiratory disease is through promotion of reactive oxygen species (ROS).18,19 The transcription factor nuclear factor erythroid 2-related factor (Nrf2) is activated by oxidative stress and leads to the transcription of antioxidant genes, such as glutathione Stransferase-pi 1 (GSTP1) and glutathione S-transferase-mu 1 (GSTM1). Therefore, ROS-related genes and polymorphisms may result in different responses to PM and ETS.20 The influence of genetic variation on the association between prenatal exposure to indoor PM and/or ETS and susceptibility to RTIs in infancy remains to be studied.
Epigenetic modifications are one of the mechanisms by which prenatal exposures can affect disease later in life. DNA methylation is a well-characterized epigenetic modification, and there is evidence that it may modulate the lifelong effect of prenatal smoke exposure.21,22,23
To address these issues, a prospective birth cohort study was performed. The effect of prenatal indoor PM and/or ETS exposure on the susceptibility of RTIs in infancy was evaluated. The influence of ROS-related gene polymorphisms on RTI susceptibility in infancy was also assessed. Furthermore, whether prenatal indoor PM and ETS exposure can alter DNA methylation was investigated.
MATERIALS AND METHODS
Study design
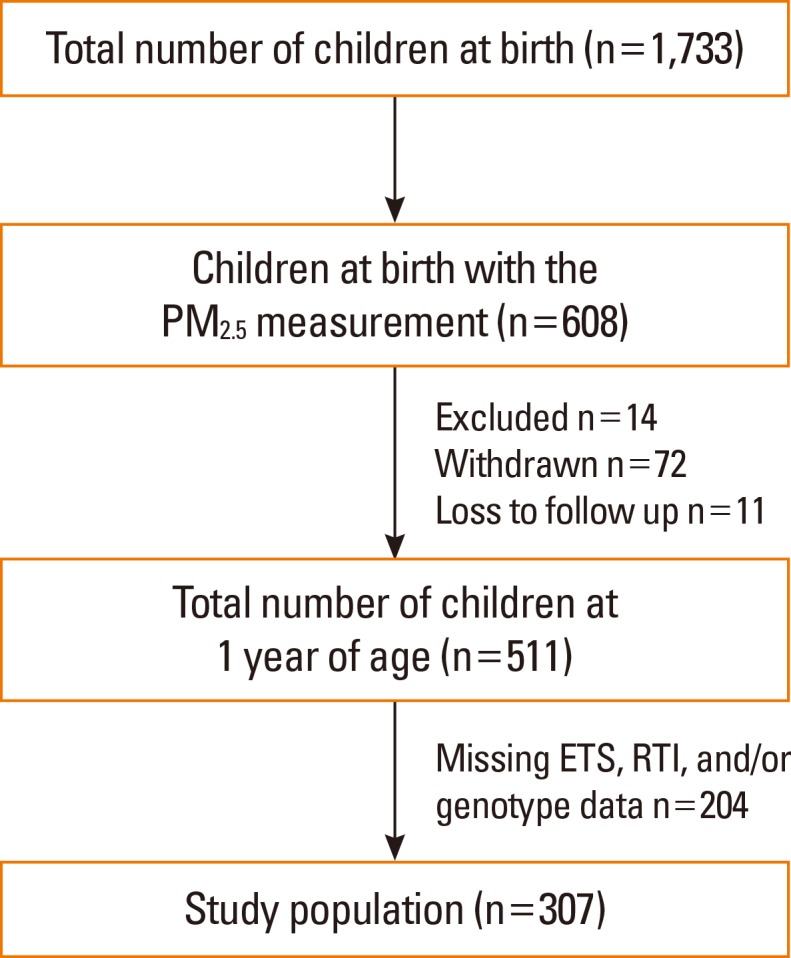
Healthy newborns (n=1,733) were recruited between November 2007 and December 2013. This prospective, general population-based, birth cohort was designated as the COhort for Childhood Origin of Asthma and Allergic Diseases (COCOA); follow-up and further recruitment of this cohort is ongoing. The study methods have been detailed elsewhere.24,25 The indoor level of fine particulate matter (PM2.5) has been measured since 2009 for the applicants. In 608 infants, the indoor levels of PM2.5 were evaluated between 26 and 36 weeks of pregnancy. Of these, based on the complete PM2.5, ETS exposure, RTIs, and genotype data, 307 infants were finally included in the study (Fig. 1). Whether the 6- and 12-month-old infants had had any RTIs was determined by parental report of physician-diagnosed RTIs: "Has a doctor diagnosed RTIs in your child during the last 6 months?" Bronchiolitis, tracheobronchitis, and/or pneumonia were considered as LRTIs and common cold, sinusitis, otitis media, and/or croup as upper RTIs (URTIs).
Fig. 1. Flow chart of the study population. Of infants with prenatal indoor PM2.5 measurements (n=608), 14 met the exclusion criteria, 72 were withdrawn from the study, and 11 were lost to follow-up. An additional 204 infants were then excluded because the RTIs, prenatal ETS exposure, or genotype data were missing. ETS, environmental tobacco smoke; PM2.5, fine particulate matter; RTIs, respiratory tract infections.
Exposure assessment
Starting in May 2009, indoor PM2.5 samples were collected by specialists during home visit between 26 and 36 weeks of pregnancy. In addition, PM2.5 samples at 6 months after birth were collected in the subgroup (n=75) for the applicants. PM2.5 concentrations were measured 3 times in the parents' bed room by using a particle discriminator (Model GT-331; SIBATA Co., Japan) with a laser light-scattering optical particle counter for 5 minutes. The mean value of 3 measurements was used for evaluation. The indoor PM2.5 values were log-transformed and dichotomized to high or low by using the median value before being entered into the regression models. Mothers were asked the following questions about their ETS exposure at home: "Have you been regularly exposed to passive smoking during your current pregnancy?"
The groups were stratified by exposure time. This led to 4 study population groups receiving the following combination of prenatal/postnatal exposures: prenatal ETS/prenatal PM2.5, prenatal ETS/postnatal PM2.5, postnatal ETS/prenatal PM2.5, and postnatal ETS/postnatal PM2.5. To assess whether the 2 indoor pollutants acted additively to increase RTI susceptibility in infancy, each group was divided into 4 groups according to their ETS exposure and whether the indoor PM2.5 levels were high or low.
Genotyping
Genomic DNA was prepared from heparinized newborn umbilical cord blood by using a G-DEX II kit (Intron, Seoul, Korea). ROS-related genes were analyzed as follows. The Nrf2 (rs6726395) and GSTP1 (rs1695) polymorphisms were genotyped by using a TaqMan assay (ABI, Foster City, CA, USA). The GSTM1 copy number was measured by real-time polymerase chain reaction (PCR). The genotyping method is detailed in the Supplemental Material.
Bisulfite conversion and genome-wide methylation array
Nine subjects were selected from the study population to undergo genome-wide methylation analysis of cord blood genomic DNA. Bisulfite conversion was performed by using the EZ DNA methylation kit (Zymo Research, Irvine, CA, USA) according to the manufacturer's instructions. The bisulfite-converted genomic DNA was analyzed by using the Infinium Human Methylation 450 Beadchip (Illumina, San Diego, CA, USA), with >450,000 probes covering 99% of reference sequence genes, following the Illumina Infinium HD Methylation protocol. The 9 subjects and the methylation array are described in the Supplemental Material.
