

Abstract
Vitamin A and its derivatives, retinoids, have been widely studied for their use as cancer chemotherapeutic agents. With respect to colorectal cancer (CRC), several critical mutations dysregulate pathways implicated in progression and metastasis, resulting in aberrant Wnt/β-catenin signaling, gain-of-function mutations in K-ras and phosphatidylinositol-3-kinase/Akt, cyclooxygenase-2 over-expression, reduction of peroxisome proliferator-activated receptor γ activation, and loss of p53 function. Dysregulation leads to increased cellular proliferation and invasion and decreased cell-cell interaction and differentiation. Retinoids affect these pathways by various mechanisms, many involving retinoic acid receptors (RAR). RAR bind to all-trans-retinoic acid (ATRA) to induce the transcription of genes responsible for cellular differentiation. Although most research concerning the chemotherapeutic efficacy of retinoids focuses on the ability of ATRA to decrease cancer cell proliferation, increase differentiation, or promote apoptosis; as CRC progresses, RAR expression is often lost, rendering treatment of CRCs with ATRA ineffective. Our laboratory focuses on the ability of dietary vitamin A to decrease CRC cell proliferation and invasion via RAR-independent pathways. This review discusses our research and others concerning the ability of retinoids to ameliorate the defective signaling pathways listed above and decrease tumor cell proliferation and invasion through both RAR-dependent and RAR-independent mechanisms.
Keywords: Colorectal cancer, Retinoid, Vitamin A, β-catenin, Phosphatidylinositol-3-kinase, K-ras, Cyclooxygenase-2, Peroxisome proliferator-activated receptor γ, P53, Phosphatase and tensin homolog deleted on chromosome 10
Core tip: Vitamin A and its derivatives, the retinoids, have been widely studied in many types of cancer for their ability to increase cell differentiation and decrease cell proliferation. This review focuses on the ability of retinoids to affect signaling pathways commonly disrupted in colorectal cancer. We discuss vitamin A metabolism and signaling, how this process becomes aberrant as colorectal cancer progresses, and how treatment with both dietary vitamin A and exogenous retinoids can alter these dysregulated signaling pathways to decrease colorectal cancer cell proliferation and invasion.
INTRODUCTION
Colorectal cancer (CRC) is the third most commonly diagnosed cancer in men and the second most commonly diagnosed cancer in women worldwide[1,2]. An estimated 1.2 million cases occurred worldwide in 2008, with the highest incidence rates occurring in developed countries including North America, Australia, New Zealand, Japan and Europe[1]. Global trends reflect an overall increase in the incidence of CRC, with the highest increases observed throughout Asia and Europe[1]. About 608700 deaths occurred as a result of CRC in 2008, accounting for 8% of all cancer-related deaths worldwide[1]. Approximately 50% of those patients diagnosed with CRC will experience metastasis to the liver, which is the primary site of CRC metastasis[3]. Risk factors for CRC are both genetic and environmental. A personal or family history of CRC and a personal history of chronic inflammatory bowel disease increase the risk for CRC[4]. Physical inactivity, obesity, smoking, and dietary patterns such as high red and processed meat consumption as well as moderate-to-heavy alcohol use also increase the risk for CRC[4]. Retinoids have long been studied for their effects on organismal development and cellular differentiation, particularly with respect to cancer. Retinoids are currently used as chemotherapies against cancers of epithelial origin, including basal and squamous cell carcinomas. Furthermore, retinoids (whose metabolism is shown in Figure 1) are known to affect signaling pathways frequently altered which result in the development and progression of CRC (Figure 2 and Table 1). CRC is highly influenced by diet, therefore it stands to reason that direct contact with retinoids from supplemented diets or exogenous retinoids administered as medication may have chemotherapeutic effects on CRC tumors.
Figure 1.
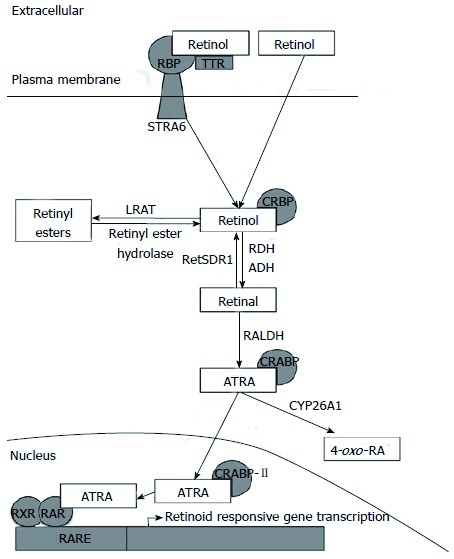
Retinoid metabolism. Vitamin A circulates as retinol bound to RBP and TTR. Retinol can be absorbed into cells via STRA6 or diffusion through the cell membrane. Intracellularly, retinol can be stored as retinyl esters or converted to ATRA. ATRA travels to the nucleus where it binds RAR to induce the transcription of retinoid-responsive genes. RBP: Retinol binding protein; TTR: Transthyretin; STRA6: Stimulated by retinoic acid 6; CRBP: Cellular retinol binding protein; LRAT: Lecithin retinol acyltransferase; RALDH: Retinaldehyde dehydrogenase; CRABP: Cellular retinoic acid binding protein; CYP26A1: Cytochrome P450 26A1; 4-oxo-RA: 4-oxo-retinoic acid; ATRA: All-trans-retinoic acid; RXR: Retinoid X receptor; RAR: Retinoic acid receptor; RARE: Retinoic acid response element.
Figure 2.
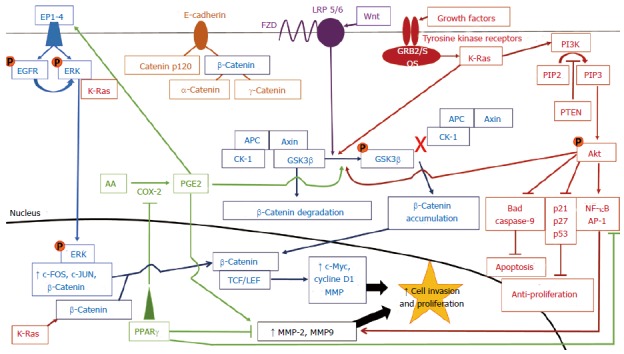
Crosstalk between signaling pathways that lead to colorectal cancer progression. Each pathway is indicated by a specific color. Orange circles represent phosphate groups. β-Catenin is found at the cell membrane, complexed with E-cadherin, in the cytosol, and in the nucleus. Cytosolic β-catenin can be targeted for proteosomal degradation by GSK3β when GSK3β is not phosphorylated and is complexed with APC, Axin, and CK-1. Nuclear β-catenin induces gene transcription when complexed with TCF/LEF transcription factors. Ultimately, all pathways increase the transcription of genes favoring cellular proliferation (c-Myc, cyclin D1) and invasion (MMPs), most via increasing β-catenin-mediated gene transcription. CRC: Colorectal cancer; EP1-4: E-prostanoid receptor types 1-4; EGFR: Epidermal growth factor receptor; ERK: Extracellular signal-regulated kinase; K-Ras: Kirsten rat sarcoma viral oncogene homolog; FZD: Frizzled; LRP: Lipoprotein related receptor proteins 5/6; GRB2/SOS: Growth factor receptor-bound protein 2/son of sevenless; PI3K: Phosphatidylinositol-3-kinase; PIP2: Phosphatidylinositol-4,5-bisphosphate; PIP3: Phosphatidylinositol-3,4,5-triphosphate; PTEN: Phosphatase and tensin homolog deleted on chromosome 10; APC: Adenomatous polyposis coli; CK-1: Casein kinase 1; GSK3β: Glycogen synthase kinase 3β; PGE2: Prostaglandin E2; COX2: Cyclooxygenase 2; AA: Arachidonic acid; PPARγ: Peroxisome proliferator-activated receptor γ; TCF/LEF: T-cell factor/lymphoid enhancer factor; MMP: Matrix metalloproteinase; NF-κB: Nuclear factor-kappa B; AP-1: Activator protein 1.
Table 1.
Summary of pathways dsyregulated in colorectal cancer and the effect of retinoids on these pathways in both colorectal cancer and other tumor types
| Protein | Mutation rate | Result of gene mutation | Response to retinoid treatment |
| APC | 80%[57,65] | Loss of β-catenin degradation[58]; constitutive activation of the Wnt/β-catenin pathway[59]; decreased RDH levels inhibiting formation of ATRA[42] | Not determined |
| β-Catenin | 5%[56] | Loss of β-catenin degradation[56]; constitutive activation of the Wnt/β-catenin pathway[56]; increased CYP26A1 levels resulting in increased degradation of ATRA | Increased degradation of β-catenin via RXR-mediated pathway[23,24] |
| PI3K | 30%-50%[77,78] | Activation of Akt and loss of GSK3β function[80,82]; increased cancer metastasis[88], partially through NF-κB activation and increased expression of MMP-2 and -9[87,89,90]; positive cell cycle progression through cyclin D1[105]; loss of cell-cell adhesion by Snail accumulation to repress E-cadherin[106] | Decrease MMP-2 and MMP-9 activity[28]; increase TIMP-1 expression[28]; decrease the phosphorylation of GSK3β, decrease cellular proliferation, and increase the expression of pro-apoptotic proteins in human leiomyoma and myometrial cells[115]; CRBP-I inhibits PI3K/Akt activation in breast cancer cells[116]; inhibit PI3K activity to decrease CRC cell invasion in vitro and metastasis in vivo[25] |
| PTEN | 20%-40%[80] | Loss of PI3K/Akt inhibition[80]; correlation with tumor aggressiveness and invasiveness[109-111] | Suppression of cellular proliferation and enhanced apoptosis by increasing PTEN expression in smooth muscle cells, neuroblastoma and glioblastoma cells, promyelocytes, leukemia cells, fibroblasts, and breast, endometrial, and hepatocellular carcinoma cells[119-128] |
| COX-2 | 80%-90%[134-136] | Increased PGE2 signaling[133,137,138], ERK activation[140], PI3K/Akt signaling through increased EGFR[133,140,141], β-catenin stabilization[142,143], and MMP-2 and MMP-9 expression to promote cellular proliferation[144,145] | Decrease COX-2 expression[146], PGE2, β-catenin levels, and MMP-9[135,144]; inhibition of cell growth[151]; increased apoptosis and RARβ expression[152] |
| PPARγ | 8%[161] | Loss of inhibitory action of gene transcription of pro-survival and growth amplification genes[155,162-165]; increased expression of COX-2[154] | Suppress COX-2 and MMP-7 expression and induction of cell cycle arrest and apoptosis[171]; induce expression of RARβ mRNA in breast cancer cells[175]; increase apoptosis in glioblastoma cells[176]; stimulate PTEN expression in leukemia cells and fibroblasts[121,128] |
| p53 | 50%[177,178] | Loss of anti-growth and apoptotic activity; loss of p53/Siah-1-mediated β-catenin degradation[187] | Increase retinyl ester storage through transcription of retSDR1[54]; enhance p53-mediated cell cycle inhibition and apoptosis through activation of AP-2α and p21 in breast cancer cells[192], caspases in keratinocytes[188], Btg2 and CRABP-II in breast cancer cells[191]; STRA6 induction in ovarian cancer cells, fibroblasts, and CRC cells[193] |
APC: Adenomatous polyposis coli; RDH: Retinol dehydrogenase; ATRA: All-trans-retinoic acid; CYP26A1: Cytochrome P450 26A1; RXR: Retinoid X receptor; PI3K: Phosphatidylinositol-3-kinase; GSK3β: Glycogen synthase kinase 3β; NF-κB: Nuclear factor-kappa B; MMP: Matrix metalloproteinase; TIMP-1: Tissue inhibitor of matrix metalloproteinase 1; CRBP: Cellular retinol binding protein; CRC: Colorectal cancer; PTEN: Phosphatase and tensin homolog deleted on chromosome 10; COX2: Cyclooxygenase 2; PGE2: Prostaglandin E2; ERK: Extracellular signal-regulated kinase; EGFR: Epidermal growth factor receptor; RARβ: Retinoic acid receptor β; PPARγ: Peroxisome proliferator-activated receptor γ; AP-2α: Activator protein 2α; Btg2: Beta cell translocation gene 2; CRABP-II: Cellular retinoic acid binding protein II; STRA6: Stimulated by retinoic acid 6.
VITAMIN A METABOLISM
Vitamin A (retinol) and its derivatives, the retinoids, are a group of fat-soluble compounds composed of a similar structure in which a hydrophobic β-ionone ring is joined to a hydrophilic polar moiety by a conjugated tetraene linear chain[5]. Retinol is also able to be synthesized from some types of fat-soluble, antioxidant carotenoids found in fruits and vegetables. While there are several different carotenoid molecules found in plants, only β-carotene, α-carotene, and β-cryptoxanthin have provitamin A activity[6,7]. In the diet, these carotenoids are consumed primarily through carrots, cantaloupes, sweet potatoes, and spinach[6]. Theoretically, cleaving the β-carotene molecule would yield two retinal molecules, each with a β-ionone ring, which can then be converted to two retinol molecules for cellular use[6]. However, this conversion occurs at a much lower rate in vivo, with the retinol activity equivalent of β-carotene being much lower than a 1:2 ratio of β-carotene:retinol[6]. Both α-carotene and β-cryptoxanthin only contain one β-ionone ring each and thus have about 50% of the provitamin A activity of β-carotene[6].
Retinol is derived from retinyl esters found in animal sources such as butter, eggs, and meats[8,9]. During digestion in the intestinal lumen, the long-chain fatty acids are cleaved from the retinyl esters via hydrolysis, yielding free retinol[10]. The free retinol is then absorbed into the mucosal cells where it is bound by cellular retinol binding protein-II (CRBP-II), which facilitates the re-esterification of retinol by lethicin retinol acyltransferase (LRAT)[10]. Once re-esterified with long-chain fatty acids such as palmitate, the resulting retinyl esters are incorporated into chylomicrons and secreted into the lymphatic circulation[10]. After draining into the general circulation and transferring their lipid contents into peripheral cells, the remaining chylomicron remnants containing the retinyl esters are taken up by hepatocytes[5]. Depending on bodily needs, the liver either stores the retinyl esters in stellate cells or hydrolyzes the retinyl esters to once again yield free retinol, which binds to retinol binding protein (RBP)[5]. The resulting RBP-retinol complex is released into circulation, where it binds to a small protein, transthyretin (TTR), which prevents the retinol from being excreted by the kidneys[5]. This RBP-retinol-TTR complex circulates in the plasma, until retinol dissociates from the protein complex to enter target cells[11]. The transport of retinol into the cell and its intracellular fate is shown in Figure 1. Because retinol is lipophilic, the molecule can freely diffuse through the plasma membrane of cells[11]. In some cells or during vitamin A deficiency, retinol may be taken up by cells through the RBP receptor, STRA6 (stimulated by retinoic acid 6’)[5,11,12]. Cellular uptake of retinol via STRA6 is highly preserved in ocular cells, in which the loss of STRA6 leads to visual impairments[13]. However, in STRA6-null mice, retinoid homeostasis was only moderately affected, with physiological functions that critically depend on all-trans-retinoic acid (ATRA) in both the adult and embryo remaining intact[14]. This indicates that while the receptor functions to assist cells in taking up retinol, STRA6 is not necessary to sustain normal function in cells other than those in the eyes. After diffusion into cells, the internalized free retinol is bound to CRBP or is oxidized to retinal by retinol dehydrogenases (RDH) or alcohol dehydrogenases (ADH) and then to ATRA by retinaldehyde dehydrogenases (RALDH)[5]. ATRA then binds to cellular retinoic acid binding proteins (CRABPs)[5]. CRABP-II shuttles ATRA to the nucleus of the cell, where ATRA serves as a ligand for retinoic acid receptors (RAR).
