

Abstract
Key points
Endothelial function in resistance vessels entails Ca2+ and electrical signalling to promote vasodilatation and increase tissue blood flow. Whether membrane potential (V m) governs intracellular calcium concentration ([Ca2+]i) of the endothelium remains controversial.
[Ca2+]i and V m were evaluated simultaneously during intracellular current injection using intact endothelial tubes freshly isolated from mouse skeletal muscle resistance arteries.
[Ca2+]i did not change during hyperpolarization or depolarization under resting conditions. However in the presence of 100 nM ACh (∼EC50), [Ca2+]i increased during hyperpolarization and decreased during depolarization. These responses required extracellular Ca2+ and were attenuated by half with genetic ablation of TRPV4 channels.
In native microvascular endothelium, half‐maximal stimulation of muscarinic receptors enables V m to govern [Ca2+]i by activating Ca2+‐permeable channels in the plasma membrane. This effect of V m is absent at rest and can be masked during maximal receptor stimulation.
Abstract
In resistance arteries, coupling a rise of intracellular calcium concentration ([Ca2+]i) to endothelial cell hyperpolarization underlies smooth muscle cell relaxation and vasodilatation, thereby increasing tissue blood flow and oxygen delivery. A controversy persists as to whether changes in membrane potential (V m) alter endothelial cell [Ca2+]i. We tested the hypothesis that V m governs [Ca2+]i in endothelium of resistance arteries by performing Fura‐2 photometry while recording and controlling V m of intact endothelial tubes freshly isolated from superior epigastric arteries of C57BL/6 mice. Under resting conditions, [Ca2+]i did not change when V m shifted from baseline (∼−40 mV) via exposure to 10 μM NS309 (hyperpolarization to ∼−80 mV), via equilibration with 145 mm [K+]o (depolarization to ∼−5 mV), or during intracellular current injection (±0.5 to 5 nA, 20 s pulses) while V m changed linearly between ∼−80 mV and +10 mV. In contrast, during the plateau (i.e. Ca2+ influx) phase of the [Ca2+]i response to approximately half‐maximal stimulation with 100 nm ACh (∼EC50), [Ca2+]i increased as V m hyperpolarized below −40 mV and decreased as V m depolarized above −40 mV. The magnitude of [Ca2+]i reduction during depolarizing current injections correlated with the amplitude of the plateau [Ca2+]i response to ACh. The effect of hyperpolarization on [Ca2+]i was abolished following removal of extracellular Ca2+, was enhanced subtly by raising extracellular [Ca2+] from 2 mm to 10 mm and was reduced by half in endothelium of TRPV4−/− mice. Thus, during submaximal activation of muscarinic receptors, V m can modulate Ca2+ entry through the plasma membrane in accord with the electrochemical driving force.
Abbreviations
- ACh
acetylcholine
- BKCa
large‐conductance Ca2+‐activated K+ channel
- [Ca2+]i
intracellular Ca2+ concentration
- [Ca2+]o
extracellular Ca2+ concentration
- EC
endothelial cell
- EC50
drug concentration giving half‐maximal response
- EK
Nernst equilibrium potential for K+
- ER
endoplasmic reticulum
- FCCP
carbonyl cyanide p‐trifluoromethoxyphenylhydrazone
- GSK101
GSK1016790A
- GSK219
GSK2193874
- ID
internal diameter
- [K+]o
extracellular K+ concentration
- NO
nitric oxide
- OD
outer diameter
- PSS
physiological salt solution
- SEA
superior epigastric artery
- SKCa/IKCa
small‐ and intermediate‐conductance Ca2+‐activated K+ channels
- SMC
smooth muscle cell
- TRP
transient receptor potential
- TRPV4
transient receptor potential vanilloid type 4 channel
- TRPV4−/−
TRPV4 knockout
- Vm
membrane potential
Introduction
A key role for the endothelium of resistance vessels entails the regulation of intracellular Ca2+ ([Ca2+]i) and membrane potential (V m) to govern smooth muscle cell (SMC) relaxation, vasodilatation and tissue blood flow (Busse et al. 2002; Ledoux et al. 2006; Bagher & Segal, 2011; Garland et al. 2011). The activation of Gq protein‐coupled muscarinic (M3) receptors stimulates the production of inositol 1,4,5‐trisphosphate and diacylglycerol. Through binding to its receptor on the endoplasmic reticulum (ER), inositol 1,4,5‐trisphosphate evokes the release of Ca2+ from internal stores as reflected by the initial peak of the Ca2+ response (Himmel et al. 1993). In turn, emptying intracellular Ca2+ stores stimulates the activation of Ca2+ permeable channels in the plasma membrane (which include an array of transient receptor potential (TRP) channels; Yue et al. 2015) and the ensuing plateau phase of the [Ca2+]i response (see Dora & Garland, 2013; Ruhle & Trebak, 2013 for details regarding the regulation Ca2+ influx). The rise in [Ca2+]i activates small‐ and intermediate‐conductance Ca2+‐activated K+ channels (SKCa/IKCa) in the plasma membrane as reflected by endothelium‐dependent hyperpolarization in response to muscarinic receptor stimulation. While the ensuing efflux of K+ results in a more negative cell interior, it remains controversial as to whether hyperpolarization enhances Ca2+ entry into endothelial cells (ECs) by increasing its electrical driving force (Dora & Garland, 2013). With production of nitric oxide (NO) as a vasodilator also governed by a rise in [Ca2+]i (Busse & Mulsch, 1990), the regulation of Ca2+ entry in the endothelium of resistance vessels is integral to the control of tissue blood flow and oxygen delivery.
The influx of Ca2+ is driven by an ∼20,000‐fold concentration gradient from the extracellular fluid, electronegativity of the cell interior and the open probability of Ca2+‐permeable ion channels (Clapham, 2007). Previous endeavours to assess whether V m impacts Ca2+ influx into native ECs have used several approaches to control V m including maximal stimulation of muscarinic receptors (e.g. with ACh or methacholine), manipulating extracellular K+ concentration ([K+]o), activating or inhibiting K+ channels, and electrically ‘clamping’ V m (Cohen & Jackson, 2005; Dora & Garland, 2013). Complementary experiments have used ECs grown in culture. However, caveats inherent to earlier studies include limited dynamic range of [Ca2+]i responses, the time course used for controlling V m, and altered expression of proteins integral to Ca2+ signalling. For example, muscarinic receptors can be lost from native ECs when grown in culture (Tracey & Peach, 1992). Cultured ECs may in turn express large‐conductance Ca2+‐activated K+ channels (BKCa) which are otherwise present in SMCs (Sandow & Grayson, 2009). Unlike SKCa/IKCa, BKCa is governed by voltage as well as [Ca2+]i (Nelson & Quayle, 1995; Jackson, 2005). As determined using intracellular current injection to control V m throughout the physiological range (∼−80 mV to +10 mV), endothelial ‘tubes’ freshly isolated from the mouse superior epigastric artery (SEA; a resistance artery supplying abdominal skeletal muscle; in vivo diameter, ∼150 μm) lack voltage‐gated ion channels and thereby provide a valuable model for addressing whether changes in V m can modulate Ca2+ flux across the plasma membrane. Thus, intracellular injection of negative or positive current enables rigorous evaluation of how changing V m under prescribed conditions may evoke alterations in [Ca2+]i.
