

Abstract
Hepatocellular carcinoma (HCC) is one of the most common malignancies worldwide. The main etiologies of HCC are hepatitis B virus and hepatitis C virus (HCV), and non-hepatitis B/non-hepatitis C HCC (NBNC-HCC) has also been identified as an etiological factor. Although the incidence of HCV-related HCC in Japan has decreased slightly in recent years, that of NBNC-HCC has increased. The onset mechanism of NBNC-HCC, which has various etiologies, remains unclear; however, nonalcoholic steatohepatitis (NASH), a severe form of nonalcoholic fatty liver disease, is known to be an important risk factor for NBNC-HCC. Among the different advanced glycation end-products (AGEs) formed by the Maillard reaction, glyceraldehyde-derived AGEs, the predominant components of toxic AGEs (TAGE), have been associated with NASH and NBNC-HCC, including NASH-related HCC. Furthermore, the expression of the receptor for AGEs (RAGE) has been correlated with the malignant progression of HCC. Therefore, TAGE induce oxidative stress by binding with RAGE may, in turn, lead to adverse effects, such as fibrosis and malignant transformation, in hepatic stellate cells and tumor cells during NASH or NASH-related HCC progression. The aim of this review was to examine the contribution of the TAGE-RAGE axis in NASH-related HCC.
Keywords: Hepatocellular carcinoma, Nonalcoholic steatohepatitis, Advanced glycation end-products, Toxic advanced glycation end-products, Receptor for advanced glycation end-products, Hepatic stellate cells
Core tip: Expression of the receptor for advanced glycation end-products (RAGE), which is a multi-ligand cell surface receptor, is correlated with the poor therapeutic outcomes and malignancy of hepatocellular carcinoma (HCC). The synthesis of toxic advanced glycation end-products (TAGE), ligands of RAGE, is increased in nonalcoholic steatohepatitis (NASH) as well as in NASH-related HCC. Interactions between TAGE and RAGE induce oxidative stress, which may, in turn, lead to adverse effects in tumor cells and hepatic stellate cells during NASH or NASH-related HCC progression. Therefore, these findings prompted us to suggest that the TAGE-RAGE axis may be a treatment target in NASH-related HCC.
INTRODUCTION
Hepatocellular carcinoma (HCC), which accounts for approximately 90% of all primary liver cancers, is one of the most common malignancies in men and women, and is the third leading cause of cancer-related mortality worldwide[1-3]. Hepatitis B virus (HBV) and hepatitis C virus (HCV) are known to be the main risk factors for HCC, accounting for over 75% of HCC worldwide[4]. The incidence of HCC is particularly high in Asia[3,4], and HBV infection is endemic in eastern/south-eastern Asia, while HCV infection is prevalent in Japan[5-7]. Among diagnosed HCC patients in Japan in 2006-2009, 84.1% had virus-related HCC (HCV: 66.3%, HBV: 14.1%, HCV + HBV: 3.7%) and 15.9% had non-hepatitis B/non-hepatitis C HCC (NBNC-HCC) [alcoholic: 7.2%, etiology unknown: 5.1%, nonalcoholic fatty liver disease (NAFLD): 2.0%, Others: 1.6%][7,8]. Although the incidence of HCV-related HCC has decreased slightly in recent years, that of NBNC-HCC has increased[9,10]. Nonalcoholic steatohepatitis (NASH), a severe form of NAFLD, has been identified as an important risk factor among the etiological factors of NBNC-HCC. The incidence of NASH-related HCC is expected to increase in the future as the number of patients with NAFLD is increasing worldwide.
Advanced glycation end-products (AGEs) formed by the Maillard reaction, a nonenzymatic reaction between the ketone or aldehyde groups of sugars and the amino groups of proteins, have been implicated in aging and diabetes-related pathological complications[11,12]. This reaction begins with the conversion of reversible Schiff base adducts to more stable covalently bound Amadori rearrangement products. Over the course of days to weeks, these Amadori products undergo further rearrangement reactions to form irreversibly bound moieties known as AGEs[13]. Recent studies have suggested that AGEs are formed not only from sugars, but also from carbonyl compounds produced as a result of the autoxidation of sugars and from other metabolic pathways[14,15]. There is evidence to suggest that glyceraldehyde-derived AGEs (Glycer-AGEs), the predominant components of toxic AGEs (TAGE), are closely associated with insulin resistance, obesity, hypertension, diabetes complications, cardiovascular diseases, dementia, NASH, and cancer[16-24]. We recently demonstrated that TAGE were present at significantly higher concentrations in the sera of patients with NASH than in those with simple steatosis or healthy controls, and that TAGE accumulated in the livers of patients with NASH[25]. Extracellular TAGE induce oxidative stress by binding with the receptor for AGEs (RAGE), which, in turn, causes adverse effects in various types of cells[26-31]. The TAGE-RAGE axis has been shown to increase the malignancy of various types of cancer cells[18,32-35].
These findings suggest that TAGE play an important role in the development and progression of NASH and NASH-related HCC. In this review, we discuss the contribution of the TAGE-RAGE axis in NASH-related HCC.
BACKGROUND OF NASH AND NBNC-HCC
NAFLD, which is the most common liver disease worldwide, is a disease that ranges from simple steatosis to NASH[36-41]. Approximately 20%-30% of the population has evidence of fatty liver disease attributed to NAFLD, and approximately 10% of patients with NAFLD progress to NASH[42]. NASH, which is a disease that has the typical histopathological findings of alcoholic liver disease in patients without a history of significant alcohol abuse, is recognized as a component of the metabolic syndrome and has been closely associated with insulin resistance as well as glucose and lipid metabolic disorders[43-46]. Although simple steatosis appears to be a benign and non-progressive condition, NASH is a potentially progressive disease that can lead to fibrosis, cirrhosis, and HCC[47,48]. Approximately 8%-26% of patients with NASH progress to cirrhosis, and approximately 10% of patients with cirrhotic NASH transform to HCC after 5 years[47,49]. Several case series have recently been published on NASH-related HCC[50,51]. Furthermore, NASH was shown to increase the risk of HCC without the development of cirrhosis[52]. While NASH is a risk factor for HCC, the cirrhosis caused by NASH is also considered to be an important risk factor.
The etiology of NBNC-HCC is often cryptogenic cirrhosis (CC). Most cases of CC are considered to be end-stage NASH because the prevalence of obesity and diabetes among patients with CC is similar to that of patients with NASH[53]. In addition, patients who undergo orthotopic liver transplantation for CC often develop NAFLD and NASH after transplant[54]. However, the histopathological features of NASH often disappear when cirrhosis is established[55]. Marrero et al[56] reported that HCV (51%) and CC (29%) were the first and second most common etiologies among 105 patients with HCC in the United States, respectively, that 50% of patients with CC had a prior histological diagnosis of NASH or clinical features associated with NAFLD, and that NAFLD-related CC accounted for 13% of patients with HCC. These findings suggested the existence of NASH-related HCC.
ALTERNATIVE ROUTES FOR THE FORMATION OF AGES IN VIVO
The formation of AGEs, which occurs through a non-enzymatic glycation reaction, is known to result from not only glucose, but also the actions of various metabolites that are primarily located intracellularly[13,15,57].
We previously reported the contribution of fructose, α-hydroxyaldehydes (glyceraldehyde and glycolaldehyde), and dicarbonyl compounds (methylglyoxal, glyoxal, and 3-deoxyglucosone) as well as glucose in the glycation of proteins. Seven immunochemically distinct classes of AGEs (Glu-AGEs, glucose-derived AGEs; Fru-AGEs, fructose-derived AGEs; Glycer-AGEs, glyceraldehyde-derived AGEs; Glycol-AGEs, glycolaldehyde-derived AGEs; MGO-AGEs, methylglyoxal-derived AGEs; GO-AGEs, glyoxal-derived AGEs; and 3-DG-AGEs, 3-deoxyglucosone-derived AGEs) were detected in the sera of type 2 diabetic subjects undergoing hemodialysis[13,58-61]. These findings suggested that all seven forms of AGEs were synthesized in vivo (Figure 1).