Statistical analysis
Chi-square and t tests were used to assess the significance of differences between the groups, as appropriate. The associations between prenatal indoor PM2.5 and/or ETS exposure and the incidence of RTIs at 12 months of age were analyzed by using multiple logistic regression. Adjustments were made for potential confounding factors, namely, maternal age at delivery, maternal body mass index, maternal educational degree, gestational age, delivery mode, infant sex, and family history of allergic diseases. The results are expressed as adjusted odds ratios (aORs) and 95% confidence intervals (CIs). All statistical analyses were performed by using SPSS 18.0 software (SPSS Inc., Chicago, IL, USA), with a P value <0.05 considered statistically significant.
Ethics statement
The study was approved by the Institutional Review Board of Asan Medical Center (IRB No. 2008-0616), Samsung Medical Center (IRB No. 2009-02-021), Yonsei University (IRB No. 4-2008-0588), and CHA Medical Center (IRB No. 2010-010).
RESULTS
Study population characteristics
Table 1 summarizes the characteristics of the study population (n=307). The study population consisted of 171 boys and 136 girls. In total, 61.6% had been prenatally exposed to maternal ETS, and the mean indoor PM2.5 level during pregnancy was 6.08 ± 7.64 µg/m3. The frequencies of the Nrf2 (rs6726395) GG, GSTP1 (rs1695) AG or GG, and GSTM1 null genotypes were 40.1%, 36.9%, and 56.7%, respectively. The distribution of the 2 polymorphisms was in Hardy-Weinberg equilibrium. The incidences of LRTIs and URTIs by the age of 12 months were 16.3% and 76.2%, respectively.
Table 1. Characteristics of infants who were included and excluded from the study.
| Participants (307) | Non-participants (1,426) | P value | |||
|---|---|---|---|---|---|
| Number | Mean±SD or % | Number | Mean±SD or % | ||
| Sex (male, %) | 171 | 55.7 | 673 | 51.8 | 0.228 |
| Gestational age (week) | 303 | 39.27±1.21 | 1,257 | 39.10±1.27 | 0.042 |
| Maternal age at birth (year) | 307 | 32.69±3.44 | 1,296 | 32.56±3.43 | 0.535 |
| Cesarean section delivery (%) | 90/269 | 33.5 | 377/1,117 | 33.8 | 0.943 |
| Maternal body mass index (kg/m2) | 307 | 20.53±2.24 | 1,352 | 20.66±2.63 | 0.369 |
| Maternal education state | 307 | 1,332 | 0.999 | ||
| ≤High school (%) | 21 | 6.8 | 90 | 6.8 | |
| University or college (%) | 218 | 71.0 | 947 | 71.1 | |
| Graduate school (%) | 68 | 22.2 | 295 | 22.2 | |
| Parental history of allergic disease (%) | 178/295 | 60.3 | 615/1,085 | 56.7 | 0.288 |
| Maternal ETS exposure during pregnancy (%) | 189/307 | 61.6 | 668/1,084 | 61.6 | 1.000 |
| Infantile ETS exposure at 1 year (%) | 75/241 | 31.1 | 196/600 | 32.7 | 0.684 |
| PM2.5 during pregnancy (µg/m3) | 307 | 6.08±7.64 | 307 | 5.70±4.97 | 0.461 |
| PM2.5 at 6 months of age (µg/m3) | 75 | 5.11±7.05 | 245 | 5.40±5.33 | 0.699 |
| Nrf2 GG genotype (%) | 123/307 | 40.1 | 351/913 | 38.4 | 0.636 |
| GSTP1 AG or GG genotype (%) | 113/306 | 36.9 | 295/919 | 32.1 | 0.124 |
| GSTM1 null genotype (%) | 173/305 | 56.7 | 490/911 | 53.8 | 0.388 |
| Incidence of LRTIs* at 1 year (%) | 49/300 | 16.3 | 111/637 | 17.4 | 0.917 |
| Incidence of URTIs† at 1 year (%) | 234/307 | 76.2 | 450/668 | 67.4 | 0.005 |
*Lower respiratory tract infections: tracheobronchitis, pneumonia, and bronchiolitis; †Upper respiratory tract infections: common cold, sinusitis, otitis media, and croup.
ETS, environmental tobacco smoke; GSTM1, glutathione S-transferase-mu 1; GSTP1, glutathione S-transferase-pi 1; Nrf2, nuclear factor erythroid 2-related factor; PM2.5, fine particulate matter; LRTIs, lower respiratory tract infections; URTIs, upper respiratory tract infections.
Table 1 also shows the characteristics of the infants who were not included for reasons shown in Fig. 1. There were no significant differences between participants and non-participants, except in gestational age and the incidence of URTIs.
Risk factors for RTIs in infancy
High PM2.5 levels during pregnancy were independent risk factors for LRTIs in infants (aOR=2.11; 95% CI: 1.12, 3.99) (Table 2). However, none of the early-life environmental factors increased the risk of URTIs. When we compared indoor PM2.5 levels according to RTIs and exposure time, prenatal indoor PM2.5 levels were higher in infants with LRTIs than in those without (mean=7.21 vs 5.71, respectively; 95% CI: 4.99, 9.44 vs 4.97, 6.45, respectively; P=0.119, data not shown). These differences were not distinct according to postnatal PM2.5 levels or the presence of URTIs.
Table 2. Risk factors for respiratory tract infections in infancy.
| LRTIs | URTIs | |||||
|---|---|---|---|---|---|---|
| aOR* | 95% CI | P value | aOR* | 95% CI | P value | |
| Sex (male) | 1.53 | (1.01, 2.31) | 0.046 | 1.04 | (0.75, 1.45) | 0.799 |
| Parental history of allergic disease | 0.87 | (0.58, 1.30) | 0.500 | 0.97 | (0.70, 1.35) | 0.863 |
| Gestational age | 0.94 | (0.80, 1.10) | 0.450 | 0.94 | (0.83, 1.08) | 0.393 |
| Cesarean section delivery | 1.14 | (0.74, 1.75) | 0.550 | 0.75 | (0.53, 1.06) | 0.098 |
| Higher maternal education state | 1.08 | (0.74, 1.58) | 0.683 | 1.12 | (0.82, 1.54) | 0.468 |
| Maternal age at birth | 0.99 | (0.93, 1.05) | 0.713 | 1.06 | (1.01, 1.11) | 0.019 |
| Maternal body mass index | 1.04 | (0.96, 1.12) | 0.363 | 0.94 | (0.88, 1.01) | 0.078 |
| Higher PM2.5 during pregnancy | 2.11 | (1.12, 3.99) | 0.021 | 1.23 | (0.71, 2.15) | 0.463 |
| Higher PM2.5 at 6 months of age | 0.89 | (0.37, 2.18) | 0.801 | 1.00 | (0.52, 1.94) | 0.999 |
| Maternal ETS exposure during pregnancy | 1.33 | (0.87, 2.04) | 0.193 | 1.15 | (0.82, 1.62) | 0.414 |
| Infantile ETS exposure at 1 year | 0.87 | (0.53, 1.42) | 0.576 | 0.79 | (0.53, 1.17) | 0.241 |
| Nrf2 GG genotype | 1.27 | (0.82, 1.97) | 0.290 | 0.55 | (0.39, 0.78) | 0.001 |
| GSTP1 AG or GG genotype | 1.53 | (0.98, 2.38) | 0.062 | 0.89 | (0.62, 1.28) | 0.523 |
| GSTM1 null genotype | 1.69 | (1.07, 2.67) | 0.024 | 0.91 | (0.64, 1.30) | 0.612 |
*Odds ratios were adjusted for maternal age, maternal body mass index, maternal educational state, infant sex, gestational age, delivery mode, and family history of allergy.
ETS, environmental tobacco smoke; GSTM1, glutathione S-transferase-mu 1; GSTP1, glutathione S-transferase-pi 1; Nrf2, nuclear factor erythroid 2-related factor; PM2.5, fine particulate matter; LRTIs, lower respiratory tract infections; URTIs, upper respiratory tract infections.