In the present study, we tested the hypothesis that V m governs [Ca2+]i of native microvascular ECs. Fura‐2 dye was used to monitor [Ca2+]i while controlling V m in endothelial tubes freshly isolated from the mouse SEA. The relationship between V m and [Ca2+]i was investigated under resting conditions and during half‐maximal stimulation with ACh (EC50; 100 nm) to increase (but not maximize) Ca2+ permeability of the plasma membrane. Our findings illustrate that during resting conditions, [Ca2+]i was not affected by changing V m through a range spanning ∼−80 mV to +10 mV. However, during submaximal stimulation with ACh, [Ca2+]i increased as V m hyperpolarized below −40 mV and [Ca2+]i decreased as V m depolarized above −40 mV. These effects of altering V m in the presence of ACh did not occur in the absence of extracellular Ca2+ and were attenuated by half in endothelium isolated from TRP vanilloid type 4 channel knockout (TRPV4−/−) mice. Thus, V m can modulate Ca2+ influx according to the electrical driving force during submaximal activation of Ca2+‐permeable channels in the plasma membrane of microvascular endothelium.
Methods
Animal care and use
All animal care and experimental procedures were approved by the Animal Care and Use Committee of the University of Missouri and performed in accord with the National Research Council's Guide for the Care and Use of Laboratory Animals (8th edn, 2011). Mice were housed in an enriched environment maintained on a 12:12 h light–dark cycle at ∼23°C with fresh tap water and standard chow available ad libitum. Experiments were performed on male C57BL/6 mice (3–6 months old; n = 47) obtained from the National Institute on Aging colonies at Charles River Laboratories (Wilmington, MA, USA). In complementary experiments, male TRPV4−/− mice (C57BL/6 background, 3–6 months old, n = 5) bred at the University of Missouri (breeders obtained from GlaxoSmithKline, King of Prussia, PA, USA) were used to investigate the role of TRPV4 (Thorneloe et al. 2008). Each mouse was anaesthetized with pentobarbital sodium (60 mg kg−1, intraperitoneal injection) and abdominal fur was removed by shaving. Upon completion of tissue removal, the anaesthetized mouse was killed by exsanguination.
Solutions
All solutions were used at pH 7.4. Control physiological salt solution (PSS) contained the following (in mmol l−1): 2 CaCl2, 140 NaCl, 5 KCl, 1 MgCl2, 10 Hepes, 10 glucose. During microdissection of SEAs, CaCl2 was absent from the PSS to relax SMCs. During dissociation of SMCs to obtain endothelial tubes, PSS contained 0.62 mg ml−1 papain (≥6 units), 1.5 mg ml−1 collagenase (≥15 units), 1.0 mg ml−1 dithioerythritol, 0.1% bovine serum albumin (USB Corp., Cleveland, OH, USA) and 0.1 mmol l−1 CaCl2. For experiments with nominally zero [Ca2+]o, CaCl2 was replaced with 2 mm MgCl2 (final [MgCl2] = 3 mm). For experiments with 10 mm [Ca2+]o, NaCl was replaced isosmotically with CaCl2. For experiments with 145 mm [K+]o, NaCl was replaced on an equimolar basis with KCl. Reagents were obtained from Sigma‐Aldrich (St Louis, MO, USA) unless indicated otherwise.
Surgery and microdissection
A ventral midline incision was made through the skin from the sternum to the pubis to expose the abdominal musculature. While viewing through a stereo microscope (SMZ800; Nikon, Tokyo, Japan), fat and connective tissue superficial to the sternum were removed to expose the proximal end of SEAs bilaterally; each was ligated together with its adjacent vein (6‐0 silk suture; Ethicon, Somerville, NJ, USA) to maintain blood in the lumen and thereby facilitate visualization during further dissection. The abdominal musculature was removed from the mouse and separated along the linea alba and each half was pinned onto transparent silicone rubber (Sylgard 184; Dow Corning, Midland, MI, USA) in control PSS maintained at 4°C. The SEA was dissected free of surrounding tissue from its proximal end to the first branch point (segment length: ∼2 cm) and then cannulated at one end to flush blood from the lumen with PSS. Cannulae were made from borosilicate glass capillaries (G150T‐4; Warner Instruments, Hamden, CT, USA) pulled horizontally (P‐97; Sutter Instrument Co., Novato, CA, USA), then shaped and heat‐polished at one end (tip outer diameter (OD): 50–80 μm) using a custom‐built microforge.
Endothelial cell tube isolation and superfusion
Endothelial tubes were prepared as described (Socha & Segal, 2013). Briefly, each SEA was cut into segments 3–5 mm long and incubated in dissociation PSS for 30 min at 37°C. Vessel segments were transferred to a tissue chamber (RC‐27N; Warner) containing dissociation PSS at room temperature. To dissociate SMCs, a vessel segment was gently triturated using aspiration and ejection from a micropipette during visual inspection at 200×. Dissociation pipettes (tip internal diameter (ID): ∼80 μm) were prepared from borosilicate glass capillary tubes (1.0 mm OD, 0.58 mm ID; World Precision Instruments (WPI), Sarasota, FL, USA) pulled (P‐97; Sutter) and heat‐polished at one end. Following dissociation, the tissue chamber containing the endothelial tube was secured to an aluminium platform (width: 14.5 cm, length: 24 cm, thickness: 0.4 cm). A micromanipulator (DT3‐100; Siskiyou Corp., Grants Pass, OR, USA) mounted at each end of the platform held a blunt‐ended heat‐polished micropipette (OD, 60–100 μm) used to position and secure the endothelial tube (width: ∼60 μm, length: ∼1 mm) against the bottom (coverslip) of the tissue chamber. The platform was secured on an inverted microscope (Eclipse TS100, Nikon) mounted on a vibration‐isolated table (Technical Manufacturing Corp., Peabody, MA, USA) and superfused at 3–4 ml min−1 with control PSS along the axis of the endothelial tube. Throughout experiments, temperature was maintained at 32ºC using an in‐line heater (SH‐27B; Warner) and heating platform (PH6; Warner) coupled to a temperature controller (TC‐344B; Warner). Preparations were stable for the duration of all experiments (1–2 h) under these conditions.
