

Abstract
Increased awareness about the importance of protein folding and trafficking to the etiology of gain-of-function diseases has driven extensive efforts to understand the cell and molecular biology underlying the life cycle of normal secretory pathway proteins and the detrimental effects of abnormal proteins. In this regard, the quality-control machinery in the endoplasmic reticulum (ER) has emerged as a major mechanism by which cells ensure that secreted and transmembrane proteins either adopt stable secondary, tertiary, and quaternary structures or are retained in the ER and degraded. Here we examine cellular and molecular aspects of ER retention in transfected fibroblasts expressing missense mutations in the Proteolipid Protein-1 (PLP1) gene that cause mild or severe forms of neurodegenerative disease in humans. Mild mutations cause protein retention in the ER that is partially dependent on the presence of a cytoplasmically exposed heptapeptide, KGRGSRG. In contrast, retention associated with severe mutations occurs independently of this peptide. Accordingly, the function of this novel heptapeptide has a significant impact on pathogenesis and provides new insight into the functions of the two splice isoforms encoded by the PLP1 gene, PLP1 and DM-20.
Keywords: spastic paraplegia, Pelizaeus-Merzbacher disease, unfolded protein response, forward transport, ERAD, myelin, oligodendrocytes
The concept of intracellular protein misfolding/aggregation as the cellular basis of disease is reasonably postulated by many groups to underlie the pathophysiology of a number of neurodegenerative disorders, including Alzheimer’s, Parkinson’s, and Charcot-Marie-Tooth diseases; spinocerebellar ataxias; and amyotrophic lateral sclerosis (Zoghbi and Botas, 2002; Forman et al., 2003; Gow and Sharma, 2003). Our own studies implicate protein misfolding in the etiology of two neurodegenerative disorders, Pelizaeus-Merzbacher disease (PMD) and spastic paraplegia type II (SPG2), which are caused by missense mutations in the Proteolipid Protein-1 (PLP1) gene that give rise to a broad range in disease severity (Gow et al., 1994b, 1998; Southwood et al., 2002). Thus, mildly affected patients exhibit lower limb spasticity and have a relatively normal life span, whereas severely affected individuals never learn to talk or walk and have a markedly shortened life span (Garbern, 2005). PMD is also caused by PLP1 gene duplications and deletions, but mechanisms underlying the pathophysiology are distinct (Gow and Sharma, 2003).
The PLP1 gene is abundantly expressed by oligodendrocytes in the central nervous system (CNS) and yields two major transcripts by alternative splicing in exon 3. Inclusion of exon 3A and B yields an mRNA encoding PLP1, whereas exclusion of exon 3B specifies DM-20. The absence of exon 3B (105 nucleotides) does not alter the reading frame in exons 4–7, so the primary structures of both proteins differ only by the 35 amino acids of the PLP1-specific peptide in the central cytoplasmic loop domain of these polytopic membrane proteins. Most missense mutations in the PLP1 gene occur outside of exon 3B and are present in both PLP1 and DM-20 (for review see Hudson and Nadon, 1992).
At the heart of our investigations into the cellular and molecular mechanisms underlying PLP1 missense mutations is a transfected COS-7 cell assay used to examine PLP1 and DM-20 intracellular trafficking (Gow and Lazzarini, 1996; Gow, 2002). The wild-type proteins are synthesized on the endoplasmic reticulum (ER), traverse the secretory pathway to the cell surface, and enter the endocytic pathway en route to lysosomes (Gow et al., 1994a). In contrast, missense mutations associated with severe disease cause PLP1 and DM-20 to misfold and accumulate in the ER (Gow et al., 1994b). ER accumulation of mutant proteins is commonly observed in eukaryotes, where molecular chaperones and other mediators of the quality control machinery serve to ensure that only stably folded secretory pathway proteins gain access to other intracellular compartments (Ellgaard and Helenius, 2003). Paradoxically, mild mutations cause PLP1 to accumulate in the ER, whereas DM-20 reaches all major intracellular compartments (Gow and Lazzarini, 1996). These data suggest that mechanisms in addition to misfolding are responsible for ER accumulation of PLP1, such as retention signals that bind to ER-resident receptors, as has been described for several proteins (Michelsen et al., 2005).
Herein we focus on the mechanism underlying ER retention of PLP1 and explore three missense mutations that cause severe or mild forms of disease. Deletion-series and mutagenesis experiments demonstrate that the alternatively spliced PLP1-specific peptide contains a novel signal that regulates accumulation in the ER for mutations causing mild disease. This heptapeptide is distinct from ER retention signals that have been characterized in other transmembrane proteins.