Effects of prenatal exposure to both indoor PM2.5 and ETS on RTIs susceptibility in infancy
Prenatal high indoor PM2.5 and ETS exposure acted additively to increase the risk of LRTIs (aOR=6.56; 95% CI: 2.02, 21.24) in infants (Fig. 2A and Table S1). Such an additive effect was not replicated when examining the effects of exposure to prenatal PM2.5/postnatal ETS, postnatal PM2.5/prenatal ETS, and postnatal PM2.5/postnatal ETS on any respiratory outcomes in infants. In addition, such additive effects were not observed for URTIs risk, regardless of exposure time (Fig. 2B and Table S1).
Fig. 2. Effect of combined exposure to both PM2.5 and ETS according to exposure time on susceptibility to (A) lower and (B) upper respiratory tract infections in infancy. The groups were stratified by exposure time: (a) prenatal PM2.5/prenatal ETS; (b) prenatal PM2.5/postnatal ETS; (c) postnatal PM2.5/prenatal ETS; and (d) postnatal PM2.5/postnatal ETS. Prenatal high indoor PM2.5 and ETS exposure acted additively to increase the risk of lower respiratory tract infections. *Adjustments for maternal age at delivery, maternal body mass index, maternal educational degree, gestational age, delivery mode, infant sex, and family history of allergy. ETS, environmental tobacco smoke; PM2.5, fine particulate matter.

Effect of GSTM1, GSTP1, and Nrf2 genotypes on the relationship between prenatal indoor PM2.5/ETS exposure and RTIs in infancy
Prenatal exposure to both high indoor PM2.5 and ETS increased LRTIs risk in the GSTM1 null, GSTP1 AG or GG, and Nrf2 GG genotypes (aOR=8.18, 95% CI: 1.53, 43.72; aOR=7.37, 95% CI: 1.12, 48.66; and aOR=23.69, 95% CI: 2.13, 263.07, respectively). Such gene-environment interactions were not observed for URTIs (Tables 3,4,5).
Table 3. Influence of the glutathione S-transferase-mu 1 (GSTM1) copy number variation on the results of prenatal exposure to both PM2.5 and ETS.
| GSTM1 present | GSTM1 null | GSTM1 present | GSTM1 null | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LRTIs | LRTIs | URTIs | URTIs | |||||||||
| No (n=112) | Yes (n=17) | No (n=137) | Yes (n=32) | No (n=36) | Yes (n=87) | No (n=37) | Yes (n=150) | |||||
| n | n | aOR* (95% CI) | n | n | aOR* (95% CI) | n | n | aOR* (95% CI) | n | n | aOR* (95% CI) | |
| PM2.5 (Low) | 29 | 2 | 1.00 | 34 | 3 | 1.00 | 6 | 26 | 1.00 | 10 | 27 | 1.00 |
| ETS (No) | ||||||||||||
| PM2.5 (Low) | 37 | 7 | 1.68 | 45 | 12 | 4.52 | 11 | 33 | 0.51 | 12 | 48 | 2.02 |
| ETS (Yes) | (0.24, 11.79) | (0.87, 23.60) | (0.14, 1.83) | (0.68, 6.01) | ||||||||
| PM2.5 (High) | 13 | 2 | 2.26 | 28 | 4 | 1.65 | 4 | 12 | 0.69 | 9 | 25 | 1.17 |
| ETS (No) | (0.26, 19.36) | (0.24, 11.29) | (0.13, 3.76) | (0.38, 3.57) | ||||||||
| PM2.5 (High) | 33 | 6 | 3.75 | 30 | 13 | 8.18 | 9 | 32 | 0.57 | 11 | 33 | 1.86 |
| ETS (Yes) | (0.52, 27.10) | (1.53, 43.72) | (0.14, 2.35) | (0.60, 5.75) | ||||||||
*Odds ratios were adjusted for maternal age, maternal body mass index, maternal educational state, infant sex, gestational age, delivery mode, and family history of allergy.
aOR, adjusted odds ratio; CI, confidence interval; ETS, environmental tobacco smoke; PM2.5, fine particulate matter; GSTM1, glutathione S-transferase-mu 1; LRTIs, lower respiratory tract infections; URTIs, upper respiratory tract infections.
Table 4. Influence of the glutathione S-transferase-pi 1 (GSTP1) (rs1695) polymorphism on the results of prenatal exposure to both PM2.5 and ETS.
| GSTP1 AA | GSTP1 AG+GG | GSTP1 AA | GSTP1 AG+GG | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LRTIs | LRTIs | URTIs | URTIs | |||||||||
| No (n=159) | Yes (n=29) | No (n=91) | Yes (n=20) | No (n=45) | Yes (n=151) | No (n=27) | Yes (n=86) | |||||
| n | n | aOR* (95% CI) | n | n | aOR* (95% CI) | n | n | aOR* (95% CI) | n | n | aOR* (95% CI) | |
| PM2.5 (Low) | 38 | 3 | 1.00 | 25 | 2 | 1.00 | 9 | 33 | 1.00 | 7 | 20 | 1.00 |
| ETS (No) | ||||||||||||
| PM2.5 (Low) | 59 | 12 | 3.57 | 24 | 7 | 4.04 | 15 | 58 | 1.14 | 9 | 23 | 1.18 |
| ETS (Yes) | (0.66, 19.17) | (0.62, 26.30) | (0.41, 3.15) | (0.29, 4.71) | ||||||||
| PM2.5 (High) | 26 | 3 | 2.82 | 14 | 3 | 1.70 | 9 | 23 | 0.75 | 3 | 14 | 1.43 |
| ETS (No) | (0.40, 19.77) | (0.19, 15.01) | (0.24, 2.34) | (0.28, 7.30) | ||||||||
| PM2.5 (High) | 36 | 11 | 6.32 | 28 | 8 | 7.37 | 12 | 37 | 0.83 | 8 | 29 | 1.80 |
| ETS (Yes) | (1.18, 33.94) | (1.12, 48.66) | (0.28, 2.43) | (0.41, 7.97) | ||||||||
*Odds ratios were adjusted for maternal age, maternal body mass index, maternal educational state, infant sex, gestational age, delivery mode, and family history of allergy.
aOR, adjusted odds ratio; CI, confidence interval; ETS, environmental tobacco smoke; PM2.5, fine particulate matter; GSTP1, glutathione S-transferase-pi 1, LRTIs, lower respiratory tract infections; URTIs, upper respiratory tract infections.
Table 5. Influence of the nuclear factor erythroid 2-related factor (Nrf2) (rs6726395) on the results of prenatal exposure to both PM2.5 and ETS.
| Nrf2 GG | Nrf2 GA+AA | Nrf2 GG | Nrf2 GA+AA | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LRTIs | LRTIs | URTIs | URTIs | |||||||||
| No (n=98) | Yes (n=22) | No (n=153) | Yes (n=27) | No (n=36) | Yes (n=87) | No (n=37) | Yes (n=150) | |||||
| n | n | aOR* (95% CI) | n | n | aOR* (95% CI) | n | n | aOR* (95% CI) | n | n | aOR* (95% CI) | |
| PM2.5 (Low) | 31 | 2 | 1.00 | 32 | 3 | 1.00 | 9 | 24 | 1.00 | 7 | 29 | 1.00 |
| ETS (No) | ||||||||||||
| PM2.5 (Low) | 26 | 10 | 9.66 | 57 | 9 | 1.19 | 9 | 27 | 1.20 | 15 | 54 | 1.09 |
| ETS (Yes) | (0.99, 94.03) | (0.27, 5.18) | (0.36, 3.94) | (0.34, 3.53) | ||||||||
| PM2.5 (High) | 20 | 2 | 4.27 | 21 | 4 | 1.19 | 9 | 15 | 0.66 | 4 | 22 | 1.65 |
| ETS (No) | (0.31, 58.04) | (0.19, 7.31) | (0.19, 2.29) | (0.36, 7.52) | ||||||||
| PM2.5 (High) | 21 | 8 | 23.69 | 43 | 11 | 2.82 | 9 | 21 | 0.81 | 11 | 45 | 1.75 |
| ETS (Yes) | (2.13, 263.07) | (0.65, 12.31) | (0.23, 2.94) | (0.48, 6.42) | ||||||||
*Odds ratios were adjusted for maternal age, maternal body mass index, maternal educational state, infant sex, gestational age, delivery mode, and family history of allergy.
aOR, adjusted odds ratio; CI, confidence interval; ETS, environmental tobacco smoke; PM2.5, fine particulate matter; Nrf2, nuclear factor erythroid 2-related factor; LRTIs, lower respiratory tract infections; URTIs, upper respiratory tract infections.