Ca2+ photometry
Ca2+ photometry was performed using an IonOptix system (Milford, MA, USA) as described (Socha et al. 2011; Behringer et al. 2012). Briefly, prior to loading Fura‐2 dye, the preparation was maintained at room temperature for 10 min while autofluorescence was recorded at 510 nm during alternate excitation at 340 and 380 nm (10 Hz). Fura‐2 AM dye (5 μm; F14185; Life Technologies, Eugene, OR, USA) was loaded for 20 min followed by a 20 min washout to allow for intracellular de‐esterification. Temperature was raised to 32°C during the final 10 min of washout. Autofluorescence (averaged values over 30 s acquisition) during excitation at 340 and 380 nm was subtracted from respective recordings at 510 nm. The photometric window was 140 μm × 50 μm using a 40× objective (Nikon S Fluor; numerical aperture, 0.90) and encompassed ∼50 ECs (Fig. 1). All experiments were performed with 2 mm [Ca2+]o unless stated otherwise.
Figure 1.
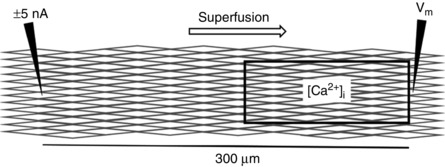
Diagram of simultaneous [Ca2+]i and Vm measurements in an endothelial tube
A window 140 μm × 50 μm (encompassing ∼50 ECs) monitored [Ca2+]i using Fura‐2 dye photometry. For simultaneous measurements, the window was located adjacent to a microelectrode recording V m (right) located 300 μm from a microelectrode injecting current (−5 to +5 nA; left) to control (i.e. clamp) V m at a designated level.
To enable approximation of [Ca2+]i in accord with the Fura‐2 ratio, minimum and maximum F 340/F 380 values (R min, 0.43 ± 0.02 and R max, 5.79 ± 0.92, n = 3) were determined in endothelial tubes using a previous protocol with modification (Socha et al. 2011). Briefly, endothelial tubes were superfused with PSS containing 0 mm Ca2+, 5 mm EGTA, 1 μm thapsigargin, 1 μm carbonyl cyanide p‐trifluoromethoxyphenylhydrazone (FCCP) and 3 μm ionomycin for 1 h to deplete intracellular Ca2+ (R min), followed by replacement with PSS containing 3 μm ionomycin and 10 mm Ca2+ to maximize intracellular Ca2+ (R max). Data acquired during respective conditions were also used to calculate the value of β (i.e. F min/F max at 380 nm = 7.40 ± 1.09). The F 340/F 380 (R) values were converted to [Ca2+]i (in nm) using the equation [Ca2+]i = K dβ(R − R min)/(R max − R) (Grynkiewicz et al. 1985) assuming an intracellular Fura‐2 K d of 282 nm (Knot & Nelson, 1998).
Intracellular recording and current microinjection
Microelectrodes were pulled (P‐97; Sutter) from glass capillary tubes (GC100F‐10; Warner) and backfilled with 2 mol l−1 KCl (tip resistance, ∼150 MΩ). Using one electrode, current (±0.5 to 5 nA, 20 s pulses) was delivered using an Axoclamp electrometer (2B; Molecular Devices, Sunnyvale, CA, USA) driven by a function generator (CFG253; Tektronix, Beaverton, OR, USA). A second electrode was positioned in an EC located 300 μm downstream (with respect to the direction of PSS superfusion) to record V m using a second amplifier (IE‐210; Warner). An Ag–AgCl pellet placed in effluent PSS served as a reference electrode. Amplifier outputs were connected to an analog‐to‐digital converter (Digidata 1322A; Molecular Devices) and data were recorded at 1000 Hz on a Dell personal computer using Axoscope 10.1 software (Molecular Devices). Individual cells were penetrated along the midline of the endothelial tube where distance was defined with reference to a calibrated eyepiece reticle while viewing at 400× magnification. Alterations in V m at the downstream electrode in response to current injected at the upstream electrode verified effective intercellular coupling through gap junctions while ‘clamping’ V m in accord with the level of current injection (Behringer & Segal, 2012 b; Behringer et al. 2012, 2013).
Simultaneous Ca2+ photometry and electrophysiology
For these experiments, the photometric window (as above) was positioned adjacent to the microelectrode used for recording V m 300 μm downstream from the site of current microinjection (Fig. 1). Two experimental approaches were used to determine whether (and if so, under what conditions) V m influenced [Ca2+]i in accord with its electrochemical driving force across the plasma membrane. In the first approach, NS309 (10 μm) was added to the PSS to open SKCa/IKCa maximally and thereby hyperpolarize ECs to approximate the Nernst equilibrium potential for K+ (E K; ∼−90 mV) (Strobaek et al. 2004; Li et al. 2009; Behringer & Segal, 2012 b). Alternatively, PSS containing 145 mm [K+]o was used to clamp V m in a depolarized state by altering E K (to ∼0 mV). Respective treatments approximated the boundaries of the physiological range of EC V m (i.e. ∼−80 to +10 mV) (Ledoux et al. 2008) and were administered alone for 2 min then maintained for two additional minutes during exposure to 100 nm ACh. Respective treatments were paired to stimulation with ACh alone (Fig. 2).
Figure 2.
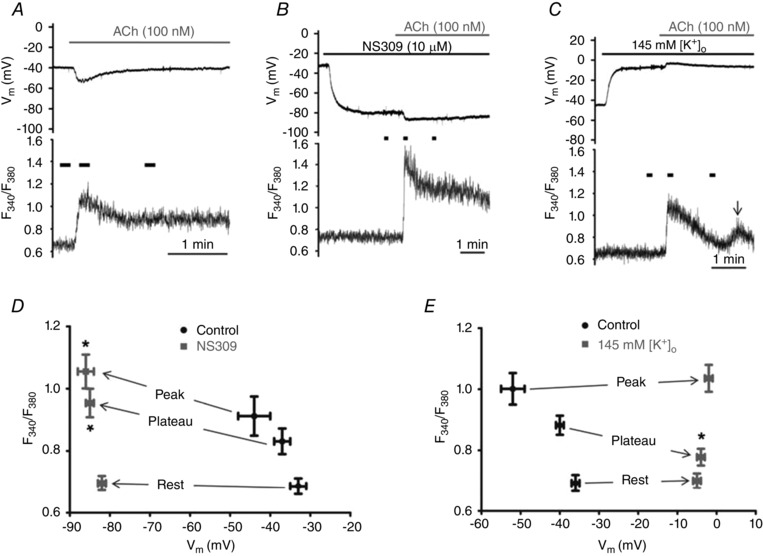
Effects of 10 μm NS309 and 145 mm [K+]o on Vm and [Ca2+]i before and during stimulation with ACh
A, simultaneous V m (top) and F 340/F 380 (bottom) recordings during 100 nm ACh under control conditions. B, as in A with 10 μm NS309 pretreatment (2 min) to activate SKCa/IKCa preceding ACh. Top: note hyperpolarization to ∼80 mV with NS309; ACh increased V m to ∼−85 mV. Bottom: note lack of F 340/F 380 response until ACh is introduced. C, as in A with 145 mm [K+]o pretreatment for 2 min. Depolarization to ∼−5 mV had no effect on F 340/F 380 until the addition of ACh. Note diminished F 340/F 380 during plateau phase versus control (compare with A). Arrow indicates a transient secondary rise in F 340/F 380 ∼2 min after initial peak response to ACh. Short horizontal bars above F 340/F 380 traces (bottom panels of A–C) indicate periods of data acquisition for Rest, Peak and Plateau (90 s after Peak) values, respectively; note difference in time scales. D, summary of F 340/F 380 at corresponding V m before (Rest) and during ACh (Peak and Plateau) for paired experiments before and during NS309 (n = 10 endothelial tubes from 10 mice). E, as in D for paired experiments during ACh alone and during ACh with 145 mm [K+]o (n = 9 endothelial tubes from 9 mice). *P < 0.05, NS309 or 145 mm KCl versus respective Control values.