MATERIALS AND METHODS
Mutagenesis Constructs
Constructs are generated by PCR-based mutagenesis using one of two approaches. First, the original PLP1-specific peptide deletion series constructs were each generated in two parts by PCR using a circular plasmid template comprising of 1.4-kb human PLP1 cDNA cloned into the Eco RI site of pUC8 (Puckett et al., 1987). The vector primer M13R is used with different antisense primers in exon 3 to amplify the cDNA upstream of the intended deletions. The vector primer M13F is used with different sense primers in exon 3 to amplify the cDNA downstream of deletions. All exon 3 primers carry an Hae II site at the 5′ end to allow ligation of upstream and downstream PCR products together. Upstream and downstream PCR products are also digested with Eco RI and Sal I (New England Biolabs, Beverly, MA), respectively, to allow reconstruction of the cDNA (minus the intended deletion) in pSP64 (Promega, Madison, WI). cDNAs are cloned into the Eco RI site of pCMV5 (Lorence et al., 1990) for transfections. The second approach pertains to all other constructs, and we use Quik-change mutagenesis (Stratagene, La Jolla, CA) as recommended by the manufacturers. In primer design, we use mammalian codons to make the necessary amino acid changes and include additional silent nucleotide changes to generate unique 6-base restriction sites to track the amino acid changes. These changes form the core of the primers and are flanked on either side by 11 or 12 bases of wild-type sequence for primer binding. We mutagenize pUC8- or pCMV5-based plasmids containing PLP1 or DM-20 cDNAs. All plasmids are bidirectionally sequenced across the coding regions.
Transfections, Immunocytochemistry, and Quantification
Plasmids for transfection are purified on CsCl gradients (Gow and Lazzarini, 1996) or with Qiagen maxiprep kits (Valencia, CA). We use Fugene 6 to transfect 60-mm dishes of log-phase COS-7 cells with 4 μg of supercoiled DNA for 24 hr, as recommended by the manufacturer. Cells are then split into duplicate 35-mm dishes and fixed after 24 hr in 2% paraformaldehyde in DMEM at 37°C for 30 min and labeled with rat anti-PLP1/DM-20 antibodies (clone AA3) as detailed previously (Gow et al., 1997). Lysosomes are visualized by using mouse anti-LAMP-2 antibodies (Hybridoma Bank). All secondary antibodies (diluted 1:100 in block) are from Vector Laboratories (Burlingame, CA) and Southern Biotechnology (Birmingham, AL).
Quantification of PLP1 and DM-20 Trafficking in COS-7 Cells
Analysis of PLP1 and DM-20 trafficking through the secretory pathway is performed by using a blinded analysis. Cells are assessed for protein accumulation in the ER or trafficking to the cell surface and lysosomes with a Leica DMRA2 epifluorescence microscope. Random fields from duplicate dishes are counted, 50 transfected cells/dish, and the data are combined. The number of cells with cell surface and lysosome labeling is expressed as a proportion of transfected cells counted. Results are expressed as mean ± standard deviation from three or four independent transfections (n = 3 or 4).
RESULTS
A Protein Misfolding Conundrum
The shared topology and four-transmembrane organization of PLP1 and DM-20 are illustrated in Figure 1 (top) along with the PLP1-specific peptide (gray ellipse) and the locations of three missense mutations (Gow et al., 1997). In previous studies, we have examined the intra-cellular trafficking of a large number of mutant PLPs and DM-20s in transfected COS-7 cells (Gow et al., 1994a,b; Gow and Lazzarini, 1996; Tosic et al., 1996, 1997).
Fig. 1.

Trafficking of PLP1 gene products through the secretory pathway of transfected cells. Schematics of PLP1 (top left) and DM-20 (top right) illustrating topology, sites of missense mutation relevant to the current study, and location of the PLP1-specific peptide. Confocal extended-focus series through transfected COS-7 cells labeled with antibodies against PLP1 (green in a,c,e,g) or DM-20 (green in b,d,f,h) and the lysosomal marker LAMP2 (red).
Wild-type PLP1 and DM-20 traffic through the secretory and endocytic pathways of transfected COS-7 cells (Fig. 1a,b, respectively). Mutations associated with severe disease, represented by msd in Figure 1c,d, disrupt the trafficking of PLP1 and DM-20 and cause accumulation in the ER that is most easily interpreted as protein misfolding. Indeed, the unfolded protein response (UPR) is activated in vivo in oligodendrocytes from PMD patients and in Plp1 mutant mice (Gow and Lazzarini, 1996; Gow et al., 1998; Southwood et al., 2002). Furthermore, mutant PLP1 stably associates with molecular chaperones (Swanton et al., 2003).
Mutations causing mild disease, represented by rsh and V218F, disrupt the trafficking of PLP1 (Fig. 1e,g, respectively) but not DM-20 (Fig. 1f,h, respectively). In these instances, we may interpret the accumulation of the mutant PLP1s as misfolding, but how do we interpret the trafficking of the DM-20s throughout the cell? Thus, disparate effects of the rsh and V218F mutations on PLP1 and DM-20 reveal a conundrum: if a mutation causes PLP1 to misfold, we might expect that the same mutation should not be permissive for DM-20 trafficking throughout the cell.