Relationship between DNA methylation patterns and prenatal indoor PM2.5/ETS exposure
Prenatal indoor PM2.5/ETS exposure was associated specifically with 15 CpG sites, 6 of which were located in intergenic regions. Of the remaining 9 CpG sites, 5 were hypomethylated and 4 were hypermethylated by PM2.5/ETS exposure. This analysis is described in the Supplemental Material.
DISCUSSION
The present study showed that indoor PM2.5 and ETS exposure may have an effect on RTIs in infants and revealed that the adverse effect may depend on the timing of the exposure. The ability of indoor PM2.5 and/or ETS to increase susceptibility to LRTIs appeared to be stronger when the exposure occurred during the prenatal period rather than the postnatal period. This study also showed that the genetic polymorphisms GSTM1, GSTP1 (rs1695), and/or Nrf2 (rs6726395) were further associated with the increased susceptibility of LRTIs in indoor PM2.5/ETS-exposed infants. Thus, the susceptibility of LRTIs in infancy may be shaped by gene-environment interactions between ROS-related genes and prenatal indoor PM2.5/ETS exposure. To our knowledge, this is the first study to evaluate the association between combined exposure to indoor PM2.5/ETS and infants' susceptibility to LRTIs that is associated with exposure time and genetic susceptibility.
Most people spend as much as 90% of their time indoors, especially pregnant women and infants. Chronic exposure to indoor pollutants at home or school can increase air pollutant inhalation and significantly impact health.26,27 The interaction between PM and ETS, the most important indoor air pollutants, modify their individual harmful effects on respiratory outcomes.7,8 However, studies about the indoor PM concentration that would have an adverse health outcome and an interaction between PM and ETS, especially prenatal exposure, are scarce. Our study revealed the effect of indoor PM2.5 even in the low concentration and the additive effect of indoor PM2.5/ETS exposure during the prenatal period on the development of LRTIs in infancy.
The fetal period is critical for lung and immune development. Although some epidemiologic studies showed that prenatal PM or ETS exposure increases the risk of wheezing, asthma, and respiratory infections,28,29,30,31 studies comparing the effect according to exposure time are limited. A few studies revealed that prenatal exposure has a stronger effect on respiratory outcomes than postnatal exposure.32,33,34 We also found its stronger associations with combined exposures to indoor PM2.5/ETS during the prenatal period than the postnatal period. However, this result must be interpreted with caution because it is difficult to clearly separate exposure periods.
Air pollutants and tobacco smoke exert their harmful effects on health by inducing oxidative stress in exposed cells and tissues.18,19 Air pollutants can be directly absorbed to the fetal circulation and produce ROS, ultimately inducing inflammatory and oxidative stress responses in the fetal lung.35 The fetus can also be affected indirectly by the oxidative stress and inflammatory cytokine production induced in the placenta by the pollutants.36 Of particular interest in this regard are several intracellular antioxidant enzymes, including GSTM1 and GSTP1, which defend the airway epithelium from damage caused by oxidants and inflammation. These enzymes are regulated by the transcription factor Nrf2, which translocates to the nucleus after oxidative stress induction.37 These enzymes in respiratory disease pathogenesis after pollutant exposure is provided by results showing that children with the GSTM1 null genotype are more likely to develop asthma and wheezing after prenatal ETS exposure than children with the GSTM1 present genotype.20 Similarly, our study showed that while both prenatal indoor PM2.5 and ETS exposure greatly increased the incidence of LRTIs in infants, this effect was particularly marked in the infants with the GSTM1 null, GSTP1 (rs1695) AG or GG, or Nrf2 (rs6726395) GG genotypes.
ROS reacts with lipids, proteins, and DNA, resulting in cell membrane damage, alteration of gene and protein expression, and even cell death.18,37 Secondary mediators generated by oxidant reactions with lipids, proteins, and other biomolecules contribute to the toxic effects of pollutants. Oxidative stress also induces MAP kinase and NF-κB activation, which may ultimately produce a variety of proinflammatory mediators. Proinflammatory mediators from the airway epithelium play a critical role in the pathogenesis of several pulmonary diseases. Previous experimental studies supported the association between air pollutants and oxidative stress by demonstrating that antioxidant pretreatment attenuates oxidative stress and airway epithelial cell injury induced by air pollutants.38,39,40
Prenatal exposure to environmental factors may affect disease susceptibility later in life by inducing epigenetic changes. A cross-sectional study of children under 18 years of age revealed that air pollutants and ETS both associate with significantly increased DNA methylation and decreased transcription of interferon gamma (IFN-γ) in T-effector cells and forkhead box transcription factor 3 (Foxp3) in T-regulatory cells.41 Interestingly, GSTM1 and GSTP1 polymorphisms alter the ability of prenatal tobacco smoke exposure to induce global DNA methylation.21 Although the sample size in our experiment was too small to make firm conclusions, our data suggest that the ability of prenatal indoor PM2.5 and ETS exposure to promote LRTIs in infancy may be due to DNA methylation alterations; the 9 CpG sites whose methylation was significantly altered by PM2.5/ETS exposure were in subjects with LRTIs (Table S2 and Fig. S1). Further studies on this issue are required.
This study has several limitations. First, it was not possible to clearly distinguish between the effects of prenatal and postnatal exposure or exposure that persisted during both the preand postnatal periods. Further studies on the effect of indoor PM and ETS exposure during specific prenatal and postnatal periods may help identify the mechanisms involved. Second, the RTIs and ETS data were derived from questionnaires, and the indoor PM2.5 levels were measured only 1 day between 26 and 36 weeks of pregnancy and at 6 months of age. Therefore, an information bias could not be excluded. Although questionnaires may misclassify ETS exposure, previous studies have shown a fairly good correlation between self-reported ETS exposure and biomarkers of ETS exposure.5,42,43,44 Future studies may gain greater sensitivity by using more objective and precise measures of RTIs, and smoke and indoor PM exposure. The third limitation is the relatively small study population, which is because the indoor PM2.5 measurements started later in the COCOA study, and these data were thus only available for about one-third of the whole COCOA cohort. However, it is unlikely that the addition of indoor PM2.5 measurements to the protocol introduced a selection bias because the study participants and non-participants did not differ significantly in terms of their characteristics. An increase in the sample size would be likely to lead to more consistent and significant data. The fourth limitation is that we only selected 1 polymorphism from each gene. However, these polymorphisms have been shown in several studies to contribute to asthma susceptibility.20,45,46,47
An important strength of our study is its prospective design: the indoor PM2.5 and ETS exposure data and the data on many potential confounders were collected before the children were born. This is likely to have markedly reduced the study bias. An additional strength is that the PM2.5 measurement was performed at home. This direct measurement of residential indoor PM2.5 probably estimates the actual exposure levels more accurately than other indirect methods. Finally, we investigated genotypic data for GSTM1, GSTP1, and Nrf2 to determine gene-environment interactions between both PM2.5/ETS exposure and LRTIs. These results imply that air pollutant exposure should be reduced, especially in genetically susceptible infants, and support a mechanism for oxidative stress in inducing adverse respiratory outcomes by air pollutants.