Figure 1.
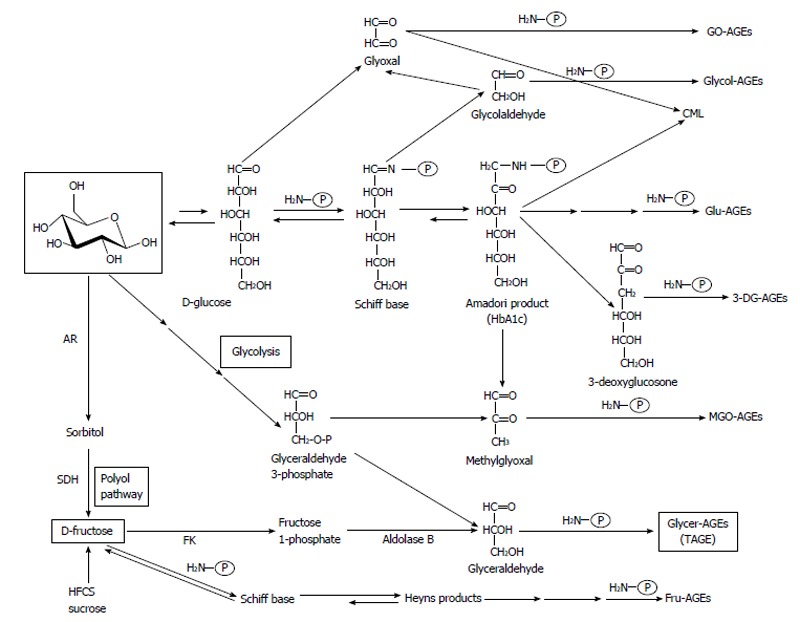
Alternative routes for the formation of advanced glycation end-products in vivo. Reducing sugars, such as glucose, fructose, and glyceraldehyde are known to react non-enzymatically with the amino groups of proteins to form reversible Schiff bases and Amadori product/Heyns products. These early glycation products undergo further complex reactions such as rearrangement, dehydration, and condensation to become irreversibly cross-linked, heterogeneous fluorescent derivatives, termed advanced glycation end-products (AGEs). Glu-AGEs: Glucose-derived AGEs; Fru-AGEs: Fructose-derived AGEs; Glycer-AGEs: Glyceraldehyde-derived AGEs; Glycol-AGEs: Glycolaldehyde-derived AGEs; MGO-AGEs: Methylglyoxal-derived AGEs; GO-AGEs: Glyoxal-derived AGEs; 3-DG-AGEs: 3-deoxyglucosone-derived AGEs; CML: Nε-(carboxymethyl)lysine; P-NH2: Free amino residue of a protein; AR: Aldose reductase; SDH: Sorbitol dehydrogenase; FK: Fructokinase; HFCS: High-fructose corn syrup; HbA1c: Hemoglobin A1c; TAGE: Toxic advanced glycation end-products.
PATHWAY FOR THE IN VIVO FORMATION OF TAGE
Glyceraldehyde, a precursor of TAGE, is produced by two pathways (the glycolytic pathway and fructose metabolic pathway)[17,21,22,24,62]. In the glycolytic pathway (glycolysis), the intermediate glyceraldehyde-3-phosphate (G-3-P) is metabolized by glyceraldehyde-3-phosphate dehydrogenase (GAPDH), G-3-P accumulates intracellularly due to a decrease in GAPDH enzyme activity. Accumulated G-3-P then shifts to another metabolic route, and the amount of glyceraldehyde is increased. In the fructose metabolic pathway (fructolysis), fructose is mainly metabolized in the liver. Fructose is phosphorylated to fructose-1-phosphate (F-1-P) by a fructokinase, and liver aldolase B cleaves F-1-P to produce dihydroxyacetone phosphate and glyceraldehyde. The accumulated glyceraldehyde due to a metabolic disorder is then transported or leaks passively across the plasma membrane, thereby promoting the intracellular and extracellular formation of TAGE (Figure 2).
Figure 2.
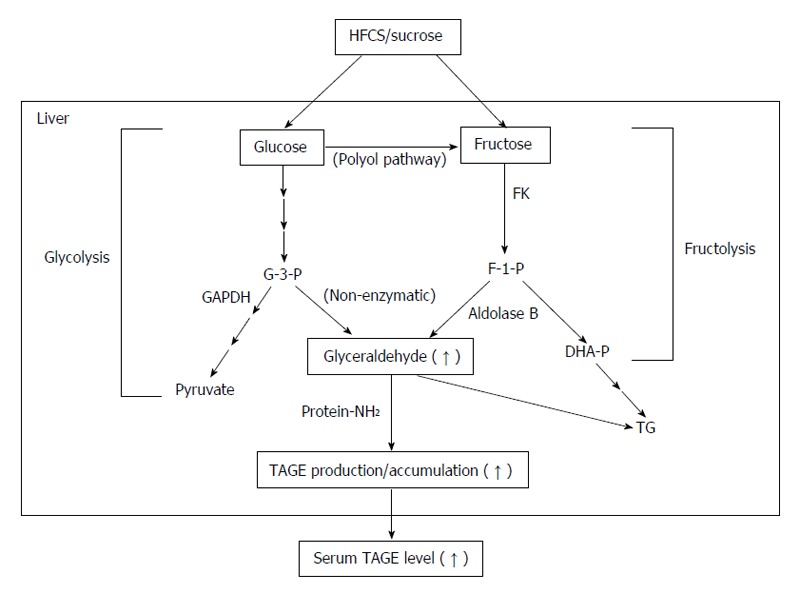
In vivo production routes of glycer-advanced glycation end-products (toxic advanced glycation end-products). The chronic and excessive ingestion of sugar-sweetened beverages (HFCS/sucrose) increases the levels of the sugar metabolite, glyceraldehyde in the liver. The glycolytic intermediate glyceraldehyde-3-phosphate (G-3-P) is normally catabolized (glycolysis) by the enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH). G-3-P accumulates intracellularly with a decline in GAPDH activity. The metabolism of G-3-P then shifts to another route, resulting in an increase in the amount of glyceraldehyde, which promotes the formation of glycer-advanced glycation end-products (AGEs) (TAGE). Fructose from the daily diet and polyol pathway is phosphorylated to fructose-1-phosphate (F-1-P) by fructokinase and is then catabolized to glyceraldehyde and dihydroxyacetone phosphate by aldolase B (fructolysis). The newly synthesized glyceraldehyde is then transported or leaks passively across the plasma membrane. Glyceraldehyde promotes the formation of TAGE both intracellularly and extracellularly. DHA-P: Dihydroxyacetone-phosphate; FK: Fructokinase; HFCS: High-fructose corn syrup; TAGE: Toxic advanced glycation end-products; TG: Triglyceride; Protein-NH2: Free amino residue of a protein.
SERUM TAGE LEVELS IN NASH AND NBNC-HCC
AGEs were originally characterized by their ability to form cross-links with and between amino groups and have a yellow brown fluorescent color. However, this term is now used for a broad range of advanced products of the glycation process, including Nε-(carboxymethyl)lysine (CML), Nε-(carboxyethyl)lysine, and pyrraline, which are not cross-linked proteins and do not have color or fluorescence[13,63-67]. CML was previously shown to be formed from precursors such as glycolaldehyde and glyoxal via the intra-molecular Cannizzaro reaction, a process that is largely independent of glucose autoxidation[14]. CML may also be formed independently of the presence of fructose-lysine during the metal-catalyzed oxidation of low-density lipoproteins and peroxidation of polyunsaturated fatty acids[68]. A recently supported concept is that CML is a marker of oxidation rather than glycation.