In the second approach, defined levels of intracellular current injection were used to clamp V m incrementally and thereby test how gradations in V m affected [Ca2+]i at rest (Fig. 3) and in the presence of 100 nm ACh (Figs 4 and 5). The first series of these experiments evaluated the effect of clamping V m on [Ca2+]i before and then during stimulation with ACh. Thus, a designated level of current injection was begun 20 s prior to and maintained throughout 2 min of exposure to 100 nm ACh (see Fig. 4 A, −3 nA corresponds to V m of ∼−65 mV; −5 to +5 nA corresponds to V m ranging from −80 to +10 mV). At ∼30 s following the peak [Ca2+]i response to ACh (i.e. during the sustained phase of Ca2+ influx), current injection was stopped and then resumed in 20 s intervals to evaluate the [Ca2+]i response to a given level of current injection; data were collected ∼90 s into the stimulation protocol. Following a 3 min washout of ACh to restore resting conditions, this procedure was repeated up to 12 times with respective levels of current injection randomized in magnitude and polarity across experiments. Our previous studies have confirmed that ACh evokes reproducible peak and plateau [Ca2+]i responses for at least 12 applications repeated in such manner (Behringer et al. 2012; Socha et al. 2012). The second series of these experiments investigated the effect of changing V m incrementally from −80 to +10 mV on [Ca2+]i during the sustained plateau phase of continuous exposure to 100 nm ACh (see Fig. 5 A). Thus, starting at ∼30 s after the peak [Ca2+]i response to ACh, each level of current (−5 to +5 nA) was injected for 20 s followed by 20 s of rest, alternating through the entire range of current amplitude and polarity (randomized as above) during ∼10 min of continuous exposure to ACh.
Figure 3.
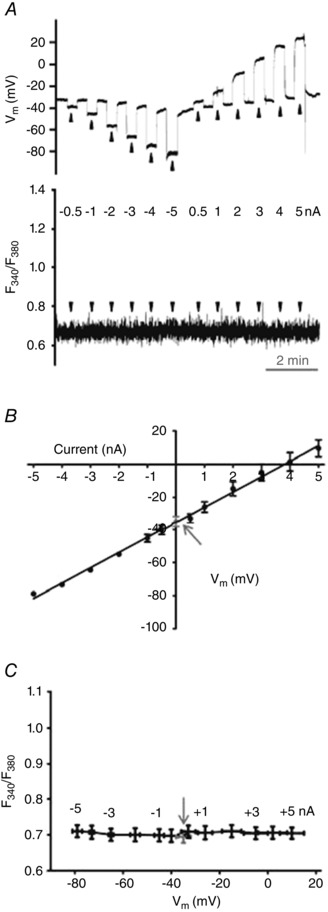
Changing Vm with current injection does not alter resting [Ca2+]i
A, simultaneous V m (top) and F 340/F 380 (bottom) recordings during control conditions at rest (V m, −35 ± 3 mV; F 340/F 380, 0.69 ± 0.02) and during current injection (−5 to +5 nA; 20 s pulses at arrowheads). While V m (top) responded in a stepwise manner F 340/F 380 (bottom) did not change. B, linear regression of V m versus injected current (r 2 = 0.990 ± 0.002). C, summary data illustrating stability of F 340/F 380 throughout range of V m during respective levels of current injection (shown near data points for reference). Arrows in B and C indicate resting V m and F 340/F 380, respectively. Summary data binned according to level of current injection for n = 7 endothelial tubes from 6 mice.
Figure 4.
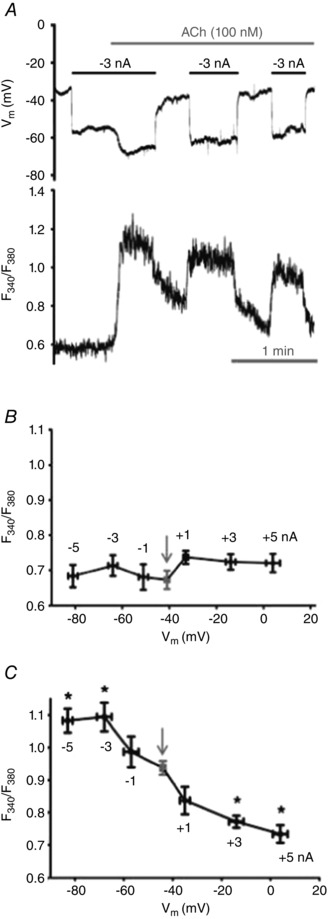
Changing Vm with intracellular current injection alters [Ca2+]i only during stimulation with ACh
A, simultaneous recordings of V m (top) and F 340/F 380 (bottom) with −3 nA current pulses before and during 100 nm ACh. Note lack of effect of hyperpolarization on [Ca2+]i until ACh is introduced. B, summary of F 340/F 380 values at designated values of V m throughout levels of current injection (shown near data points) prior to ACh. Resting F 340/F 380 was not altered from control (arrow) while V m ranged from −82 ± 2 mV to 4 ± 3 mV. C, summary of F 340/F 380 values at designated values of V m throughout range of current injection during plateau of [Ca2+]i response to ACh; one current level studied during each ACh stimulation. *P < 0.05, F 340/F 380 during current injection versus zero current (arrow; elevated F 340/F 380 versus B due to ACh). Summary data in B and C binned according to level of current injection for n = 6–7 endothelial tubes from 6–7 mice.