It should be noted that there was an important difference between previous studies and ours, namely, that maternal ETS exposure was considered in our study. It was not possible to evaluate the effect of maternal active smoking because the active smoking rate of Korean women is low: only 11.4% of the COCOA cohort mothers had smoked before their pregnancy, of whom only 1 continued to smoke during pregnancy. Thus, maternal ETS exposure was more likely to be an important source of pollutant exposure in our cohort than maternal smoking.
CONCLUSIONS
Indoor PM2.5 and ETS exposure increases susceptibility to LRTIs in infants. This effect was particularly marked when the exposure occurred in the prenatal period. Moreover, the effect was modified by ROS-related gene polymorphisms. Along with studies suggesting that acute LRTIs in early life is associated with a long-standing susceptibility to all forms of lung disease, including asthma,15,16,17 our study highlights the importance of health intervention strategies that focus on the indoor environment in the prenatal period. Additional analyses of genetic and epigenetic variants may help individualize such strategies. Further studies of gene-environment interactions and epigenetic mechanisms that shape the effect of air pollutants on susceptibility to LRTIs and chronic lung diseases are warranted, along with studies assessing the association between LRTIs in infancy and the development of chronic lung diseases, such as asthma.
ACKNOWLEDGMENTS
The authors thank Kyung-Shin Lee, Jin-Ah Park, and Hee-Suk Kim for organizing the data. We would also like to express our gratitude to Ja-Young Kwon, Suk-Joo Choi, Soo-Young Oh, Kyung-Ju Lee, and Hey-Sung Won for helping collect the obstetric data. We also thank Sung-Ok Kwon, Se-Young Oh, Kyung-Sook Lee, Yee-Jin Shin, Jong-Hwan Lim, Whan-Cheol Kim, and Ho Kim for their participation in this study.
Footnotes
This research was supported by funds (2008-E33030-00, 2009-E33033-00, 2011-E33021-00, 2012-E33012-00, and 2013-E51011-00) from the Research of Korea Centers for Disease Control and Prevention.
There are no financial or other issues that might lead to conflict of interest.
Supplementary Materials
Summary of the genome-wide methylation array data. Cord blood of 3 healthy unexposed controls (i.e., infants with neither lower respiratory tract infection [LRTIs] nor prenatal exposure to fine particulate matter or environmental tobacco smoke [ETS]; designated as controls), 3 infants with LRTIs but no prenatal PM2.5 or ETS exposure (designated as LRTIs only), and 3 infants with both LRTIs and exposure (designated as LRTIs plus exposure) was subjected to genome-wide methylation array analysis. (A-C) Heat maps demonstrating the methylation differences between control and LRTIs plus exposure (A), between control and LTRIs only (B), and between LTRIs plus exposure and LRTIs only (C). (D) A Venn diagram depicting the overlap of differentially methylated CpG sites between the control, LRTIs only, and LRTIs plus exposure groups. The red number 15 indicates methylation differences caused by PM2.5 and ETS exposure.
Supplementary Table 1. Effect of exposure to both PM2.5 and ETS according to exposure time on susceptibility to respiratory tract infections in infancy.
| LRTIs | URTIs | |||||
|---|---|---|---|---|---|---|
| No | Yes | aOR* (95% CI) | No | Yes | aOR* (95% CI) | |
| n (%) | n (%) | n (%) | n (%) | |||
| Prenatal PM2.5 (Low) | 68 (22.8) | 5 (8.3) | 1.00 | 17 (22.1) | 57 (19.5) | 1.00 |
| Prenatal ETS (No) | ||||||
| Prenatal PM2.5 (Low) | 97 (32.6) | 22 (36.7) | 2.68 (0.84, 8.61) | 27 (35.1) | 96 (32.9) | 1.06 (0.50, 2.23) |
| Prenatal ETS (Yes) | ||||||
| Prenatal PM2.5 (High) | 54 (18.1) | 10 (26.7) | 2.27 (0.62, 8.34) | 13 (16.9) | 54 (18.5) | 1.14 (0.48, 2.70) |
| Prenatal ETS (No) | ||||||
| Prenatal PM2.5 (High) | 79 (26.5) | 23 (38.3) | 6.56 (2.02, 21.24) | 20 (26.0) | 85 (29.1) | 1.46 (0.65, 3.30) |
| Prenatal ETS (Yes) | ||||||
| Total | 298 | 60 | 77 | 292 | ||
| Prenatal PM2.5 (Low) | 86 (37.2) | 17 (31.5) | 1.00 | 22 (40.0) | 85 (35.1) | 1.00 |
| Postnatal ETS (No) | ||||||
| Prenatal PM2.5 (Low) | 41 (17.7) | 6 (11.1) | 0.88 (0.30, 2.59) | 9 (16.4) | 40 (16.5) | 1.21 (0.47, 3.08) |
| Postnatal ETS (Yes) | ||||||
| Prenatal PM2.5 (High) | 73 (31.6) | 22 (40.7) | 1.92 (0.85, 4.36) | 17 (30.9) | 81 (33.5) | 1.40 (0.64, 3.04) |
| Postnatal ETS (No) | ||||||
| Prenatal PM2.5 (High) | 31 (13.4) | 9 (16.7) | 2.05 (0.69, 6.08) | 7 (12.7) | 36 (14.9) | 1.25 (0.43, 3.67) |
| Postnatal ETS (Yes) | ||||||
| Total | 231 | 54 | 55 | 242 | ||
| Postnatal PM2.5 (Low) | 43 (22.4) | 7 (21.2) | 1.00 | 16 (22.9) | 36 (22.5) | 1.00 |
| Prenatal ETS (No) | ||||||
| Postnata PM2.5 (Low) | 56 (29.2) | 11 (33.3) | 2.06 (0.54, 7.86) | 24 (34.3) | 43 (26.9) | 1.18 (0.46, 3.07) |
| Prenatal ETS (Yes) | ||||||
| Postnata PM2.5 (High) | 34 (17/7) | 7 (21.2) | 2.09 (0.49, 9.02) | 13 (18.6) | 29 (18.1) | 0.79 (0.28, 2.25) |
| Prenatal ETS (No) | ||||||
| Postnata PM2.5 (High) | 59 (30.7) | 8 (24.2) | 1.12 (0.27, 4.63) | 17 (24.3) | 52 (32.5) | 1.29 (0.49, 3.38) |
| Prenatal ETS (Yes) | ||||||
| Total | 192 | 33 | 70 | 160 | ||
| Postnatal PM2.5 (Low) | 51 (31.9) | 8 (29.6) | 1.00 | 19 (31.7) | 41 (31.8) | 1.00 |
| Postnatal ETS (No) | ||||||
| Postnatal PM2.5 (Low) | 31 (19.4) | 6 (22.2) | 2.54 (0.60, 10.83) | 17 (28.3) | 20 (15.5) | 0.59 (0.20, 1.73) |
| Postnatal ETS (Yes) | ||||||
| Postnatal PM2.5 (High) | 54 (33.8) | 8 (29.6) | 1.15 (0.28, 4.64) | 20 (33.3) | 42 (32.6) | 0.65 (0.26, 1.63) |
| Postnatal ETS (No) | ||||||
| Postnatal PM2.5 (High) | 24 (15.0) | 5 (18.5) | 2.29 (0.49, 10.78) | 4 (6.7) | 26 (20.2) | 1.95 (0.44, 8.72) |
| Postnatal ETS (Yes) | ||||||
| Total | 160 | 27 | 60 | 129 | ||
*Odds ratios were adjusted for maternal age, maternal body mass index, maternal educational state, infant sex, gestational age, delivery mode, and family history of allergy.
aOR, adjusted odds ratio; CI, confidence interval; ETS, environmental tobacco smoke; PM2.5, fine particulate matter; LRTIs, lower respiratory tract infections; URTIs, upper respiratory tract infections.