The RAR and retinoid X receptors (RXR) belong to the nuclear hormone receptor superfamily and are ligand-dependent transcription factors[15]. Each receptor occurs in three subtypes: RARα, -β, and -γ; and RXRα, -β, and -γ. Further, seven different splice variants of RARα (RARα1-7), four different splice variants of RARβ (RARβ1-4), and seven different splice variants of RARγ (RARγ1-7) have been identified[16]. Two different splice variants of each RXR subtype have also been identified that RXRα1 and 2, RXRβ1 and 2, and RXRγ1 and 2[17]. ATRA binds to and activates all subtypes of RAR with a high affinity[15,17]. While the only known retinoid ligand for RXR is 9-cis-RA, there has been a general inability to detect this retinoid isomer in vivo[18,19]. Recently, 9-cis-RA was detected in pancreatic tissue, but the ability of 9-cis-RA to act as a ligand for RXR in cells other than pancreatic cells remains controversial[20]. In the absence of ATRA, the RAR/RXR heterodimer binds to RA response elements (RARE) present on DNA promoter regions of ATRA-target genes[21]. The RAR/RXR complex recruits co-repressor proteins, which in turn recruit histone deacetylases (HDAC) to the DNA region[21]. HDAC remove acetyl groups from histone proteins, changing the chromatin structure and negatively regulating gene transcription[21]. By the binding of ATRA, RAR undergoes a conformational change to release inhibitory co-repressor proteins and recruit co-activator proteins, such as histone acetyl transferases, to enhance transcriptional activity[22]. The vast majority of research regarding the ability of retinoids to prevent cancer progression has focused on ATRA and RAR-mediated phenomena. However, as discussed below, cells become resistant to the effects of ATRA on cellular proliferation and differentiation as tumors progress[8,15]. To this end, our laboratory has shown that retinol has non-genomic effects, exclusive of ATRA, such as interference with pathways involving phosphatidylinositol 3-kinase (PI3K) and β-catenin, which play key roles in the progression of cancer[23-29].
ABBERANT VITAMIN A SIGNALING AND METABOLISM IN COLORECTAL CANCER
The luminal side of the colon is an epithelial layer of tissue which is composed of a single sheet of columnar epithelial cells which are folded into finger-like invaginations that are supported by the lamina propria to form a functional unit called a Lieberkuhn’s crypt[30]. Different types of epithelial cells line the crypt, including epithelial colonocytes, goblet cells, and endocrine cells[31]. The cells at the bottom of the crypt are stem cells that differentiate into the various epithelial cell types as they move upward to the top of the crypt in a process known as “upward migration”[31]. As the cells migrate upwards, they become terminally differentiated and stop proliferating[31]. Once the cells reach the top of the crypt, they undergo apoptosis and are sloughed off into the lumen[31]. When these cells mutate to retain their proliferative capacity and avoid apoptosis once they reach the top of the crypt, they have the potential to form an adenomatous polyp[31]. These abnormalities may result as a process of inherited genetic mutations, replicative mistakes, or epigenetic changes. If undetected, these polyps may progress into a cancerous lesion[31].
The growth and differentiation of epithelial cells is strongly controlled by retinoid-activated genes. Genes involved in transcription, cell signaling, and tumor suppression contain RAREs in their promoter regions, indicating the importance of ATRA in gene expression[18]. In many epithelial-derived adenomas and carcinomas, the expression of one or more RAR is lost and the cell loses its ability to regulate normal growth[17,32]. This phenomenon is termed “ATRA-resistance”. The RARs themselves contain RAREs in their regulatory regions and are thus RA-inducible genes[21,33]. Treatment of patients with premalignant oral lesions with 13-cis-RA, a synthetic retinoid, increased the expression of RARβ, which correlated with clinical response, signifying the beneficial effects of retinoid treatment in increasing anti-tumor gene activity in cancers[33,34]. However, the loss of tumor-suppressive RARβ is common in premalignant and malignant tissues and cells, as reviewed in Xu[33]. Loss of RAR has been shown to be partly due to epigenetic changes such as histone modification and DNA methylation becoming aberrant during carcinogenesis, silencing RAR gene expression[33,35-38]. The loss of RARβ2 in the HCT-116 colon cancer cell line has been suggested to originate as a result of hypermethylation and the ensuing loss of RARα, which is an upstream regulator of RARβ2[39]. Restoration of RARα by a DNA methylation inhibitor resulted in the re-establishment of RARβ2 expression, indicating a potential role for the combined chemotherapeutic action of DNA methylation inhibitors and retinoids[39]. In contrast, Lee et al[32] demonstrated that treatment of RA-sensitive and RA-resistant human colon cancer cell lines with ATRA induced the expression of RARα in all cell lines while only increasing the expression of RARβ in colon cancer cell lines sensitive to RA. Over-expression of RARβ in the RA-resistant colon cancer cell line, DLD-1, resulted in the re-acquisition of RA-sensitivity, inducing growth inhibition and apoptosis in this cell line with ATRA treatment[32]. Over-expression of RARβ in LoVo cells, another RA-resistant human colon cancer cell line, showed similar results in which treatment with ATRA resulted in retinoid-mediated growth inhibition[40].
In addition to the loss of RAR expression and the consequential ATRA resistance, as CRC progresses, colorectal tumor cells appear to lose the ability to produce ATRA[26,41,42] while, at the same time, increasing ATRA degradation via the cytochrome P450 enzyme, CYP26A1[43]. Recently, Kropotova et al[41] found that all genes involved in ATRA synthesis were decreased in CRC tumors and colorectal cell lines. The researchers also found that ADH IB and IC, the most abundant retinol oxidizing enzymes, exhibited decreased gene expression when adenomas were compared to more advanced carcinomas. Similarly, mRNA levels for RDH-5 and L were decreased in colon tumors and CRC cell lines when compared to normal colon cells[42]. As a result, the CRC cell lines produced only small amounts of ATRA from retinol, a phenomenon our group also observed with the ATRA-resistant CRC cell lines HCT-116, SW620 and WiDR[26]. Loss of adenomatous polyposis coli (APC) function, as seen in the SW620 cell line[44], inhibits RDH expression, the enzyme which converts retinol to retinaldehyde[42]. Interestingly, transfection of APC into an APC-deficient cell line increased the expression of RDH-L and the formation of ATRA, indicating crosstalk between Wnt/β-catenin signaling and retinoid metabolism[42]. To elaborate, APC mediates the proteosomal degradation of C-terminal binding protein 1 (CtBP1). Loss of APC increases the levels of CtBP1. Increased CtBP1, in turn, decreases RDH levels, inhibiting the production of ATRA[45]. Loss of ATRA ultimately leads to less colonocyte differentiation, as ATRA is necessary for epithelial cell differentiation[46]. In fact, homozygous loss of APC causes failed intestinal cell differentiation independent of catenin-mediated gene transcription but dependent upon CtBP1, leading to the hypothetical two-step model of colon adenoma initiation and progression[47]. In this model, APC loss and the resulting increase in CtBP1 leads to adenoma initiation, successive K-ras activation, and the nuclear translocation of β-catenin causing progression to a carcinoma. An incongruity with this model is that administration of ATRA to ApcMin mice, which are heterozygous for a dysfunctional APC mutation, did not prevent tumor formation[48]. Shelton et al[43] found that CYP26A1 was increased in tumors from APCMin mice, spontaneous human CRC, and in tumors from patients with familial adenomatous polyposis coli (FAP). These researchers also showed that CYP26A1 expression was dependent upon β-catenin-induced gene expression[43]. Finally, retinoid storage may be altered in cancer. Lecithin retinol acyltransferase (LRAT) esterifies retinol to retinyl esters, the storage form of vitamin A while retSDR1 converts retinal to retinol. The promoter of the LRAT gene is hypermethylated in CRC cell lines and tumors when compared to normal tissue[49]. This hypermethylation would decrease LRAT gene expression, potentially decreasing the availability of intracellular retinoids; however, the role of LRAT in cancer progression is controversial with some studies in non-CRC models showing that decreased LRAT levels are protective against carcinogens and correlate with better patient outcomes[50-52]. Proteins in the p53 family have also been shown to affect retinoid metabolism by modulating the expression of retinal short-chain dehydrogenase/reductase (retSDR1). The retSDR1 enzyme is important in regulating retinoid metabolism and storage in many different cell types[53]. Treatment of neuroblastoma cells with physiological concentrations of retinol leads to the accumulation and storage of retinyl esters through the induction of retSDR1 enzyme levels[53]. The overexpression of p53 in the colorectal adenocarcinoma cell line DLD-1 and the CRC cell line HCT-116 yielded a strong induction of both retSDR1 mRNA expression and protein level, even in cells with truncated reporters[54]. The binding of p53 to the retSDR1 promoter was further increased following DNA damage to the cells[54,55]. Importantly, retSDR1 mRNA was shown to be elevated in CRC tumor tissues when compared with healthy samples from the same individuals[54]. These results signify that one mechanism by which p53 acts as a tumor suppressor is by inducing retSDR1 expression in carcinomas to work against tumor progression by supporting retinoid metabolism in these cells[54].
In summary, colorectal tumors often (1) lack RAR, the receptors for ATRA; (2) lose the ability to synthesize ATRA, the RAR ligand, from vitamin A; (3) exhibit increased degradation of ATRA via CYP26A1 to 4-oxo-retinoic acid (4-oxo-RA) and (4) may have altered retinoid storage. The regulation of retinoid metabolism is controlled by proteins such as APC, β-catenin, and p53 that play crucial roles in the promotion and progression of CRC as we elaborate below.
THE WNT/β-CATENIN SIGNALING PATHWAY
The Wnt/β-catenin signaling pathway is an important process that regulates the proliferation, differentiation, and motility of cells in normal intestinal epithelium[3,56]. This pathway, and others affecting CRC progression, are shown in Figure 2. During normal intestinal functioning, the APC protein forms a cytoplasmic complex with Axin, another protein present in the cytosol. Both proteins contain binding sites for other members of their functional complex[57]. Together, the APC-Axin complex recruits other functional members, the serine and threonine kinases glycogen synthase kinase 3β (GSK3β) and casein kinase 1 (CK-1)[57]. Together, these proteins form what is known as the β-catenin “destruction complex”[57]. β-catenin, when present in the cytosol, is sequentially bound and phosphorylated by these kinases and thus earmarked for degradation through an ubiquitin-proteasome-mediated pathway[57].
β-catenin performs a dual function in the cell, where it acts as both a transcription factor in the nucleus and as a cell adhesion stabilizer at the cell membrane. When in the cytosol, β-catenin binds to E-cadherin, a transmembrane protein responsible for the formation and maintenance of intercellular adherens junctions formed when epithelial cells come into contact[58]. E-cadherin binds to catenin p120 and β-catenin, which then binds to α-catenin and γ-catenin to anchor E-cadherin to the actin cytoskeleton[58,59]. Together, these proteins form a functional unit termed the E-caderhin-catenin unit (ECCU), in which β-catenin plays the role of an intermediary protein connecting E-cadherin to the α- and γ-catenin proteins that bind to the actin cytoskeleton[58]. The loss of E-cadherin function is thought to occur late in carcinogenesis and leads to the destruction of the ECCU, which causes a loss of the adherens junction and subsequent increase in cell motility and migration[58]. While the function of APC results in the degradation of β-catenin and β-catenin is necessary to form the ECCU, APC and E-cadherin compete for binding of β-catenin and work together to maintain the equilibrium of β-catenin concentration in the cell[58]. Loss of APC function results in E-cadherin saturation and the consequent accumulation of cytosolic β-catenin, which then translocates to the nucleus to enhance the transcription of genes important in cell growth and motility[58,59]. Thus, loss of APC function leads to a disruption in the equilibrium of β-catenin concentration and increased Wnt signaling[58,59]. Similarly, truncation of APC may result in β-catenin binding but not degradation, making β-catenin unavailable for E-cadherin binding[58]. While the over-expression of β-catenin is an important step in early tumorigenesis, later stages of carcinogenesis and loss of tumor differentiation may lead to loss of both β-catenin and E-cadherin expression, leading to the loss of ECCU formation and increased ability to metastasize[58].
Because β-catenin is both degraded and sequestered to the cell membrane during normal APC and E-cadherin function, it is unable to accumulate in the cytosol and translocate to the nucleus, where it binds to proteins of the T-cell factor/lymphoid enhancer factor (TCF/LEF) families[56,57]. If allowed to form a complex with TCF/LEF proteins, β-catenin acts as a transcription co-factor to allow TCF/LEF transcription factors to bind to the regulatory regions of genes regulating cell differentiation, proliferation, and migration such as c-Myc, matrix metalloproteinase-7 (MMP-7), and cyclin D1[3,57,60,61]. Ligand-bound RARs have been shown to compete with TCF in breast cancer cells to decrease β-catenin-mediated gene transcription[62]. In contrast, others have shown that overexpression of RARγ in cholangiocarcinoma cells increases the nuclear translocation of β-catenin[63], indicating that the effect of RARs on β-catenin varies with tumor type. In phosphorylating β-catenin and thus marking it for ubiquitin-mediated proteasomal degradation, APC and its protein complex constituents act as negative regulators of the Wnt/β-catenin signaling pathway and maintain the homeostasis of intestinal crypt cells and stem cells[3,57,60,64].
Due to its importance in negatively regulating the Wnt/β-catenin signaling pathway, mutations resulting in the loss of APC function are generally thought to be the earliest step in CRC tumorigenesis[56,57]. As a result, APC mutations are found in approximately 80% of human CRCs while mutations involving β-catenin are found in about 5% of all human CRCs[56,57,65]. This APC mutation can be due to an inherited mutation, as in the case of FAP, or due to environmentally-regulated hypermethylation or dysregulation of the APC gene[61,66]. In loss-of-function APC mutations, the ability to degrade β-catenin is lost, allowing the Wnt/β-catenin signaling pathway to become constitutively active and upregulate the transcription of oncogenes important in tumor cell proliferation and metastasis[56]. The mutation of the APC gene leads to the inability of the APC protein to be exported from the nucleus into the cytoplasm, where APC normally forms a complex with the other proteins involved in the β-catenin destruction complex[61]. The loss of APC results in the increased ability of Wnt proteins to bind to membrane-bound receptors in the Frizzled (FZD) and low density lipoprotein receptor-related families to activate kinases that phosphorylate GSK3β[60,61]. The phosphorylation of GSK3β causes the cytosolic β-catenin destruction complex to become de-stabilized, allowing for the accumulation of β-catenin in the cytosol and its subsequent translocation to the nucleus[60]. When Wnt[66] receptors are not engaged, CK-1 and GSK3β are available to phosphorylate β-catenin to mark it for degradation.
K-RAS MUTATIONS AND CROSSTALK WITH OTHER PATHWAYS
While the APC mutation is found in most colon tumors and is generally regarded to be the earliest step in carcinogenesis, doubt has been placed on its ability to single-handedly cause neoplastic formation. In 30%-50% of CRC tumors, mutation of the K-ras gene has also been found, implicating its co-involvement in tumorigenesis[3,60,65,67]. K-ras is responsible for the transduction of mitogenic signals from growth factor receptors on the cell surface to the nucleus[65]. K-ras acts as a molecular switch to regulate the extracellular signal-regulated kinase (ERK) and PI3K/Akt signaling pathways[3]. During K-ras activation, the binding of growth factors to receptor tyrosine kinases causes the recruitment of the growth factor receptor-bound protein 2/son of sevenless (GRB2/SOS) protein complex to the inner cell membrane[60]. This protein complex activates the G-protein Ras (rat sarcoma), resulting in the phosphorylated ERK translocation to the nucleus[60]. In the nucleus, ERK interacts with transcription factors to induce the transcription of target genes such as c-FOS and c-JUN, which regulate proliferation, differentiation, and apoptosis[60].
Additionally, K-ras activation results in the increased transcription of β-catenin, resulting in the increased accumulation of β-catenin in the cytosol[60]. Mutations of K-ras destroy the GTPase activity of K-ras and fix K-ras in its GTP-bound active forms to permanently activate K-ras and increase ERK signaling[3,60,65,67]. The K-ras mutation interacts with the Wnt/β-catenin signaling pathway by causing the phosphorylation of GSK3β through activation of PI3K[60]. As previously discussed, inactivation of GSK3β leads to de-stabilization of the destruction complex and the resultant stabilization and mobilization of cytosolic β-catenin to the nucleus[60]. Normal activity of GSK3β contributes to negative regulation of both the K-ras and Wnt/β-catenin signaling pathways by phosphorylating K-ras, contributing to its degradation[64]. Thus, GSK3β plays an important role in regulation of both the K-ras and Wnt/β-catenin signaling pathways by degrading key intermediates of each pathway and preventing the transcription of genes important in tumor promotion[64].