Figure 5.
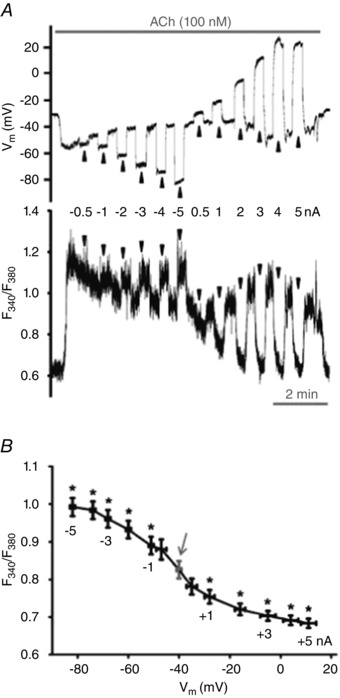
[Ca2+]i changes with Vm during stimulation with ACh
A, simultaneous V m (top) and F 340/F 380 (bottom) recordings during current injection (−5 to +5 nA; 20 s pulses at arrowheads) during 100 nm ACh. B, summary of F 340/F 380 values for corresponding values of V m at rest (arrow) and during 100 nm ACh; data binned according to level of current injection (shown near data points). This relationship is sigmoidal (R 2 = 0.974 ± 0.005) with V m at half‐maximal F 340/F 380 = −44 ± 2 mV. Note increase in F 340/F 380 with magnitude of hyperpolarization and decrease in F 340/F 380 (to baseline levels) as V m approximates 0 mV. Summary data represent n = 30 endothelial tubes from 25 mice. *P < 0.05, F 340/F 380 during current injection versus F 340/F 380 without current injection (arrow).
Pharmacology
Throughout experiments, a half‐maximal ACh stimulus (100 nm; EC50) (Behringer et al. 2012) was used to open Ca2+‐permeable ion channels which underlie [Ca2+]i influx during sustained activation of muscarinic receptors (Busse et al. 2002). Our rationale for half‐maximal stimulation with ACh was to (1) avoid saturating Ca2+ signalling pathways (e.g. as occurs during maximal stimulation with 3 μm ACh; Behringer et al. 2012) to thereby allow for dynamic [Ca2+]i responses (e.g. Ca2+ flux through TRP channels) during changes in V m; and (2) limit the hyperpolarization and current leakage that accompany maximal SKCa/IKCa activation during maximal stimulation with ACh (Behringer & Segal, 2012 b). Other drugs used were NS309 (SKCa/IKCa activator), obtained from Tocris Bioscience (Bristol, UK), the TRPV4 agonist GSK1016790A (GSK101) and the TRPV4 antagonist GSK2193874 (GSK219) (Thorneloe et al. 2008) obtained from Sigma. As internal controls for the role of TRPV4, complementary experiments were performed using endothelial tubes isolated from SEAs of TRPV4−/− mice in which the expression of TRPV4 was deleted genetically (Thorneloe et al. 2008).
Data analyses
Data analyses included (1) fluorescence emission collected at 510 nm and expressed as the ratio during excitation at 340 nm and 380 nm (F 340/F 380); (2) change in F 340/F 380 ratio (∆F 340/F 380) = peak response F 340/F 380 − preceding baseline F 340/F 380; (3) resting V m (mV); and (4) change in V m (ΔV m) = peak response V m − preceding baseline V m. All summary data reflect values averaged over 10 s during stable recordings. Statistical analyses (GraphPad Software, Inc., La Jolla, CA, USA) included linear regression, sigmoidal fits, paired and unpaired Student's t tests, and one‐way repeated measures analysis of variance with Tukey's post hoc comparisons. Differences were accepted as statistically significant with P < 0.05. Summary data are presented as means ± SEM. Typically, one endothelial tube was studied per mouse for a given protocol; in some cases a second tube was prepared for study on the same day from the contralateral abdominal muscle (kept in PSS at 4°C) of the same animal. Values of n are the number of endothelial tubes studied using a given protocol. With the exception of TRPV4−/− versus wild‐type preparations, all comparisons are paired (i.e. control versus treatment) within a given experimental protocol. Sampling conditions were not altered across preparations and background fluorescence of Fura‐2 was collected and subtracted for each experiment.
Results
Our goal was to determine whether V m can govern endothelial [Ca2+]i using an experimental strategy that enhanced the open‐probability of Ca2+‐permeable channels in the plasma membrane and did so without maximizing this signalling pathway. In native microvascular endothelium this condition was evoked via stimulation of muscarinic receptors using a concentration of ACh that approximated the EC50 for [Ca2+]i and V m (Behringer et al. 2012). From the control baseline, 100 nm ACh evoked a rise in F 340/F 380 (Fig. 2 A). The initial peak reflects intracellular release of Ca2+ from the ER, with the sustained plateau attributable to Ca2+ influx through the plasma membrane (Cohen & Jackson, 2005; Socha et al. 2012). Consistent with original findings (Busse et al. 1988), these changes in [Ca2+]i mirrored the dynamics of V m (Fig. 2 A). Indeed, this inverse correspondence between respective signalling events is what led to questioning the relationship(s) between [Ca2+]i and V m (Busse et al. 2002; Garland et al. 2011; Dora & Garland, 2013).
Hyperpolarization with NS309 enhances Ca2+ influx during stimulation with ACh
We altered V m by activating SKCa/IKCa with NS309 before and during stimulation of muscarinic receptors (Behringer et al. 2012). Thus, NS309 (10 μm) hyperpolarized cells from −34 ± 3 mV under control conditions to a stable value of −82 ± 1 mV, yet had no significant effect on F 340/F 380 (Fig. 2 B). However, the addition of 100 nm ACh evoked a prompt and robust elevation in peak and plateau [Ca2+]i (Fig. 2 B) with responses that were ∼52% and ∼67% greater (P < 0.05) than control conditions, respectively (Fig. 2 A, B and D, and Table 1).
Table 1.