Sebeková et al[69] initially suggested that the catabolism and clearance of circulating CML was impaired by various liver diseases. In their study, plasma CML levels were measured in 51 patients with liver cirrhosis (five of whom were followed for 36 mo after liver transplantation) and 19 healthy controls. The main findings obtained were that: (1) plasma CML levels were markedly elevated in patients with liver cirrhosis and positively correlated with severity of the disease; (2) plasma CML levels were inversely associated with residual liver function in patients, as estimated by serum albumin and plasma bilirubin levels; and (3) plasma CML levels were markedly decreased (to approximately 50% of those before treatment) within 3 mo of liver transplantation. These findings suggested that the liver may play an important role in the removal of circulating CML and that the hepatic clearance of circulating CML may be impaired due to liver cirrhosis. Yagmur et al[70] also reported that serum CML levels were significantly higher in patients with liver cirrhosis than in patients without cirrhosis, and were positively associated with the severity of cirrhosis defined by the Child-Pugh score. These findings suggested that circulating CML levels may be a useful biomarker for evaluating residual liver function. However, Moy et al[71] recently reported that serum CML levels were inversely correlated with the risk of HCC. They measured serum CML levels in 145 patients with HCC and 340 control patients, who were male Finnish smokers, and found that high serum CML levels correlated with a lower risk of HCC. Furthermore, this relationship did not change in the case of NBNC-HCC. Therefore, the relationship between circulating CML levels and liver disease needs to be examined in more detail in future studies.
Our clinical data indicated that TAGE played a role in the etiology of NASH and that serum TAGE levels were a useful clinical tool for discriminating between NASH-related HCC, NASH, and simple steatosis[25,72,73]. We measured serum AGE levels (CML, Glu-AGEs, and TAGE) in 66 NASH patients without cirrhosis, 10 patients with simple steatosis, and 30 control patients. We found that serum TAGE levels were significantly higher in NASH patients than in those with simple steatosis and the controls; however, no significant difference was observed in CML and Glu-AGE levels between the groups[25]. We measured serum TAGE levels in 43 NASH patients with dyslipidemia in order to determine whether they played a role in the treatment of NASH. Serum TAGE levels were measured and clinical laboratory tests were performed periodically during the administration of atorvastatin (10 mg daily), a hydroxymethylglutaryl-CoA reductase inhibitor, for 12 mo. This treatment significantly decreased serum TAGE levels, and significantly improved biochemical and histological findings[72]. We also measured serum TAGE levels in 90 patients with NBNC-HCC, 56 NASH patients without HCC, and 27 control patients, and found that serum TAGE levels were significantly higher in NBNC-HCC patients than in those with NASH without HCC and controls. Among the patients with NBNC-HCC, 10 had NASH-related HCC, 49 alcoholic-related HCC, and 31 etiology unknown HCC, and no significant differences were observed in serum TAGE levels between these groups[73]. These findings suggested that the formation of TAGE, but not CML, was enhanced by the development and progression of NASH, and that enhanced TAGE may influence the development and progression of NBNC-HCC. Brenner et al[74] previously reported that increases in AGE-RAGE-mediated inflammation in patients with end-stage liver diseases including HCC following liver transplantation were dependent on reactive carbonyl species-derived AGEs, but not CML. Therefore, TAGE may play a more important role in the development and progression of NASH-related HCC than CML[24,75].
RAGE EXPRESSION IN HCC
RAGE, a multi-ligand cell surface receptor, interacts with distinct molecules that have been implicated in homeostasis, development, and inflammation. RAGE binding by ligands such as AGEs, high mobility group box 1, and S100/calgranulins has been shown to trigger the activation of key cell signaling pathways, thereby reprogramming cellular properties[76]. Previous studies suggested that the expression of RAGE was associated with the malignant progression of cancer [77-80].
In the liver, RAGE is expressed in hepatocytes and hepatic stellate cells (HSCs)[81,82]. Liver damage caused by various factors such as inflammation, drugs, and hepatic ischemia/reperfusion (I/R) is known to increase the expression of RAGE, which induces further liver failure[83-89]. For example, Kuhla et al[89] reported that exposure to galactosamine/lipopolysaccharides induced RAGE expression, leading to inflammation, and a pre- or post-treatment with an anti-RAGE antibody attenuated enhanced inflammation, apoptosis, and necrosis. Therefore, RAGE is crucially involved in the exacerbation of liver disease and may also play a role in HCC, which occurs following liver failure. A previous study reported that the expression of RAGE mRNA was higher in the liver cells of hepatitis and HCC patients than in normal liver cells[81], and RAGE expression correlated with the poor therapeutic outcomes and malignancy of HCC[90,91]. Ito et al[90] investigated the relationship between RAGE expression and clinical outcomes in 65 patients who underwent initial hepatectomy for HCC. The number of patients that expressed RAGE was significantly higher among those with well and poorly differentiated HCC than those with moderately differentiated HCC, and the 5-year survival rate was significantly lower in the RAGE-positive group than in the RAGE-negative group. Yang et al[91] investigated the relationship between RAGE expression and clinicopathological features in 75 patients with HCC. HCC tissues expressed significantly higher levels of RAGE than non-cancerous tissues, and the expression of RAGE was closely associated with pathological staging and lymph-vascular space invasion. Furthermore, recent studies suggested that RAGE expression increased under hypoxic conditions in HCC cell lines[79,81]. The survival of RAGE-transfected cells was significantly prolonged under hypoxic conditions, and an anti-RAGE siRNA treatment eliminated this influence[81]. Therefore, inhibitors of RAGE expression may be effective as new HCC therapeutic drugs. Koh et al[88] reported that losartan, a peroxisome proliferator-activated receptor-γ (PPAR-γ) activator, attenuated the enhanced expression of RAGE in I/R and attenuated liver damage-inducing factors such as aspartate or alanine aminotransferase, tumor necrosis factor-α, and interleukin-6. Yang et al[91] demonstrated that pioglitazone, a PPAR-γ agonist, decreased the expression of RAGE in HCC cell lines, suppressed cell proliferation and cell invasion, and induced apoptosis and cell cycle arrest. We also previously reported that telmisartan may down-regulate the expression of RAGE through its PPAR-γ-modulating activity in the human HCC cell line, Hep3B. Telmisartan, but not candesartan, decreased RAGE mRNA and protein expression levels, and GW9662, an inhibitor of PPAR-γ, blocked the inhibitory effects of telmisartan on RAGE mRNA and protein expression. Troglitazone and ciglitazone, which are full agonists of PPAR-γ, mimicked the effects of telmisartan[92]. Therefore, PPAR-γ activators may become important targets as inhibitors of RAGE expression in treatment strategies for HCC.
Full-length RAGE, which is generally referred to as RAGE, induces adverse effects, whereas the soluble form of RAGE (sRAGE) attenuates these effects. sRAGE, a circulating isoform of RAGE, has been detected in plasma and consists of an endogenous secretory RAGE (esRAGE), which is a splice variant, as well as a proteolytically cleaved isoform of cell surface RAGE. sRAGE, including esRAGE, is known to act as a decoy receptor of RAGE by binding with AGEs and other ligands competitively[93,94]. Zeng et al[83] reported that the survival of mice treated with sRAGE after hepatic I/R injury was better than that of mice treated with PBS, and that the blockade of RAGE signaling by sRAGE attenuated hepatic I/R injury. In order to elucidate the relationship between serum sRAGE levels and NASH, Yilmaz et al[95] measured serum sRAGE levels in 48 patients with NASH (definite NASH, n = 40, and borderline NASH, n = 8) and 14 control patients, and found that serum sRAGE levels were significantly lower in patients with NASH than in controls. Furthermore, regarding the relationship between serum sRAGE levels and HCC, Moy et al[71] reported that serum sRAGE levels inversely correlated with the risk of HCC or NBNC-HCC, in addition to serum CML levels, as discussed above. Kohles et al[96] also recently demonstrated that serum sRAGE levels were significantly lower in patients with progressive HCC than in patients without progressive HCC. On the other hand, we, along with others, recently found that serum sRAGE levels were positively, rather than inversely, associated with serum TAGE levels in non-diabetic and diabetic subjects[97,98]. These findings suggested that since TAGE up-regulates the expression of RAGE, increases in sRAGE levels may reflect the expression of full-length RAGE[99]. However, the relationship between sRAGE and TAGE in NASH or NASH-related HCC has not yet been elucidated in detail; therefore, further analyses will be necessary in the future.