Supplementary Table 2. Different methylation sites depending on prenatal exposure to indoor PM2.5 and ETS.
| ID | Chromosome | Gene symbol | Full name | Functional location |
|---|---|---|---|---|
| Loci hypermethylated by indoor pollutants | ||||
| cg27202913 | 16 | CDH15 | cadherin 15, type 1, M-cadherin | Gene body |
| cg21771569 | X | CDKL5 | cyclin-dependent kinase-like 5 | 5'UTR |
| cg13279926 | 7 | PTPRN2 | protein tyrosine phosphatase, receptor type, N polypeptide 2 | Gene body |
| cg17002719 | 7 | VWDE | von Willebrand factor D and EGF domains | Promoter |
| Loci hypomethylated by indoor pollutants | ||||
| rs6546473 | 2 | ANTXR1 | anthrax toxin receptor 1 | Gene body |
| cg16558994 | 1 | KIF17 | kinesin family member 17 | Gene body |
| cg23737713 | 3 | NDUFB4 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 4 | Gene body |
| cg09219280 | 1 | NFIA | nuclear factor I/A | Gene body |
| cg04930858 | X | PIM2 | pim-2 oncogene, serine/threonine protein kinase pim-2 | Promoter |
References
- 1.Hasheminassab S, Daher N, Shafer MM, Schauer JJ, Delfino RJ, Sioutas C. Chemical characterization and source apportionment of indoor and outdoor fine particulate matter (PM(2.5)) in retirement communities of the Los Angeles Basin. Sci Total Environ. 2014;490:528–537. doi: 10.1016/j.scitotenv.2014.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clougherty JE, Houseman EA, Levy JI. Source apportionment of indoor residential fine particulate matter using land use regression and constrained factor analysis. Indoor Air. 2011;21:53–66. doi: 10.1111/j.1600-0668.2010.00682.x. [DOI] [PubMed] [Google Scholar]
- 3.Wallace LA, Mitchell H, O'Connor GT, Neas L, Lippmann M, Kattan M, et al. Particle concentrations in inner-city homes of children with asthma: the effect of smoking, cooking, and outdoor pollution. Environ Health Perspect. 2003;111:1265–1272. doi: 10.1289/ehp.6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guarnieri M, Balmes JR. Outdoor air pollution and asthma. Lancet. 2014;383:1581–1592. doi: 10.1016/S0140-6736(14)60617-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosa MJ, Jung KH, Perzanowski MS, Kelvin EA, Darling KW, Camann DE, et al. Prenatal exposure to polycyclic aromatic hydrocarbons, environmental tobacco smoke and asthma. Respir Med. 2011;105:869–876. doi: 10.1016/j.rmed.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller RL, Garfinkel R, Horton M, Camann D, Perera FP, Whyatt RM, et al. Polycyclic aromatic hydrocarbons, environmental tobacco smoke, and respiratory symptoms in an inner-city birth cohort. Chest. 2004;126:1071–1078. doi: 10.1378/chest.126.4.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sonnenschein-van der Voort AM, de Kluizenaar Y, Jaddoe VW, Gabriele C, Raat H, Moll HA, et al. Air pollution, fetal and infant tobacco smoke exposure, and wheezing in preschool children: a population-based prospective birth cohort. Environ Health. 2012;11:91. doi: 10.1186/1476-069X-11-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rabinovitch N, Silveira L, Gelfand EW, Strand M. The response of children with asthma to ambient particulate is modified by tobacco smoke exposure. Am J Respir Crit Care Med. 2011;184:1350–1357. doi: 10.1164/rccm.201010-1706OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicolai T, Carr D, Weiland SK, Duhme H, von Ehrenstein O, Wagner C, et al. Urban traffic and pollutant exposure related to respiratory outcomes and atopy in a large sample of children. Eur Respir J. 2003;21:956–963. doi: 10.1183/09031936.03.00041103a. [DOI] [PubMed] [Google Scholar]
- 10.Jedrychowski W, Bendkowska I, Flak E, Penar A, Jacek R, Kaim I, et al. Estimated risk for altered fetal growth resulting from exposure to fine particles during pregnancy: an epidemiologic prospective cohort study in Poland. Environ Health Perspect. 2004;112:1398–1402. doi: 10.1289/ehp.7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hertz-Picciotto I, Park HY, Dostal M, Kocan A, Trnovec T, Sram R. Prenatal exposures to persistent and non-persistent organic compounds and effects on immune system development. Basic Clin Pharmacol Toxicol. 2008;102:146–154. doi: 10.1111/j.1742-7843.2007.00190.x. [DOI] [PubMed] [Google Scholar]
- 12.Herr CE, Ghosh R, Dostal M, Skokanova V, Ashwood P, Lipsett M, et al. Exposure to air pollution in critical prenatal time windows and IgE levels in newborns. Pediatr Allergy Immunol. 2011;22:75–84. doi: 10.1111/j.1399-3038.2010.01074.x. [DOI] [PubMed] [Google Scholar]
- 13.Latzin P, Frey U, Armann J, Kieninger E, Fuchs O, Röösli M, et al. Exposure to moderate air pollution during late pregnancy and cord blood cytokine secretion in healthy neonates. PLoS One. 2011;6:e23130. doi: 10.1371/journal.pone.0023130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hertz-Picciotto I, Herr CE, Yap PS, Dostál M, Shumway RH, Ashwood P, et al. Air pollution and lymphocyte phenotype proportions in cord blood. Environ Health Perspect. 2005;113:1391–1398. doi: 10.1289/ehp.7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gern JE. Viral respiratory infection and the link to asthma. Pediatr Infect Dis J. 2008;27:S97–S103. doi: 10.1097/INF.0b013e318168b718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sigurs N, Aljassim F, Kjellman B, Robinson PD, Sigurbergsson F, Bjarnason R, et al. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax. 2010;65:1045–1052. doi: 10.1136/thx.2009.121582. [DOI] [PubMed] [Google Scholar]
- 17.Holt PG, Sly PD. Viral infections and atopy in asthma pathogenesis: new rationales for asthma prevention and treatment. Nat Med. 2012;18:726–735. doi: 10.1038/nm.2768. [DOI] [PubMed] [Google Scholar]
- 18.Ciencewicki J, Trivedi S, Kleeberger SR. Oxidants and the pathogenesis of lung diseases. J Allergy Clin Immunol. 2008;122:456–468. doi: 10.1016/j.jaci.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim BJ, Lee SY, Kim HB, Lee E, Hong SJ. Environmental changes, microbiota, and allergic diseases. Allergy Asthma Immunol Res. 2014;6:389–400. doi: 10.4168/aair.2014.6.5.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilliland FD, Li YF, Dubeau L, Berhane K, Avol E, McConnell R, et al. Effects of glutathione S-transferase M1, maternal smoking during pregnancy, and environmental tobacco smoke on asthma and wheezing in children. Am J Respir Crit Care Med. 2002;166:457–463. doi: 10.1164/rccm.2112064. [DOI] [PubMed] [Google Scholar]
- 21.Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med. 2009;180:462–467. doi: 10.1164/rccm.200901-0135OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guerrero-Preston R, Goldman LR, Brebi-Mieville P, Ili-Gangas C, Lebron C, Witter FR, et al. Global DNA hypomethylation is associated with in utero exposure to cotinine and perfluorinated alkyl compounds. Epigenetics. 2010;5:539–546. doi: 10.4161/epi.5.6.12378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flom JD, Ferris JS, Liao Y, Tehranifar P, Richards CB, Cho YH, et al. Prenatal smoke exposure and genomic DNA methylation in a multiethnic birth cohort. Cancer Epidemiol Biomarkers Prev. 2011;20:2518–2523. doi: 10.1158/1055-9965.EPI-11-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim HB, Ahn KM, Kim KW, Shin YH, Yu J, Seo JH, et al. Cord blood cellular proliferative response as a predictive factor for atopic dermatitis at 12 months. J Korean Med Sci. 2012;27:1320–1326. doi: 10.3346/jkms.2012.27.11.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang HJ, Lee SY, Suh DI, Shin YH, Kim BJ, Seo JH, et al. The Cohort for Childhood Origin of Asthma and allergic diseases (COCOA) study: design, rationale and methods. BMC Pulm Med. 2014;14:109. doi: 10.1186/1471-2466-14-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pawankar R, Canonica GW, Holgate ST, Lockey RF. World Allergy Organization (WAO) white book on allergy. Milwaukee (WI): World Allergy Organization; 2011. [Google Scholar]