Effects of hyperpolarization (10 μm NS309) and depolarization (145 mm [K+]o on [Ca2+]i and Vm responses to ACh
| F 340/F 380 | V m (mV) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Condition | Rest | Peak | Peak ∆ | Plateau | Plateau ∆ | Rest | Peak | Peak ∆ | Plateau | Plateau ∆ | n |
| Control | 0.69 ± 0.03 | 0.91 ± 0.06 | 0.23 ± 0.07 | 0.83 ± 0.04 | 0.15 ± 0.04 | −33 ± 2 | −44 ± 4 | −11 ± 4 | −37 ± 2 | −4 ± 1 | 10 |
| NS309 (10 μm) | 0.70 ± 0.03 | 1.06 ± 0.05* | 0.35 ± 0.05* | 0.95 ± 0.05* | 0.25 ± 0.04* | −82 ± 1* | −86 ± 2* | −4 ± 1* | −85 ± 1* | −4 ± 1 | 10 |
| Control | 0.69 ± 0.03 | 1.00 ± 0.05 | 0.31 ± 0.06 | 0.88 ± 0.06 | 0.19 ± 0.04 | −36 ± 1 | −52 ± 3 | −16 ± 4 | −40 ± 1 | −4 ± 1 | 9 |
| 145 mm [K+]o | 0.70 ± 0.02 | 1.04 ± 0.04 | 0.34 ± 0.05 | 0.78 ± 0.03* | 0.08 ± 0.01* | −5 ± 1* | −2 ± 1* | 3 ± 1* | −4 ± 1* | 2 ± 1* | 9 |
Summary of values for Resting, Peak and Plateau phases for [Ca2+]i (F 340/F 380, ∆F 340/F 380) and membrane potential (V m, ∆V m) of endothelial tubes during stimulation with 100 nm ACh (see Fig. 2). [Ca2+]o was 2 mm for all experiments. ACh was present under all conditions except ‘Rest’.
*P < 0.05, treatment versus paired Control.
Depolarization with 145 mm KCl reduces Ca2+ influx during stimulation with ACh
To investigate the effect of depolarization on [Ca2+]i, [K+]o was raised to 145 mm, lowering V m from −36 ± 2 mV (rest) to −5 ± 1 mV (n = 9). Despite no effect on resting F 340/F 380 or the peak [Ca2+]i response to 100 nm ACh (Fig. 2 C and E), the plateau F 340/F 380 response was ≈58% lower (P < 0.05) than the control response with 5 mm [K+]o (Fig. 2 A, C and E, and Table 1). The presence of 145 mm [K+]o also resulted in a transient secondary rise in [Ca2+]i at 130 ± 10 s following the initial peak response to ACh (Fig. 2 C). The amplitude of this secondary [Ca2+]i transient was not different from the plateau F 340/F 380 response to 100 nm ACh under control conditions for the same preparations (Table 1).
Intracellular current injection does not alter [Ca2+]i until stimulation with ACh
Under control conditions, neither hyperpolarizing nor depolarizing current injections (V m range: −79 ± 2 mV to 10 ± 5 mV) altered resting [Ca2+]i (Figs. 3 A and C, and 4 B). Finding that V m was related linearly to the magnitude and polarity of the injected current (Fig. 3 B) confirms the lack of voltage‐gated ion channels in native ECs (Cohen & Jackson, 2005; Jackson, 2005; Ledoux et al. 2008; Behringer et al. 2012). The independence of [Ca2+]i from V m was maintained throughout the range of current injection (−5 to +5 nA; Figs. 3 C and 4 B). In contrast, during a given level of hyperpolarization (e.g. −3 nA), addition of 100 nm ACh promptly increased F 340/F 380 (Fig. 4 A). Further, ensuing current pulses during the plateau phase of the [Ca2+]i response to ACh changed F 340/F 380 in a manner that increased with the magnitude of hyperpolarization and decreased with the magnitude of depolarization (Fig. 4 C).
[Ca2+]i responds bi‐directionally to changes in V m during stimulation with ACh
Despite a large electrochemical gradient for Ca2+ entry, the opening of channels permeable to Ca2+ is essential for extracellular Ca2+ to access the cell interior (Clapham, 2007). In the presence of 100 nm ACh, stepwise current injections (−5 to +5 nA) altered V m from −82 ± 1 mV to 11 ± 3 mV (Fig. 5 A, top and Fig. 5 B). As evidenced by concomitant changes in F 340/F 380, hyperpolarization increased [Ca2+]i while depolarization decreased [Ca2+]i (Fig. 5 A, bottom and Fig. 5 B). Sigmoid fits to these summary data indicate that V m at half‐maximal F 340/F 380 corresponds to −44 ± 2 mV (Fig. 5 B). In accord with F 340/F 380 values, estimated [Ca2+]i concentrations (see Methods) were 104 ± 5 nm at rest and 168 ± 11 nm during the ACh plateau, where injection of −5 nA increased [Ca2+]i to 246 ± 12 nm and +5 nA decreased [Ca2+]i to 104 ± 5 nm. Thus, while not manifested under control conditions, [Ca2+]i responds bidirectionally to changing V m during half‐maximal stimulation of muscarinic receptors.
In 30 endothelial tubes from 25 mice, exposure to 100 nm ACh resulted in a plateau [Ca2+]i response that varied across experiments. Regression analyses of the ΔF 340/F 380 in response to ACh versus the ΔF 340/F 380 during current injection revealed no correlation during hyperpolarizing current injections. Thus, while the rise in [Ca2+]i during hyperpolarization tended to increase with the level of negative current injection, it was independent
of the plateau [Ca2+]i response to ACh (Fig. 6 A). In contrast, there were significant negative correlations between respective [Ca2+]i responses during depolarizing current injections (Fig. 6 B). Thus, reductions in [Ca2+]i during depolarization increased with the magnitude of the plateau [Ca2+]i response to ACh, such that +3 nA and +5 nA (corresponding V m: −5 ± 3 and 11 ± 3 mV) consistently lowered plateau F 340/F 380 to resting levels that preceded stimulation with ACh (see Fig. 5 A).
Figure 6.
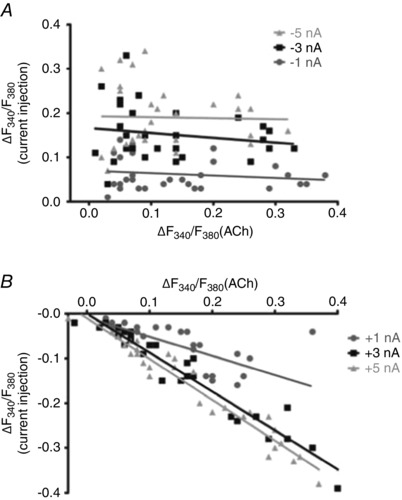
Plateau [Ca2+]i responses to ACh correlate with [Ca2+]i responses to depolarization
A, scatter plot illustrating magnitude of ∆F 340/F 380 during negative current injections of −1, −3 and −5 nA during the plateau ∆F 340/F 380 response to 100 nm ACh (Y‐axis) versus the plateau ∆F 340/F 380 response (from resting baseline) to 100 nm ACh (X‐axis). Note lack of correlation (R 2 < 0.035). B, as in A during positive current injections of +1, +3 and +5 nA. Note negative correlations between the decreased [Ca2+]i during depolarization and the plateau [Ca2+]i response to ACh (R 2 > 0.55). Individual data points correspond to summary data in Fig. 5 B.
