

Abstract
Chronic hepatitis C virus (HCV) infection is a public health issue that often progresses to life-threatening complications, including liver cirrhosis, fibrosis, and hepatocellular carcinoma. Impaired immune responses to HCV are key features of chronic HCV infection. Therefore, intervention strategies usually involve enhancing the immune responses against HCV. Cytotoxic CD8+ T lymphocytes (CTLs) play a critical role in the control of HCV infection. However, their cytolytic function can be impaired by the expression of co-inhibitory molecules. Programmed death-1 (PD-1) receptor and its ligand PD-L1 function in a T cell co-inhibitory pathway, which either blocks the function of CTLs or the differentiation of CD8+ T cells. During chronic HCV infection, the immune inhibitory receptor PD-1 is upregulated on dysfunctional HCV-specific CD8+ T cells. As such, blockade of the PD-1/PD-L1 pathway in these CD8+ T cells might restore their functional capabilities. Indeed, clinical trials using therapies to block this pathway have shown promise in the fostering of anti-HCV immunity. Understanding how chronic HCV infection induces upregulation of PD-1 on HCV specific T cells and how the PD-1/PD-L1 interaction develops HCV specific T cell dysfunction will accelerate the development of an efficacious prophylactic and therapeutic vaccination against chronic HCV infections, which will significantly improve HCV treatments and patient survival. In this review, we discuss the relationship between PD-1 expression and clinical responses and the potential use of PD-1 blockade for anti-HCV therapy.
Keywords: Hepatitis C virus, Programmed death-1, Hepatitis C virus immunotherapy, Exhausted T cells, Hepatitis C virus immune escape
Core tip: The programmed death-1 (PD-1)/PD-L1 pathway is an attractive target for anti-hepatitis C virus (HCV) immunotherapy because it restores the functional capacities of HCV-specific T cells. This is an extremely promising development in anti-HCV vaccines research since restoration of exhausted anti-HCV T cells is a major challenge when developing either prophylactic or therapeutic vaccines. This review will discuss the correlation between PD-1 expression and the clinical outcome in HCV patients and how this information can be potentially applied to block PD-1/PD-L1 pathway for HCV immunotherapy.
INTRODUCTION
Chronic viral infections, including hepatitis C virus (HCV), hepatitis B virus (HBV), and human immunodeficiency virus (HIV), are among the main causes of death worldwide[1]. While most viral infections prompt successful T cell responses that remove the infections, HCV, HBV, and HIV have acquired mechanisms to avoid immune elimination, permitting them to persist in many, if not all, infected individuals. These escape mechanisms lower the responsiveness of patients to anti-viral therapy.
HCV is found in nearly every region of the world, affecting an estimated 170 million patients and 1%-2% of the overall population in most infected countries[2]. HCV not only causes hepatitis C, but it also provides the perfect infection setting to study viral evasion mechanisms, since the infection persists in most infected individuals while 25% of infected patients effectively clear the virus. This allows for the comparison of immune responses between responders and non-responders. How these immune responses determine whether a patient eliminates infection or develops a chronic infection is not completely understood. Accordingly, viral escape from immune cells has been suggested as a contributing factor to HCV as well as HIV and HBV infection.
In the acute stage of HCV infection, 20%-40% of patients improve spontaneously[3], and this recovery is associated with a robust, HCV-specific T cell responses[4-6]. The discriminating role of the HCV specific CD8+ cells responses in the unprompted recovery of acute HCV infection was demonstrated in chimpanzees[5,6], the only animal model for the study of HCV. Even in chimpanzees with chronically developing, acute HCV infection, intrahepatic infusion of CD8+ T cells promoted a partial decline in the HCV load[7,8]. In chimpanzees, vaccination with an experimental prophylactic vaccine induced HCV-specific CD8+ cell responses and suppression of acute HCV infection[9]. These studies led to the hypothesis that recovery from HCV may be due to induction of HCV-specific T cell responses. Hence, research efforts for the development of novel treatments for chronic HCV infection have focused on T cell responses.
Dysfunction of virus-specific CD8+ cells is a fundamental property of persistent viral infections like HCV; consequently, restoration of T cell capacity is a major aim in the generation of immune-based therapies for persistent infection of viruses[10]. Many factors are known to contribute to T cell dysfunction, including inhibitory cytokines, regulatory T cells, and inhibitory receptors expressed on T cells[10]. Accordingly, removal or blockade of these inhibitory factors may be a promising approach for the treatment of persistent viral infection.
Overall, the mechanisms that have been proposed to date to explain impaired immunity in chronic HCV infection are summarized as follows: (1) HCV escapes immune responses by developing mutations; (2) primary T cell exhaustion after an extensive response; (3) impaired antigen presentation of dendritic cells (DCs); (4) impaired natural killer (NK) cell activities; (5) skewing the Th1 type cytokine to a Th2 type; (6) suppression by HCV proteins; (7) impaired T cell maturation; (8) suppression by regulatory T cells; (9) the nature of the tolerogenic environment in the liver; and (10) the expression of co-inhibitory molecules on immune cells[11].
An important inhibitory receptor that downregulates T cell function is programmed death-1 (PD-1)[12]. PD-1, with its two known ligands B7-H1/PD-L1 and B7-DC/PD-L2, has recently been shown to be upregulated on HCV- and HIV-specific CD8+ cells, indicating that PD-1 upregulation may be an essential mechanism for viral immune escape in chronic HCV and HIV infections[13-17].
Here, we provide up to date review on the role of PD-1 in HCV immune evasion and the potential use of PD-1 blockade for anti-HCV therapy. Understanding the relationship between PD-1/PD-L1 and T cell dysfunction and its role in HCV persistence will accelerate the development of an efficacious prophylactic and therapeutic vaccination against chronic HCV infection.