K-ras mutations develop after APC loss during progression and metastasis of CRCs, enhancing neoplastic growth[3]. This enhancement of neoplastic growth is achieved by enhanced activation of Wnt/β-catenin signaling[3]. In many cancers, simultaneous activation of K-ras- and β-catenin-dependent pathways are often seen[60]. In human CRC cells and CRC mouse models, gain-of-function K-ras mutations coupled with loss-of-function APC mutations were associated with increased nuclear β-catenin levels and increased size, number, and incidence of tumors when compared to cells or mice with K-ras or APC mutations alone[3]. The resulting tumors displayed an increased migration rate and invasive capability through the increased activity of cyclin D1, which promotes cell cycle progression[3,60]. This evidence results in the theory that carcinogenesis in colon cells requires APC loss with an additional K-ras mutation[3]. Administration of ATRA to mice treated with the carcinogen deoxycholic acid (DCA) decreased colon tumor incidence, but ATRA did not affect the rate of K-ras mutation due to DCA administration[68]. Although we are not aware of any additional research regarding the ability of retinoids to affect K-ras expression or function in CRC, our laboratory and others have shown that retinoids can decrease β-catenin levels and thereby β-catenin-dependent gene transcription as described below.
Table 1 summarizes the effect of retinoids on proteins that affect CRC progression. Although retinoids do not appear to directly alter APC or K-ras activity, they do directly affect β-catenin levels. β-catenin degradation has been shown to be mediated by the activity of three pathways: (1) the APC/GSK3β pathway; (2) the p53/Siah-1 pathway; and (3) an RXRα-dependent pathway. The RXR-mediated pathway was discovered when Xiao et al[69] showed that RXR agonists caused the degradation of RXRα and reduced β-catenin-mediated activation of gene transcription and cell proliferation. Additional work has shown that there is a direct interaction between RXRα and β-catenin[70]. Specifically, in the RXRα-dependent pathway, RXRα binds to nuclear β-catenin and facilitates the transport of β-catenin back into the cytosol where β-catenin is ubiquitinated and degraded by the proteosome. Interestingly, RXRα expression is decreased in advanced CRC when compared to normal adjacent tissue and this decrease is associated with aberrant β-catenin expression[71]. Retinoids increase β-catenin degradation in a variety of tumor types. For example, N-(4 hydroxyphenyl)retinamide (fenretinide) induced the degradation of β-catenin in prostate cancer cells[72] and ATRA decreased β-catenin levels in head and neck cancer stem cells[73]. With respect to CRC, our laboratory has shown that retinol treatment increased β-catenin degradation in ATRA resistant CRC cell lines via a RXR-mediated pathway[23,24].
PHOSPHATIDYLINOSITOL 3-KINASE/AKT SIGNALING
The PI3K/protein kinase B (Akt) signaling pathway is another important pathway, the activation of which induces cellular transformation, proliferation, migration, and survival, all of which work together to promote tumor progression[74-76]. Mutations resulting in aberrant activation of this pathway have been implicated in 30%-50% of all human CRCs[77,78]. This dysregulation occurs via three mechanisms: (1) activating mutations in exons 9 and 20 on the PIK3CA gene; (2) overexpression of Akt itself or activating mutations in the Akt PH domain to increase signaling; and (3) loss of function or expression of the negative regulator phosphatase and tensin homolog deleted on chromosome 10 (PTEN)[79-81]. PI3K belongs to a family of lipid kinases, and is characterized by its ability to phosphorylate the inositol rings of phospholipids on the inner cell membrane[82]. PI3K is present on the cell membrane as a heterodimer, consisting of one of four catalytic p110 subunits and one of two regulatory subunits[80,82]. P110α (PIK3CA) and p110β (PIK3CB) are ubiquitously expressed, with PIK3CA commonly being the more abundant catalytic subunit[82]. PIK3CA and PIK3CB bind to one of two regulatory subunits: p85α or p85β[82]. Class I PI3K enzymes bind Akt via pleckstrin homology (PH) domain-containing proteins and are activated mainly by receptor tyrosine kinases, such as those belonging to the epidermal growth factor receptor (EGFR) family, which accept a variety of extracellular signals necessary to stimulate cellular proliferation[80,82]. Once activated, PI3K catalyzes the phosphorylation of membrane-bound phosphatidylinositol-4,5-bisphosphate (PIP2) to generate the second messenger phosphatidylinositol-3,4,5-triphosphate (PIP3)[82]. The generation of PIP3 allows for the recruitment of PH domain-containing proteins to the inner plasma membrane[80]. Most notably, the PH domains of 3-phosphoinositide-dependent protein kinase 1 (PDK1) and Akt are drawn together, and PDK1 mediates the phosphorylation of Akt at the threonine 308 site[80,83].
Activating mutations in the Akt1 gene are rare, occurring in less than 2% of all CRCs[80]. Activating mutations in PDK1 are even rarer, occurring in less than 1% of all CRCs[80]; however, because these proteins are immediately downstream of PI3K, over-activation of PI3K due either to activating mutations of the PI3K gene or due to mutations of PTEN, the PI3K inhibitor, ultimately results in the over-activation of Akt. Akt occurs in three isoforms: Akt1, 2, and 3, with Akt1 being most broadly expressed[82]. Akt contains two phosphorylation sites, both of which are required to be phosphorylated for full Akt activation[84]. Phosphorylation of Akt at the threonine 308 site by PDK1 partially activates Akt, whereas full activation requires conjunctive phosphorylation of the serine 473 site by other kinases, such as the mammalian target of rapamycin (mTOR) complex 2 (mTORC2)[83,85]. Full activation of Akt enables Akt to modulate the activity of pathways and expression of genes involved in the regulation of cell survival and proliferation as well as metastasis[86]. As reviewed in Fresno Vara et al[82] and Danielsen et al[77], Akt prevents the anti-proliferative activities of tumor suppressor genes p21, p27, and p53. Akt also blocks apoptosis in cancer cells by inactivating signals produced by Bcl-2 associated-death promoter (Bad) and caspase-9 proteins, and activates nuclear factor-kappa B (NF-κB), a transcription factor involved in the transcription of genes important in maintaining cell survival and increasing cell invasion[77,82,87]. The mechanism by which Akt activation promotes metastasis is incompletely understood, but elevated Akt phosphorylation has been shown to be correlated with the invasiveness of cancer in human CRC tissues[88]. Specifically, increased levels of phosphorylated Akt are associated with venous invasion of colorectal carcinomas, tumor depth, and the presence of lymph node metastases[88].
One possible mechanism linking Akt activity to cell invasion relies on the activation of NF-κB. NF-κB upregulates the transcription of matrix metalloproteinases (MMPs), which are a class of zinc-dependent enzymes responsible for the degradation of the extracellular matrix[87,89,90]. Specifically, MMP-2 (gelatinase A) and MMP-9 (gelatinase B) belong to a family of gelatinase enzymes that degrade the collagen component of the extracellular matrix[90,91]. Both MMP-2 and MMP-9 are overexpressed in many colon carcinomas when compared with non-cancerous tissue and are associated with increased invasiveness of cancers, advanced tumor stage, and poor survival[87,89,91,92]. Relevant to this review, MMP-9 and MMP-2 have been shown to be overexpressed in colorectal carcinomas, but not adenomas, indicating their importance in tumor promotion and progression[93]. MMP-2 and -9 are present in the cytosol in inactive pro forms, and cleavage of MMP-2 and -9 by membrane-type matrix metalloproteinases (MT-MMP), such as MT1-MMP, convert inactive pro-MMP-2 and -9 to active MMP-2 and -9[94,95]. This cleavage is inhibited by tissue inhibitors of metalloproteinases (TIMPs), specifically TIMP-1 and -2, which interact with the intermediate (inactive) MMP-9 and -2, respectively, before the proteases are fully activated[94,96]. TIMP-1 expression is regulated by activator protein-1 (AP-1), a transcription factor regulated by the activation of the mitogen-activated protein kinase (MAPK) pathway[90]. Thus, it has been suggested that both PI3K/Akt and MAPK signaling activation must occur simultaneously to regulate MMP-2 and -9 activity and thereby cell invasion[90]. ATRA has been shown to decrease MMP-2 and -9 activity as well as protein and mRNA levels and increase TIMP-1 in a variety of cancers[97-101]. With respect to CRC, our laboratory has shown that treatment of the ATRA-resistant human CRC cancer cell lines HCT-116 and SW620 with retinol resulted in decreased MMP-9 mRNA levels[28]. MMP-2 mRNA levels were decreased in SW620 cells but not in HCT-116 cells[28]. Importantly, the reduction of MMP-2 and MMP-9 mRNA was matched by a reduction in MMP activity[28]. Retinol treatment of HCT-116 and SW620 cells also increased the expression of TIMP-1, potentiating the inhibition of MMP-9 activity in these cells[28].
While TIMP-1 and MMP-2 and 9 expression are regulated by AP-1 and AP-1 activity is in turn repressed by retinoids, this is not thought to be the mechanism by which retinoids affect TIMP-1 and MMP-2 and 9 expression. AP-1 is composed of the proto-oncogenes c-JUN and c-FOS and its activity is associated with cellular proliferation and invasion[102]. Suppression of AP-1 by 9-cis-RA led to the inhibition of cyclin D1 and MMP-2 and 9 in breast cancer cells, however this effect was not matched in SW480 CRC cells, which have low AP-1 activity[102]. Instead, the trans-repressive effects of the cyclin D1 promoter, which contains AP-1 and TCF sites, was independent of the AP-1 site in these CRC cells and required the involvement of a TCF binding element[103]. This data shows that while AP-1 activity is involved in cellular proliferation and invasion, retinoids appear to exert their repressive effects on MMP levels through their interaction with pathways that decrease β-catenin, as β-catenin forms a transactivation complex with TCF/LEF transcription factors. However, promising research involving novel synthetic retinoid derivatives may better target AP-1 for tumor suppression. Um et al[104] developed the synthetic retinoid 4-amino-2-(butyrylamino)phenyl-(2E,4E,6E,8E)-3,7-dimethyl-9-(2,6,6-trimethyl-1-cyclohexenyl)-2,4,6,8-nonatetraenoate (ABPN), which greatly inhibited AP-1 activity in HCT-116 cells. ABPN suppressed c-JUN activity, which led to a decrease in MMP-2 expression, by directly affecting AP-1[104].
It is widely accepted that cross-talk between the PI3K/Akt pathway and the Wnt/β-catenin signaling pathway occurs with GSK3β. Activated Akt phosphorylates GSK3β, inactivating GSK3β and causing a loss of function[82]. Without GSK3β to phosphorylate cytosolic β-catenin and mark it for degradation, stabilized β-catenin can accumulate in the cytosol and eventually translocate to the nucleus to act as a co-factor for gene transcription, as discussed previously[82,86]. Additionally, it has been shown that GSK3β phosphorylation of cyclin D1 stimulates cyclin D1 degradation[105]. Therefore, in tumor cells with increased Akt signaling and loss of GSK3β activation, cyclin D1 remains stable and able to positively regulate cell cycle progression[105]. The loss of GSK3β functioning also results in the increased accumulation of Snail, a zinc-finger transcriptional repressor of E-cadherin[106]. Active, unphosphorylated GSK3β binds to Snail and activates its degradation[107]. Loss of GSK3β function by Akt hyperactivation permits Snail to act as a transcription factor to repress E-cadherin transcription, decreasing cell-cell adhesion through E-cadherin loss[106,107]. As discussed, Akt activation also increases NF-κB transcriptional activity, which in turn increases Snail expression in epithelial cells[106]. Alternatively, it has also been proposed that 3%-5% of total cellular GSK3β is stably bound to Axin to form a complex reserved specifically for Wnt signaling[108]. One study conducted in prostate and breast cancer cell lines and C. elegans has shown that inhibition of PI3K by the PI3K inhibitor, wortmannin, does not affect GSK3β phosphorylation[108]. Thus, Wnt signaling by PI3K inhibition remains unchanged, refuting the common theory that there is cross-talk between the two pathways[108]. Instead, this evidence suggests that CRC presents with activating mutations in both the Wnt/β-catenin pathway and the PI3K/Akt pathway simultaneously, creating the notion that cross-talk between the two pathways occurs with a common GSK3β protein[108].
PTEN functions as a negative regulator of PI3K signaling by dephosphorylating the second messenger PIP3 to convert PIP3 back to PIP2[109,110]. PTEN exists in the cell as a cytoplasmic protein in an inactive, phosphorylated state[110]. Phosphorylation of PTEN serine and threonine residues stabilizes the protein in a closed state[110]. Upon activation, dephosphorylated PTEN contains an active phosphatase domain[110]. However, this active site leaves PTEN in an unstable conformation susceptible to proteasomal degradation[110]. In this way, the normal negative feedback loop of PI3K signaling and PTEN inhibition can proceed[110]. When active, PTEN is recruited to the plasma membrane where it binds to PIP3 and dephosphorylates the second messenger, inhibiting the downstream Akt signaling[110]. The loss of PTEN expression results in the accumulation of PIP3 at the plasma membrane, resulting in increased recruitment of Akt to the plasma membrane and increased Akt activation[80]. Because of this negative regulation of PI3K/Akt signaling, PTEN is associated with inhibition of cell cycle progression, induction of cell death, modulation of cell cycle arrest signals, and stimulation of angiogenesis[110].
PTEN mutations and loss of PTEN expression have been shown to occur in a high number of CRCs, with this loss correlating with tumor aggressiveness and invasiveness[109-111]. This correlation might be explained by the involvement of PTEN with maintaining normal cell polarity[109]. Loss of PTEN results in a loss of cell polarity, leading to increased epidermal-to-mesenchymal transition (EMT) of cancer cells and loss of tight junctions[109]. Similarly, reduced expression of PTEN and loss of PTEN are shown to indicate more advanced stages and metastasis of CRC[111]. Loss of PTEN occurs due to loss of chromosomal heterozygosity in CRC tumors with chromosomal instability and is estimated to occur in about 20%-40% of CRCs, while PTEN mutations in tumors without chromosomal instability occur much less frequently, in less than 5% of cases[80,81,110,111]. PTEN expression itself is regulated by peroxisome proliferator activated receptor γ (PPARγ) and p53 activity, both of which are implicated in CRC and will be discussed in further detail later in this review[110].
Due to PTEN interaction with the PI3K/Akt signaling pathway, it has been proposed that loss of PTEN expression and mutations in PIK3CA may work synergistically to increase the activity of both PI3K/Akt and Wnt/β-catenin signaling[79]. However, data obtained from the European Prospective Investigation of Cancer Norfolk Study showed that loss of PTEN expression and PIK3CA mutations occurred independently of one another in CRCs[81]. Further mechanistic studies involving CRC tumors supported these results and showed activating PIK3CA mutations to occur in about 30% of tumors, independent of PTEN loss[80].
As mentioned previously, there is cross-talk between the PI3K/Akt pathway and the Wnt/β-catenin pathway. Investigation into PIK3CA mutations in CRC revealed that in human CRC cells carrying APC mutations and showing constitutive Wnt pathway activation, PI3K inhibition led to no change in the subcellular localization of β-catenin[79]. Interestingly, although the nuclear localization of β-catenin was unaffected by PI3K inhibition, the concentration of β-catenin phosphorylated at the putative Akt serine 552 phosphorylation site was lower in cells in which PI3K activity was inhibited[79]. β-catenin/LEF/TCF-mediated gene transcription was also lower in the PI3K-inhibited cells, resulting in decreased expression of Wnt target genes c-Myc, cyclin D1, and LEF-1[79]. As a component of the β-catenin transcriptional complex, the decrease in LEF-1 expression indicates a further decrease in the transcriptional activity of β-catenin[79]. Taken together, these results demonstrate that the nuclear localization of β-catenin and its transcriptional activity are independent processes, but are linked by PI3K[79].