An Activity Conferred by the PLP1-Specific Peptide
To be sure, we have previously demonstrated altered conformations for DM-20 harboring the rsh (DM-20rsh) and V218F (DM-20V218F) mutations by using three conformationally sensitive antibodies (Gow et al., 1997; Southwood and Gow, 2001). These data indicate that DM-20rsh and DM-20V218F do, in fact, misfold but that the nonnative conformations are stable and allow these proteins to dissociate from molecular chaperones and exit the ER in transfected fibroblasts and oligodendrocytes in vivo (Gow and Lazzarini, 1996; Thomson et al., 1997; Gow et al., 1998). In contrast, PLP1rsh and PLP1V218F accumulate in the ER of almost all transfected cells and oligodendrocytes, which indicates that retention must, somehow, involve the PLP1-specific peptide.
We hypothesize, in this light, that the PLP1-specific peptide confers a unique sensitivity on PLP1 to conformational changes arising from missense mutations throughout the protein, including extracellular domains. This notion does not necessarily imply physical interaction of other regions of the protein with the PLP1-specific peptide but rather that this domain exhibits an activity, for example, an interaction motif, that directly or indirectly confers the sensitivity. If so, then suppression of this activity should cause PLP1 to exhibit DM-20-like insensitivity toward missense mutations such as rsh and V218F.
Most Internal Deletions of Exon 3 in PLP1 Do Not Cause Trafficking Defects
To identify functional motifs in the PLP1-specific peptide, we generated successive inframe deletions of exon 3 for expression studies in transfected COS-7 cells. Figure 2A illustrates salient features of wild-type PLP1 and six deletion constructs, in which 20 cytoplasmically exposed amino acids encoded by exon 3A (Δ3A), or portions of exon 3B encoding 5, 12, 22, 31, or 35 amino acids are removed (constructs Δ3B1-4 and DM-20, respectively). Importantly, most constructs in this series encode proteins that traverse the secretory pathway to the cell surface in >95% of transfected cells (Fig. 2B), which indicates that PLP1 is recalcitrant to small and moderate-sized internal deletions in the cytoplasmic loop domain. However, the Δ3B4:PLP1 mutant traverses the secretory pathway in only 50% of transfected cells, which is consistent with the generation of protein with an unstable conformation that is detected by the quality-control machinery.
Fig. 2.
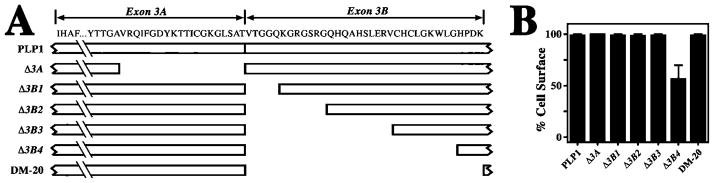
Quantification of protein trafficking to the cell surface for the deletion series mutants. A: Schematic showing the primary structure of PLP1 encoded by exon 3, along with a series of deletions in this region. B: Trafficking of the deletion series constructs in A. The constructs are quantified by the proportion of transfected cells in which the mutants traverse the secretory pathway to the cell surface.
Demonstrating Inactivation of the PLP1-Specific Peptide
To determine whether the activity of the PLP1-specific peptide is disrupted by any of the cytoplasmic loop domain deletions in Figure 2A, we generated a series of compound mutant constructs in which these deletions are coupled with the rsh, V218F, or msd missense mutations. An overview of these compound mutants is given in Figure 3A, where internal exon 3 deletions are represented generically by the broken line in the PLP1-specific peptide (gray oval), and the positions of PMD-causing missense mutations in the second extracellular domain (rsh, V218F, or msd) are represented by black dots. Importantly, each PLP1 compound mutant harbors a single PLP1-specific peptide deletion and a single missense mutation.
Fig. 3.

Quantification of protein trafficking to the cell surface for missense:deletion series compound mutants. A: Schematic showing the location of the PLP1-specific peptide deletions in the cytoplasmic loop domain and missense mutations in the second extracellular domain. B–D: Trafficking of the PLP1 deletion series mutations specified in Figure 2A coupled with the msd, rsh, and V218F missense mutations in the second extracellular domain, respectively. The constructs are quantified by the proportion of transfected cells in which the mutants traverse the secretory pathway to the cell surface.
Intracellular trafficking of PLP1-specific peptide deletion: msd compound mutants is shown in Figure 3B. For all constructs tested, the compound mutants are retained in the ER in 100% of the transfected cells. These data are consistent with Figure 1c,d as well as with our previous studies (Gow and Lazzarini, 1996; Gow et al., 1998) and indicate that accumulation of these proteins in the ER occurs independently of the activity of the PLP1-specific peptide.