HCV TREATMENT AND FAILURE
Combination therapy with pegylated interferon (PEG-IFN) and ribavirin (RBV) is the current standard therapy for individuals with chronic HCV infection[18-21]. Treatment duration is 48 wk for HCV genotypes 1 and 4, and 24 wk for genotypes 2 and 3. The dominant majority of treated patients, especially those with HCV genotypes 2 or 3, show a significant virologic response. Almost 66% of patients with HCV genotype 2 or 3 accomplish rapid virologic response (RVR), characterized by untraceable HCV RNA within 4 wk of starting treatment, and 97% have undetectable HCV RNA within 12-24 wk of starting treatment. Seventy-six percent attain sustained virologic response (SVR)[21,22]. Unfortunately, only approximately half of all patients accomplish SVR with 24-48 wk of therapy with PEG-IFN and RBV[18,20]. Factors that contribute to non-responsiveness, other than genotype, are high baseline HCV viral-load, high fibrosis stage in the liver, male gender, old age, race, obesity, alcohol intake, insulin resistance, liver steatosis, and alterations in the host immune response, such as high interleukin (IL)-8 and IL-10 serum levels[23-25].
Current IFN-based therapy does not work in many patients, possibly due to a combination of viral and host factors. Innate immunity to HCV is activated by cellular sensors that identify the presence of pathogen-associated molecular patterns (PAMPs). Key fundamental cellular sensors for HCV infection are toll-like receptor 3 (TLR3), which recognizes double-stranded RNA (dsRNA), and the RNA helicase retinoic acid-inducible gene 1 (RIG-1). The HCV PAMP sensors TLR3 and RIG-1 signal through the adaptor proteins TIR-domain-containing adapter-inducing interferon-β (TRIF) and Cardif, respectively. Remarkably, the HCV NS3-4A serine protease relieves both Cardif[26,27] and TRIF[28] to disable signals initiated by RIG-1 and TLR3. In addition to blockade of the upstream events of IFN-β transcription by NS3-4A, there is evidence that other HCV proteins block IFN signaling downstream of the IFN-α/β receptor. Overexpression of HCV core protein causes activation of suppressors of cytokine signaling (SOCS) 3 protein[29], which in turn hinders signal transducer and activator of transcription 1 (STAT1) phosphorylation by janus kinase 1 (Jak1). These mechanisms support viral persistence even in the face of IFN-based therapies. Further understanding of the molecular mechanisms underlying HCV resistance to the host immune response will lead to generation of novel therapeutic strategies. Moreover, host factors, such as insulin resistance and race, have considerable effects on treatment responsiveness. Adjustment of adverse host factors, whenever possible, may be a feasible alternative for the optimization of HCV therapy.
PEG-IFN is contraindicated in decompensated cirrhosis[30] and is associated with constitutional, autoimmune, neuropsychiatric, and hematological side effects[31], whereas RBV is contraindicated in renal failure[32] and is associated with rash, cough, hemolysis, and teratogenesis[31]. Therefore, many patients are ineligible for or intolerant to PEG-IFN and RBV therapy. However, the approval of sofosbuvir (Sovaldi®, Gilead Sciences), which is a direct acting pyrimidine nucleotide analog that represents the first NS5B HCV polymerase inhibitor, is considered a key step towards a new era in chronic hepatitis C therapy. It was among the first approved antiviral agents with strong activity and high genetic barrier against all HCV genotypes. Additionally, its safety profile is highly favorable, even when it is prescribed to patients with very advanced liver disease and high risk of complications (e.g., cirrhosis with portal hypertension and liver transplant recipients).
ANTI-HCV IMMUNITY
Impaired immune responses to HCV are hallmarks of chronic HCV infection. Therefore, intervention approaches commonly include those that can boost the immune responses against HCV. These immunotherapies for chronic HCV infections include anti-HCV neutralizing antibodies, antagonists of T cell inhibitory factors, therapeutic vaccines, agonists for TLRs, and cytokines[33]. These therapies can be utilized alone or in combination with other antiviral drugs for chronic HCV therapy.
To date, immune-based therapies have not demonstrated satisfactory efficacy. In general, a virologic response was shown only in a small group of patients, and in these cases, the effect was marginal and transient. A critical reason for the poor efficacy is the inadequate activation and stimulation of immune responses. It should be mentioned, however, that the virologic responders showed the strongest T cell responses in a late study that tested the peptide vaccine IC41[34]. This observation demonstrated that a sufficient virologic response might be accomplished by sufficient activation and stimulation of the immune system. Thus, enhancement of the protocol/regimen is needed to improve the efficacy of immune-based therapies.
One possible critical mechanism underlying the inability of HCV patients to resolve the infection is the imbalance between the stimulatory and regulatory immune cells[35]. The poor adequacy of immune-based therapies may be due to various factors. First, many individuals with chronic HCV infection have delayed impairment of the anti-HCV immune response, and it seems unlikely that longstanding immune dysfunction can be repaired by immune-based therapies. Second, HCV advances quickly, and persistent HCV infection brings about the specific survival of viruses that are most proficient at evading host immune responses. Accordingly, these viruses may have the capacity to resist clearance despite the improvement of immune responses by immunotherapies. Finally, the poor adequacy may be credited, in part, to the selection of patients.
Combination therapy might be an efficient strategy for improving the efficiency of immunotherapies For instance, the impact of therapeutic vaccines could be enhanced by combining them with antagonists of T cell inhibitory factors and/or agonists of TLRs. Interestingly, combining antagonists of IL-10R or PD-1 with a therapeutic vaccine strengthened the effects in a murine model of persistent lymphocytic choriomeningitis virus (LCMV) infection[36]. In general, immunotherapies have been well-endured and have not been associated with severe adverse effects. However, improvements in immunotherapies aiming to prompt stronger immune responses may aggravate liver injury and cause severe hepatitis in extreme situations. In this regard, it would be useful to demonstrate the differences between cytotoxic virus-clearing and tissue-damaging T cell responses. A better understanding of the cellular and molecular mechanisms implicated in T cell dysfunction will pave the way for highly efficacious immunotherapies for chronic HCV.
PD-1 EXPRESSION ON IMMUNE CELLS IN HCV PATIENTS
PD-1 and its ligands play a critical role in the inhibition of the immune system by banning the activation of T-cells, which subsequently decreases autoimmunity and advances self-tolerance. The inhibitory effect of PD-1 is achieved through a dual mechanism of inducing apoptosis in antigen specific T-cells in lymph nodes and decreasing apoptosis regulatory T cells (Tregs) (Figure 1). New classes of drugs that block PD-1, such as Nivolumab, Pembrolizumab, Pidilizumab, and BMS-936559, activate the immune system to attack cancers and are used to treat tumors.
Figure 1.