Interestingly, retinoid treatment in some cancer cell lines has been shown to upregulate the activity of the PI3K/Akt signaling pathway, increasing cell proliferation and invasion to promote tumor growth[112-114]. However, in other cancer cell lines, treatment with retinoids has been shown to inhibit PI3K/Akt signaling[115-118]. These retinoid effects have mostly been shown to be mediated through RAR-mediated pathways involving ATRA binding to receptors[115,116]. Specifically, ATRA has been shown to decrease the phosphorylation of GSK3β, decrease cellular proliferation, and increase the expression of pro-apoptotic proteins in human leiomyoma and myometrial cells[115]. In addition, CRBP-I inhibits PI3K/Akt activation in breast cancer cells through a RAR-mediated pathway by decreasing the heterodimerization of p85 and p110[116]. To our knowledge, our laboratory is the only laboratory to investigate retinoid inhibition of the PI3K/Akt signaling pathway in CRC. Furthermore, because retinoid receptor activity is often down-regulated in CRC, our laboratory studied the effects of retinol, the dietary form of vitamin A, on the PI3K/Akt signaling pathway in human CRC cells exhibiting ATRA-resistance[29]. We have shown that PI3K activity is inhibited by retinol in a dose-dependent manner independent of RAR signaling or inhibition of p85/p110 heterodimerization[29]. We recently showed that it is the ability of retinol to inhibit PI3K activity that confers the ability of vitamin A to decrease CRC cell invasion in vitro and metastasis in vivo[25]. Specifically, by comparing the effects of retinol treatment on parental HCT-116 cells, expressing one allele of constitutively active PI3K (caPI3K), to mutant HCT-116 cells expressing two alleles of caPI3K, we showed that retinol treatment decreased in vitro cell invasion in parental HCT-116 cells, but not in mutant HCT-116 cells[25]. Retinol treatment also decreased total MMP-9 protein levels and active MMP-9 levels in parental HCT-116 cells, while these levels remained unchanged in HCT-116 cells expressing two alleles of caPI3K[25]. Finally, dietary vitamin A supplementation tended to result in a lower incidence of hepatic metastases in mice intrasplenically injected with parental HCT-116 cells but not in mice intrasplenically injected with mutant HCT-116 cells.
More research is needed to determine the mechanism by which vitamin A inhibits PI3K activity in CRC, but one possible mechanism is by the up-regulation of PTEN. Although the effect of retinoids on PTEN activity has not been examined in CRC to our knowledge, retinoids have been shown to alter PTEN activity in smooth muscle cells, neuroblastoma and glioblastoma cells, promyelocytes, leukemia cells, fibroblasts, and breast, endometrial, and hepatocellular carcinoma cells[119-128]. In particular, ATRA treatment of breast cancer cells reduced the methylation of the PTEN gene promoter to activate PTEN transcription[122]. Suppression of growth factors by ATRA in hepatocellular carcinoma cells increases PTEN levels and synchronously decreases the presence of phosphorylated Akt[123]. Increases of PTEN and consequent decreases of Akt occur with retinoid treatment of neuroblastoma and glioblastoma cells and of smooth muscle cells as well[119,126,127]. By increasing PTEN, cellular proliferation is suppressed and apoptosis is induced, perhaps partially through the inhibition of NF-κB transcriptional activity[126,127]. Concurrent activation of PPARγ with retinoid treatment may also be helpful in synergistically reducing carcinogenesis, which will be discussed further in the following section.
CYCLOOXYGENASE-2 AND PEROXISOME PROLIFERATOR ACTIVATED RECEPTOR-γ
The use of non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin reduces the incidence of CRC and other cancers of the gastrointestinal (GI) tract[129,130]. Chronic NSAID use has been shown to reduce the risk of CRC by as much as 40%-50%, as well as decrease the multiplicity and size of tumors presenting with APC loss[131,132]. These drugs mediate their effects through inhibition of cyclooxygenase (COX) enzymes. COX-2 is an inducible enzyme expressed in the presence of inflammatory cytokines, growth factors, and tumor promoters[133]. In the presence of these factors, COX-2 converts free arachidonic acid to prostaglandin H2 (PGH2), which is the precursor to other prostaglandins, specifically prostaglandin E2 (PGE2)[133,134]. COX-2 over-expression is associated with more aggressive tumors of the GI tract and increased levels of COX-2 mRNA are present in 80%-90% of CRCs[134-136]. This over-expression of COX-2 results in the increased levels of PGE2. Elevated PGE2 is present in high levels in cancer tissues and increases the carcinogenic process by stimulating cell proliferation, suppressing apoptosis, increasing cell motility, and promoting angiogenesis[133,137,138]. The biological effects of PGE2 are mediated by E-prostanoid (EP) G-protein coupled receptor subtypes 1-4 which are present in high levels in CRCs[133,139]. The loss of these EP receptors is associated with decreased PGE2 signaling and decreased cancer malignancy[139]. It should be noted that carcinoma cells that do not display increased COX-2 expression may still receive paracrine signals by PGE2 through EP receptors and thus still exhibit the growth stimulatory effects of PGE2 as well as increased cell motility and activation of ERK signaling[140]. PGE2 binding to EP receptors results in increased phosphorylation of EGFR and the downstream mediator ERK, which induces the expression of c-FOS, a gene involved in promoting cell proliferation[133,140,141].
While activation of EGFR contributes to increased PI3K/Akt signaling, COX-2 over-expression also results in the dissociation of GSK3β from the β-catenin destruction complex, leading to the stabilization of β-catenin for translocation to the nucleus[142,143]. PGE2 treatment in human CRC cells led to rapid phosphorylation of GSK3β on its serine 9 residue by Akt, inhibiting the kinase activity of GSK3β[143]. This action was, however, dependent on the loss of APC function in CRC because β-catenin stabilization by PGE2 occurs downstream of APC loss[143]. Inhibition of PGE2 in zebrafish embryos and human CRC cells demonstrating APC loss increased the degradation of β-catenin, with COX-2 knockdown reducing the levels of β-catenin[144]. ATRA treatment of zebrafish embryos and human CRC cells decreased the levels of β-catenin by a mechanism that requires the attenuation of COX-2 expression and subsequent decrease in PGE2 accumulation[144]. β-catenin reduction as a result of ATRA treatment also led to the decreased expression of MMP-9[144]. Furthermore, PGE2 led to the increased expression of TCF-4, a component of the β-catenin transactivation complex, resulting in increased transcription of genes downstream of β-catenin[142]. PGE2 thus leads to the expression of cyclin D1 and vascular endothelial growth factor (VEGF) in vitro and in vivo, which contribute to the increased formation of intestinal polyps[142]. This effect by PGE2 is synergistically perpetuated by mutated β-catenin[142].
COX-2 over-expression in CRC is also correlated with an increased expression of MMP-2 and MMP-9, both of which contribute to CRC motility and metastasis[145]. Suppression of COX-2 by selective inhibitors in mouse CRC cells decreased proliferation associated with cyclin D1 and inhibited cell migration and motility with an associated decrease in both MMP-2 and MMP-9[135]. This suppression of COX-2 also decreased tumor growth both in vitro and in vivo, while also slowing liver metastasis[135]. This process may be particularly important when considering metastasis of CRC, as COX-2 expression has been shown to be even higher in metastatic liver tumors[135]. Broad spectrum MMP inhibitors decreased the number of adenomas in mice lacking APC function by decreasing proliferation, inhibiting angiogenesis, and stimulating apoptosis, with a synergistic effect seen when combined with COX-2 inhibitors[145].
Moreover, the lack of a functional APC protein is correlated with the elevated expression of COX-2[146]. APC controls ATRA biosynthesis through the activity of RDH enzymes in human CRC, with this loss of RDH correlating with the increased expression of COX-2[146]. In zebrafish embryos and human CRC cells presenting with a functional loss of APC, this over-expression of COX-2 was attenuated by treatment with ATRA[146]. This attenuation of COX-2 expression was the result of a mechanism involving ATRA inhibition of the levels of CCAAT/enhancer-binding protein (C/EBP) cis-acting elements, which are present in the promoter region of the COX-2 gene[146]. ATRA treatment decreased the expression of C/EBP-β, which leads to the decreased expression of COX-2[146].
The suppression of COX-2 by retinoids has been demonstrated in a variety of human epithelial carcinomas[147-150]. This suppression has been shown to be mediated by a multitude of factors, some of which have been described above, and which also includes a RARα-dependent pathway to limit the amount of CREB-binding protein (CBP)/p300 histone acetyltransferase activity available for AP-1 induction of COX-2[148]. In human CRC cells, treatment with the retinoid analogue fenretinide decreased COX-2 mRNA and inhibited PGE2 expression, resulting in inhibition of cell growth[151]. Therapy with the selective COX-2 inhibitor celecoxib enhanced the growth inhibitory effects of ATRA in both COX-2-high-expressing HT-29 human CRC cells and COX-2-low-expressing SW480 human CRC cells, resulting in increased apoptosis and elevated RARβ expression through COX-2-independent mechanisms[152]. RARβ2 methylation was inversely associated with COX-2 expression, with increased methylation of RARβ2 in CRC tumors also presenting with high COX-2 expression[153]. These tumors correlated with a worse patient prognosis, proposing the importance of both COX-2 and RARβ2 expression in colorectal carcinogenesis[153]. Overall, COX-2 is over-expressed in CRC tumors, leading to elevated PGE2 and β-catenin and the resulting cellular proliferation and tumor metastasis. Treatment with retinoids inhibits this over-expression of COX-2, suppressing the tumor growth-inducing effects of COX-2.
COX-2 expression is regulated in part by PPARγ. Specifically, the activation of PPARγ decreases COX-2 expression by up to 90% and induces caspase-3-dependent apoptosis in human CRC cells[154]. The COX-2 gene contains a peroxisome proliferator response element (PPRE) in its promoter, which allows the binding of PPARγ-RXRα heterodimers to inhibit COX-2 gene transcription[155,156]. PPARγ belongs to the nuclear hormone receptor superfamily of ligand-dependent transcription factors[157]. Ligands existing for PPARγ include prostaglandins, polyunsaturated fatty acids (PUFAs), NSAIDs, and thiazolidinediones (TZDs)[158]. TZDs are a class of PPARγ agonist medications, used in diabetic patients to regulate lipid and glucose metabolism via PPARγ activation[158,159]. Upon ligand binding, PPARγ changes conformation to release corepressor proteins and recruit coactivator proteins, such as PPARγ-coactivator-1 (PGC-1)[160]. PPARγ then forms an obligate heterodimer with RXRα, and the resulting heterodimer binds to PPREs in the promoter regions of target genes to regulate expression[156]. In CRC, mutations of PPARγ occur in about 8% of cases, indicating its potential role as a tumor suppressor[161]. Many studies in CRC cell lines and animal models have demonstrated this effect, with PPARγ activation resulting in growth inhibition, apoptotic cell death, and decreased cell invasion[155,162-165]. However, the opposite effect has been observed in mice lacking APC function, with PPARγ activation resulting in tumor promotion[166,167]. In rats fed a high-fat diet, PPARγ and RARβ mRNA expression was suppressed, concomitant with an increase in COX-2 and β-catenin levels and in the number of aberrant crypt foci (ACF)[168]. Supplementing diets with retinyl esters or ATRA attenuated the increases in COX-2 and β-catenin expression and inhibited the formation of ACF[168]. This data indicates that dietary factors, such as lipids and retinoids, are strongly influential in protein expression and tumor formation.
The mechanisms by which PPARγ act on tumor formation are still unknown, yet the evidence presented thus far suggests the importance of PPARγ in tumor growth inhibition. PPRE-independent mechanisms may also be involved, as PPARγ activation has also been shown to interfere with NF-κB and AP-1 to inhibit the transcription of pro-survival and growth amplification genes[157,158,169]. As mentioned, the activation of PPARγ by ligand binding results in the suppression of COX-2 expression in human CRC cells with an ensuing decrease in PGE2 accumulation[156,170]. Additionally, PPARγ agonists lead to a decrease in both MMP-2 and MMP-9 and an increase in TIMP-1 and TIMP-2[156,159]. Treatment with ATRA and synthetic RXR ligands synergistically enhanced this effect, which ultimately led to a decrease in cell proliferation, invasion, and an increase in apoptosis[156,171]. Treatment of HCT-15 cells with ATRA and the TZD rosiglitazone synergistically suppressed COX-2 and MMP-7 expression and induced cell cycle arrest and apoptosis[171]. The growth suppressing effects of PPARγ in CRC have been shown to occur by modulating the transcription of genes regulating cell cycle progression. Treatment of human CRC cells with PPARγ agonists induced apoptosis in cells by halting cell cycling progression and inhibiting the expression of genes such as cyclin D1 and c-Myc[157,158,172]. Adding synthetic RXR ligands to treatment with PPARγ agonists can augment cell growth inhibition and induce terminal differentiation by increasing the interaction of PPARγ and RXRα and their ability to form a heterodimer[169]. However, treatment of human CRC cells with RXR ligands alone does not cause PPARγ-RXRα heterodimer formation in the absence of PPARγ activation[156,172]. Therefore, dual treatment with synthetic rexinoid RXR ligands and PPARγ agonists may work together to inhibit the growth and metastasis of colonic tumors. As synthetic RXR ligands, rexinoids are not true retinoids. True retinoids bind RAR and are the focus of this review. Research regarding PPARγ and retinoids in CRC is lacking, as PPARγ only heterodimerizes with RXRα and not RAR. Yet, expression of RARβ mRNA can be induced by PPARγ activation in other cancers such as lung, breast, liver, and brain cancers[173-176]. ATRA alone and a combination of PPARγ and RXR ligands induced RARβ expression in ATRA-resistant breast cancer cells in the presence of HDAC inhibitors[175]. This induction of RARβ expression was reduced in the presence of a PPARγ antagonist, indicating the involvement of PPARγ/RXR heterodimer activity in RARβ transcription[175]. Treatment of breast and lung cancer cells with PPARγ and RXR ligands also induced apoptosis in these cells[175]. Apoptotic glioblastoma cells showed an increased level of RARβ expression when undergoing apoptosis, and PPARγ agonists induced RARβ mRNA in glioblastoma cells, suggesting that PPARγ activation may mediate apoptosis through RARβ activity[176]. Furthermore, treatment of leukemia cells with a combination of ATRA and the PPARγ agonist, ciglitazone, synergistically increased PTEN levels and inhibited the growth and proliferation of these cells by inducing cell cycle arrest[121]. Both 9-cis-RA and PPARγ activation in fibroblasts stimulated PTEN expression, which led to a decrease in Akt phosphorylation[128]. Because PTEN expression is regulated in part by PPARγ activation, PPARγ ligands have been shown to decrease proliferation of endometrial cancer cells via PTEN induction and the inhibition of VEGF secretion[120]. Taken together, this research proposes that retinoid treatment in conjunction with PPARγ activation may be helpful in overcoming ATRA-resistance, inhibiting tumor growth, and promoting cancer cell death in CRC.
P53/SIAH-1 SIGNALING
Mutations of the tumor suppressor gene p53 are the most common mutations found in human cancers, with p53 absence or mutations present in 50% of CRC cases[177,178]. As a tumor suppressor gene, p53 is activated in response to genotoxic stimuli in healthy cells, to which p53 responds by arresting cell cycle progression and inducing apoptosis[179]. In healthy cells, p53 suppression is necessary for normal growth and is thus present at low concentrations, its expression is regulated through ubiquitin-dependent degradation most notably by the ubiquitin ligase, MDM2[179]. MDM2 is phosphorylated by kinases such as Akt, after which the activated MDM2 localizes to the nucleus and ubiquinates p53[179]. The ubiquitinated p53 is then exported from the nucleus, where it is degraded in the cytosol to maintain cell proliferative activity[179]. Up-regulation of MDM2 activity and transcription also occurs downstream of other oncogenic pathways to inhibit p53 activity, such as ERK and K-ras signaling[179]. Similarly, MDM2 is a p53 target gene, creating a negative feedback loop to control p53 expression and activity[179]. In response to genotoxic damage, p53 is activated by kinases, which phosphorylate p53 in its MDM2 binding region, stabilizing p53 and allowing it to accumulate and bind to DNA to induce the transcription of genes such as cyclin kinase-dependent cell cycle inhibitor p21 and pro-apoptotic Bcl-2 associated x protein (BAX)[178-181]. P53 also directly inhibits anti-apoptotic proteins such as B-cell CLL/lymphoma-2 (Bcl-2) and Bcl-2 like isoform 1 (Bcl-xL), which inhibit the release of cytochrome c from the mitochondria to prevent the cell from initiating apoptosis[180]. Silencing of Bcl-2 in CRC cells leads to major p53-mediated apoptosis, demonstrating that Bcl-2 inhibits apoptosis in cells by also inhibiting p53 activity[180]. In CRC cells with mutant p53, transfection with wild-type p53 induces apoptosis and inhibits colony formation in vitro and inhibits tumor formation in vivo[182].