[Ca2+]i responses to changing V m during stimulation with ACh require extracellular Ca2+
In the absence of extracellular Ca2+, the plateau F 340/F 380 response to ACh was effectively abolished (Table 2). Under these conditions, current injections that altered V m from −62 ± 4 mV to 13 ± 6 mV had no significant effect on F 340/F 380 (Fig. 7). In the same preparations, including 2 mm [Ca2+]o resulted in [Ca2+]i responses consistent with those illustrated in Fig. 5 (Fig. 7 B). Thus, the rise in [Ca2+]i during hyperpolarizing current injections reflects Ca2+ influx from the extracellular fluid. We then tested whether F 340/F 380 responses to changing V m in the presence of ACh would be enhanced by elevating [Ca2+]o to 10 mm. Through the range of −5 nA to +5 nA current injection (with V m ranging from −90 ± 2 to 2 ± 9 mV), V m at half‐maximal F 340/F 380 shifted to the left from −44 ± 3 mV (with 2 mm [Ca2+]o) to −50 ± 4 mV (with 10 mm [Ca2+]o) and F 340/F 380 was enhanced by ∼7–10% at V m ≥ −80 mV (P < 0.05; Fig. 8). Further, plateau F 340/F 380 responses to ACh were significantly higher (P < 0.05) in the presence of 10 mm [Ca2+]o compared to control with 2 mm [Ca2+]o (Table 2). Thus, in the presence of ACh, increasing [Ca2+]o can augment Ca2+ entry during hyperpolarization.
Table 2.
Effect of extracellular Ca2+ on [Ca2+]i and Vm responses to ACh
| F 340/F 380 | V m (mV) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Condition | Rest | Peak | Peak ∆ | Plateau | Plateau ∆ | Rest | Peak | Peak ∆ | Plateau | Plateau ∆ | n |
| Control | 0.72 ± 0.01 | 0.96 ± 0.07 | 0.24 ± 0.07 | 0.82 ± 0.03 | 0.11 ± 0.03 | −39 ± 4 | −49 ± 5 | −10 ± 4 | −42 ± 4 | −3 ± 1 | 8 |
| 0 [Ca2+]o | 0.69 ± 0.02 | 0.81 ± 0.05* | 0.12 ± 0.04* | 0.71 ± 0.02* | 0.03 ± 0.01* | −25 ± 2* | −31 ± 2* | −6 ± 2 | −24 ± 2* | −1 ± 1* | 8 |
| Control | 0.68 ± 0.01 | 0.86 ± 0.04 | 0.18 ± 0.04 | 0.79 ± 0.03 | 0.10 ± 0.03 | −36 ± 3 | −41 ± 3 | −5 ± 1 | −38 ± 3 | −2 ± 1 | 10 |
| 10 mm [Ca2+]o | 0.71 ± 0.02 | 1.12 ± 0.04* | 0.41 ± 0.04* | 0.93 ± 0.02* | 0.22 ± 0.03* | −52 ± 4* | −63 ± 3* | −11 ± 2* | −57 ± 4* | −5 ± 2 | 10 |
Summary of values for Resting, Peak and Plateau phases for [Ca2+]i (F 340/F 380, ∆F 340/F 380) and membrane potential (V m, ∆V m) of endothelial tubes during stimulation with 100 nm ACh when [Ca2+]o was nominally 0 (Ca2+‐free PSS) or elevated to 10 mm and compared to respective Controls with [Ca2+]o = 2 mm. ACh was present under all conditions except ‘Rest’.
Note that removal of [Ca2+]o was associated with depolarization while elevating [Ca2+]o to 10 mm was associated with hyperpolarization. *P < 0.05, treatment versus paired Control.
Figure 7.
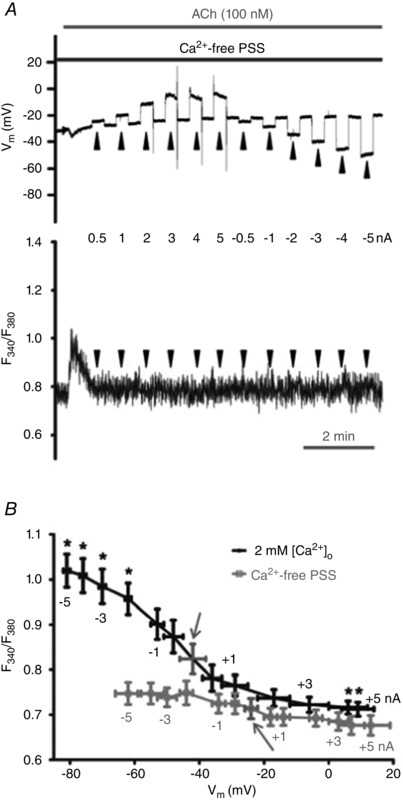
[Ca2+]i responses to changing Vm during stimulation with ACh require extracellular Ca2+
A, simultaneous V m (top) and F 340/F 380 (bottom) recordings during current injection (−5 to +5 nA; 20 s pulses at arrowheads) during 100 nm ACh in Ca2+‐free PSS. Note absence of sustained elevation (i.e. plateau phase) in [Ca2+]i response. B, overlay of summary data for F 340/F 380 values at corresponding levels of V m (binned according to level of current injection) during 100 nm ACh with 2 mm [Ca2+]o and with Ca2+‐free PSS. Arrows indicate V m and F 340/F 380 under respective conditions with zero current. Summary data are paired experiments for n = 8 endothelial tubes from 8 mice. *P < 0.05, F 340/F 380 during current injection versus F 340/F 380 with zero current.
Figure 8.
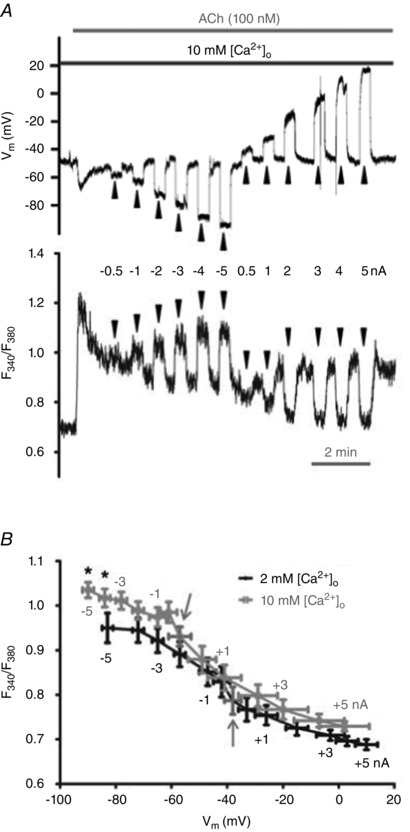
Increasing [Ca2+]o increases [Ca2+]i with maximal hyperpolarization during stimulation with ACh
A, simultaneous V m (top) and F 340/F 380 (bottom) recordings during current injection (−5 to +5 nA; 20 s pulses at arrowheads) during 100 nm ACh; [Ca2+]o = 10 mm. B, overlay of summary data for F 340/F 380 values at corresponding levels of V m (binned according to level of current injection). Arrows indicate V m and F 340/F 380 during plateau response to ACh with zero current. Summary data are paired experiments for n = 10 endothelial tubes from 10 mice. *P < 0.05, F 340/F 380 during 10 mm [Ca2+]o versus 2 mm [Ca2+]o.