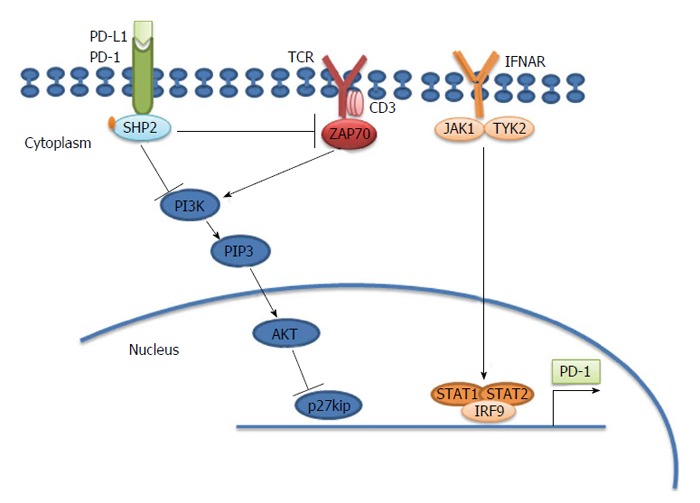
Programmed death-1 causes T cell exhaustion. Programmed death-1 (PD-1) inhibits the T cell receptor (TCR) signaling pathway through src homology 2-containing protein-tyrosine phosphatase 2 (SHP2). PD-1 is located in the immune synapse at the T cell-antigen presenting cell (APC) interface. When its physiological ligand (PD-L1 or PD-L2) binds, PD-1 suppresses the activation and function of T cells through the recruitment of SHP-2, which dephosphorylates and inactivates ZAP70, a major integrator of TCR-mediated signaling. In chronically activated (“exhausted”) T cells, interferon (IFN)-α causes overexpression of PD-1 through the binding of the transcription factor IRF9 to the signal transducer and activator of transcription (STAT)1 and STAT2 promoters. PD-1 also results in accumulation of p27kip1, which is an inhibitor of cyclin dependent kinases to block cell cycle and proliferation[84]. ZAP70: Zeta-chain (TCR) associated protein kinase 70 kDa; IRF9: Interferon regulatory factor 9; JAK1: Janus kinase 1.
PD-1 has two ligands-PD-L1 (B7-H1)[37,38], which is largely expressed on both hematopoietic and parenchymal cells, and PD-L2 (B7-DC)[39,40], which is mainly expressed on macrophages and DCs. Barber et al[12] found that PD-L1 was expressed at very high levels in splenocytes from persistently infected mice, particularly on virally infected cells. Consequently, not only did the exhausted cytotoxic T cells express high levels of PD-1, but its ligand was upregulated on infected cells (Figure 1). PD ligands are differentially regulated, where IFN-γ primarily stimulates PD-L1 expression and IL-4 stimulates PD-L2 expression[41,42]. Recent studies showed that antibody-mediated interference with PD-1 caused regression of several tumor types, including melanoma, renal-cell cancer, and non-small-cell lung cancer, in some patients[43,44]. The inhibitory effect of PD-1 is achieved through a dual mechanism that involves simultaneous induction of apoptosis in antigen specific T-cells in lymph nodes and decreasing apoptosis in regulatory T cells[45,46] (Figure 1).
In the acute stage of HCV infection, HCV specific T cells have been shown to be inadequately functional regardless of the final outcome of the disease[47-51]. A possible mechanism directing this behavior of the HCV-specific T response is exhaustion. When T cells are chronically exposed to high antigen loads, the PD-1/PD-L1 ligand pathway may play a role in T-cell exhaustion. Blocking the PD-1/PD-L1 interaction can permit restoration of exhausted T cells[12,52-55]. These studies indicated that high expression of the inhibitory PD-1 receptor appears to be a signature of functional T cell exhaustion.
Kasprowicz et al[17] have demonstrated elevated PD-1 expression on almost all HCV specific CD8+ and CD4+ T cells through the early phase of acute infection, irrespective of clinical outcome or viral load. They also showed that PD-1 expression is reliant on the tissue microenvironment, where the T cells execute their antiviral functions. Interestingly, the overall PD-1 expression levels of infiltrating CD8+ and CD4+ T lymphocytes in the liver were significantly higher compared to peripheral blood[17]. The mean PD-1 expression level on most of CD8+ T liver-residing lymphocytes was 71%, while the median expression on peripheral blood CD8+ T cells from the same subjects was 33%. For liver-derived CD4+ T lymphocytes, the median PD-1 expression level was 53%, while PD-1 expression for cells in the peripheral blood was only 25%[17].
It has been shown that chronic HCV infection has a wide effect on PD-1 expression. For example, PD-1 is highly expressed on peripheral B cells and monocytes, since it is induced upon activation[56]. In patients with chronic HCV, CD56high NK cells expressed greater levels of PD-1, convenient considering their greater functional deficiency and less mature CD56low differentiation state[57]. In addition, PD-1 is expressed on Kupffer cells in the liver, other monocyte-derived cells, as well as epithelial, endothelial, and tumor cells[52,58,59].
Although HCV-specific CD8+ T-cells are generally dysfunctional in HCV persistence, their level of impairment varied considerably among patients depending on PD-1 expression. Within an individual patient, the function and PD-1 expression of HCV-specific CD8+ T-cells varied between the liver and peripheral blood[60]. Additional studies on the expression patterns of diverse splice variants of PD-1, PD-L1, and receptor-ligand interactions in diseased tissue will be important in determining a more comprehensive estimation of the level of the inhibitory signal and its effect on the outcome of human infection. Such studies will not only provide a superior mechanistic understanding of the PD-1 pathway in controlling T cell responses but will also encourage specific manipulation of this pathway therapeutically.