Missense mutations occur in 80% of all p53 mutations, resulting in a stable protein that accumulates inside the nucleus of tumor cells but lacks its specific DNA-binding activity and, therefore, lacks transcriptional activity[183]. As a result, an accumulation of p53 in the cell is generally thought to be mutagenic, although it is important to distinguish this mutant p53 accumulation in tumor cells from wild-type p53 expression[183]. The accumulation of mutant p53 in CRC patients is strongly correlated with increased metastasis and poor prognosis, further implicating the importance of p53 involvement in cell cycle regulation and stimulation of apoptosis in tumor cells[177]. Most p53 mutations occur in the later stages of adenoma-to-carcinoma progression, after which time many other pathways such as K-ras and the Wnt/β-catenin signaling pathway may already be dysregulated[184]. This point is particularly interesting to consider when looking at p53 involvement in β-catenin degradation. Siah-1 is a p53-inducible protein that binds ubiquitin-conjugating enzymes and targets proteins for degradation to ultimately result in tumor suppression[185]. Specifically, Siah-1 binds to the carboxyl terminus of APC and decreases β-catenin via a degradation pathway independent of GSK3β phosphorylation[185]. While Siah-1 does not affect APC levels, Siah-1 influence on β-catenin levels are dependent upon Siah-1 binding to APC[185]. In CRC cells with truncated APC, Siah-1 is unable to decrease β-catenin levels, making this process ineffective in cells expressing APC mutations[186]. Siah-1-mediated degradation of both mutant and wild-type β-catenin in CRC cells was supported by a decrease in TCF/LEF reporter activity and the consequent reduction of β-catenin target genes cyclin D1 and c-Myc to result in cell cycle arrest[185-187]. Increased p53 expression in CRC cells resulted in increased degradation of β-catenin and a decrease in TCF/LEF activity only in the presence of Siah-1, indicating that p53 degradation of β-catenin is dependent on Siah-1 activity[185,187]. Because Siah-1 expression is regulated by p53, the loss of p53 transcriptional activity inhibits Siah-1 expression and activity, preventing the p53/Siah-1 pathway activity to cause β-catenin degradation[187].
In addition to affecting retinoid metabolism and storage, retinoid treatment in many different cell types induces p53 mRNA and protein expression to inhibit cell cycle progression and promote apoptosis[188-193]. ATRA treatment of keratinocytes led to an increase in p53 mRNA and protein levels and a corresponding increase in caspase-3, 6, 7, and 9 enzyme levels, which are responsible for mediating apoptosis[188]. Apoptosis and growth inhibition of mammary carcinoma cells is controlled by RA-induced p53 activity increase, which in turn upregulates the expression of the anti-proliferative B-cell translocation gene, member 2 (Btg2)[191]. Btg2 inhibits cell cycle progression by down-regulating the expression of cyclin D1, and this effect is further augmented by the over-expression of CRABP-II, which transports RA to nuclear RAR, to induce the transcription of RA-responsive genes[191]. In murine embryonic stem cells, ATRA caused neural differentiation and apoptosis through increasing p53 mRNA and protein levels to instigate cell cycle arrest[189]. The up-regulation of p21 protein concentration is an important effect of p53 activation as shown in human mammary epithelial cells, of which treatment with 9-cis-RA, ATRA, and fenretinide increases p21 expression and thus, cell growth, in a p53-dependent manner[190]. Furthermore, p21 expression in breast cancer cells and HCT-116 CRC cells is increased by p53 interaction with the tumor suppressor activating enhancer-binding protein-2 α (AP-2α), a RA-inducible gene that regulates apoptosis, cell growth, and differentiation[192]. AP-2α interaction with p53 resulted in enhanced binding to the promoter of p21, which led to cell cycle arrest in these cells[192]. The induction of STRA6, the RBP receptor, by p53 has also been shown to mediate apoptosis in ovarian cancer cells, normal human fibroblasts, and HCT-116 cells expressing wild type p53[193]. Transfection of these with STRA6 increased apoptosis, and inhibition of STRA6 severely compromised p53-induced apoptosis[193]. While the effects of retinoids on p53 expression and activity have not been widely studied with regard to CRC, the known results are summarized in Table 1. In general, retinoid treatment of CRC cells appears to enhance the expression and activity of p53 to further increase tumor suppressor p21 levels, ultimately leading to cell cycle arrest and the initiation of apoptosis.
CONCLUSION
Retinoids decrease signaling via the major pathways that promote CRC progression. Ultimately, each pathway is followed to its conclusion, retinoids decrease levels of MMPs, cyclin D1, and other factors that induce cellular invasion or proliferation. Often, β-catenin is an intermediate in these pathways, reflecting the central role of β-catenin in CRC progression. Overall pathway interactions are illustrated in Figure 2, and effects of mutations on CRC progression and the effects of retinoids on these mutated proteins are summarized in Table 1. Because retinoids inhibit critical pathways to decrease CRC progression, dietary vitamin A supplementation or retinoid chemotherapy, alone or in combination with other medications, may prove beneficial for the prevention of the progression and metastasis of CRC.
Footnotes
Conflict-of-interest statement: Neither Catherine C Applegate nor Michelle A Lane have any conflicts of interest related to this manuscript. Neither author has received fees for serving as a speaker, a consultant, or an advisory board member. Michelle A Lane has research funding from Texas State University and the Heather Custer Memorial Fund. Both authors are employees of Texas State University. Michelle A Lane has a diversified stock portfolio as part of her retirement plan offered by her employer, Texas State University. These stocks/shares do not present a conflict of interest. Neither author owns a patent.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: April 24, 2015
First decision: June 1, 2015
Article in press: September 16, 2015
P- Reviewer: Panarelli NC, Sipos F S- Editor: Qiu S L- Editor: A E- Editor: Wu HL
References
- 1.American Cancer Society. Global Cancer Facts and Figures. 2nd ed. Atlanta: American Cancer Society; 2011. [Google Scholar]
- 2.International Agency for Research on Cancer. Colorectal cancer statistics World Cancer Research Fund International. GLOBOCAN. v1.0, Cancer Incidence and Mortality Worldwide. Atlanta: American Cancer Society; 2012. p. IARC CancerBase No. 11 [Internet]. Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Lyon: International Agency for Research on Cancer, 2014 [cited 2015 Jan 16]. Available from: http://globocan.iarc.fr. [Google Scholar]
- 3.Moon BS, Jeong WJ, Park J, Kim TI, Min do S, Choi KY. Role of oncogenic K-ras in cancer stem cell activation by aberrant Wnt/β-catenin signaling. J Natl Cancer Inst. 2014;106:djt373. doi: 10.1093/jnci/djt373. [DOI] [PubMed] [Google Scholar]
- 4.American Cancer Society. Colorectal Cancer Facts and Figures 2014-2016. Atlanta: American Cancer Society; 2014. [Google Scholar]
- 5.Das BC, Thapa P, Karki R, Das S, Mahapatra S, Liu TC, Torregroza I, Wallace DP, Kambhampati S, Van Veldhuizen P, et al. Retinoic acid signaling pathways in development and diseases. Bioorg Med Chem. 2014;22:673–683. doi: 10.1016/j.bmc.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harrison EH. Mechanisms of digestion and absorption of dietary vitamin A. Annu Rev Nutr. 2005;25:87–103. doi: 10.1146/annurev.nutr.25.050304.092614. [DOI] [PubMed] [Google Scholar]
- 7.Reboul E. Absorption of vitamin A and carotenoids by the enterocyte: focus on transport proteins. Nutrients. 2013;5:3563–3581. doi: 10.3390/nu5093563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bushue N, Wan YJ. Retinoid pathway and cancer therapeutics. Adv Drug Deliv Rev. 2010;62:1285–1298. doi: 10.1016/j.addr.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alizadeh F, Bolhassani A, Khavari A, Bathaie SZ, Naji T, Bidgoli SA. Retinoids and their biological effects against cancer. Int Immunopharmacol. 2014;18:43–49. doi: 10.1016/j.intimp.2013.10.027. [DOI] [PubMed] [Google Scholar]
- 10.Harrison EH. Mechanisms involved in the intestinal absorption of dietary vitamin A and provitamin A carotenoids. Biochim Biophys Acta. 2012;1821:70–77. doi: 10.1016/j.bbalip.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noy N. Signaling by retinol and its serum binding protein. Prostaglandins Leukot Essent Fatty Acids. 2015;93:3–7. doi: 10.1016/j.plefa.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laursen KB, Kashyap V, Scandura J, Gudas LJ. An alternative retinoic acid-responsive Stra6 promoter regulated in response to retinol deficiency. J Biol Chem. 2015;290:4356–4366. doi: 10.1074/jbc.M114.613968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amengual J, Zhang N, Kemerer M, Maeda T, Palczewski K, Von Lintig J. STRA6 is critical for cellular vitamin A uptake and homeostasis. Hum Mol Genet. 2014;23:5402–5417. doi: 10.1093/hmg/ddu258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berry DC, Jacobs H, Marwarha G, Gely-Pernot A, O’Byrne SM, DeSantis D, Klopfenstein M, Feret B, Dennefeld C, Blaner WS, et al. The STRA6 receptor is essential for retinol-binding protein-induced insulin resistance but not for maintaining vitamin A homeostasis in tissues other than the eye. J Biol Chem. 2013;288:24528–24539. doi: 10.1074/jbc.M113.484014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amann PM, Eichmüller SB, Schmidt J, Bazhin AV. Regulation of gene expression by retinoids. Curr Med Chem. 2011;18:1405–1412. doi: 10.2174/092986711795029618. [DOI] [PubMed] [Google Scholar]
- 16.Parrado A, Despouy G, Kraïba R, Le Pogam C, Dupas S, Choquette M, Robledo M, Larghero J, Bui H, Le Gall I, et al. Retinoic acid receptor alpha1 variants, RARalpha1DeltaB and RARalpha1DeltaBC, define a new class of nuclear receptor isoforms. Nucleic Acids Res. 2001;29:4901–4908. doi: 10.1093/nar/29.24.4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.di Masi A, Leboffe L, De Marinis E, Pagano F, Cicconi L, Rochette-Egly C, Lo-Coco F, Ascenzi P, Nervi C. Retinoic acid receptors: from molecular mechanisms to cancer therapy. Mol Aspects Med. 2015;41:1–115. doi: 10.1016/j.mam.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 18.Al Tanoury Z, Piskunov A, Rochette-Egly C. Vitamin A and retinoid signaling: genomic and nongenomic effects. J Lipid Res. 2013;54:1761–1775. doi: 10.1194/jlr.R030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolf G. Is 9-cis-retinoic acid the endogenous ligand for the retinoic acid-X receptor? Nutr Rev. 2006;64:532–538. doi: 10.1111/j.1753-4887.2006.tb00186.x. [DOI] [PubMed] [Google Scholar]
- 20.Kane MA. Analysis, occurrence, and function of 9-cis-retinoic acid. Biochim Biophys Acta. 2012;1821:10–20. doi: 10.1016/j.bbalip.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 21.Soprano DR, Qin P, Soprano KJ. Retinoic acid receptors and cancers. Annu Rev Nutr. 2004;24:201–221. doi: 10.1146/annurev.nutr.24.012003.132407. [DOI] [PubMed] [Google Scholar]
- 22.le Maire A, Bourguet W. Retinoic acid receptors: structural basis for coregulator interaction and exchange. Subcell Biochem. 2014;70:37–54. doi: 10.1007/978-94-017-9050-5_3. [DOI] [PubMed] [Google Scholar]
- 23.Dillard AC, Lane MA. Retinol decreases beta-catenin protein levels in retinoic acid-resistant colon cancer cell lines. Mol Carcinog. 2007;46:315–329. doi: 10.1002/mc.20280. [DOI] [PubMed] [Google Scholar]
- 24.Dillard AC, Lane MA. Retinol Increases beta-catenin-RXRalpha binding leading to the increased proteasomal degradation of beta-catenin and RXRalpha. Nutr Cancer. 2008;60:97–108. doi: 10.1080/01635580701586754. [DOI] [PubMed] [Google Scholar]
- 25.Lengyel JN, Park EY, Brunson AR, Pinali D, Lane MA. Phosphatidylinositol 3-kinase mediates the ability of retinol to decrease colorectal cancer cell invasion. Nutr Cancer. 2014;66:1352–1361. doi: 10.1080/01635581.2014.956258. [DOI] [PubMed] [Google Scholar]
- 26.Park EY, Dillard A, Williams EA, Wilder ET, Pepper MR, Lane MA. Retinol inhibits the growth of all-trans-retinoic acid-sensitive and all-trans-retinoic acid-resistant colon cancer cells through a retinoic acid receptor-independent mechanism. Cancer Res. 2005;65:9923–9933. doi: 10.1158/0008-5472.CAN-05-1604. [DOI] [PubMed] [Google Scholar]
- 27.Park EY, Pinali D, Lindley K, Lane MA. Hepatic vitamin A preloading reduces colorectal cancer metastatic multiplicity in a mouse xenograft model. Nutr Cancer. 2012;64:732–740. doi: 10.1080/01635581.2012.687425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park EY, Wilder ET, Chipuk JE, Lane MA. Retinol decreases phosphatidylinositol 3-kinase activity in colon cancer cells. Mol Carcinog. 2008;47:264–274. doi: 10.1002/mc.20381. [DOI] [PubMed] [Google Scholar]
- 29.Park EY, Wilder ET, Lane MA. Retinol inhibits the invasion of retinoic acid-resistant colon cancer cells in vitro and decreases matrix metalloproteinase mRNA, protein, and activity levels. Nutr Cancer. 2007;57:66–77. doi: 10.1080/01635580701268238. [DOI] [PubMed] [Google Scholar]
- 30.Ricci-Vitiani L, Fabrizi E, Palio E, De Maria R. Colon cancer stem cells. J Mol Med (Berl) 2009;87:1097–1104. doi: 10.1007/s00109-009-0518-4. [DOI] [PubMed] [Google Scholar]
- 31.Fredericks E. Colorectal carcinogenesis: Molecular aspects. South African Gastroen Rev. 2013;11:11–18. [Google Scholar]
- 32.Lee MO, Han SY, Jiang S, Park JH, Kim SJ. Differential effects of retinoic acid on growth and apoptosis in human colon cancer cell lines associated with the induction of retinoic acid receptor beta. Biochem Pharmacol. 2000;59:485–496. doi: 10.1016/s0006-2952(99)00355-x. [DOI] [PubMed] [Google Scholar]