THE TAGE-RAGE AXIS IN HCC AND HSCS
We previously described the effects of the TAGE-RAGE axis in HCC[100]. TAGE induced the expression of C-reactive protein (CRP), an inflammatory marker, via the activation of Rac-1 in Hep3B, and its induction was attenuated by a pretreatment with anti-RAGE anti-serum. Signal transducer and activator of transcription 3 - and nuclear factor-kappa B-dependent pathways and a reactive oxygen species (ROS)-dependent pathway exist in this signaling pathway, and have been suggested to participate with each other in the early and late stages of CRP induction[100]. A previous study reported a relationship between increases in the expression of CRP and the malignancy of HCC. Kinoshita et al[101] analyzed the relationship between serum CRP levels and poor prognoses in 186 patients with HCC, and demonstrated that serum CRP levels correlated with a poor prognosis in HCC patients. Furthermore, Kim et al[102] examined serum CRP levels in 83 HCC patients with malignant portal vein invasion and 1056 HCC patients without portal vein invasion who underwent liver resection. They found that CRP levels were significantly higher in HCC patients with malignant portal vein invasion than in HCC patients without portal vein invasion, and that CRP levels correlated with the risk of tumor recurrence in HCC patients with malignant portal vein invasion. TAGE significantly enhanced cell proliferation in the human HCC cell line HuH7, which expressed RAGE on the cell surface, but not in the human HCC cell line HepG2, which does not express RAGE. Flow cytometry with anti-RAGE antibody staining demonstrated the expression of membrane-bound RAGE in both HuH7 and HepG2 cells at 24.3% and 6.2%, respectively. Furthermore, MK615, an extract of the Japanese apricot, was shown to suppress TAGE-induced cell proliferation by decreasing the expression of RAGE on the cell surface[34]. The expression of vascular endothelial growth factor mRNA and protein was significantly greater in Hep3B cells treated with TAGE than with the control non-glycated bovine serum albumin (BSA). Furthermore, the proliferation and migration of as well as tube formation by human umbilical vein endothelial cells was significantly greater with the conditioned medium of TAGE-treated Hep3B cells than with the conditioned medium of control non-glycated BSA-treated Hep3B cells. On the other hand, TAGE did not influence HepG2 cells[34]. This may have been due to differences in the expression of RAGE on cell surfaces. Glu-AGEs were also found to have no influence on Hep3B or HepG2 cells because they are known to have lower binding affinity with RAGE than TAGE. The findings suggested that the TAGE-RAGE axis played an important role in the malignant transformation of HCC (Figure 3).
Figure 3.
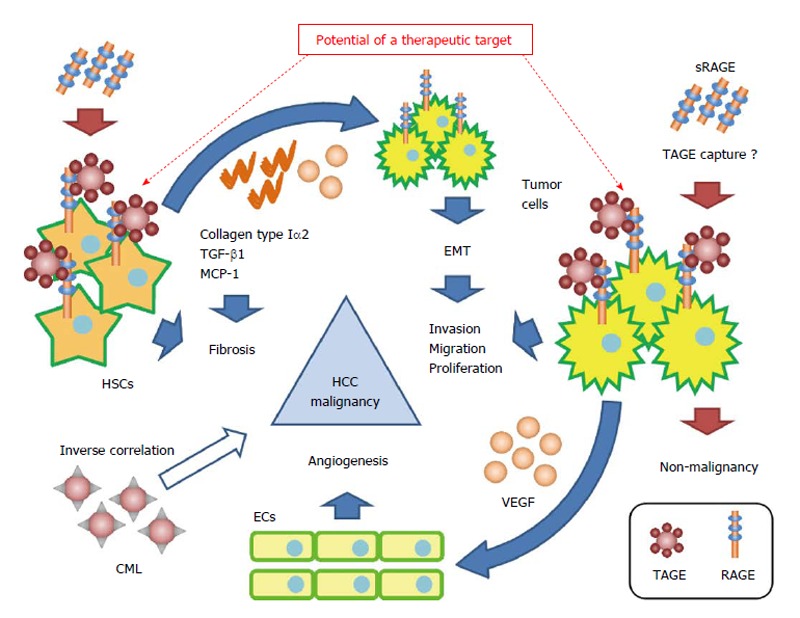
Proposed model for the contribution of the toxic advanced glycation end-products-receptor axis in nonalcoholic steatohepatitis-related hepatocellular carcinoma. The interaction between TAGE and RAGE alters intracellular signaling in tumor cells and hepatic stellate cells, and induces angiogenesis, invasion, migration, proliferation, and fibrosis. This cooperation by the TAGE-RAGE axis may lead to the malignant progression of nonalcoholic steatohepatitis (NASH)-related hepatocellular carcinoma (HCC). CML and sRAGE inversely correlate with the risk of HCC, and sRAGE, which plays the role of a decoy receptor of RAGE, prevents the malignant progression of HCC. The TAGE-RAGE axis may become a treatment target in NASH and NASH-related HCC. CML: Nε-(carboxymethyl)lysine; ECs: Endothelial cells; EMT: Epithelial mesenchymal transition; HSCs: Hepatic stellate cells; MCP-1: Monocyte chemoattractant protein-1; RAGE: Receptor for advanced glycation end-products; sRAGE: Soluble receptor for advanced glycation end; TAGE: Toxic advanced glycation end-products; TGF-β1: Transforming growth factor-β1; VEGF: Vascular endothelial growth factor.
We previously reported the effects of the TAGE-RAGE axis in HSCs. TAGE induced the expression of transforming growth factor-β1 and collagen type Iα2, which are fibrogenic factors, as well as that of monocyte chemoattractant protein-1, an inflammatory factor, via the generation of NADPH oxidase-derived ROS in the human stellate cell line LI90[103]. The activation of HSCs, which mainly produce the extracellular matrix, has been shown to play a pivotal role in liver fibrogenesis[104], and promotes the onset and progression of HCC[105,106]. Amann et al[107] found that activated HSCs increased the malignancy of HCC. The main findings of their study were: (1) the conditioned medium of activated HSCs significantly increased the proliferation and migration of human HCC cell lines (HepG2, Hep3B, and PLC); (2) activated HSCs significantly increased the volumes of spheroids formed in the three-dimensional coculture of HSCs and HCC, and these spheroids showed smaller central necrotic areas than those of spheroids formed only with HCC; and (3) tumor size and invasion ability were significantly greater following the co-implantation of HSCs and HepG2 into nude mice than the implantation of HepG2 alone in vivo. Furthermore, Yang et al[108] reported that collagen type I, which is secreted by HSCs, enhanced the metastatic ability of HCC via epithelial mesenchymal transition. These findings suggested that the TAGE-RAGE axis in HSCs indirectly caused the malignant transformation of HCC (Figure 3).
CONCLUSION
Slight increases in the incidence of NBNC-HCC in recent years have changed the etiology of HCC. The onset mechanism of NBNC-HCC, the etiology of which is varied, currently remains unclear, and, as a consequence, has led to its later diagnosis and larger NBNC-HCC tumors than virus-related HCC tumors. However, the survival rate of early stage NBNC-HCC patients was previously reported to be higher than that of virus-related HCC patients[109-111]. The clinical data suggest that NBNC-HCC can be resolved by early pathogenesis-based treatment, because most of patients diagnosed with NBNC-HCC may have NASH, an important etiological factor of NBNC-HCC. We herein indicated that TAGE, enhanced by NASH, contributes to the malignancy of NASH-related HCC via RAGE. Therefore, the TAGE-RAGE axis may become an important treatment target in NASH-related HCC, and warrants further study.
Footnotes
P- Reviewer: Ampuero J, Gallego-Duran R, Lukacs-Kornek V S- Editor: Yu J L- Editor: Webster JR E- Editor: Liu SQ
Supported by The Japan Society for the Promotion of Science (JSPS) KAKENHI, Grant No. 22300264 and No. 25282029 (to Takeuchi M); the Ministry of Education, Culture, Sports, Science; Technology (MEXT), Regional Innovation Strategy Support Program (to Takeuchi M); and Kanazawa Medical University, No. SR2012-04.