In Figure 3C, PLP1rsh accumulates in the ER in almost 100% of transfected cells, and removal of exon 3A or the first five amino acids of the PLP1-specific peptide (i.e., Δ3A:PLP1rsh and Δ3B1:PLP1rsh, respectively) acts similarly. However, deletion of 12 amino acids from the PLP1-specific peptide enables Δ3B2:PLP1rsh to escape the ER to the cell surface and into lysosomes in more than 60% of transfected cells. Thus, we appear to have disrupted the activity of the PLP1-specific peptide by deleting the heptapeptide KGRGSRG. On the other hand, deletion of 22 or 31 amino acids from the PLP1-specific peptide causes ER retention of the compound mutants. These data seem at odds with data for the Δ3B1:PLP1rsh construct; however, in view of the instability of the Δ3B4:PLP1 mutant observed in Figure 2B, we speculate that these compound mutants may be too unstable for efficient trafficking. Finally, deleting all 35 amino acids encoded by exon 3B enables trafficking of mutant DM-20s to the surface in 90% of cells (data not shown).
Similarly to the rsh constructs, the Δ3A:PLP1V218F and Δ3B1:PLP1V218F proteins accumulate in the ER (Fig. 3D). Deleting 12 or 22 amino acids from the PLP1-speific domain enables Δ3B2:PLP1V218F and Δ3B3:PLP1V218F to reach the cell surface in a significant proportion of cells. Removing 31 amino acids causes the protein to accumulate in the ER, as expected from Figure 2B and the rsh data in Figure 3C. Finally, DM-20V218F reaches the surface in 89% of cells (data not shown).
KGRGSRG Heptapeptide Is Necessary But May Not Be Sufficient for ER Retention of Mutant PLP1
To refine further the activity-conferring region of the PLP1-specific peptide, we generated the Δ3B5 mutant in which the KGRGSRG heptapeptide has been removed from the PLP1-specific peptide (Fig. 4A). Quantification is shown in Figure 4B, where 30–50% of Δ3B5:PLP1rsh and Δ3B5:PLP1V218F compound mutants (black) reach the cell surface. White bars show the data for the PLP1rsh and PLP1V218F transfections from Figure 3 for comparison. Thus, the KGRGSRG heptapeptide is necessary for the activity of the PLP1-specific peptide.
Fig. 4.

Quantification of protein trafficking to the cell surface for missense:heptapeptide compound mutants. A: Schematic showing the primary structure of PLP1 encoded by exon 3, along with deletions and insertions of the heptapeptide, KGRGSRG (gray rectangle). B: Trafficking of PLP1 compound mutants harboring the heptapeptide deletion and the rsh, V218F, or msd missense mutations. The constructs are quantified by the proportion of transfected cells in which the mutants traverse the secretory pathway to the cell surface. C: Trafficking of DM-20 compound mutants harboring the heptapeptide insertions and the rsh missense mutation. The constructs are quantified by the proportion of transfected cells in which the mutants traverse the secretory pathway to the cell surface.
To determine whether the heptapeptide is sufficient to confer the activity of the PLP1-specific peptide, we inserted KGRGSRG into two regions of the central cytoplasmic domain of DM-20, as shown in Figure 4A for constructs 5′+3B5 and 3′+3B5. Insertion of this peptide into the 5′ location (Fig. 4C) does not suppress the trafficking of either 5′+3B5:DM-20 or the 5′+3B5:DM-20rsh compound mutant to the cell surface, which indicates that the heptapeptide is not an autonomous ER retention signal. In contrast, insertion of the heptapeptide into the 3′ location potently inhibits trafficking of the 3′+3B5:DM-20rsh compound mutant. These data indicate that the heptapeptide is sufficient to confer PLP1-specific peptide activity; however, trafficking is disrupted in 25% of cells expressing the 3′+3B5:DM-20 mutant, and we cannot formally exclude the possibility that ER retention of the 3′+3B5:DM-20rsh compound mutant is linked to protein misfolding. Thus, we conclude that the heptapeptide is necessary but may not be sufficient to confer activity of the PLP1-specific peptide.
Canonical ER Retention Signals Are Absent in the Heptapeptide
Several recently characterized motifs in cytoplasmic domains of transmembrane proteins have been shown to confer ER retention. Interestingly, these peptide sequences are often encoded by alternatively spliced exons (for review see Michelsen et al., 2005). Masking of these signals by subunit assembly or covalent modification enables a protein to exit the ER to other compartments, and the best characterized of these motifs include cytoplasmic di-arginine motifs or serine/threonine-containing peptides that are phosphorylated. Interestingly, the PLP1 heptapeptide contains an RxxR tetrapeptide and a serine residue, and we speculated that the PLP1-specific activity might be associated with one of these signals. Accordingly, we generated several PLP1 mutants by separately mutating the arginine residues to alanine, the serine to alanine, or the serine to aspartic acid, which mimics phosphorylation (Fig. 5A). As indicated in Figure 5B,C, mutating the arginine residues or the serine does not perturb wild-type PLP1 trafficking. In contrast, the rsh compound mutants are efficiently retained in the ER, indicating that the PLP1-specific peptide remains active. Thus, the heptapeptide does not conform to known ER retention motifs.