- 33.Xu XC. Tumor-suppressive activity of retinoic acid receptor-beta in cancer. Cancer Lett. 2007;253:14–24. doi: 10.1016/j.canlet.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lotan R, Xu X-C, Lippman SM, Ro JY, Lee JS, Lee JJ, Hong WK. Suppression of retinoic acid receptor-beta in premalignant oral lesions and its up-regulation by isotretinoin. N Engl J Med. 1995;(21):1405. doi: 10.1056/NEJM199505253322103. [DOI] [PubMed] [Google Scholar]
- 35.Lai ZL, Tsou YA, Fan SR, Tsai MH, Chen HL, Chang NW, Cheng JC, Chen CM. Methylation-associated gene silencing of RARB in areca carcinogens induced mouse oral squamous cell carcinoma. Biomed Res Int. 2014;2014:378358. doi: 10.1155/2014/378358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schenk T, Stengel S, Zelent A. Unlocking the potential of retinoic acid in anticancer therapy. Br J Cancer. 2014;111:2039–2045. doi: 10.1038/bjc.2014.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urvalek A, Laursen KB, Gudas LJ. The roles of retinoic acid and retinoic acid receptors in inducing epigenetic changes. Subcell Biochem. 2014;70:129–149. doi: 10.1007/978-94-017-9050-5_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mongan NP, Gudas LJ. Diverse actions of retinoid receptors in cancer prevention and treatment. Differentiation. 2007;75:853–870. doi: 10.1111/j.1432-0436.2007.00206.x. [DOI] [PubMed] [Google Scholar]
- 39.Moison C, Senamaud-Beaufort C, Fourrière L, Champion C, Ceccaldi A, Lacomme S, Daunay A, Tost J, Arimondo PB, Guieysse-Peugeot AL. DNA methylation associated with polycomb repression in retinoic acid receptor β silencing. FASEB J. 2013;27:1468–1478. doi: 10.1096/fj.12-210971. [DOI] [PubMed] [Google Scholar]
- 40.Nicke B, Riecken EO, Rosewicz S. Induction of retinoic acid receptor beta mediates growth inhibition in retinoid resistant human colon carcinoma cells. Gut. 1999;45:51–57. doi: 10.1136/gut.45.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kropotova ES, Zinovieva OL, Zyryanova AF, Dybovaya VI, Prasolov VS, Beresten SF, Oparina NY, Mashkova TD. Altered expression of multiple genes involved in retinoic acid biosynthesis in human colorectal cancer. Pathol Oncol Res. 2014;20:707–717. doi: 10.1007/s12253-014-9751-4. [DOI] [PubMed] [Google Scholar]
- 42.Jette C, Peterson PW, Sandoval IT, Manos EJ, Hadley E, Ireland CM, Jones DA. The tumor suppressor adenomatous polyposis coli and caudal related homeodomain protein regulate expression of retinol dehydrogenase L. J Biol Chem. 2004;279:34397–34405. doi: 10.1074/jbc.M314021200. [DOI] [PubMed] [Google Scholar]
- 43.Shelton DN, Sandoval IT, Eisinger A, Chidester S, Ratnayake A, Ireland CM, Jones DA. Up-regulation of CYP26A1 in adenomatous polyposis coli-deficient vertebrates via a WNT-dependent mechanism: implications for intestinal cell differentiation and colon tumor development. Cancer Res. 2006;66:7571–7577. doi: 10.1158/0008-5472.CAN-06-1067. [DOI] [PubMed] [Google Scholar]
- 44.Bordonaro M, Mariadason JM, Aslam F, Heerdt BG, Augenlicht LH. Butyrate-induced apoptotic cascade in colonic carcinoma cells: modulation of the beta-catenin-Tcf pathway and concordance with effects of sulindac and trichostatin A but not curcumin. Cell Growth Differ. 1999;10:713–720. [PubMed] [Google Scholar]
- 45.Nadauld LD, Chidester S, Shelton DN, Rai K, Broadbent T, Sandoval IT, Peterson PW, Manos EJ, Ireland CM, Yost HJ, et al. Dual roles for adenomatous polyposis coli in regulating retinoic acid biosynthesis and Wnt during ocular development. Proc Natl Acad Sci USA. 2006;103:13409–13414. doi: 10.1073/pnas.0601634103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baltes S, Nau H, Lampen A. All-trans retinoic acid enhances differentiation and influences permeability of intestinal Caco-2 cells under serum-free conditions. Dev Growth Differ. 2004;46:503–514. doi: 10.1111/j.1440-169x.2004.00765.x. [DOI] [PubMed] [Google Scholar]
- 47.Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, Broadbent T, Sarkar S, Burt RW, Jones DA. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell. 2009;137:623–634. doi: 10.1016/j.cell.2009.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Møllersen L, Paulsen JE, Olstørn HB, Knutsen HK, Alexander J. Dietary retinoic acid supplementation stimulates intestinal tumour formation and growth in multiple intestinal neoplasia (Min)/+ mice. Carcinogenesis. 2004;25:149–153. doi: 10.1093/carcin/bgg176. [DOI] [PubMed] [Google Scholar]
- 49.Cheng YW, Pincas H, Huang J, Zachariah E, Zeng Z, Notterman DA, Paty P, Barany F. High incidence of LRAT promoter hypermethylation in colorectal cancer correlates with tumor stage. Med Oncol. 2014;31:254. doi: 10.1007/s12032-014-0254-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hassel JC, Amann PM, Schadendorf D, Eichmüller SB, Nagler M, Bazhin AV. Lecithin retinol acyltransferase as a potential prognostic marker for malignant melanoma. Exp Dermatol. 2013;22:757–759. doi: 10.1111/exd.12236. [DOI] [PubMed] [Google Scholar]
- 51.Amann PM, Luo C, Owen RW, Hofmann C, Freudenberger M, Schadendorf D, Eichmüller SB, Bazhin AV. Vitamin A metabolism in benign and malignant melanocytic skin cells: importance of lecithin/retinol acyltransferase and RPE65. J Cell Physiol. 2012;227:718–728. doi: 10.1002/jcp.22779. [DOI] [PubMed] [Google Scholar]
- 52.Shirakami Y, Gottesman ME, Blaner WS. Diethylnitrosamine-induced hepatocarcinogenesis is suppressed in lecithin: retinol acyltransferase-deficient mice primarily through retinoid actions immediately after carcinogen administration. Carcinogenesis. 2012;33:268–274. doi: 10.1093/carcin/bgr275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cerignoli F, Guo X, Cardinali B, Rinaldi C, Casaletto J, Frati L, Screpanti I, Gudas LJ, Gulino A, Thiele CJ, et al. retSDR1, a short-chain retinol dehydrogenase/reductase, is retinoic acid-inducible and frequently deleted in human neuroblastoma cell lines. Cancer Res. 2002;62:1196–1204. [PubMed] [Google Scholar]
- 54.Kirschner RD, Rother K, Müller GA, Engeland K. The retinal dehydrogenase/reductase retSDR1/DHRS3 gene is activated by p53 and p63 but not by mutants derived from tumors or EEC/ADULT malformation syndromes. Cell Cycle. 2010;9:2177–2188. doi: 10.4161/cc.9.11.11844. [DOI] [PubMed] [Google Scholar]
- 55.Zhou H, van Bokhoven H. Regulation of vitamin metabolism by p53 and p63 in development and cancer. Cell Cycle. 2010;9:2709. [PubMed] [Google Scholar]
- 56.Voloshanenko O, Erdmann G, Dubash TD, Augustin I, Metzig M, Moffa G, Hundsrucker C, Kerr G, Sandmann T, Anchang B, et al. Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat Commun. 2013;4:2610. doi: 10.1038/ncomms3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Burgess AW, Faux MC, Layton MJ, Ramsay RG. Wnt signaling and colon tumorigenesis--a view from the periphery. Exp Cell Res. 2011;317:2748–2758. doi: 10.1016/j.yexcr.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 58.Ilyas M, Tomlinson IP. The interactions of APC, E-cadherin and beta-catenin in tumour development and progression. J Pathol. 1997;182:128–137. doi: 10.1002/(SICI)1096-9896(199706)182:2<128::AID-PATH839>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 59.Pellón-Cárdenas O, Schweitzer J, D’Souza-Schorey C. Endocytic trafficking and Wnt/β-catenin signaling. Curr Drug Targets. 2011;12:1216–1222. doi: 10.2174/138945011795906552. [DOI] [PubMed] [Google Scholar]
- 60.Zeller E, Hammer K, Kirschnick M, Braeuning A. Mechanisms of RAS/β-catenin interactions. Arch Toxicol. 2013;87:611–632. doi: 10.1007/s00204-013-1035-3. [DOI] [PubMed] [Google Scholar]
- 61.Wu WKK, Wang XJ, Cheng ASL, Luo MXM, Ng SSM, To KF, Chan FKL, Cho CH, Sung JJY, Yu J. Dysregulation and crosstalk of cellular signaling pathways in colon carcinogenesis. Crit Rev Oncol Hematol. 2013;86:251–277. doi: 10.1016/j.critrevonc.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 62.Easwaran V, Pishvaian M, Byers S, Byers S. Cross-regulation of B-catenin-LEF/TCF and retinoid signaling pathways. Curr Biol. 1999;9:1415–1418. doi: 10.1016/s0960-9822(00)80088-3. [DOI] [PubMed] [Google Scholar]
- 63.Huang GL, Luo Q, Rui G, Zhang W, Zhang QY, Chen QX, Shen DY. Oncogenic activity of retinoic acid receptor γ is exhibited through activation of the Akt/NF-κB and Wnt/β-catenin pathways in cholangiocarcinoma. Mol Cell Biol. 2013;33:3416–3425. doi: 10.1128/MCB.00384-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jeong WJ, Yoon J, Park JC, Lee SH, Lee SH, Kaduwal S, Kim H, Yoon JB, Choi KY. Ras stabilization through aberrant activation of Wnt/β-catenin signaling promotes intestinal tumorigenesis. Sci Signal. 2012;5:ra30. doi: 10.1126/scisignal.2002242. [DOI] [PubMed] [Google Scholar]
- 65.Raskov H, Pommergaard HC, Burcharth J, Rosenberg J. Colorectal carcinogenesis--update and perspectives. World J Gastroenterol. 2014;20:18151–18164. doi: 10.3748/wjg.v20.i48.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sancho E, Batlle E, Clevers H. Signaling pathways in intestinal development and cancer. Annu Rev Cell Dev Biol. 2004;20:695–723. doi: 10.1146/annurev.cellbio.20.010403.092805. [DOI] [PubMed] [Google Scholar]
- 67.Goel S, Huang J, Klampfer L. K-ras, intestinal homeostasis and colon cancer. Curr Clin Pharmacol. 2015;10:73–81. doi: 10.2174/1574884708666131111204440. [DOI] [PubMed] [Google Scholar]
- 68.Narahara H, Tatsuta M, Iishi H, Baba M, Uedo N, Sakai N, Yano H, Ishiguro S. K-ras point mutation is associated with enhancement by deoxycholic acid of colon carcinogenesis induced by azoxymethane, but not with its attenuation by all-trans-retinoic acid. Int J Cancer. 2000;88:157–161. doi: 10.1002/1097-0215(20001015)88:2<157::aid-ijc2>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 69.Xiao JH, Ghosn C, Hinchman C, Forbes C, Wang J, Snider N, Cordrey A, Zhao Y, Chandraratna RA. Adenomatous polyposis coli (APC)-independent regulation of beta-catenin degradation via a retinoid X receptor-mediated pathway. J Biol Chem. 2003;278:29954–29962. doi: 10.1074/jbc.M304761200. [DOI] [PubMed] [Google Scholar]
- 70.Han A, Tong C, Hu D, Bi X, Yang W. A direct protein-protein interaction is involved in the suppression of beta-catenin transcription by retinoid X receptor alpha in colorectal cancer cells. Cancer Biol Ther. 2008;7:454–459. doi: 10.4161/cbt.7.3.5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang F, Meng F, Li H, Dong Y, Yang W, Han A. Suppression of retinoid X receptor alpha and aberrant β-catenin expression significantly associates with progression of colorectal carcinoma. Eur J Cancer. 2011;47:2060–2067. doi: 10.1016/j.ejca.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 72.Benelli R, Monteghirfo S, Venè R, Tosetti F, Ferrari N. The chemopreventive retinoid 4HPR impairs prostate cancer cell migration and invasion by interfering with FAK/AKT/GSK3beta pathway and beta-catenin stability. Mol Cancer. 2010;9:142. doi: 10.1186/1476-4598-9-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lim YC, Kang HJ, Kim YS, Choi EC. All-trans-retinoic acid inhibits growth of head and neck cancer stem cells by suppression of Wnt/β-catenin pathway. Eur J Cancer. 2012;48:3310–3318. doi: 10.1016/j.ejca.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 74.Xu L, Zhang Y, Wang H, Zhang G, Ding Y, Zhao L. Tumor suppressor miR-1 restrains epithelial-mesenchymal transition and metastasis of colorectal carcinoma via the MAPK and PI3K/AKT pathway. J Transl Med. 2014;12:244. doi: 10.1186/s12967-014-0244-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kobayashi M, Nagata S, Iwasaki T, Yanagihara K, Saitoh I, Karouji Y, Ihara S, Fukui Y. Dedifferentiation of adenocarcinomas by activation of phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1999;96:4874–4879. doi: 10.1073/pnas.96.9.4874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen J, Shao R, Li L, Xu ZP, Gu W. Effective inhibition of colon cancer cell growth with MgAl-layered double hydroxide (LDH) loaded 5-FU and PI3K/mTOR dual inhibitor BEZ-235 through apoptotic pathways. Int J Nanomedicine. 2014;9:3403–3411. doi: 10.2147/IJN.S61633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Danielsen SA, Eide PW, Nesbakken A, Guren T, Leithe E, Lothe RA. Portrait of the PI3K/AKT pathway in colorectal cancer. Biochim Biophys Acta. 2015;1855:104–121. doi: 10.1016/j.bbcan.2014.09.008. [DOI] [PubMed] [Google Scholar]
- 78.Bauer TM, Patel MR, Infante JR. Targeting PI3 kinase in cancer. Pharmacol Ther. 2015;146:53–60. doi: 10.1016/j.pharmthera.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 79.Ormanns S, Neumann J, Horst D, Kirchner T, Jung A. WNT signaling and distant metastasis in colon cancer through transcriptional activity of nuclear β-catenin depend on active PI3K signaling. Oncotarget. 2014;5:2999–3011. doi: 10.18632/oncotarget.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ihle NT, Powis G, Kopetz S. PI-3-Kinase inhibitors in colorectal cancer. Curr Cancer Drug Targets. 2011;11:190–198. doi: 10.2174/156800911794328448. [DOI] [PubMed] [Google Scholar]
- 81.Naguib A, Cooke JC, Happerfield L, Kerr L, Gay LJ, Luben RN, Ball RY, Mitrou PN, McTaggart A, Arends MJ. Alterations in PTEN and PIK3CA in colorectal cancers in the EPIC Norfolk study: associations with clinicopathological and dietary factors. BMC Cancer. 2011;11:123. doi: 10.1186/1471-2407-11-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 83.Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012;4:a011189. doi: 10.1101/cshperspect.a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Saji M, Ringel MD. The PI3K-Akt-mTOR pathway in initiation and progression of thyroid tumors. Mol Cell Endocrinol. 2010;321:20–28. doi: 10.1016/j.mce.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 86.Setia S, Nehru B, Sanyal SN. The PI3K/Akt pathway in colitis associated colon cancer and its chemoprevention with celecoxib, a Cox-2 selective inhibitor. Biomed Pharmacother. 2014;68:721–727. doi: 10.1016/j.biopha.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 87.Kim D, Kim S, Koh H, Yoon SO, Chung AS, Cho KS, Chung J. Akt/PKB promotes cancer cell invasion via increased motility and metalloproteinase production. FASEB J. 2001;15:1953–1962. doi: 10.1096/fj.01-0198com. [DOI] [PubMed] [Google Scholar]