CORRELATION OF PD-1 EXPRESSION AND CLINICAL RESPONSES
The identification of cellular and molecular factors predicting clinical response to immunotherapy is strongly desirable, not only to aid in the design of therapies that overcome and enhance the inhibitory and stimulatory mechanisms, but also to preselect patients most likely to benefit from therapy and spare others from unnecessary exposure to possible side effects. Similar to its inhibitory role in anti-cancer immunity, the PD-1 signaling pathway has also been found to shape the overall immunity in HCV infection. For instance, PD-1 expression in acute HCV infection was found to be a signature of functional HCV-specific CD8 T cell exhaustion[16]. In this study, and as reported for the acute infection of HBV[61], the expression of PD-1 by HCV specific cytotoxic T cells was decreased in self-limited infections after the acute stage of infection in conjunction with CD8+ T cell differentiation towards a memory CD127 phenotype[61]. In contrast, HCV specific CD8+ T cells maintained high levels of PD-1 in patients with chronic advancement of infection and remained functionally impeded with no change from an effector to a memory phenotype[16]. Other studies, however, reported high levels of PD-1 expression (60%-100%) on all HCV specific CD8+ and CD4+ cells during the early stage of acute infection, irrespective of the clinical outcome or viral load[17]. Taken together, these results suggested that a role for the PD-1/PD-L1 interaction in regulating CD8+ T cell function may exist under conditions of continuous high levels of HCV antigen stimulation.
Consistent with this suggestion, another study showed that the level of PD-1 expression in early phases of HCV infection was significantly more on HCV-specific T cells from patients who advanced to chronic HCV infection than from those who eliminated infection; and this correlation was independent of HCV RNA titer levels[62]. The reason for this difference is unclear, but it may be due to differences between the routes of infection between the two studies. This suggests that some of the biological differences that cause the development of symptoms likewise affect PD-1 expression. Additionally, the duration of infection in patients defined as acutely infected could be different between studies. Table 1 provides a list of studies involving PD-1 expression and the role of PD-1 in HCV infection.
Table 1.
Differences between the studies made on the correlation between programmed death-1 expression and clinical outcome
| Ref. | No. of patients | Mode of transfection | Symptoms | PD-1 expression levels in acute infection | PD-1 expression levels in resolved patients |
| Urbani et al[16] | 19 | Sexually transmission | Symptomatic | High | Decreased |
| Kasprowicz et al[17] | 37 | Sexually transmission | Symptomatic | High | Irrespective |
| Rutebemberwa et al[62] | 20 | Injection drug use | Asymptomatic | High | Decreased |
PD-1: Programmed death-1.
PD-1 expression was investigated in 72 treatment- naïve patients with persistent HCV[57]. In this study, PD-1 expression was upregulated significantly not only on CD4+ and CD8+ T cells but also on NK cells, connecting with failed early and persistent virologic response to therapy. In contrast, patients with SVR demonstrated decreases in PD-1 after therapy completion, demonstrating that PD-1 expressed by NK cells is critical in persistent HCV[57].
PD-1+ HCV-specific CD8+ T cells in chronic HCV infection have a tendency to co-express Tim-3[63], 2B4, CD160 and other inhibitory molecules[64], particularly in the liver[65] since intrahepatic T cells showed a more exhausted phenotype than in the blood. In addition, the level of TIM-3 from patients with persistent HCV infection was greater than those who resolved the infection.
The results from other studies, however, are inconsistent regarding the differences in levels of PD-1/PD-L1 on HCV-specific CD8+ cells contrasts between those who clear HCV infection and those with persistent infection[16,17,62]. Most of these studies though concluded that PD-1 expression levels are elevated on HCV-specific T cells vs naïve CD8+ T cells or on T cells specific for some control antigens in the acute stage of infection, regardless of the outcome.
RESTORATION OF ANTI-HCV RESPONSES IN VITRO BY BLOCKING PD-1
During the acute phase of HCV infection, HCV specific T cells have been characterized as poorly functional, regardless of the outcome of the disease[47-51]. A possible explanation for this behavior of the HCV-specific CD8+ T response is exhaustion, which is supported by the initial rapid kinetics of HCV spread, replication after infection, and later on, by the continuous exposure of CD8+ T cells to high antigen load. Several studies demonstrated that the PD-1/PD-L1 pathway plays a role in T-cell exhaustion when CD8+ cells are chronically exposed to high level antigen loads. Blockade of this pathway can permit restoration of exhausted CD8+ T cells[12,52-55] and results in expansion of HCV specific T cell proliferation[15,16,66] (Figure 2).
Figure 2.
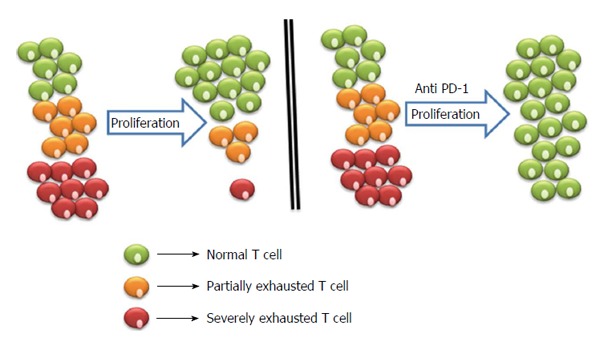
Proliferation of exhausted T cells and blockage strategies to reverse exhaustion. Severely exhausted T cells (red cells) proliferate poorly in comparison to partially exhausted and normal T cells (yellow and green cells). Antibody blockade of the pathway with PD-1 and its ligand reverses exhaustion and restores the functional capacities of exhausted T cells. PD-1: Programmed death-1.
For instance, PD-L1 blockade improved HCV-specific T cell proliferation in a dose-dependent manner. However, proliferation of cytomegalovirus (CMV)-specific CD8+ T cells was not affected by PD-1/PD-L1 blockade, consistent with their low expression levels of PD-1. Similar to its effects on CD8+ T cells in HCV, blocking the PD-1/PD-L1 interaction in vitro restored effector function and enhanced the proliferative ability of exhausted CD8+ T cells in many chronic infections, including HIV, HBV, simian immunodeficiency virus (SIV), LCMV, and Epstein-Barr virus (EBV)[13,15,67-71].
This functional T cell restoration by blocking the PD-1/PD-L1 pathway is a hierarchical phenomenon that appears to reflect the different sensitivities to exhaustion of the diverse T cell functions[72]. Restoration of proliferation capacity is relatively faster than the restoration of IFN-γ and IL-2 production but with no effect on cytotoxicity. In line with this, treatment with anti-PD-L1 antibodies does not always predict the expected positive effect of PD-1 blockade[73], suggesting that PD-L1 binds at least one additional receptor other than PD-1 to mediate its costimulatory function. Moreover, exhaustion was more effectively overcomed in HCV, than in HIV[68] by PD-1/PD-L1 blockade, as demonstrated by the increased capability of HCV-specific CD8+ T cells to expand and to produce IFN and IL-2 after incubation with anti PD-L1 antibodies[15].