Fig. 5.

Quantification of protein trafficking to the cell surface for missense:heptapeptide mutagenesis compound mutants. A: Schematic showing the primary structure of PLP1 encoded by exon 3, along with targeted mutagenesis sites (asterisks) in the heptapeptide KGRGSRG (gray rectangle). B–D: Trafficking of PLP1 compound mutants harboring the rsh missense mutation coupled with the heptapeptide arginine-to-alanine mutations (B), the heptapeptide glycine-to-glutamine mutations (C), or the heptapeptide serine-to-alanine and serine-to-aspartic acid mutations (D). The constructs are quantified by the proportion of transfected cells in which the mutants traverse the secretory pathway to the cell surface.
Disease-Causing Missense Mutations in Exon 3B Do Not Disrupt the Activity of the PLP1-Specific Peptide
In view of the suggestion from Figure 4 that other peptides present in exon 3 may contribute to the activity of the PLP1-specific peptide, we sought additional mutations in exon 3. Several missense mutations associated with SPG2 have been reported in exon 3B, and we hypothesized that such mutations might cause disease by disrupting the activity of the PLP1-specific peptide. To test this hypothesis, we generated two mutations found in patients (Saugier-Veber et al., 1994; Cailloux et al., 2000) that cause histidine-to-tyrosine changes in exon 3B (Fig. 6A). One of these mutations (H129Y) is adjacent to the carboxyl-terminal end of the heptapeptide, and the other (H139Y) is 10 amino acids farther downstream.
Fig. 6.

Quantification of protein trafficking to the cell surface for missense:SPG2 compound mutants. A: Schematic showing the primary structure of PLP1 encoded by exon 3, along with the mutation sites identified in two families with SPG2 (asterisks). The location of the heptapeptide KGRGSRG is shown (gray rectangle). B,C: Trafficking of PLP1 compound mutants harboring the rsh, V218F, and msd missense mutations coupled with either the H129Y mutation (B) or the H139Y mutation (C). The constructs are quantified by the proportion of transfected cells in which the mutants traverse the secretory pathway to the cell surface.
In Figure 6B, trafficking of the PLP1H129Y mutant is unperturbed compared with controls. However, the H129Y:rsh and H129Y:V218F compound mutants accumulate efficiently in the ER, thereby indicating that the PLP1-specific peptide is functional. As expected, the H129Y:msd compound mutants also accumulate in the ER. The results shown in Figure 6C are similar, and together these data indicate that the activity of the PLP1-specific peptide is largely intact.
DISCUSSION
In the years since we proposed the PLP1 misfolding hypothesis to account for disease in PMD and SPG2 patients (Gow et al., 1994a,b), we have continued to examine the clinical phenotypes of patients from additional families (Gow and Lazzarini, 1996). Our data indicate that the transfected COS-7 cell assay has considerable potential for use as a prognostic tool in evaluating disease severity of novel missense mutations. Indeed, this assay correlates with disease severity for more than 70% of the pedigrees we have examined to date (unpublished data). In the current study, we explore the molecular basis of the transfected COS-7 cell assay to develop a deeper understanding of the mechanisms underlying mild forms of disease.
Our previous demonstrations in transfected fibroblasts that the rsh and V218F missense mutations disrupt native protein conformation (Gow et al., 1997; Southwood and Gow, 2001), imply that ER accumulation of PLP1rsh and PLP1V218F stems from protein misfolding and that these mutants are retained in the ER through stable associations with molecular chaperones. However, data presented in the current study suggest an alternate mechanism, that the presence or absence of the KGRGSRG heptapeptide is a significant determinant for ER retention or trafficking of these mutants. Although our past and current studies evoke different conclusions regarding the effect of mild mutations on PLP1 folding, the mechanisms are not necessarily incompatible. Most importantly from the perspective of pathogenesis, these data are congruent, because the outcome in either case is accumulation of protein in the ER, which activates the unfolded protein response (Southwood et al., 2002).