- 88.Itoh N, Semba S, Ito M, Takeda H, Kawata S, Yamakawa M. Phosphorylation of Akt/PKB is required for suppression of cancer cell apoptosis and tumor progression in human colorectal carcinoma. Cancer. 2002;94:3127–3134. doi: 10.1002/cncr.10591. [DOI] [PubMed] [Google Scholar]
- 89.Cheng JC, Chou CH, Kuo ML, Hsieh CY. Radiation-enhanced hepatocellular carcinoma cell invasion with MMP-9 expression through PI3K/Akt/NF-kappaB signal transduction pathway. Oncogene. 2006;25:7009–7018. doi: 10.1038/sj.onc.1209706. [DOI] [PubMed] [Google Scholar]
- 90.Qiu Q, Yang M, Tsang BK, Gruslin A. EGF-induced trophoblast secretion of MMP-9 and TIMP-1 involves activation of both PI3K and MAPK signalling pathways. Reproduction. 2004;128:355–363. doi: 10.1530/rep.1.00234. [DOI] [PubMed] [Google Scholar]
- 91.Wilson CL, Heppner KJ, Labosky PA, Hogan BL, Matrisian LM. Intestinal tumorigenesis is suppressed in mice lacking the metalloproteinase matrilysin. Proc Natl Acad Sci USA. 1997;94:1402–1407. doi: 10.1073/pnas.94.4.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen JS, Wang Q, Fu XH, Huang XH, Chen XL, Cao LQ, Chen LZ, Tan HX, Li W, Bi J, et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol Res. 2009;39:177–186. doi: 10.1111/j.1872-034X.2008.00449.x. [DOI] [PubMed] [Google Scholar]
- 93.Heslin MJ, Yan J, Johnson MR, Weiss H, Diasio RB, Urist MM. Role of matrix metalloproteinases in colorectal carcinogenesis. Ann Surg. 2001;233:786–792. doi: 10.1097/00000658-200106000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 95.Hwang YP, Yun HJ, Kim HG, Han EH, Lee GW, Jeong HG. Suppression of PMA-induced tumor cell invasion by dihydroartemisinin via inhibition of PKCalpha/Raf/MAPKs and NF-kappaB/AP-1-dependent mechanisms. Biochem Pharmacol. 2010;79:1714–1726. doi: 10.1016/j.bcp.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 96.Hornebeck W, Lambert E, Petitfrère E, Bernard P. Beneficial and detrimental influences of tissue inhibitor of metalloproteinase-1 (TIMP-1) in tumor progression. Biochimie. 2005;87:377–383. doi: 10.1016/j.biochi.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 97.Liu H, Zang C, Fenner MH, Possinger K, Elstner E. PPARgamma ligands and ATRA inhibit the invasion of human breast cancer cells in vitro. Breast Cancer Res Treat. 2003;79:63–74. doi: 10.1023/a:1023366117157. [DOI] [PubMed] [Google Scholar]
- 98.Lateef H, Stevens MJ, Varani J. All-trans-retinoic acid suppresses matrix metalloproteinase activity and increases collagen synthesis in diabetic human skin in organ culture. Am J Pathol. 2004;165:167–174. doi: 10.1016/S0002-9440(10)63285-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Benbow U, Schoenermark MP, Mitchell TI, Rutter JL, Shimokawa K, Nagase H, Brinckerhoff CE. A novel host/tumor cell interaction activates matrix metalloproteinase 1 and mediates invasion through type I collagen. J Biol Chem. 1999;274:25371–25378. doi: 10.1074/jbc.274.36.25371. [DOI] [PubMed] [Google Scholar]
- 100.Nwankwo JO. Anti-metastatic activities of all-trans retinoic acid, indole-3-carbinol and (+)-catechin in Dunning rat invasive prostate adenocarcinoma cells. Anticancer Res. 2002;22:4129–4135. [PubMed] [Google Scholar]
- 101.Andela VB, Rosier RN. The proteosome inhibitor MG132 attenuates retinoic acid receptor trans-activation and enhances trans-repression of nuclear factor kappaB. Potential relevance to chemo-preventive interventions with retinoids. Mol Cancer. 2004;3:8. doi: 10.1186/1476-4598-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shah S, Pishvaian MJ, Easwaran V, Brown PH, Byers SW. The role of cadherin, beta-catenin, and AP-1 in retinoid-regulated carcinoma cell differentiation and proliferation. J Biol Chem. 2002;277:25313–25322. doi: 10.1074/jbc.M203158200. [DOI] [PubMed] [Google Scholar]
- 103.Shah S, Hecht A, Pestell R, Byers SW. Trans-repression of beta-catenin activity by nuclear receptors. J Biol Chem. 2003;278:48137–48145. doi: 10.1074/jbc.M307154200. [DOI] [PubMed] [Google Scholar]
- 104.Um SJ, Han HS, Kwon YJ, Park SH, Rho YS, Sin HS, Park JS. Novel retinoic acid derivative ABPN has potent inhibitory activity on cell growth and apoptosis in cancer cells. Int J Cancer. 2003;107:1038–1046. doi: 10.1002/ijc.11489. [DOI] [PubMed] [Google Scholar]
- 105.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol. 2005;168:29–33. doi: 10.1083/jcb.200409067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Qiao M, Sheng S, Pardee AB. Metastasis and AKT activation. Cell Cycle. 2008;7:2991–2996. doi: 10.4161/cc.7.19.6784. [DOI] [PubMed] [Google Scholar]
- 108.Ng SS, Mahmoudi T, Danenberg E, Bejaoui I, de Lau W, Korswagen HC, Schutte M, Clevers H. Phosphatidylinositol 3-kinase signaling does not activate the wnt cascade. J Biol Chem. 2009;284:35308–35313. doi: 10.1074/jbc.M109.078261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Langlois MJ, Bergeron S, Bernatchez G, Boudreau F, Saucier C, Perreault N, Carrier JC, Rivard N. The PTEN phosphatase controls intestinal epithelial cell polarity and barrier function: role in colorectal cancer progression. PLoS One. 2010;5:e15742. doi: 10.1371/journal.pone.0015742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Molinari F, Frattini M. Functions and Regulation of the PTEN Gene in Colorectal Cancer. Front Oncol. 2013;3:326. doi: 10.3389/fonc.2013.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Waniczek D, Śnietura M, Młynarczyk-Liszka J, Pigłowski W, Kopeć A, Lange D, Rudzki M, Arendt J. PTEN expression profiles in colorectal adenocarcinoma and its precancerous lesions. Pol J Pathol. 2013;64:15–20. doi: 10.5114/pjp.2013.34598. [DOI] [PubMed] [Google Scholar]
- 112.García-Regalado A, Vargas M, García-Carrancá A, Aréchaga-Ocampo E, González-De la Rosa CH. Activation of Akt pathway by transcription-independent mechanisms of retinoic acid promotes survival and invasion in lung cancer cells. Mol Cancer. 2013;12:44. doi: 10.1186/1476-4598-12-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Uruno A, Sugawara A, Kanatsuka H, Kagechika H, Saito A, Sato K, Kudo M, Takeuchi K, Ito S. Upregulation of nitric oxide production in vascular endothelial cells by all-trans retinoic acid through the phosphoinositide 3-kinase/Akt pathway. Circulation. 2005;112:727–736. doi: 10.1161/CIRCULATIONAHA.104.500959. [DOI] [PubMed] [Google Scholar]
- 114.López-Carballo G, Moreno L, Masiá S, Pérez P, Barettino D. Activation of the phosphatidylinositol 3-kinase/Akt signaling pathway by retinoic acid is required for neural differentiation of SH-SY5Y human neuroblastoma cells. J Biol Chem. 2002;277:25297–25304. doi: 10.1074/jbc.M201869200. [DOI] [PubMed] [Google Scholar]
- 115.Ben-Sasson H, Ben-Meir A, Shushan A, Karra L, Rojansky N, Klein BY, Levitzki R, Ben-Bassat H. All-trans-retinoic acid mediates changes in PI3K and retinoic acid signaling proteins of leiomyomas. Fertil Steril. 2011;95:2080–2086. doi: 10.1016/j.fertnstert.2011.01.155. [DOI] [PubMed] [Google Scholar]
- 116.Farias EF, Marzan C, Mira-y-Lopez R. Cellular retinol-binding protein-I inhibits PI3K/Akt signaling through a retinoic acid receptor-dependent mechanism that regulates p85-p110 heterodimerization. Oncogene. 2005;24:1598–1606. doi: 10.1038/sj.onc.1208347. [DOI] [PubMed] [Google Scholar]
- 117.So PL, Wang GY, Wang K, Chuang M, Chiueh VC, Kenny PA, Epstein EH. PI3K-AKT signaling is a downstream effector of retinoid prevention of murine basal cell carcinogenesis. Cancer Prev Res (Phila) 2014;7:407–417. doi: 10.1158/1940-6207.CAPR-13-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Baba A, Shimizu M, Ohno T, Shirakami Y, Kubota M, Kochi T, Terakura D, Tsurumi H, Moriwaki H. Synergistic growth inhibition by acyclic retinoid and phosphatidylinositol 3-kinase inhibitor in human hepatoma cells. BMC Cancer. 2013;13:465. doi: 10.1186/1471-2407-13-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tran-Lundmark K, Tannenberg P, Rauch BH, Ekstrand J, Tran PK, Hedin U, Kinsella MG. Perlecan Heparan Sulfate Is Required for the Inhibition of Smooth Muscle Cell Proliferation by All-trans-Retinoic Acid. J Cell Physiol. 2015;230:482–487. doi: 10.1002/jcp.24731. [DOI] [PubMed] [Google Scholar]
- 120.Nickkho-Amiry M, McVey R, Holland C. Peroxisome proliferator-activated receptors modulate proliferation and angiogenesis in human endometrial carcinoma. Mol Cancer Res. 2012;10:441–453. doi: 10.1158/1541-7786.MCR-11-0233. [DOI] [PubMed] [Google Scholar]
- 121.Lee YR, Yu HN, Noh EM, Kim JS, Song EK, Han MK, Kim BS, Lee SH, Park J. Peroxisome proliferator-activated receptor gamma and retinoic acid receptor synergistically up-regulate the tumor suppressor PTEN in human promyeloid leukemia cells. Int J Hematol. 2007;85:231–237. doi: 10.1532/IJH97.A30615. [DOI] [PubMed] [Google Scholar]
- 122.Stefanska B, Salamé P, Bednarek A, Fabianowska-Majewska K. Comparative effects of retinoic acid, vitamin D and resveratrol alone and in combination with adenosine analogues on methylation and expression of phosphatase and tensin homologue tumour suppressor gene in breast cancer cells. Br J Nutr. 2012;107:781–790. doi: 10.1017/S0007114511003631. [DOI] [PubMed] [Google Scholar]
- 123.Li M, Li H, Li C, Wang S, Jiang W, Liu Z, Zhou S, Liu X, McNutt MA, Li G. Alpha-fetoprotein: a new member of intracellular signal molecules in regulation of the PI3K/AKT signaling in human hepatoma cell lines. Int J Cancer. 2011;128:524–532. doi: 10.1002/ijc.25373. [DOI] [PubMed] [Google Scholar]
- 124.Janardhanan R, Banik NL, Ray SK. N-Myc down regulation induced differentiation, early cell cycle exit, and apoptosis in human malignant neuroblastoma cells having wild type or mutant p53. Biochem Pharmacol. 2009;78:1105–1114. doi: 10.1016/j.bcp.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, Pandolfi PP. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature. 2008;455:813–817. doi: 10.1038/nature07290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhang R, Banik NL, Ray SK. Combination of all-trans retinoic acid and interferon-gamma upregulated p27(kip1) and down regulated CDK2 to cause cell cycle arrest leading to differentiation and apoptosis in human glioblastoma LN18 (PTEN-proficient) and U87MG (PTEN-deficient) cells. Cancer Chemother Pharmacol. 2008;62:407–416. doi: 10.1007/s00280-007-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang R, Banik NL, Ray SK. Combination of all-trans retinoic acid and interferon-gamma suppressed PI3K/Akt survival pathway in glioblastoma T98G cells whereas NF-kappaB survival signaling in glioblastoma U87MG cells for induction of apoptosis. Neurochem Res. 2007;32:2194–2202. doi: 10.1007/s11064-007-9417-7. [DOI] [PubMed] [Google Scholar]
- 128.Lee SJ, Yang EK, Kim SG. Peroxisome proliferator-activated receptor-gamma and retinoic acid X receptor alpha represses the TGFbeta1 gene via PTEN-mediated p70 ribosomal S6 kinase-1 inhibition: role for Zf9 dephosphorylation. Mol Pharmacol. 2006;70:415–425. doi: 10.1124/mol.106.022954. [DOI] [PubMed] [Google Scholar]
- 129.Sandler RS, Halabi S, Baron JA. Daily Aspirin Use Was Associated with a Reduced Incidence of Colorectal Adenomas. Annals of Internal Medicine. 2004;141:378–379. [Google Scholar]
- 130.Baron JA, Cole BF, Sandler RS, Haile RW, Ahnen D, Bresalier R, McKeown-Eyssen G, Summers RW, Rothstein R, Burke CA, et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med. 2003;348:891–899. doi: 10.1056/NEJMoa021735. [DOI] [PubMed] [Google Scholar]
- 131.Peek RM. Prevention of colorectal cancer through the use of COX-2 selective inhibitors. Cancer Chemother Pharmacol. 2004;54 Suppl 1:S50–S56. doi: 10.1007/s00280-004-0887-x. [DOI] [PubMed] [Google Scholar]
- 132.Jacoby RF, Seibert K, Cole CE, Kelloff G, Lubet RA. The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the min mouse model of adenomatous polyposis. Cancer Res. 2000;60:5040–5044. [PubMed] [Google Scholar]
- 133.Wu WK, Sung JJ, Lee CW, Yu J, Cho CH. Cyclooxygenase-2 in tumorigenesis of gastrointestinal cancers: an update on the molecular mechanisms. Cancer Lett. 2010;295:7–16. doi: 10.1016/j.canlet.2010.03.015. [DOI] [PubMed] [Google Scholar]
- 134.Roelofs HM, Te Morsche RH, van Heumen BW, Nagengast FM, Peters WH. Over-expression of COX-2 mRNA in colorectal cancer. BMC Gastroenterol. 2014;14:1. doi: 10.1186/1471-230X-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yao M, Lam EC, Kelly CR, Zhou W, Wolfe MM. Cyclooxygenase-2 selective inhibition with NS-398 suppresses proliferation and invasiveness and delays liver metastasis in colorectal cancer. Br J Cancer. 2004;90:712–719. doi: 10.1038/sj.bjc.6601489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chen WS, Wei SJ, Liu JM, Hsiao M, Kou-Lin J, Yang WK. Tumor invasiveness and liver metastasis of colon cancer cells correlated with cyclooxygenase-2 (COX-2) expression and inhibited by a COX-2-selective inhibitor, etodolac. Int J Cancer. 2001;91:894–899. doi: 10.1002/1097-0215(200102)9999:9999<894::aid-ijc1146>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 137.Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55:115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Brown JR, DuBois RN. COX-2: a molecular target for colorectal cancer prevention. J Clin Oncol. 2005;23:2840–2855. doi: 10.1200/JCO.2005.09.051. [DOI] [PubMed] [Google Scholar]
- 139.Wu CH, Shih YW, Chang CH, Ou TT, Huang CC, Hsu JD, Wang CJ. EP4 upregulation of Ras signaling and feedback regulation of Ras in human colon tissues and cancer cells. Arch Toxicol. 2010;84:731–740. doi: 10.1007/s00204-010-0562-4. [DOI] [PubMed] [Google Scholar]
- 140.Sheng H, Shao J, Washington MK, DuBois RN. Prostaglandin E2 increases growth and motility of colorectal carcinoma cells. J Biol Chem. 2001;276:18075–18081. doi: 10.1074/jbc.M009689200. [DOI] [PubMed] [Google Scholar]
- 141.Fujimura T, Ohta T, Oyama K, Miyashita T, Miwa K. Role of cyclooxygenase-2 in the carcinogenesis of gastrointestinal tract cancers: a review and report of personal experience. World J Gastroenterol. 2006;12:1336–1345. doi: 10.3748/wjg.v12.i9.1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shao J, Jung C, Liu C, Sheng H. Prostaglandin E2 Stimulates the beta-catenin/T cell factor-dependent transcription in colon cancer. J Biol Chem. 2005;280:26565–26572. doi: 10.1074/jbc.M413056200. [DOI] [PubMed] [Google Scholar]