Collectively, these in vitro data suggest that PD-1 signaling on T cells is a significant inhibitory pathway during chronic HCV infection. Consequently, the possibility of partially restoring CD8+ T cell function by blocking PD-1/PD-L1 interaction may provide a valuable tool for the enhancement of available therapies to cure chronic hepatitis C.
POTENTIAL USE OF IN VIVO PD-1 BLOCKING FOR ANTI-HCV THERAPY
As demonstrated above, in vitro blockade of PD-1 can restore the functionality of HCV-specific T cells. Blockade of PD-1 signaling was tested in vivo in both chimpanzees[74] and in patients with chronic HCV infection[75]. In the chimpanzee study, an increase in HCV specific CD8+ cell responses and a considerable, although transient, reduction in HCV viremia was only seen in one of three chimpanzees. This chimpanzee had the strongest and broadest CD4+ and CD8+ T cell response before the development of chronic infection, which suggested that PD-1 blockade alone is not sufficient to attain viral clearance[74]. In the patient study, a single dose (10 mg/kg) of the PD-1 blocking antibody BMS-936558 was followed by a greater than 0.5 log10 IU/mL decrease in HCV RNA titer in five of 45 (11%) patients. At the highest dose given (10 mg/kg), a > 4 log10 IU/mL decrease in HCV RNA titer was seen in three of 20 (15%) patients. This decrease of HCV replication continued for more than 8 wk in most patients[75].
Interestingly, in vivo PD-L1 blockade does not appear to affect IL-10 level during chronic infection, although it downregulated IL-10 and upregulated IL-2 and IFN-γ during in vitro stimulation[76,77]. Another ex vivo study showed that intrahepatic T cells were significantly dysfunctional and insusceptible to PD-1 blockade[60]. Therefore, the effect of PD-1 blockade is characterized by T cell compartmentalization. Additionally, PD-1 expression on HCV specific CD8+ cells is affected by viral immune-evasion in chronic HCV infections[62].
Indeed, an efficient helper T cell function is compulsory for the development of virus specific CD8 cells[78]. Thus, the synergistic effect of CD8- and CD4-mediated T cell functions enhanced by anti-PD-L1 may represent a strategy to enhance the effect of available anti-HCV drugs. Consequently, it is conceivable that utilizing PD-1/PD-L1 blockade can enhance the antiviral effect of IFN/RBV therapy. Additional studies are required, however, to survey whether the enhancement of the T cell function induced by PD-1/PD-L1 blockade and favored by IFN-α therapy in patients with a recent HCV infection can be also accomplished in chronic infections of longer duration, where the effect of long lasting exhaustion may be more difficult to succeed. Therefore, the potential of partially restoring CD8+ cells function by blocking PD-1/PD-L1 interaction could provide an additional tool to enhance available therapies to cure chronic HCV.
CONCLUSION
In spite of much progress in our understanding of T cell responses against HCV over the past decade, many critical research questions remain to be answered. It will be important to determine whether there is a causal correlation between the outcome of HCV infection and PD-1 expression. Functionally deficient exhausted HCV specific T cells are a significant cause and outcome of chronic viral infection. This is a great challenge to vaccine designs that either aim to eliminate or prevent such infections by interceding the T cell response. Whether developing a therapeutic vaccine to eliminate disease in infected patients or a prophylactic vaccine to prevent HCV in healthy individuals, it is imperative to keep this considerable obstacle in mind.
The possibility of restoring the function of HCV specific T cells by blocking the PD-1/PDL-1 pathway and reverting T-cell dysfunction in chronic HCV seems a worthy direction for anti-HCV immunotherapy. While the therapeutic application of this strategy in HCV infection is constrained by the recent, ongoing development of highly efficacious new treatments, it is promising that further investigation of PD-1 pathway blockade during antiviral therapies is warranted.
However, the systemic administration of PD-L1/PD-1 blocking antibodies carries the high risk of violating peripheral tolerance. Most tissues depend on PD-L1 expression to decrease T-cell effector activities that might cause autoimmune attack. To demonstrate this, knocking out PD-1 or PD-L1 pathways in mouse models causes severe, deadly autoimmunity[79,80]. In humans, single-nucleotide polymorphisms (SNP) of the PDCD1 gene (encoding PD-1) are connected with systemic lupus erythematosus[81]. Therefore, efforts have to be made to enhance this promising strategy to maximize anti-HCV therapeutic activities while minimizing toxicity. We propose that one possible attractive alternative to the systemic blockade of PD-1 for HCV immunotherapy is to target the suppression of PD-1/PD-L1 co-stimulation during antigen presentation. This was demonstrated in mouse antigen presenting cells (APCs)[82,83]. This local and transient blockade may provide the positive effects needed to adequately boost anti-HCV immunity while restricting possible side effects to a minimum.
Overall, continuing work to understand better how the PD-1/PD-L1 pathway functions is imperative and will encourage development of new vaccination approaches that can overcome HCV specific T cell exhaustion.
Footnotes
P- Reviewer: Jin B S- Editor: Ma YJ L- Editor: A E- Editor: Liu SQ
Supported by Science and Technology Development Fund (STDF; grants No. 1469 and No. 5245); and Tanta University Fund, Egypt to Mohamed L Salem, the Principal investigator of these projects.