Several ER retention motifs have been identified in the cytoplasmic domains of diverse transmembrane proteins that form multimeric complexes, but the best characterized is the di-arginine motif (Michelsen et al., 2005). In some instances, this motif is present in every subunit, such as in the ATP-sensitive inwardly rectifying potassium channel; in others, only one subunit harbors the motif, as in the neurotransmitter receptor GABAB. Masking the motif leads to ER export and has been shown to occur by several mechanisms, including subunit assembly into multimeric complexes, serine/threonine phosphorylation, or interference with the binding of ER-resident retention factors. In the case of PLP1, a di-arginine and a serine residue are located in the heptapeptide; however, mutating these amino acids to alanine or changing the serine to aspartic acid does not inactivate heptapeptide activity. Thus, the PLP1-specific peptide must effect ER retention through a novel mechanism.
The data in Figure 4 show that the PLP1 heptapeptide is necessary for PLP1-specific peptide activity but may not be sufficient, which suggests the existence of other retention signals. What might these signals be? Palmitoylation is known to regulate ER retention for at least some transmembrane proteins (Drisdel et al., 2004), and we note the presence of three cysteine residues in the vicinity of the heptapeptide that are palmitoylated in vivo (Weimbs and Stoffel, 1992). However, mutation of all of these cysteine residues to alanine is without effect on PLP1-specific peptide activity (data not shown). Indeed, we can also rule out the involvement of peptide motifs that require any of the cysteine residues for function, as well as involvement of the two histidine residues in the PLP1-specific peptide that are associated with SPG2. The presence of either H129Y or H139Y does not reduce ER accumulation of PLP1rsh compound mutants, indicating that the PLP1-specific peptide is active. Parenthetically, trafficking of the PLP1H129Y and PLP1H139Y mutants through the secretory pathway to all major compartments in COS-7 cells indicates that these mutations do not disrupt the higher ordered structure of PLP1. Thus, these mutations must cause disease through a novel mechanism that is distinct from all of the missense mutations we have studied outside of the PLP1-specific peptide and, presumably, is one that does not involve the unfolded protein response.
The relevance of utilizing transfected COS-7 cells to examine the trafficking of PLP1 and DM-20 mutants through the secretory pathway, as in the current study, is a matter of continued debate, because oligodendrocytes are thought to be more appropriate. We discount this view and argue that fibroblasts are no less relevant than cultured oligodendrocytes on several grounds. First, the investigation of protein trafficking is an issue of cell biology not of myelin biology; the processes and pathways governing protein trafficking are conserved from yeast to mammals, and this understanding is firmly seated in the protein trafficking literature. Second, we have demonstrated that wild-type PLP1 gene products modify the trafficking of missense gene products (Gow and Lazzarini, 1996), and transfection of primary oligodendrocytes or derived cell lines to coexpress endogenous wild-type PLP1 gene products and transfected PLP1 mutants will likely yield artifactual data. Oligodendrocytes in vivo express a single PLP1 allele, and a mix of mutant and wild-type proteins in a single cell is rarely encountered. Transfection of PLP1 null oligodendrocytes is not a useful alternative, as these cells cause Pelizaeus-Merzbacher disease and cannot be considered normal. Third, it is a bold assumption that cell lines such as CG4 and OLI-neu are fully differentiated oligodendrocytes. Moreover, it is rank speculation that these cells even express the molecular machinery to process PLP1 identically to differentiated oligodendrocytes in vivo. Thus, there is no logical basis for using oligodendrocyte-lineage cells in preference to other cell types for the purpose of examining the trafficking of PLP1 mutants.
Finally, an important issue relevant to myelin biology pertains to the necessity of an ER retention motif for PLP1 but not for DM-20. The PLP1 gene is a member of the lipophilin family, which comprises three genes (Gow, 1997). The archetypal member of this family dates back at least 550 million years (Stecca et al., 2000) and expanded to three genes in marine vertebrates (Kitagawa et al., 1993). However, all members lack the PLP1-specific peptide, which appeared in amphibians approximately 300 million years before present. Importantly, the appearance of this peptide coincides with the ascension of PLP1 as the dominant structural protein of CNS myelin in terrestrial vertebrates (Yoshida and Colman, 1996). Thus, our earlier finding that DM-20 cannot recapitulate the function of PLP1 in vivo (Stecca et al., 2000) supports the argument that the function of the PLP1-specific peptide is closely related to the unique structural role that PLP1 plays in myelin.
In view of the data indicating that ER retention motifs serve to abrogate exit from the ER prior to multimeric complex assembly for a number of proteins (Michelsen et al., 2005), we speculate that the PLP1-specific peptide functions similarly. Indeed, early analytical ultracentrifugation studies indicate that PLP1 purified from bovine white matter is hexameric (Gow et al., 1985), which may be critical for myelin stability. If so, does the absence of a retention signal in DM-20 imply that this protein remains monomeric? Probably not, because DM-20 can at least form heteromeric complexes with wild-type PLP1 in vivo (McLaughlin et al., 2002). In addition, DM-20 can extricate misfolded PLP1msd from the ER of transfected cells (Gow and Lazzarini, 1996), which suggests a chaperone function for DM-20. Whatever the case, we can be certain that the presence of DM-20 in terrestrial vertebrate myelin is not a consequence of insufficient evolutionary time to mutate the splice donor site in exon 3A. To the contrary, current evidence shows that, after acquisition of the PLP1-specific peptide by the DMα gene, amphibians lost the ability to generate transcripts encoding DM-20, even though the requisite splice donor site is intact in these animals (Venkatesh et al., 2001). However, alternative splicing in exon 3 reappears in lizards, indicative of positive selection, and is observed in all other terrestrial vertebrates.