Conflict-of-interest statement: The authors declare no conflict of interest associated with this manuscript.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 18, 2015
First decision: April 23, 2015
Article in press: September 7, 2015
References
- 1.Yu AS, Keeffe EB. Management of hepatocellular carcinoma. Rev Gastroenterol Disord. 2003;3:8–24. [PubMed] [Google Scholar]
- 2.Bosch FX, Ribes J, Díaz M, Cléries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 4.Bosetti C, Turati F, La Vecchia C. Hepatocellular carcinoma epidemiology. Best Pract Res Clin Gastroenterol. 2014;28:753–770. doi: 10.1016/j.bpg.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 5.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 6.Flores A, Marrero JA. Emerging trends in hepatocellular carcinoma: focus on diagnosis and therapeutics. Clin Med Insights Oncol. 2014;8:71–76. doi: 10.4137/CMO.S9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tokushige K, Hashimoto E, Kodama K. Hepatocarcinogenesis in non-alcoholic fatty liver disease in Japan. J Gastroenterol Hepatol. 2013;28 Suppl 4:88–92. doi: 10.1111/jgh.12239. [DOI] [PubMed] [Google Scholar]
- 8.Tokushige K, Hashimoto E, Horie Y, Taniai M, Higuchi S. Hepatocellular carcinoma in Japanese patients with nonalcoholic fatty liver disease, alcoholic liver disease, and chronic liver disease of unknown etiology: report of the nationwide survey. J Gastroenterol. 2011;46:1230–1237. doi: 10.1007/s00535-011-0431-9. [DOI] [PubMed] [Google Scholar]
- 9.Hatanaka K, Kudo M, Fukunaga T, Ueshima K, Chung H, Minami Y, Sakaguchi Y, Hagiwara S, Orino A, Osaki Y. Clinical characteristics of NonBNonC- HCC: Comparison with HBV and HCV related HCC. Intervirology. 2007;50:24–31. doi: 10.1159/000096309. [DOI] [PubMed] [Google Scholar]
- 10.Nagaoki Y, Hyogo H, Aikata H, Tanaka M, Naeshiro N, Nakahara T, Honda Y, Miyaki D, Kawaoka T, Takaki S, et al. Recent trend of clinical features in patients with hepatocellular carcinoma. Hepatol Res. 2012;42:368–375. doi: 10.1111/j.1872-034X.2011.00929.x. [DOI] [PubMed] [Google Scholar]
- 11.al-Abed Y, Kapurniotu A, Bucala R. Advanced glycation end products: detection and reversal. Methods Enzymol. 1999;309:152–172. doi: 10.1016/s0076-6879(99)09013-8. [DOI] [PubMed] [Google Scholar]
- 12.Vlassara H, Palace MR. Diabetes and advanced glycation endproducts. J Intern Med. 2002;251:87–101. doi: 10.1046/j.1365-2796.2002.00932.x. [DOI] [PubMed] [Google Scholar]
- 13.Takeuchi M, Makita Z. Alternative routes for the formation of immunochemically distinct advanced glycation end-products in vivo. Curr Mol Med. 2001;1:305–315. doi: 10.2174/1566524013363735. [DOI] [PubMed] [Google Scholar]
- 14.Glomb MA, Monnier VM. Mechanism of protein modification by glyoxal and glycolaldehyde, reactive intermediates of the Maillard reaction. J Biol Chem. 1995;270:10017–10026. doi: 10.1074/jbc.270.17.10017. [DOI] [PubMed] [Google Scholar]
- 15.Thornalley PJ, Langborg A, Minhas HS. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J. 1999;344 Pt 1:109–116. [PMC free article] [PubMed] [Google Scholar]
- 16.Takeuchi M, Yamagishi S. TAGE (toxic AGEs) hypothesis in various chronic diseases. Med Hypotheses. 2004;63:449–452. doi: 10.1016/j.mehy.2004.02.042. [DOI] [PubMed] [Google Scholar]
- 17.Sato T, Iwaki M, Shimogaito N, Wu X, Yamagishi S, Takeuchi M. TAGE (toxic AGEs) theory in diabetic complications. Curr Mol Med. 2006;6:351–358. doi: 10.2174/156652406776894536. [DOI] [PubMed] [Google Scholar]
- 18.Abe R, Yamagishi S. AGE-RAGE system and carcinogenesis. Curr Pharm Des. 2008;14:940–945. doi: 10.2174/138161208784139765. [DOI] [PubMed] [Google Scholar]
- 19.Hyogo H, Yamagishi S. Advanced glycation end products (AGEs) and their involvement in liver disease. Curr Pharm Des. 2008;14:969–972. doi: 10.2174/138161208784139701. [DOI] [PubMed] [Google Scholar]
- 20.Takeuchi M, Yamagishi S. Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimer’s disease. Curr Pharm Des. 2008;14:973–978. doi: 10.2174/138161208784139693. [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi M, Yamagishi S. Involvement of toxic AGEs (TAGE) in the pathogenesis of diabetic vascular complications and Alzheimer’s disease. J Alzheimers Dis. 2009;16:845–858. doi: 10.3233/JAD-2009-0974. [DOI] [PubMed] [Google Scholar]
- 22.Takeuchi M, Takino J, Yamagishi S. Involvement of the toxic AGEs (TAGE)-RAGE system in the pathogenesis of diabetic vascular complications: a novel therapeutic strategy. Curr Drug Targets. 2010;11:1468–1482. doi: 10.2174/1389450111009011468. [DOI] [PubMed] [Google Scholar]
- 23.Yamagishi S, Maeda S, Matsui T, Ueda S, Fukami K, Okuda S. Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochim Biophys Acta. 2012;1820:663–671. doi: 10.1016/j.bbagen.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 24.Takeuchi M, Takino J, Sakasai-Sakai A, Takata T, Ueda T, Tsutsumi M, Hyogo H, Yamagishi S. Involvement of the TAGE-RAGE system in non-alcoholic steatohepatitis: Novel treatment strategies. World J Hepatol. 2014;6:880–893. doi: 10.4254/wjh.v6.i12.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hyogo H, Yamagishi S, Iwamoto K, Arihiro K, Takeuchi M, Sato T, Ochi H, Nonaka M, Nabeshima Y, Inoue M, et al. Elevated levels of serum advanced glycation end products in patients with non-alcoholic steatohepatitis. J Gastroenterol Hepatol. 2007;22:1112–1119. doi: 10.1111/j.1440-1746.2007.04943.x. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt AM, Stern D. Atherosclerosis and diabetes: the RAGE connection. Curr Atheroscler Rep. 2000;2:430–436. doi: 10.1007/s11883-000-0082-4. [DOI] [PubMed] [Google Scholar]
- 27.Wendt T, Bucciarelli L, Qu W, Lu Y, Yan SF, Stern DM, Schmidt AM. Receptor for advanced glycation endproducts (RAGE) and vascular inflammation: insights into the pathogenesis of macrovascular complications in diabetes. Curr Atheroscler Rep. 2002;4:228–237. doi: 10.1007/s11883-002-0024-4. [DOI] [PubMed] [Google Scholar]
- 28.Rahbar S, Figarola JL. Novel inhibitors of advanced glycation endproducts. Arch Biochem Biophys. 2003;419:63–79. doi: 10.1016/j.abb.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 29.Yamagishi S, Takeuchi M, Inagaki Y, Nakamura K, Imaizumi T. Role of advanced glycation end products (AGEs) and their receptor (RAGE) in the pathogenesis of diabetic microangiopathy. Int J Clin Pharmacol Res. 2003;23:129–134. [PubMed] [Google Scholar]
- 30.Yamagishi S, Imaizumi T. Diabetic vascular complications: pathophysiology, biochemical basis and potential therapeutic strategy. Curr Pharm Des. 2005;11:2279–2299. doi: 10.2174/1381612054367300. [DOI] [PubMed] [Google Scholar]
- 31.Takenaka K, Yamagishi S, Matsui T, Nakamura K, Imaizumi T. Role of advanced glycation end products (AGEs) in thrombogenic abnormalities in diabetes. Curr Neurovasc Res. 2006;3:73–77. doi: 10.2174/156720206775541804. [DOI] [PubMed] [Google Scholar]