- 143.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 144.Eisinger AL, Nadauld LD, Shelton DN, Prescott SM, Stafforini DM, Jones DA. Retinoic acid inhibits beta-catenin through suppression of Cox-2: a role for truncated adenomatous polyposis coli. J Biol Chem. 2007;282:29394–29400. doi: 10.1074/jbc.M609768200. [DOI] [PubMed] [Google Scholar]
- 145.Wagenaar-Miller RA, Hanley G, Shattuck-Brandt R, DuBois RN, Bell RL, Matrisian LM, Morgan DW. Cooperative effects of matrix metalloproteinase and cyclooxygenase-2 inhibition on intestinal adenoma reduction. Br J Cancer. 2003;88:1445–1452. doi: 10.1038/sj.bjc.6600867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Eisinger AL, Nadauld LD, Shelton DN, Peterson PW, Phelps RA, Chidester S, Stafforini DM, Prescott SM, Jones DA. The adenomatous polyposis coli tumor suppressor gene regulates expression of cyclooxygenase-2 by a mechanism that involves retinoic acid. J Biol Chem. 2006;281:20474–20482. doi: 10.1074/jbc.M602859200. [DOI] [PubMed] [Google Scholar]
- 147.Mestre JR, Subbaramaiah K, Sacks PG, Schantz SP, Tanabe T, Inoue H, Dannenberg AJ. Retinoids suppress phorbol ester-mediated induction of cyclooxygenase-2. Cancer Res. 1997;57:1081–1085. [PubMed] [Google Scholar]
- 148.Subbaramaiah K, Cole PA, Dannenberg AJ. Retinoids and carnosol suppress cyclooxygenase-2 transcription by CREB-binding protein/p300-dependent and -independent mechanisms. Cancer Res. 2002;62:2522–2530. [PubMed] [Google Scholar]
- 149.Kanekura T, Higashi Y, Kanzaki T. Inhibitory effects of 9-cis-retinoic acid and pyrrolidinedithiocarbamate on cyclooxygenase (COX)-2 expression and cell growth in human skin squamous carcinoma cells. Cancer Letters. 2000;161:177–183. doi: 10.1016/s0304-3835(00)00604-2. [DOI] [PubMed] [Google Scholar]
- 150.Li M, Song S, Lippman SM, Zhang XK, Liu X, Lotan R, Xu XC. Induction of retinoic acid receptor-beta suppresses cyclooxygenase-2 expression in esophageal cancer cells. Oncogene. 2002;21:411–418. doi: 10.1038/sj.onc.1205106. [DOI] [PubMed] [Google Scholar]
- 151.Merritt G, Aliprandis ET, Prada F, Rigas B, Kashfi K. The retinoid fenretinide inhibits proliferation and downregulates cyclooxygenase-2 gene expression in human colon adenocarcinoma cell lines. Cancer Lett. 2001;164:15–23. doi: 10.1016/s0304-3835(00)00714-x. [DOI] [PubMed] [Google Scholar]
- 152.Liu JP, Wei HB, Zheng ZH, Guo WP, Fang JF. Celecoxib increases retinoid sensitivity in human colon cancer cell lines. Cell Mol Biol Lett. 2010;15:440–450. doi: 10.2478/s11658-010-0016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Miladi-Abdennadher I, Abdelmaksoud-Damak R, Ayadi L, Khabir A, Frikha F, Kallel L, Amouri A, Frikha M, Sellami-Boudawara T, Gargouri A, et al. Hypermethylation of RARβ2 correlates with high COX-2 expression and poor prognosis in patients with colorectal carcinoma. Tumour Biol. 2010;31:503–511. doi: 10.1007/s13277-010-0063-3. [DOI] [PubMed] [Google Scholar]
- 154.Yang WL, Frucht H. Activation of pparR induces apoptosis and inhibit COX-2 in human colon cancer cells. Gastroenterology. 2000;118(4, Part 1):A682. [Google Scholar]
- 155.Allred CD, Talbert DR, Southard RC, Wang X, Kilgore MW. PPARgamma1 as a molecular target of eicosapentaenoic acid in human colon cancer (HT-29) cells. J Nutr. 2008;138:250–256. doi: 10.1093/jn/138.2.250. [DOI] [PubMed] [Google Scholar]
- 156.Papi A, Rocchi P, Ferreri AM, Orlandi M. RXRγ and PPARγ ligands in combination to inhibit proliferation and invasiveness in colon cancer cells. Cancer Letters. 2010;297:65–74. doi: 10.1016/j.canlet.2010.04.026. [DOI] [PubMed] [Google Scholar]
- 157.Ban JO, Kwak DH, Oh JH, Park EJ, Cho MC, Song HS, Song MJ, Han SB, Moon DC, Kang KW, et al. Suppression of NF-kappaB and GSK-3beta is involved in colon cancer cell growth inhibition by the PPAR agonist troglitazone. Chem Biol Interact. 2010;188:75–85. doi: 10.1016/j.cbi.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 158.Theocharis S, Giaginis C, Parasi A, Margeli A, Kakisis J, Agapitos E, Kouraklis G. Expression of peroxisome proliferator-activated receptor-gamma in colon cancer: correlation with histopathological parameters, cell cycle-related molecules, and patients’ survival. Dig Dis Sci. 2007;52:2305–2311. doi: 10.1007/s10620-007-9794-4. [DOI] [PubMed] [Google Scholar]
- 159.Shen D, Deng C, Zhang M. Peroxisome proliferator-activated receptor gamma agonists inhibit the proliferation and invasion of human colon cancer cells. Postgrad Med J. 2007;83:414–419. doi: 10.1136/pmj.2006.052761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Feilchenfeldt J, Bründler MA, Soravia C, Tötsch M, Meier CA. Peroxisome proliferator-activated receptors (PPARs) and associated transcription factors in colon cancer: reduced expression of PPARγ-coactivator 1 (PGC-1) Cancer Letters. 2004;203:25–33. doi: 10.1016/j.canlet.2003.08.024. [DOI] [PubMed] [Google Scholar]
- 161.Sarraf P, Mueller E, Smith WM, Wright HM, Kum JB, Aaltonen LA, de la Chapelle A, Spiegelman BM, Eng C. Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol Cell. 1999;3:799–804. doi: 10.1016/s1097-2765(01)80012-5. [DOI] [PubMed] [Google Scholar]
- 162.Au-Yeung KK, Liu PL, Chan C, Wu WY, Lee SS, Ko JK. Herbal isoprenols induce apoptosis in human colon cancer cells through transcriptional activation of PPARgamma. Cancer Invest. 2008;26:708–717. doi: 10.1080/07357900801898656. [DOI] [PubMed] [Google Scholar]
- 163.Schwab M, Reynders V, Loitsch S, Shastri YM, Steinhilber D, Schröder O, Stein J. PPARgamma is involved in mesalazine-mediated induction of apoptosis and inhibition of cell growth in colon cancer cells. Carcinogenesis. 2008;29:1407–1414. doi: 10.1093/carcin/bgn118. [DOI] [PubMed] [Google Scholar]
- 164.Toaldo C, Pizzimenti S, Cerbone A, Pettazzoni P, Menegatti E, Daniela B, Minelli R, Giglioni B, Dianzani MU, Ferretti C, et al. PPARgamma ligands inhibit telomerase activity and hTERT expression through modulation of the Myc/Mad/Max network in colon cancer cells. J Cell Mol Med. 2010;14:1347–1357. doi: 10.1111/j.1582-4934.2009.00966.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Aires V, Brassart B, Carlier A, Scagliarini A, Mandard S, Limagne E, Solary E, Martiny L, Tarpin M, Delmas D. A role for peroxisome proliferator-activated receptor gamma in resveratrol-induced colon cancer cell apoptosis. Mol Nutr Food Res. 2014;58:1785–1794. doi: 10.1002/mnfr.201300962. [DOI] [PubMed] [Google Scholar]
- 166.Tsukahara T, Hanazawa S, Kobayashi T, Iwamoto Y, Murakami-Murofushi K. Cyclic phosphatidic acid decreases proliferation and survival of colon cancer cells by inhibiting peroxisome proliferator-activated receptor γ. Prostaglandins Other Lipid Mediat. 2010;93:126–133. doi: 10.1016/j.prostaglandins.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 167.Choi IK, Kim YH, Kim JS, Seo JH. PPAR-gamma ligand promotes the growth of APC-mutated HT-29 human colon cancer cells in vitro and in vivo. Invest New Drugs. 2008;26:283–288. doi: 10.1007/s10637-007-9108-x. [DOI] [PubMed] [Google Scholar]
- 168.Delage B, Bairras C, Buaud B, Pallet V, Cassand P. A high-fat diet generates alterations in nuclear receptor expression: prevention by vitamin A and links with cyclooxygenase-2 and beta-catenin. Int J Cancer. 2005;116:839–846. doi: 10.1002/ijc.21108. [DOI] [PubMed] [Google Scholar]
- 169.Yamazaki K, Shimizu M, Okuno M, Matsushima-Nishiwaki R, Kanemura N, Araki H, Tsurumi H, Kojima S, Weinstein IB, Moriwaki H. Synergistic effects of RXR alpha and PPAR gamma ligands to inhibit growth in human colon cancer cells--phosphorylated RXR alpha is a critical target for colon cancer management. Gut. 2007;56:1557–1563. doi: 10.1136/gut.2007.129858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cesario RM, Stone J, Yen WC, Bissonnette RP, Lamph WW. Differentiation and growth inhibition mediated via the RXR: PPARgamma heterodimer in colon cancer. Cancer Lett. 2006;240:225–233. doi: 10.1016/j.canlet.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 171.Miao R, Xu T, Liu L, Wang M, Jiang Y, Li J, Guo R. Rosiglitazone and retinoic acid inhibit proliferation and induce apoptosis in the HCT-15 human colorectal cancer cell line. Exp Ther Med. 2011;2:413–417. doi: 10.3892/etm.2011.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Shimada T, Kojima K, Yoshiura K, Hiraishi H, Terano A. Characteristics of the peroxisome proliferator activated receptor gamma (PPARgamma) ligand induced apoptosis in colon cancer cells. Gut. 2002;50:658–664. doi: 10.1136/gut.50.5.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wan YJ, Cai Y, Magee TR. Retinoic acid differentially regulates retinoic acid receptor-mediated pathways in the Hep3B cell line. Exp Cell Res. 1998;238:241–247. doi: 10.1006/excr.1997.3851. [DOI] [PubMed] [Google Scholar]
- 174.Han S, Wada RK, Sidell N. Differentiation of human neuroblastoma by phenylacetate is mediated by peroxisome proliferator-activated receptor gamma. Cancer Res. 2001;61:3998–4002. [PubMed] [Google Scholar]
- 175.James SY, Lin F, Kolluri SK, Dawson MI, Zhang XK. Regulation of retinoic acid receptor beta expression by peroxisome proliferator-activated receptor gamma ligands in cancer cells. Cancer Res. 2003;63:3531–3538. [PubMed] [Google Scholar]
- 176.Morosetti R, Servidei T, Mirabella M, Rutella S, Mangiola A, Maira G, Mastrangelo R, Koeffler HP. The PPARgamma ligands PGJ2 and rosiglitazone show a differential ability to inhibit proliferation and to induce apoptosis and differentiation of human glioblastoma cell lines. Int J Oncol. 2004;25:493–502. [PubMed] [Google Scholar]
- 177.Pancione M, Forte N, Fucci A, Sabatino L, Febbraro A, Di Blasi A, Daniele B, Parente D, Colantuoni V. Prognostic role of beta-catenin and p53 expression in the metastatic progression of sporadic colorectal cancer. Hum Pathol. 2010;41:867–876. doi: 10.1016/j.humpath.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 178.Zeestraten EC, Benard A, Reimers MS, Schouten PC, Liefers GJ, van de Velde CJ, Kuppen PJ. The prognostic value of the apoptosis pathway in colorectal cancer: a review of the literature on biomarkers identified by immunohistochemistry. Biomark Cancer. 2013;5:13–29. doi: 10.4137/BIC.S11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Vousden KH. Review: Activation of the p53 tumor suppressor protein. BBA - Reviews on Cancer. 2002;1602:47–59. doi: 10.1016/s0304-419x(02)00035-5. [DOI] [PubMed] [Google Scholar]
- 180.Jiang M, Milner J. Bcl-2 constitutively suppresses p53-dependent apoptosis in colorectal cancer cells. Genes Dev. 2003;17:832–837. doi: 10.1101/gad.252603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Huerta S, Goulet EJ, Livingston EH. Colon cancer and apoptosis. Am J Surg. 2006;191:517–526. doi: 10.1016/j.amjsurg.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 182.Shaw P, Bovey R, Tardy S, Sahli R, Sordat B, Costa J. Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line. Proc Natl Acad Sci USA. 1992;89:4495–4499. doi: 10.1073/pnas.89.10.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Soussi T. p53 alterations in human cancer: more questions than answers. Oncogene. 2007;26:2145–2156. doi: 10.1038/sj.onc.1210280. [DOI] [PubMed] [Google Scholar]
- 184.Fazeli A, Steen RG, Dickinson SL, Bautista D, Dietrich WF, Bronson RT, Bresalier RS, Lander ES, Costa J, Weinberg RA. Effects of p53 mutations on apoptosis in mouse intestinal and human colonic adenomas. Proc Natl Acad Sci USA. 1997;94:10199–10204. doi: 10.1073/pnas.94.19.10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Liu J, Stevens J, Rote CA, Yost HJ, Hu Y, Neufeld KL, White RL, Matsunami N. Siah-1 mediates a novel beta-catenin degradation pathway linking p53 to the adenomatous polyposis coli protein. Mol Cell. 2001;7:927–936. doi: 10.1016/s1097-2765(01)00241-6. [DOI] [PubMed] [Google Scholar]
- 186.Gwak J, Song T, Song JY, Yun YS, Choi IW, Jeong Y, Shin JG, Oh S. Isoreserpine promotes beta-catenin degradation via Siah-1 up-regulation in HCT116 colon cancer cells. Biochem Biophys Res Commun. 2009;387:444–449. doi: 10.1016/j.bbrc.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 187.Matsuzawa SI, Reed JC. Siah-1, SIP, and Ebi collaborate in a novel pathway for beta-catenin degradation linked to p53 responses. Mol Cell. 2001;7:915–926. doi: 10.1016/s1097-2765(01)00242-8. [DOI] [PubMed] [Google Scholar]
- 188.Mrass P, Rendl M, Mildner M, Gruber F, Lengauer B, Ballaun C, Eckhart L, Tschachler E. Retinoic acid increases the expression of p53 and proapoptotic caspases and sensitizes keratinocytes to apoptosis: a possible explanation for tumor preventive action of retinoids. Cancer Res. 2004;64:6542–6548. doi: 10.1158/0008-5472.CAN-04-1129. [DOI] [PubMed] [Google Scholar]
- 189.Sarkar SA, Sharma RP. All-trans-retinoic acid-mediated modulation of p53 during neural differentiation in murine embryonic stem cells. Cell Biol Toxicol. 2002;18:243–257. doi: 10.1023/a:1016003027850. [DOI] [PubMed] [Google Scholar]
- 190.Zhang J, Tu Y, Smith-Schneider S. Activation of p53, inhibition of telomerase activity and induction of estrogen receptor beta are associated with the anti-growth effects of combination of ovarian hormones and retinoids in immortalized human mammary epithelial cells. Cancer Cell Int. 2005;5:6. doi: 10.1186/1475-2867-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Donato LJ, Suh JH, Noy N. Suppression of mammary carcinoma cell growth by retinoic acid: the cell cycle control gene Btg2 is a direct target for retinoic acid receptor signaling. Cancer Res. 2007;67:609–615. doi: 10.1158/0008-5472.CAN-06-0989. [DOI] [PubMed] [Google Scholar]
- 192.McPherson LA, Loktev AV, Weigel RJ. Tumor suppressor activity of AP2alpha mediated through a direct interaction with p53. J Biol Chem. 2002;277:45028–45033. doi: 10.1074/jbc.M208924200. [DOI] [PubMed] [Google Scholar]
- 193.Carrera S, Cuadrado-Castano S, Samuel J, Jones GD, Villar E, Lee SW, Macip S. Stra6, a retinoic acid-responsive gene, participates in p53-induced apoptosis after DNA damage. Cell Death Differ. 2013;20:910–919. doi: 10.1038/cdd.2013.14. [DOI] [PMC free article] [PubMed] [Google Scholar]