Conflict-of-interest statement: We declare that authors have no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: April 15, 2015
First decision: June 2, 2015
Article in press: September 7, 2015
References
- 1.Weiss RA, McMichael AJ. Social and environmental risk factors in the emergence of infectious diseases. Nat Med. 2004;10:S70–S76. doi: 10.1038/nm1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alter MJ. Epidemiology of hepatitis C in the West. Semin Liver Dis. 1995;15:5–14. doi: 10.1055/s-2007-1007259. [DOI] [PubMed] [Google Scholar]
- 3.Seeff LB. Natural history of chronic hepatitis C. Hepatology. 2002;36:S35–S46. doi: 10.1053/jhep.2002.36806. [DOI] [PubMed] [Google Scholar]
- 4.Huang L, Koziel MJ. Immunology of hepatitis C virus infection. Curr Opin Gastroenterol. 2000;16:558–564. doi: 10.1097/00001574-200011000-00017. [DOI] [PubMed] [Google Scholar]
- 5.Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005;5:215–229. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- 6.Dustin LB, Rice CM. Flying under the radar: the immunobiology of hepatitis C. Annu Rev Immunol. 2007;25:71–99. doi: 10.1146/annurev.immunol.25.022106.141602. [DOI] [PubMed] [Google Scholar]
- 7.Shin EC, Capone S, Cortese R, Colloca S, Nicosia A, Folgori A, Rehermann B. The kinetics of hepatitis C virus-specific CD8 T-cell responses in the blood mirror those in the liver in acute hepatitis C virus infection. J Virol. 2008;82:9782–9788. doi: 10.1128/JVI.00475-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shin EC, Seifert U, Kato T, Rice CM, Feinstone SM, Kloetzel PM, Rehermann B. Virus-induced type I IFN stimulates generation of immunoproteasomes at the site of infection. J Clin Invest. 2006;116:3006–3014. doi: 10.1172/JCI29832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Folgori A, Capone S, Ruggeri L, Meola A, Sporeno E, Ercole BB, Pezzanera M, Tafi R, Arcuri M, Fattori E, et al. A T-cell HCV vaccine eliciting effective immunity against heterologous virus challenge in chimpanzees. Nat Med. 2006;12:190–197. doi: 10.1038/nm1353. [DOI] [PubMed] [Google Scholar]
- 10.Shin EC, Rehermann B. Taking the brake off T cells in chronic viral infection. Nat Med. 2006;12:276–277. doi: 10.1038/nm0306-276. [DOI] [PubMed] [Google Scholar]
- 11.Kanto T, Hayashi N. Immunopathogenesis of hepatitis C virus infection: multifaceted strategies subverting innate and adaptive immunity. Intern Med. 2006;45:183–191. doi: 10.2169/internalmedicine.45.1530. [DOI] [PubMed] [Google Scholar]
- 12.Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 13.Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 14.Radziewicz H, Ibegbu CC, Fernandez ML, Workowski KA, Obideen K, Wehbi M, Hanson HL, Steinberg JP, Masopust D, Wherry EJ, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. 2007;81:2545–2553. doi: 10.1128/JVI.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Penna A, Pilli M, Zerbini A, Orlandini A, Mezzadri S, Sacchelli L, Missale G, Ferrari C. Dysfunction and functional restoration of HCV-specific CD8 responses in chronic hepatitis C virus infection. Hepatology. 2007;45:588–601. doi: 10.1002/hep.21541. [DOI] [PubMed] [Google Scholar]
- 16.Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, Ferrari C. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol. 2006;80:11398–11403. doi: 10.1128/JVI.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kasprowicz V, Schulze Zur Wiesch J, Kuntzen T, Nolan BE, Longworth S, Berical A, Blum J, McMahon C, Reyor LL, Elias N, et al. High level of PD-1 expression on hepatitis C virus (HCV)-specific CD8+ and CD4+ T cells during acute HCV infection, irrespective of clinical outcome. J Virol. 2008;82:3154–3160. doi: 10.1128/JVI.02474-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hadziyannis SJ, Sette H, Morgan TR, Balan V, Diago M, Marcellin P, Ramadori G, Bodenheimer H, Bernstein D, Rizzetto M, et al. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med. 2004;140:346–355. doi: 10.7326/0003-4819-140-5-200403020-00010. [DOI] [PubMed] [Google Scholar]
- 19.Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht JK. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358:958–965. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- 20.McHutchison JG, Lawitz EJ, Shiffman ML, Muir AJ, Galler GW, McCone J, Nyberg LM, Lee WM, Ghalib RH, Schiff ER, et al. Peginterferon alfa-2b or alfa-2a with ribavirin for treatment of hepatitis C infection. N Engl J Med. 2009;361:580–593. doi: 10.1056/NEJMoa0808010. [DOI] [PubMed] [Google Scholar]
- 21.Shiffman ML, Suter F, Bacon BR, Nelson D, Harley H, Solá R, Shafran SD, Barange K, Lin A, Soman A, et al. Peginterferon alfa-2a and ribavirin for 16 or 24 weeks in HCV genotype 2 or 3. N Engl J Med. 2007;357:124–134. doi: 10.1056/NEJMoa066403. [DOI] [PubMed] [Google Scholar]
- 22.Parise E, Cheinquer H, Crespo D, Meirelles A, Martinelli A, Sette H, Gallizi J, Silva R, Lacet C, Correa E, et al. Peginterferon alfa-2a (40KD) (PEGASYS) plus ribavirin (COPEGUS) in retreatment of chronic hepatitis C patients, nonresponders and relapsers to previous conventional interferon plus ribavirin therapy. Braz J Infect Dis. 2006;10:11–16. doi: 10.1590/s1413-86702006000100003. [DOI] [PubMed] [Google Scholar]
- 23.Muir AJ, Bornstein JD, Killenberg PG. Peginterferon alfa-2b and ribavirin for the treatment of chronic hepatitis C in blacks and non-Hispanic whites. N Engl J Med. 2004;350:2265–2271. doi: 10.1056/NEJMoa032502. [DOI] [PubMed] [Google Scholar]
- 24.Poynard T, Ratziu V, McHutchison J, Manns M, Goodman Z, Zeuzem S, Younossi Z, Albrecht J. Effect of treatment with peginterferon or interferon alfa-2b and ribavirin on steatosis in patients infected with hepatitis C. Hepatology. 2003;38:75–85. doi: 10.1053/jhep.2003.50267. [DOI] [PubMed] [Google Scholar]
- 25.Gao B, Hong F, Radaeva S. Host factors and failure of interferon-alpha treatment in hepatitis C virus. Hepatology. 2004;39:880–890. doi: 10.1002/hep.20139. [DOI] [PubMed] [Google Scholar]
- 26.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 27.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci USA. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M, Lemon SM. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci USA. 2005;102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bode JG, Ludwig S, Ehrhardt C, Albrecht U, Erhardt A, Schaper F, Heinrich PC, Häussinger D. IFN-alpha antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J. 2003;17:488–490. doi: 10.1096/fj.02-0664fje. [DOI] [PubMed] [Google Scholar]