- 32.Abe R, Shimizu T, Sugawara H, Watanabe H, Nakamura H, Choei H, Sasaki N, Yamagishi S, Takeuchi M, Shimizu H. Regulation of human melanoma growth and metastasis by AGE-AGE receptor interactions. J Invest Dermatol. 2004;122:461–467. doi: 10.1046/j.0022-202X.2004.22218.x. [DOI] [PubMed] [Google Scholar]
- 33.Takino J, Yamagishi S, Takeuchi M. Cancer malignancy is enhanced by glyceraldehyde-derived advanced glycation end-products. J Oncol. 2010;2010:739852. doi: 10.1155/2010/739852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakuraoka Y, Sawada T, Okada T, Shiraki T, Miura Y, Hiraishi K, Ohsawa T, Adachi M, Takino J, Takeuchi M, et al. MK615 decreases RAGE expression and inhibits TAGE-induced proliferation in hepatocellular carcinoma cells. World J Gastroenterol. 2010;16:5334–5341. doi: 10.3748/wjg.v16.i42.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takino J, Yamagishi S, Takeuchi M. Glycer-AGEs-RAGE signaling enhances the angiogenic potential of hepatocellular carcinoma by upregulating VEGF expression. World J Gastroenterol. 2012;18:1781–1788. doi: 10.3748/wjg.v18.i15.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221–1231. doi: 10.1056/NEJMra011775. [DOI] [PubMed] [Google Scholar]
- 37.Tappy L, Lê KA. Metabolic effects of fructose and the worldwide increase in obesity. Physiol Rev. 2010;90:23–46. doi: 10.1152/physrev.00019.2009. [DOI] [PubMed] [Google Scholar]
- 38.Dekker MJ, Su Q, Baker C, Rutledge AC, Adeli K. Fructose: a highly lipogenic nutrient implicated in insulin resistance, hepatic steatosis, and the metabolic syndrome. Am J Physiol Endocrinol Metab. 2010;299:E685–E694. doi: 10.1152/ajpendo.00283.2010. [DOI] [PubMed] [Google Scholar]
- 39.Vos MB, Lavine JE. Dietary fructose in nonalcoholic fatty liver disease. Hepatology. 2013;57:2525–2531. doi: 10.1002/hep.26299. [DOI] [PubMed] [Google Scholar]
- 40.Attar BM, Van Thiel DH. Current concepts and management approaches in nonalcoholic fatty liver disease. ScientificWorldJournal. 2013;2013:481893. doi: 10.1155/2013/481893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conlon BA, Beasley JM, Aebersold K, Jhangiani SS, Wylie-Rosett J. Nutritional management of insulin resistance in nonalcoholic fatty liver disease (NAFLD) Nutrients. 2013;5:4093–4114. doi: 10.3390/nu5104093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bedogni G, Miglioli L, Masutti F, Tiribelli C, Marchesini G, Bellentani S. Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology. 2005;42:44–52. doi: 10.1002/hep.20734. [DOI] [PubMed] [Google Scholar]
- 43.Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434–438. [PubMed] [Google Scholar]
- 44.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S, Forlani G, Melchionda N. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–1850. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 45.Chitturi S, Abeygunasekera S, Farrell GC, Holmes-Walker J, Hui JM, Fung C, Karim R, Lin R, Samarasinghe D, Liddle C, et al. NASH and insulin resistance: Insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology. 2002;35:373–379. doi: 10.1053/jhep.2002.30692. [DOI] [PubMed] [Google Scholar]
- 46.Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N, Melchionda N, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–923. doi: 10.1053/jhep.2003.50161. [DOI] [PubMed] [Google Scholar]
- 47.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 48.Dam-Larsen S, Franzmann M, Andersen IB, Christoffersen P, Jensen LB, Sørensen TI, Becker U, Bendtsen F. Long term prognosis of fatty liver: risk of chronic liver disease and death. Gut. 2004;53:750–755. doi: 10.1136/gut.2003.019984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yatsuji S, Hashimoto E, Tobari M, Taniai M, Tokushige K, Shiratori K. Clinical features and outcomes of cirrhosis due to non-alcoholic steatohepatitis compared with cirrhosis caused by chronic hepatitis C. J Gastroenterol Hepatol. 2009;24:248–254. doi: 10.1111/j.1440-1746.2008.05640.x. [DOI] [PubMed] [Google Scholar]
- 50.Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–1832. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]
- 51.Takuma Y, Nouso K. Nonalcoholic steatohepatitis-associated hepatocellular carcinoma: our case series and literature review. World J Gastroenterol. 2010;16:1436–1441. doi: 10.3748/wjg.v16.i12.1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ertle J, Dechêne A, Sowa JP, Penndorf V, Herzer K, Kaiser G, Schlaak JF, Gerken G, Syn WK, Canbay A. Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. Int J Cancer. 2011;128:2436–2443. doi: 10.1002/ijc.25797. [DOI] [PubMed] [Google Scholar]
- 53.Caldwell SH, Oelsner DH, Iezzoni JC, Hespenheide EE, Battle EH, Driscoll CJ. Cryptogenic cirrhosis: clinical characterization and risk factors for underlying disease. Hepatology. 1999;29:664–669. doi: 10.1002/hep.510290347. [DOI] [PubMed] [Google Scholar]
- 54.Ong J, Younossi ZM, Reddy V, Price LL, Gramlich T, Mayes J, Boparai N. Cryptogenic cirrhosis and posttransplantation nonalcoholic fatty liver disease. Liver Transpl. 2001;7:797–801. doi: 10.1053/jlts.2001.24644. [DOI] [PubMed] [Google Scholar]
- 55.Poonawala A, Nair SP, Thuluvath PJ. Prevalence of obesity and diabetes in patients with cryptogenic cirrhosis: a case-control study. Hepatology. 2000;32:689–692. doi: 10.1053/jhep.2000.17894. [DOI] [PubMed] [Google Scholar]
- 56.Marrero JA, Fontana RJ, Su GL, Conjeevaram HS, Emick DM, Lok AS. NAFLD may be a common underlying liver disease in patients with hepatocellular carcinoma in the United States. Hepatology. 2002;36:1349–1354. doi: 10.1053/jhep.2002.36939. [DOI] [PubMed] [Google Scholar]
- 57.Thornalley PJ. Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification--a role in pathogenesis and antiproliferative chemotherapy. Gen Pharmacol. 1996;27:565–573. doi: 10.1016/0306-3623(95)02054-3. [DOI] [PubMed] [Google Scholar]
- 58.Takeuchi M, Makita Z, Yanagisawa K, Kameda Y, Koike T. Detection of noncarboxymethyllysine and carboxymethyllysine advanced glycation end products (AGE) in serum of diabetic patients. Mol Med. 1999;5:393–405. [PMC free article] [PubMed] [Google Scholar]
- 59.Takeuchi M, Makita Z, Bucala R, Suzuki T, Koike T, Kameda Y. Immunological evidence that non-carboxymethyllysine advanced glycation end-products are produced from short chain sugars and dicarbonyl compounds in vivo. Mol Med. 2000;6:114–125. [PMC free article] [PubMed] [Google Scholar]
- 60.Takeuchi M, Yanase Y, Matsuura N, Yamagishi Si S, Kameda Y, Bucala R, Makita Z. Immunological detection of a novel advanced glycation end-product. Mol Med. 2001;7:783–791. [PMC free article] [PubMed] [Google Scholar]
- 61.Takeuchi M, Iwaki M, Takino J, Shirai H, Kawakami M, Bucala R, Yamagishi S. Immunological detection of fructose-derived advanced glycation end-products. Lab Invest. 2010;90:1117–1127. doi: 10.1038/labinvest.2010.62. [DOI] [PubMed] [Google Scholar]