Acknowledgments
Contract grant sponsor: NINDS; Contract grant number: NS43783; Contract grant sponsor: National Multiple Sclerosis Society; Contract grant number: RG2891.
We thank Dr. Nick Davis for his advice, helpful discussions, and critique of the manuscript.
References
- Cailloux F, Gauthier-Barichard F, Mimault C, Isabelle V, Courtois V, Giraud G, Dastugue B, Boespflug-Tanguy O. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Clinical European Network on Brain Dysmyelinating Disease. Eur J Hum Genet. 2000;8:837–845. doi: 10.1038/sj.ejhg.5200537. [DOI] [PubMed] [Google Scholar]
- Drisdel RC, Manzana E, Green WN. The role of palmitoylation in functional expression of nicotinic alpha7 receptors. J Neurosci. 2004;24:10502–10510. doi: 10.1523/JNEUROSCI.3315-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–191. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- Forman MS, Lee VM, Trojanowski JQ. “Unfolding” pathways in neurodegenerative disease. Trends Neurosci. 2003;26:407–410. doi: 10.1016/S0166-2236(03)00197-8. [DOI] [PubMed] [Google Scholar]
- Garbern JY. Pelizaeus-Merzbacher disease: pathogenic mechanisms and insights into the roles of proteolipid protein 1 in the nervous system. J Neurol Sci. 2005;228:201–203. doi: 10.1016/j.jns.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Gow A. Redefining the lipophilin family of proteolipid proteins. J Neurosci Res. 1997;50:659–664. doi: 10.1002/(SICI)1097-4547(19971201)50:5<659::AID-JNR3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Gow A. The COS-7 cell in vitro paradigm to study myelin proteolipid protein gene mutations. In: Potter N, editor. Neurogenetics: methods and protocols. Totowa, NJ: Humana Press; 2002. pp. 263–275. [DOI] [PubMed] [Google Scholar]
- Gow A, Lazzarini RA. A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat Genet. 1996;13:422–428. doi: 10.1038/ng0896-422. [DOI] [PubMed] [Google Scholar]
- Gow A, Sharma R. The unfolded protein response in protein aggregating diseases. Neuromol Med. 2003;4:73–94. doi: 10.1385/NMM:4:1-2:73. [DOI] [PubMed] [Google Scholar]
- Gow A, Winzor DJ, Smith R. Pressure-induced dissociation of aggregates of myelin proteolipid protein. Biochim Biophys Acta. 1985;828:383–386. doi: 10.1016/0167-4838(85)90321-8. [DOI] [PubMed] [Google Scholar]
- Gow A, Friedrich VL, Lazzarini RA. Intracellular transport and sorting of the oligodendrocyte transmembrane proteolipid protein. J Neurosci Res. 1994a;37:563–573. doi: 10.1002/jnr.490370503. [DOI] [PubMed] [Google Scholar]
- Gow A, Friedrich VL, Lazzarini RA. Many naturally occurring mutations of myelin proteolipid protein impair its intracellular transport. J Neurosci Res. 1994b;37:574–583. doi: 10.1002/jnr.490370504. [DOI] [PubMed] [Google Scholar]
- Gow A, Gragerov A, Gard A, Colman DR, Lazzarini RA. Conservation of topology, but not conformation, of the proteolipid proteins of the myelin sheath. J Neurosci. 1997;17:181–189. doi: 10.1523/JNEUROSCI.17-01-00181.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A, Southwood CM, Lazzarini RA. Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus-Merzbacher disease. J Cell Biol. 1998;140:925–934. doi: 10.1083/jcb.140.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson LD, Nadon NL. Amino acid substitutions in proteolipid protein that cause dysmyelination. In: Martenson RE, editor. Myelin: biology and chemistry. Boca Raton, FL: CRC Press; 1992. pp. 677–702. [Google Scholar]
- Kitagawa K, Sinoway MP, Yang C, Gould RM, Colman DR. A proteolipid protein gene family: expression in sharks and rays and possible evolution from an ancestral gene encoding a pore-forming polypeptide. Neuron. 1993;11:433–448. doi: 10.1016/0896-6273(93)90148-k. [DOI] [PubMed] [Google Scholar]
- Lorence MC, Murry BA, Trant JM, Mason JI. Human 3β-hydroxysteroid dehydrogenase/Δ5-4 isomerase from placenta: expression in nonsteroidogenic cells of a protein that catalyzes the dehydrogenation/isomerization of C21 and C19 steroids. Endocrinology. 1990;126:2493–2498. doi: 10.1210/endo-126-5-2493. [DOI] [PubMed] [Google Scholar]