- 30.Hézode C, Fontaine H, Dorival C, Larrey D, Zoulim F, Canva V, de Ledinghen V, Poynard T, Samuel D, Bourlière M, et al. Triple therapy in treatment-experienced patients with HCV-cirrhosis in a multicentre cohort of the French Early Access Programme (ANRS CO20-CUPIC) - NCT01514890. J Hepatol. 2013;59:434–441. doi: 10.1016/j.jhep.2013.04.035. [DOI] [PubMed] [Google Scholar]
- 31.Ward RP, Kugelmas M. Using pegylated interferon and ribavirin to treat patients with chronic hepatitis C. Am Fam Physician. 2005;72:655–662. [PubMed] [Google Scholar]
- 32.Bruchfeld A, Lindahl K, Ståhle L, Söderberg M, Schvarcz R. Interferon and ribavirin treatment in patients with hepatitis C-associated renal disease and renal insufficiency. Nephrol Dial Transplant. 2003;18:1573–1580. doi: 10.1093/ndt/gfg209. [DOI] [PubMed] [Google Scholar]
- 33.Wen Y, feng LY, Jing Y, Xin L, Zhikai X. Controlling hepatitis C with immunotherapy. Lancet Infect Dis. 2009;9:652–653. doi: 10.1016/S1473-3099(09)70258-0. [DOI] [PubMed] [Google Scholar]
- 34.Klade CS, Wedemeyer H, Berg T, Hinrichsen H, Cholewinska G, Zeuzem S, Blum H, Buschle M, Jelovcan S, Buerger V, et al. Therapeutic vaccination of chronic hepatitis C nonresponder patients with the peptide vaccine IC41. Gastroenterology. 2008;134:1385–1395. doi: 10.1053/j.gastro.2008.02.058. [DOI] [PubMed] [Google Scholar]
- 35.Salem ML, El-Demellawy M, El-Azm AR. The potential use of Toll-like receptor agonists to restore the dysfunctional immunity induced by hepatitis C virus. Cell Immunol. 2010;262:96–104. doi: 10.1016/j.cellimm.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 36.Penaloza-MacMaster P, Kamphorst AO, Wieland A, Araki K, Iyer SS, West EE, O’Mara L, Yang S, Konieczny BT, Sharpe AH, et al. Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J Exp Med. 2014;211:1905–1918. doi: 10.1084/jem.20132577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 39.Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116–126. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 40.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336–347. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 41.Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, Sharpe AH. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33:2706–2716. doi: 10.1002/eji.200324228. [DOI] [PubMed] [Google Scholar]
- 42.Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci USA. 2003;100:5336–5341. doi: 10.1073/pnas.0931259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fife BT, Pauken KE. The role of the PD-1 pathway in autoimmunity and peripheral tolerance. Ann N Y Acad Sci. 2011;1217:45–59. doi: 10.1111/j.1749-6632.2010.05919.x. [DOI] [PubMed] [Google Scholar]
- 46.Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–242. doi: 10.1111/j.1600-065X.2010.00923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gruener NH, Lechner F, Jung MC, Diepolder H, Gerlach T, Lauer G, Walker B, Sullivan J, Phillips R, Pape GR, et al. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J Virol. 2001;75:5550–5558. doi: 10.1128/JVI.75.12.5550-5558.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lechner F, Wong DK, Dunbar PR, Chapman R, Chung RT, Dohrenwend P, Robbins G, Phillips R, Klenerman P, Walker BD. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med. 2000;191:1499–1512. doi: 10.1084/jem.191.9.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thimme R, Oldach D, Chang KM, Steiger C, Ray SC, Chisari FV. Determinants of viral clearance and persistence during acute hepatitis C virus infection. J Exp Med. 2001;194:1395–1406. doi: 10.1084/jem.194.10.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Urbani S, Boni C, Missale G, Elia G, Cavallo C, Massari M, Raimondo G, Ferrari C. Virus-specific CD8+ lymphocytes share the same effector-memory phenotype but exhibit functional differences in acute hepatitis B and C. J Virol. 2002;76:12423–12434. doi: 10.1128/JVI.76.24.12423-12434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Urbani S, Amadei B, Fisicaro P, Tola D, Orlandini A, Sacchelli L, Mori C, Missale G, Ferrari C. Outcome of acute hepatitis C is related to virus-specific CD4 function and maturation of antiviral memory CD8 responses. Hepatology. 2006;44:126–139. doi: 10.1002/hep.21242. [DOI] [PubMed] [Google Scholar]
- 52.Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, Greenfield EA, Freeman GJ. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257–1266. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- 53.Cai G, Karni A, Oliveira EM, Weiner HL, Hafler DA, Freeman GJ. PD-1 ligands, negative regulators for activation of naive, memory, and recently activated human CD4+ T cells. Cell Immunol. 2004;230:89–98. doi: 10.1016/j.cellimm.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 54.Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci USA. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okazaki T, Honjo T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006;27:195–201. doi: 10.1016/j.it.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 56.Riley JL, June CH. The road to recovery: translating PD-1 biology into clinical benefit. Trends Immunol. 2007;28:48–50. doi: 10.1016/j.it.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 57.Golden-Mason L, Klarquist J, Wahed AS, Rosen HR. Cutting edge: programmed death-1 expression is increased on immunocytes in chronic hepatitis C virus and predicts failure of response to antiviral therapy: race-dependent differences. J Immunol. 2008;180:3637–3641. doi: 10.4049/jimmunol.180.6.3637. [DOI] [PubMed] [Google Scholar]
- 58.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 59.Selenko-Gebauer N, Majdic O, Szekeres A, Höfler G, Guthann E, Korthäuer U, Zlabinger G, Steinberger P, Pickl WF, Stockinger H, et al. B7-H1 (programmed death-1 ligand) on dendritic cells is involved in the induction and maintenance of T cell anergy. J Immunol. 2003;170:3637–3644. doi: 10.4049/jimmunol.170.7.3637. [DOI] [PubMed] [Google Scholar]