- 27.Yoon C, Ryu K, Kim J, Lee K, Park D. New approach for particulate exposure monitoring: determination of inhaled particulate mass by 24 h real-time personal exposure monitoring. J Expo Sci Environ Epidemiol. 2012;22:344–351. doi: 10.1038/jes.2012.28. [DOI] [PubMed] [Google Scholar]
- 28.Magnusson LL, Olesen AB, Wennborg H, Olsen J. Wheezing, asthma, hayfever, and atopic eczema in childhood following exposure to tobacco smoke in fetal life. Clin Exp Allergy. 2005;35:1550–1556. doi: 10.1111/j.1365-2222.2005.02374.x. [DOI] [PubMed] [Google Scholar]
- 29.Jedrychowski W, Perera FP, Maugeri U, Mrozek-Budzyn D, Mroz E, Flak E, et al. Early wheezing phenotypes and severity of respiratory illness in very early childhood: study on intrauterine exposure to fine particle matter. Environ Int. 2009;35:877–884. doi: 10.1016/j.envint.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jedrychowski WA, Perera FP, Maugeri U, Mrozek-Budzyn D, Mroz E, Klimaszewska-Rembiasz M, et al. Intrauterine exposure to polycyclic aromatic hydrocarbons, fine particulate matter and early wheeze. Prospective birth cohort study in 4-year olds. Pediatr Allergy Immunol. 2010;21:e723–e732. doi: 10.1111/j.1399-3038.2010.01034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jedrychowski WA, Perera FP, Spengler JD, Mroz E, Stigter L, Flak E, et al. Intrauterine exposure to fine particulate matter as a risk factor for increased susceptibility to acute broncho-pulmonary infections in early childhood. Int J Hyg Environ Health. 2013;216:395–401. doi: 10.1016/j.ijheh.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raherison C, Pénard-Morand C, Moreau D, Caillaud D, Charpin D, Kopfersmitt C, et al. In utero and childhood exposure to parental tobacco smoke, and allergies in schoolchildren. Respir Med. 2007;101:107–117. doi: 10.1016/j.rmed.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 33.Singh SP, Gundavarapu S, Peña-Philippides JC, Rir-Sima-ah J, Mishra NC, Wilder JA, et al. Prenatal secondhand cigarette smoke promotes Th2 polarization and impairs goblet cell differentiation and airway mucus formation. J Immunol. 2011;187:4542–4552. doi: 10.4049/jimmunol.1101567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jedrychowski WA, Perera FP, Majewska R, Camman D, Spengler JD, Mroz E, et al. Separate and joint effects of tranplacental and postnatal inhalatory exposure to polycyclic aromatic hydrocarbons: prospective birth cohort study on wheezing events. Pediatr Pulmonol. 2014;49:162–172. doi: 10.1002/ppul.22923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kelly FJ. Oxidative stress: its role in air pollution and adverse health effects. Occup Environ Med. 2003;60:612–616. doi: 10.1136/oem.60.8.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Auten RL, Potts EN, Mason SN, Fischer B, Huang Y, Foster WM. Maternal exposure to particulate matter increases postnatal ozone-induced airway hyperreactivity in juvenile mice. Am J Respir Crit Care Med. 2009;180:1218–1226. doi: 10.1164/rccm.200901-0116OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Auerbach A, Hernandez ML. The effect of environmental oxidative stress on airway inflammation. Curr Opin Allergy Clin Immunol. 2012;12:133–139. doi: 10.1097/ACI.0b013e32835113d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu W, Peden DB, McConnell R, Fruin S, Diaz-Sanchez D. Glutathione-S-transferase M1 regulation of diesel exhaust particle-induced pro-inflammatory mediator expression in normal human bronchial epithelial cells. Part Fibre Toxicol. 2012;9:31. doi: 10.1186/1743-8977-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Messier EM, Day BJ, Bahmed K, Kleeberger SR, Tuder RM, Bowler RP, et al. N-acetylcysteine protects murine alveolar type II cells from cigarette smoke injury in a nuclear erythroid 2-related factor-2-independent manner. Am J Respir Cell Mol Biol. 2013;48:559–567. doi: 10.1165/rcmb.2012-0295OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu W, Muller R, Berhane K, Fruin S, Liu F, Jaspers I, et al. Inflammatory response of monocytes to ambient particles varies by highway proximity. Am J Respir Cell Mol Biol. 2014;51:802–809. doi: 10.1165/rcmb.2013-0265OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kohli A, Garcia MA, Miller RL, Maher C, Humblet O, Hammond SK, et al. Secondhand smoke in combination with ambient air pollution exposure is associated with increasedx CpG methylation and decreased expression of IFN-gamma in T effector cells and Foxp3 in T regulatory cells in children. Clin Epigenetics. 2012;4:17. doi: 10.1186/1868-7083-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang IJ, Hsieh WS, Wu KY, Guo YL, Hwang YH, Jee SH, et al. Effect of gestational smoke exposure on atopic dermatitis in the offspring. Pediatr Allergy Immunol. 2008;19:580–586. doi: 10.1111/j.1399-3038.2008.00759.x. [DOI] [PubMed] [Google Scholar]
- 43.Carlsten C, Dimich-Ward H, DyBuncio A, Becker AB, Chan-Yeung M. Cotinine versus questionnaire: early-life environmental tobacco smoke exposure and incident asthma. BMC Pediatr. 2012;12:187. doi: 10.1186/1471-2431-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yi O, Kwon HJ, Kim H, Ha M, Hong SJ, Hong YC, et al. Effect of environmental tobacco smoke on atopic dermatitis among children in Korea. Environ Res. 2012;113:40–45. doi: 10.1016/j.envres.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 45.Masuko H, Sakamoto T, Kaneko Y, Iijima H, Naito T, Noguchi E, et al. An interaction between Nrf2 polymorphisms and smoking status affects annual decline in FEV1: a longitudinal retrospective cohort study. BMC Med Genet. 2011;12:97. doi: 10.1186/1471-2350-12-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee E, Chang HY, Lee KS, Suh DI, Yu HS, Kang MJ, et al. The effect of perinatal anxiety on bronchiolitis is influenced by polymorphisms in ROS-related genes. BMC Pulm Med. 2014;14:154. doi: 10.1186/1471-2466-14-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang SH, Jung YH, Kim HY, Seo JH, Lee JY, Kwon JW, et al. Effect of paracetamol use on the modification of the development of asthma by reactive oxygen species genes. Ann Allergy Asthma Immunol. 2013;110:364–369.e1. doi: 10.1016/j.anai.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 48.Brasch-Andersen C, Christiansen L, Tan Q, Haagerup A, Vestbo J, Kruse TA. Possible gene dosage effect of glutathione-S-transferases on atopic asthma: using real-time PCR for quantification of GSTM1 and GSTT1 gene copy numbers. Hum Mutat. 2004;24:208–214. doi: 10.1002/humu.20074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of the genome-wide methylation array data. Cord blood of 3 healthy unexposed controls (i.e., infants with neither lower respiratory tract infection [LRTIs] nor prenatal exposure to fine particulate matter or environmental tobacco smoke [ETS]; designated as controls), 3 infants with LRTIs but no prenatal PM2.5 or ETS exposure (designated as LRTIs only), and 3 infants with both LRTIs and exposure (designated as LRTIs plus exposure) was subjected to genome-wide methylation array analysis. (A-C) Heat maps demonstrating the methylation differences between control and LRTIs plus exposure (A), between control and LTRIs only (B), and between LTRIs plus exposure and LRTIs only (C). (D) A Venn diagram depicting the overlap of differentially methylated CpG sites between the control, LRTIs only, and LRTIs plus exposure groups. The red number 15 indicates methylation differences caused by PM2.5 and ETS exposure.