- 62.Takeuchi M, Yamagishi S. Alternative routes for the formation of glyceraldehyde-derived AGEs (TAGE) in vivo. Med Hypotheses. 2004;63:453–455. doi: 10.1016/j.mehy.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 63.Bucala R, Cerami A. Advanced glycosylation: chemistry, biology, and implications for diabetes and aging. Adv Pharmacol. 1992;23:1–34. doi: 10.1016/s1054-3589(08)60961-8. [DOI] [PubMed] [Google Scholar]
- 64.Vlassara H, Bucala R, Striker L. Pathogenic effects of advanced glycosylation: biochemical, biologic, and clinical implications for diabetes and aging. Lab Invest. 1994;70:138–151. [PubMed] [Google Scholar]
- 65.Brownlee M. Advanced protein glycosylation in diabetes and aging. Annu Rev Med. 1995;46:223–234. doi: 10.1146/annurev.med.46.1.223. [DOI] [PubMed] [Google Scholar]
- 66.Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988;318:1315–1321. doi: 10.1056/NEJM198805193182007. [DOI] [PubMed] [Google Scholar]
- 67.Monnier VM, Cerami A. Nonenzymatic browning in vivo: possible process for aging of long-lived proteins. Science. 1981;211:491–493. doi: 10.1126/science.6779377. [DOI] [PubMed] [Google Scholar]
- 68.Fu MX, Requena JR, Jenkins AJ, Lyons TJ, Baynes JW, Thorpe SR. The advanced glycation end product, Nepsilon-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J Biol Chem. 1996;271:9982–9986. doi: 10.1074/jbc.271.17.9982. [DOI] [PubMed] [Google Scholar]
- 69.Sebeková K, Kupcová V, Schinzel R, Heidland A. Markedly elevated levels of plasma advanced glycation end products in patients with liver cirrhosis - amelioration by liver transplantation. J Hepatol. 2002;36:66–71. doi: 10.1016/s0168-8278(01)00232-x. [DOI] [PubMed] [Google Scholar]
- 70.Yagmur E, Tacke F, Weiss C, Lahme B, Manns MP, Kiefer P, Trautwein C, Gressner AM. Elevation of Nepsilon-(carboxymethyl)lysine-modified advanced glycation end products in chronic liver disease is an indicator of liver cirrhosis. Clin Biochem. 2006;39:39–45. doi: 10.1016/j.clinbiochem.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 71.Moy KA, Jiao L, Freedman ND, Weinstein SJ, Sinha R, Virtamo J, Albanes D, Stolzenberg-Solomon RZ. Soluble receptor for advanced glycation end products and risk of liver cancer. Hepatology. 2013;57:2338–2345. doi: 10.1002/hep.26264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kimura Y, Hyogo H, Yamagishi S, Takeuchi M, Ishitobi T, Nabeshima Y, Arihiro K, Chayama K. Atorvastatin decreases serum levels of advanced glycation endproducts (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J Gastroenterol. 2010;45:750–757. doi: 10.1007/s00535-010-0203-y. [DOI] [PubMed] [Google Scholar]
- 73.Kan H, Yamagishi SI, Ojima A, Fukami K, Ueda S, Takeuchi M, Hyogo H, Aikata H, Chayama K. Elevation of Serum Levels of Advanced Glycation End Products in Patients With Non-B or Non-C Hepatocellular Carcinoma. J Clin Lab Anal. 2014:Epub ahead of print. doi: 10.1002/jcla.21797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brenner T, Fleming TH, Spranz D, Schemmer P, Bruckner T, Uhle F, Martin EO, Weigand MA, Hofer S. Reactive metabolites and AGE-RAGE-mediated inflammation in patients following liver transplantation. Mediators Inflamm. 2013;2013:501430. doi: 10.1155/2013/501430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Takeuchi M, Sakasai-Sakai A, Takata T, Ueda T, Takino J, Tsutsumi M, Hyogo H, Yamagishi S. Serum levels of toxic AGEs (TAGE) may be a promising novel biomarker in development and progression of NASH. Med Hypotheses. 2015;84:490–493. doi: 10.1016/j.mehy.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 76.Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108:949–955. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kuniyasu H, Oue N, Wakikawa A, Shigeishi H, Matsutani N, Kuraoka K, Ito R, Yokozaki H, Yasui W. Expression of receptors for advanced glycation end-products (RAGE) is closely associated with the invasive and metastatic activity of gastric cancer. J Pathol. 2002;196:163–170. doi: 10.1002/path.1031. [DOI] [PubMed] [Google Scholar]
- 78.Bhawal UK, Ozaki Y, Nishimura M, Sugiyama M, Sasahira T, Nomura Y, Sato F, Fujimoto K, Sasaki N, Ikeda MA, et al. Association of expression of receptor for advanced glycation end products and invasive activity of oral squamous cell carcinoma. Oncology. 2005;69:246–255. doi: 10.1159/000087910. [DOI] [PubMed] [Google Scholar]
- 79.Yan W, Chang Y, Liang X, Cardinal JS, Huang H, Thorne SH, Monga SP, Geller DA, Lotze MT, Tsung A. High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology. 2012;55:1863–1875. doi: 10.1002/hep.25572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen RC, Yi PP, Zhou RR, Xiao MF, Huang ZB, Tang DL, Huang Y, Fan XG. The role of HMGB1-RAGE axis in migration and invasion of hepatocellular carcinoma cell lines. Mol Cell Biochem. 2014;390:271–280. doi: 10.1007/s11010-014-1978-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hiwatashi K, Ueno S, Abeyama K, Kubo F, Sakoda M, Maruyama I, Hamanoue M, Natsugoe S, Aikou T. A novel function of the receptor for advanced glycation end-products (RAGE) in association with tumorigenesis and tumor differentiation of HCC. Ann Surg Oncol. 2008;15:923–933. doi: 10.1245/s10434-007-9698-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fehrenbach H, Weiskirchen R, Kasper M, Gressner AM. Up-regulated expression of the receptor for advanced glycation end products in cultured rat hepatic stellate cells during transdifferentiation to myofibroblasts. Hepatology. 2001;34:943–952. doi: 10.1053/jhep.2001.28788. [DOI] [PubMed] [Google Scholar]
- 83.Zeng S, Feirt N, Goldstein M, Guarrera J, Ippagunta N, Ekong U, Dun H, Lu Y, Qu W, Schmidt AM, et al. Blockade of receptor for advanced glycation end product (RAGE) attenuates ischemia and reperfusion injury to the liver in mice. Hepatology. 2004;39:422–432. doi: 10.1002/hep.20045. [DOI] [PubMed] [Google Scholar]
- 84.Cataldegirmen G, Zeng S, Feirt N, Ippagunta N, Dun H, Qu W, Lu Y, Rong LL, Hofmann MA, Kislinger T, et al. RAGE limits regeneration after massive liver injury by coordinated suppression of TNF-alpha and NF-kappaB. J Exp Med. 2005;201:473–484. doi: 10.1084/jem.20040934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ekong U, Zeng S, Dun H, Feirt N, Guo J, Ippagunta N, Guarrera JV, Lu Y, Weinberg A, Qu W, et al. Blockade of the receptor for advanced glycation end products attenuates acetaminophen-induced hepatotoxicity in mice. J Gastroenterol Hepatol. 2006;21:682–688. doi: 10.1111/j.1440-1746.2006.04225.x. [DOI] [PubMed] [Google Scholar]
- 86.Higuchi S, Yano A, Takai S, Tsuneyama K, Fukami T, Nakajima M, Yokoi T. Metabolic activation and inflammation reactions involved in carbamazepine-induced liver injury. Toxicol Sci. 2012;130:4–16. doi: 10.1093/toxsci/kfs222. [DOI] [PubMed] [Google Scholar]
- 87.Goodwin M, Herath C, Jia Z, Leung C, Coughlan MT, Forbes J, Angus P. Advanced glycation end products augment experimental hepatic fibrosis. J Gastroenterol Hepatol. 2013;28:369–376. doi: 10.1111/jgh.12042. [DOI] [PubMed] [Google Scholar]