- 60.Nakamoto N, Kaplan DE, Coleclough J, Li Y, Valiga ME, Kaminski M, Shaked A, Olthoff K, Gostick E, Price DA, et al. Functional restoration of HCV-specific CD8 T cells by PD-1 blockade is defined by PD-1 expression and compartmentalization. Gastroenterology. 2008;134:1927–1937, 1937.e1-2. doi: 10.1053/j.gastro.2008.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boettler T, Panther E, Bengsch B, Nazarova N, Spangenberg HC, Blum HE, Thimme R. Expression of the interleukin-7 receptor alpha chain (CD127) on virus-specific CD8+ T cells identifies functionally and phenotypically defined memory T cells during acute resolving hepatitis B virus infection. J Virol. 2006;80:3532–3540. doi: 10.1128/JVI.80.7.3532-3540.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rutebemberwa A, Ray SC, Astemborski J, Levine J, Liu L, Dowd KA, Clute S, Wang C, Korman A, Sette A, et al. High-programmed death-1 levels on hepatitis C virus-specific T cells during acute infection are associated with viral persistence and require preservation of cognate antigen during chronic infection. J Immunol. 2008;181:8215–8225. doi: 10.4049/jimmunol.181.12.8215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Golden-Mason L, Palmer BE, Kassam N, Townshend-Bulson L, Livingston S, McMahon BJ, Castelblanco N, Kuchroo V, Gretch DR, Rosen HR. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol. 2009;83:9122–9130. doi: 10.1128/JVI.00639-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bengsch B, Seigel B, Ruhl M, Timm J, Kuntz M, Blum HE, Pircher H, Thimme R. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. 2010;6:e1000947. doi: 10.1371/journal.ppat.1000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kroy DC, Ciuffreda D, Cooperrider JH, Tomlinson M, Hauck GD, Aneja J, Berger C, Wolski D, Carrington M, Wherry EJ, et al. Liver environment and HCV replication affect human T-cell phenotype and expression of inhibitory receptors. Gastroenterology. 2014;146:550–561. doi: 10.1053/j.gastro.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, Rosen HR. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J Virol. 2007;81:9249–9258. doi: 10.1128/JVI.00409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, Laccabue D, Zerbini A, Cavalli A, Missale G, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol. 2007;81:4215–4225. doi: 10.1128/JVI.02844-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 69.Velu V, Kannanganat S, Ibegbu C, Chennareddi L, Villinger F, Freeman GJ, Ahmed R, Amara RR. Elevated expression levels of inhibitory receptor programmed death 1 on simian immunodeficiency virus-specific CD8 T cells during chronic infection but not after vaccination. J Virol. 2007;81:5819–5828. doi: 10.1128/JVI.00024-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ou R, Zhou S, Huang L, Moskophidis D. Critical role for alpha/beta and gamma interferons in persistence of lymphocytic choriomeningitis virus by clonal exhaustion of cytotoxic T cells. J Virol. 2001;75:8407–8423. doi: 10.1128/JVI.75.18.8407-8423.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boutboul F, Puthier D, Appay V, Pellé O, Ait-Mohand H, Combadière B, Carcelain G, Katlama C, Rowland-Jones SL, Debré P, et al. Modulation of interleukin-7 receptor expression characterizes differentiation of CD8 T cells specific for HIV, EBV and CMV. AIDS. 2005;19:1981–1986. doi: 10.1097/01.aids.0000191919.24185.46. [DOI] [PubMed] [Google Scholar]
- 72.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Salama AD, Chitnis T, Imitola J, Ansari MJ, Akiba H, Tushima F, Azuma M, Yagita H, Sayegh MH, Khoury SJ. Critical role of the programmed death-1 (PD-1) pathway in regulation of experimental autoimmune encephalomyelitis. J Exp Med. 2003;198:71–78. doi: 10.1084/jem.20022119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fuller MJ, Callendret B, Zhu B, Freeman GJ, Hasselschwert DL, Satterfield W, Sharpe AH, Dustin LB, Rice CM, Grakoui A, et al. Immunotherapy of chronic hepatitis C virus infection with antibodies against programmed cell death-1 (PD-1) Proc Natl Acad Sci USA. 2013;110:15001–15006. doi: 10.1073/pnas.1312772110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gardiner D, Lalezari J, Lawitz E, DiMicco M, Ghalib R, Reddy KR, Chang KM, Sulkowski M, Marro SO, Anderson J, et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS One. 2013;8:e63818. doi: 10.1371/journal.pone.0063818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen L, Zhang Z, Chen W, Zhang Z, Li Y, Shi M, Zhang J, Chen L, Wang S, Wang FS. B7-H1 up-regulation on myeloid dendritic cells significantly suppresses T cell immune function in patients with chronic hepatitis B. J Immunol. 2007;178:6634–6641. doi: 10.4049/jimmunol.178.10.6634. [DOI] [PubMed] [Google Scholar]
- 77.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9:562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 78.Harari A, Dutoit V, Cellerai C, Bart PA, Du Pasquier RA, Pantaleo G. Functional signatures of protective antiviral T-cell immunity in human virus infections. Immunol Rev. 2006;211:236–254. doi: 10.1111/j.0105-2896.2006.00395.x. [DOI] [PubMed] [Google Scholar]
- 79.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 80.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 81.Prokunina L, Castillejo-López C, Oberg F, Gunnarsson I, Berg L, Magnusson V, Brookes AJ, Tentler D, Kristjansdóttir H, Gröndal G, et al. A regulatory polymorphism in PDCD1 is associated with susceptibility to systemic lupus erythematosus in humans. Nat Genet. 2002;32:666–669. doi: 10.1038/ng1020. [DOI] [PubMed] [Google Scholar]
- 82.Song MY, Park SH, Nam HJ, Choi DH, Sung YC. Enhancement of vaccine-induced primary and memory CD8(+) T-cell responses by soluble PD-1. J Immunother. 2011;34:297–306. doi: 10.1097/CJI.0b013e318210ed0e. [DOI] [PubMed] [Google Scholar]
- 83.He YF, Zhang GM, Wang XH, Zhang H, Yuan Y, Li D, Feng ZH. Blocking programmed death-1 ligand-PD-1 interactions by local gene therapy results in enhancement of antitumor effect of secondary lymphoid tissue chemokine. J Immunol. 2004;173:4919–4928. doi: 10.4049/jimmunol.173.8.4919. [DOI] [PubMed] [Google Scholar]
- 84.Riley JL. PD-1 signaling in primary T cells. Immunol Rev. 2009;229:114–125. doi: 10.1111/j.1600-065X.2009.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]