Supplementary Table 1. Effect of exposure to both PM2.5 and ETS according to exposure time on susceptibility to respiratory tract infections in infancy.
| LRTIs | URTIs | |||||
|---|---|---|---|---|---|---|
| No | Yes | aOR* (95% CI) | No | Yes | aOR* (95% CI) | |
| n (%) | n (%) | n (%) | n (%) | |||
| Prenatal PM2.5 (Low) | 68 (22.8) | 5 (8.3) | 1.00 | 17 (22.1) | 57 (19.5) | 1.00 |
| Prenatal ETS (No) | ||||||
| Prenatal PM2.5 (Low) | 97 (32.6) | 22 (36.7) | 2.68 (0.84, 8.61) | 27 (35.1) | 96 (32.9) | 1.06 (0.50, 2.23) |
| Prenatal ETS (Yes) | ||||||
| Prenatal PM2.5 (High) | 54 (18.1) | 10 (26.7) | 2.27 (0.62, 8.34) | 13 (16.9) | 54 (18.5) | 1.14 (0.48, 2.70) |
| Prenatal ETS (No) | ||||||
| Prenatal PM2.5 (High) | 79 (26.5) | 23 (38.3) | 6.56 (2.02, 21.24) | 20 (26.0) | 85 (29.1) | 1.46 (0.65, 3.30) |
| Prenatal ETS (Yes) | ||||||
| Total | 298 | 60 | 77 | 292 | ||
| Prenatal PM2.5 (Low) | 86 (37.2) | 17 (31.5) | 1.00 | 22 (40.0) | 85 (35.1) | 1.00 |
| Postnatal ETS (No) | ||||||
| Prenatal PM2.5 (Low) | 41 (17.7) | 6 (11.1) | 0.88 (0.30, 2.59) | 9 (16.4) | 40 (16.5) | 1.21 (0.47, 3.08) |
| Postnatal ETS (Yes) | ||||||
| Prenatal PM2.5 (High) | 73 (31.6) | 22 (40.7) | 1.92 (0.85, 4.36) | 17 (30.9) | 81 (33.5) | 1.40 (0.64, 3.04) |
| Postnatal ETS (No) | ||||||
| Prenatal PM2.5 (High) | 31 (13.4) | 9 (16.7) | 2.05 (0.69, 6.08) | 7 (12.7) | 36 (14.9) | 1.25 (0.43, 3.67) |
| Postnatal ETS (Yes) | ||||||
| Total | 231 | 54 | 55 | 242 | ||
| Postnatal PM2.5 (Low) | 43 (22.4) | 7 (21.2) | 1.00 | 16 (22.9) | 36 (22.5) | 1.00 |
| Prenatal ETS (No) | ||||||
| Postnata PM2.5 (Low) | 56 (29.2) | 11 (33.3) | 2.06 (0.54, 7.86) | 24 (34.3) | 43 (26.9) | 1.18 (0.46, 3.07) |
| Prenatal ETS (Yes) | ||||||
| Postnata PM2.5 (High) | 34 (17/7) | 7 (21.2) | 2.09 (0.49, 9.02) | 13 (18.6) | 29 (18.1) | 0.79 (0.28, 2.25) |
| Prenatal ETS (No) | ||||||
| Postnata PM2.5 (High) | 59 (30.7) | 8 (24.2) | 1.12 (0.27, 4.63) | 17 (24.3) | 52 (32.5) | 1.29 (0.49, 3.38) |
| Prenatal ETS (Yes) | ||||||
| Total | 192 | 33 | 70 | 160 | ||
| Postnatal PM2.5 (Low) | 51 (31.9) | 8 (29.6) | 1.00 | 19 (31.7) | 41 (31.8) | 1.00 |
| Postnatal ETS (No) | ||||||
| Postnatal PM2.5 (Low) | 31 (19.4) | 6 (22.2) | 2.54 (0.60, 10.83) | 17 (28.3) | 20 (15.5) | 0.59 (0.20, 1.73) |
| Postnatal ETS (Yes) | ||||||
| Postnatal PM2.5 (High) | 54 (33.8) | 8 (29.6) | 1.15 (0.28, 4.64) | 20 (33.3) | 42 (32.6) | 0.65 (0.26, 1.63) |
| Postnatal ETS (No) | ||||||
| Postnatal PM2.5 (High) | 24 (15.0) | 5 (18.5) | 2.29 (0.49, 10.78) | 4 (6.7) | 26 (20.2) | 1.95 (0.44, 8.72) |
| Postnatal ETS (Yes) | ||||||
| Total | 160 | 27 | 60 | 129 | ||
*Odds ratios were adjusted for maternal age, maternal body mass index, maternal educational state, infant sex, gestational age, delivery mode, and family history of allergy.
aOR, adjusted odds ratio; CI, confidence interval; ETS, environmental tobacco smoke; PM2.5, fine particulate matter; LRTIs, lower respiratory tract infections; URTIs, upper respiratory tract infections.
Supplementary Table 2. Different methylation sites depending on prenatal exposure to indoor PM2.5 and ETS.
| ID | Chromosome | Gene symbol | Full name | Functional location |
|---|---|---|---|---|
| Loci hypermethylated by indoor pollutants | ||||
| cg27202913 | 16 | CDH15 | cadherin 15, type 1, M-cadherin | Gene body |
| cg21771569 | X | CDKL5 | cyclin-dependent kinase-like 5 | 5'UTR |
| cg13279926 | 7 | PTPRN2 | protein tyrosine phosphatase, receptor type, N polypeptide 2 | Gene body |
| cg17002719 | 7 | VWDE | von Willebrand factor D and EGF domains | Promoter |
| Loci hypomethylated by indoor pollutants | ||||
| rs6546473 | 2 | ANTXR1 | anthrax toxin receptor 1 | Gene body |
| cg16558994 | 1 | KIF17 | kinesin family member 17 | Gene body |
| cg23737713 | 3 | NDUFB4 | NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 4 | Gene body |
| cg09219280 | 1 | NFIA | nuclear factor I/A | Gene body |
| cg04930858 | X | PIM2 | pim-2 oncogene, serine/threonine protein kinase pim-2 | Promoter |