- 88.Koh EJ, Yoon SJ, Lee SM. Losartan protects liver against ischaemia/reperfusion injury through PPAR-γ activation and receptor for advanced glycation end-products down-regulation. Br J Pharmacol. 2013;169:1404–1416. doi: 10.1111/bph.12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kuhla A, Norden J, Abshagen K, Menger MD, Vollmar B. RAGE blockade and hepatic microcirculation in experimental endotoxaemic liver failure. Br J Surg. 2013;100:1229–1239. doi: 10.1002/bjs.9188. [DOI] [PubMed] [Google Scholar]
- 90.Ito R, Ishii Y, Wakiyama S, Shiba H, Fujioka S, Misawa T, Ishida Y, Hano H, Yanaga K. Prognostic significance of receptor for advanced glycation end products expression in hepatocellular carcinoma after hepatectomy. J Surg Res. 2014;192:503–508. doi: 10.1016/j.jss.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 91.Yang Y, Zhao LH, Huang B, Wang RY, Yuan SX, Tao QF, Xu Y, Sun HY, Lin C, Zhou WP. Pioglitazone, a PPARγ agonist, inhibits growth and invasion of human hepatocellular carcinoma via blockade of the rage signaling. Mol Carcinog. 2014:Epub ahead of print. doi: 10.1002/mc.22231. [DOI] [PubMed] [Google Scholar]
- 92.Yoshida T, Yamagishi S, Nakamura K, Matsui T, Imaizumi T, Takeuchi M, Koga H, Ueno T, Sata M. Telmisartan inhibits AGE-induced C-reactive protein production through downregulation of the receptor for AGE via peroxisome proliferator-activated receptor-gamma activation. Diabetologia. 2006;49:3094–3099. doi: 10.1007/s00125-006-0437-7. [DOI] [PubMed] [Google Scholar]
- 93.Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, Yasui K, Takeuchi M, Makita Z, Takasawa S, et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J. 2003;370:1097–1109. doi: 10.1042/BJ20021371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Geroldi D, Falcone C, Emanuele E. Soluble receptor for advanced glycation end products: from disease marker to potential therapeutic target. Curr Med Chem. 2006;13:1971–1978. doi: 10.2174/092986706777585013. [DOI] [PubMed] [Google Scholar]
- 95.Yilmaz Y, Ulukaya E, Gul OO, Arabul M, Gul CB, Atug O, Oral AY, Aker S, Dolar E. Decreased plasma levels of soluble receptor for advanced glycation endproducts (sRAGE) in patients with nonalcoholic fatty liver disease. Clin Biochem. 2009;42:802–807. doi: 10.1016/j.clinbiochem.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 96.Kohles N, Nagel D, Jüngst D, Stieber P, Holdenrieder S. Predictive value of immunogenic cell death biomarkers HMGB1, sRAGE, and DNase in liver cancer patients receiving transarterial chemoembolization therapy. Tumour Biol. 2012;33:2401–2409. doi: 10.1007/s13277-012-0504-2. [DOI] [PubMed] [Google Scholar]
- 97.Nakamura K, Yamagishi SI, Matsui T, Adachi H, Takeuchi M, Imaizumi T. Serum levels of soluble form of receptor for advanced glycation end products (sRAGE) are correlated with AGEs in both diabetic and non-diabetic subjects. Clin Exp Med. 2007;7:188–190. doi: 10.1007/s10238-007-0146-7. [DOI] [PubMed] [Google Scholar]
- 98.Yamagishi S, Adachi H, Nakamura K, Matsui T, Jinnouchi Y, Takenaka K, Takeuchi M, Enomoto M, Furuki K, Hino A, et al. Positive association between serum levels of advanced glycation end products and the soluble form of receptor for advanced glycation end products in nondiabetic subjects. Metabolism. 2006;55:1227–1231. doi: 10.1016/j.metabol.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 99.Yamagishi S, Imaizumi T. Serum levels of soluble form of receptor for advanced glycation end products (sRAGE) may reflect tissue RAGE expression in diabetes. Arterioscler Thromb Vasc Biol. 2007;27:e32; author reply e33–e34. doi: 10.1161/ATVBAHA.107.139923. [DOI] [PubMed] [Google Scholar]
- 100.Yoshida T, Yamagishi S, Nakamura K, Matsui T, Imaizumi T, Takeuchi M, Ueno T, Sata M. Pigment epithelium-derived factor (PEDF) inhibits advanced glycation end product (AGE)-induced C-reactive protein expression in hepatoma cells by suppressing Rac-1 activation. FEBS Lett. 2006;580:2788–2796. doi: 10.1016/j.febslet.2006.04.050. [DOI] [PubMed] [Google Scholar]
- 101.Kinoshita A, Onoda H, Imai N, Iwaku A, Oishi M, Tanaka K, Fushiya N, Koike K, Nishino H, Matsushima M, et al. The addition of C-reactive protein to validated staging systems improves their prognostic ability in patients with hepatocellular carcinoma. Oncology. 2014;86:308–317. doi: 10.1159/000360704. [DOI] [PubMed] [Google Scholar]
- 102.Kim JM, Kwon CH, Joh JW, Ko JS, Park JB, Lee JH, Kim SJ, Paik SW, Park CK. C-reactive protein may be a prognostic factor in hepatocellular carcinoma with malignant portal vein invasion. World J Surg Oncol. 2013;11:92. doi: 10.1186/1477-7819-11-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Iwamoto K, Kanno K, Hyogo H, Yamagishi S, Takeuchi M, Tazuma S, Chayama K. Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J Gastroenterol. 2008;43:298–304. doi: 10.1007/s00535-007-2152-7. [DOI] [PubMed] [Google Scholar]
- 104.Moreira RK. Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med. 2007;131:1728–1734. doi: 10.5858/2007-131-1728-HSCALF. [DOI] [PubMed] [Google Scholar]
- 105.Carloni V, Luong TV, Rombouts K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: more complicated than ever. Liver Int. 2014;34:834–843. doi: 10.1111/liv.12465. [DOI] [PubMed] [Google Scholar]
- 106.Coulouarn C, Clément B. Stellate cells and the development of liver cancer: therapeutic potential of targeting the stroma. J Hepatol. 2014;60:1306–1309. doi: 10.1016/j.jhep.2014.02.003. [DOI] [PubMed] [Google Scholar]
- 107.Amann T, Bataille F, Spruss T, Mühlbauer M, Gäbele E, Schölmerich J, Kiefer P, Bosserhoff AK, Hellerbrand C. Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci. 2009;100:646–653. doi: 10.1111/j.1349-7006.2009.01087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yang MC, Wang CJ, Liao PC, Yen CJ, Shan YS. Hepatic stellate cells secretes type I collagen to trigger epithelial mesenchymal transition of hepatoma cells. Am J Cancer Res. 2014;4:751–763. [PMC free article] [PubMed] [Google Scholar]
- 109.Akahoshi H, Taura N, Ichikawa T, Miyaaki H, Akiyama M, Miuma S, Ozawa E, Takeshita S, Muraoka T, Matsuzaki T, et al. Differences in prognostic factors according to viral status in patients with hepatocellular carcinoma. Oncol Rep. 2010;23:1317–1323. doi: 10.3892/or_00000766. [DOI] [PubMed] [Google Scholar]
- 110.Takuma Y, Nouso K, Makino Y, Gotoh T, Toshikuni N, Morimoto Y, Shimomura H, Yamamoto H. Outcomes after curative treatment for cryptogenic cirrhosis-associated hepatocellular carcinoma satisfying the Milan criteria. J Gastroenterol Hepatol. 2011;26:1417–1424. doi: 10.1111/j.1440-1746.2011.06775.x. [DOI] [PubMed] [Google Scholar]
- 111.Kaibori M, Ishizaki M, Matsui K, Kwon AH. Clinicopathologic characteristics of patients with non-B non-C hepatitis virus hepatocellular carcinoma after hepatectomy. Am J Surg. 2012;204:300–307. doi: 10.1016/j.amjsurg.2011.11.014. [DOI] [PubMed] [Google Scholar]