- McLaughlin M, Hunter DJ, Thomson CE, Yool D, Kirkham D, Freer AA, Griffiths IR. Evidence for possible interactions between PLP and DM20 within the myelin sheath. Glia. 2002;39:31–36. doi: 10.1002/glia.10091. [DOI] [PubMed] [Google Scholar]
- Michelsen K, Yuan H, Schwappach B. Hide and run. Arginine-based endoplasmic-reticulum-sorting motifs in the assembly of hetero-multimeric membrane proteins. EMBO Rep. 2005;6:717–722. doi: 10.1038/sj.embor.7400480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puckett C, Hudson L, Ono K, Friedrich V, Benecke J, Dubois-Dalcq M, Lazzarini RA. Myelin-specific proteolipid protein is expressed in myelinating Schwann cells but is not incorporated into myelin sheaths. J Neurosci Res. 1987;18:511–518. doi: 10.1002/jnr.490180402. [DOI] [PubMed] [Google Scholar]
- Saugier-Veber PS, Munnich A, Bonneau D, Rozet J-M, Le Merrer M, Gil R, Boespflug-Tanguy O. X-linked spastic paraplegia and Pelizaeus-Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat Genet. 1994;6:257–262. doi: 10.1038/ng0394-257. [DOI] [PubMed] [Google Scholar]
- Southwood CM, Gow A. Molecular mechanisms of disease stemming from mutations in the proteolipid protein gene. Microsc Res Techniq. 2001;52:700–708. doi: 10.1002/jemt.1054. [DOI] [PubMed] [Google Scholar]
- Southwood CM, Garbern J, Jiang W, Gow A. The unfolded protein response modulates disease severity in Pelizaeus-Merzbacher disease. Neuron. 2002;36:585–596. doi: 10.1016/s0896-6273(02)01045-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stecca B, Southwood CM, Gragerov A, Kelley KA, Friedrich VLJ, Gow A. The evolution of lipophilin genes from invertebrates to tetrapods: DM-20 cannot replace PLP in CNS myelin. J Neurosci. 2000;20:4002–4010. doi: 10.1523/JNEUROSCI.20-11-04002.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton E, High S, Woodman P. Role of calnexin in the glycan-independent quality control of proteolipid protein. EMBO J. 2003;22:2948–2958. doi: 10.1093/emboj/cdg300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson CE, Montague P, Jung M, Nave K-A, Griffiths IR. Phenotypic severity of murine Plp mutants reflects in vivo and in vitro variations in transport of PLP isoproteins. Glia. 1997;20:322–332. doi: 10.1002/(sici)1098-1136(199708)20:4<322::aid-glia5>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Tosic M, Gow A, Dolivo M, Domanska-Janik K, Lazzarini RA, Matthieu J-M. Proteolipid/DM20 proteins bearing the paralytic tremor mutation in peripheral nerves and transfected COS-7 cells. Neurochem Res. 1996;21:423–430. doi: 10.1007/BF02527706. [DOI] [PubMed] [Google Scholar]
- Tosic M, Matthey B, Gow A, Lazzarini RA, Matthieu J-M. Intracellular transport of the DM-20 bearing shaking pup (shp) mutation and its possible phenotypic consequences. J Neurosci Res. 1997;50:844–852. doi: 10.1002/(SICI)1097-4547(19971201)50:5<844::AID-JNR20>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Venkatesh B, Erdmann MV, Brenner S. Molecular synapomorphies resolve evolutionary relationships of extant jawed vertebrates. Proc Natl Acad Sci U S A. 2001;98:11382–11387. doi: 10.1073/pnas.201415598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weimbs T, Stoffel W. Proteolipid protein (PLP) of CNS myelin: positions of free, disulfide-bonded, and fatty acid thioester-linked cysteine residues and implications for the membrane topology of PLP. Biochemistry. 1992;31:12289–12296. doi: 10.1021/bi00164a002. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Colman DR. Parallel evolution and coexpression of the proteolipid proteins and protein zero in vertebrate myelin. Neuron. 1996;16:1115–1126. doi: 10.1016/s0896-6273(00)80138-5. [DOI] [PubMed] [Google Scholar]
- Zoghbi HY, Botas J. Mouse and fly models of neurodegeneration. Trends Genet. 2002;18:463–471. doi: 10.1016/s0168-9525(02)02729-4. [DOI] [PubMed] [Google Scholar]