Role of TRPV4 for Ca2+ influx during stimulation with ACh
When activated, TRPV4 are integral to Ca2+ influx across plasma membranes of vascular endothelium (Zhang et al. 2009; Sonkusare et al. 2012; Qian et al. 2014). We therefore tested the role of TRPV4 in mediating Ca2+ entry in response to changing V m using genetic and pharmacological approaches. Under resting conditions, F 340/F 380 and V m of endothelial tubes from TRPV4−/− mice (0.72 ± 0.03 and −34 ± 3 mV, respectively; n = 8) were not significantly different from those of wild‐type C57BL/6 mice (0.68 ± 0.01 and −37 ± 1 mV, respectively; n = 30; Table 3). During stimulation with 100 nm ACh, injecting −5 nA to +5 nA in TRPV4−/− altered V m from −79 ± 5 mV to 15 ± 7 mV and [Ca2+]i increased with V m ≥ ∼−65 mV (Fig. 9 A and B). When compared to C57BL/6 mice, F 340/F 380 responses in endothelial tubes from TRPV4−/− mice were reduced by ∼50% at V m ≥ −60 mV (Fig. 9 C).
Table 3.
Effect of TRPV4 on [Ca2+]i and Vm responses to ACh
| (F 340/F 380) | V m (mV) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Condition | Rest | Peak | Peak ∆ | Plateau | Plateau ∆ | Rest | Peak | Peak ∆ | Plateau | Plateau ∆ | n |
| Wild‐type | 0.68 ± 0.01 | 0.97 ± 0.03 | 0.28 ± 0.03 | 0.83 ± 0.02 | 0.14 ± 0.02 | −37 ± 1 | −50 ± 2 | −13 ± 2 | −40 ± 1 | −3 ± 1 | 30 |
| TRPV4−/− | 0.72 ± 0.03 | 0.80 ± 0.04* | 0.08 ± 0.02* | 0.76 ± 0.04 | 0.04 ± 0.01* | −34 ± 3 | −40 ± 3* | −6 ± 1* | −37 ± 2 | −2 ± 1 | 8 |
| Wild‐type Control | 0.73 ± 0.02 | 1.18 ± 0.06 | 0.45 ± 0.06 | 0.98 ± 0.04 | 0.25 ± 0.04 | −41 ± 4 | −62 ± 5 | −21 ± 5 | −51 ± 4 | −11 ± 3 | 6 |
| Wild‐type GSK219 | 0.70 ± 0.03 | 1.12 ± 0.03 | 0.42 ± 0.03 | 0.88 ± 0.04 | 0.19 ± 0.02 | −33 ± 3 | −63 ± 4 | −30 ± 6 | −44 ± 6 | −11 ± 4 | 6 |
Summary of values for Rest, Peak and Plateau phases for [Ca2+]i (F 340/F 380, ∆F 340/F 380) and membrane potential (V m, ∆V m) of endothelial tubes during stimulation with 100 nm ACh in endothelial tubes from wild‐type (C57BL/6) versus TRPV4−/− mice and from wild‐type (C57BL/6) before (Control) and during exposure to the TRPV4 antagonist GSK2193874 (GSK219; 1 μm). [Ca2+]o = 2 mm under all conditions. ACh was present under all conditions except ‘Rest’.
*P < 0.05, TRPV4−/− versus wild‐type.
Figure 9.
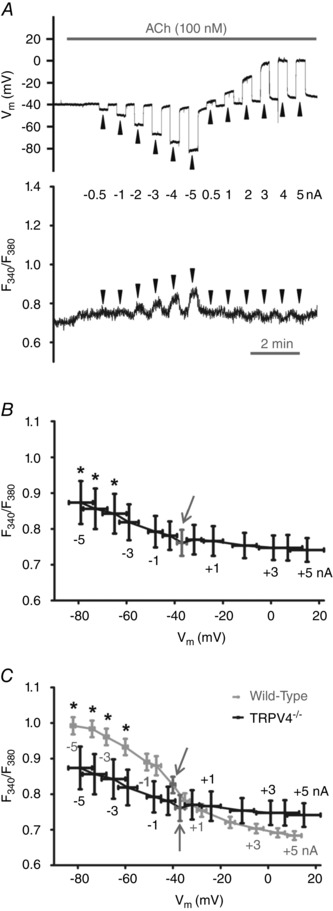
Lack of TRPV4 expression attenuates Ca2+ influx with hyperpolarization during stimulation with ACh
A, simultaneous V m (top) and F 340/F 380 (bottom) recordings in the presence of 100 nm ACh during current injection (alternating 20 s pulses at arrowheads) in endothelial tube from a TRPV4−/− mouse. B, summary data for F 340/F 380 at corresponding V m (binned according to level of current injection) during 100 nm ACh in endothelial tubes from TRPV4−/− mice. *P < 0.05, F 340/F 380 during current injection versus no current injection (arrow). Data represent 8 endothelial tubes from 5 TRPV4−/− mice. C, overlay of summary data from B with data from wild‐type (C57BL/6) mice (from Fig. 5 B) at corresponding levels of V m and current injection (all experiments with [Ca2+]o = 2 mm). Arrows indicate values with zero current. *P < 0.05, F 340/F 380 of wild‐type versus TRPV4−/− at given level of current injection.
When applied to endothelial tubes of wild‐type C57BL/6 mice, the TRPV4 antagonist GSK219 (1 μm) tended to reduce F 340/F 380 values and resting V m but these effects were not statistically significant (Table 3). However GSK219 suppressed the [Ca2+]i response to hyperpolarizing current injection during stimulation with 100 nm ACh. Thus, −3 nA increased Plateau F 340/F 380 to 1.09 ± 0.03 under control conditions and this response was reduced to 0.94 ± 0.04 (P < 0.05, n = 6) in the presence of 1 μm GSK219 despite no difference in hyperpolarization (V m = −72 ± 4 mV and −76 ± 5 mV, respectively). Application of the TRPV4 agonist GSK101 (1–10 nm) to endothelial tubes of C57BL/6 mice produced hyperpolarization (rest: −38 ± 5 mV, GSK101: −53 ± 4 mV) and increased F 340/F 380 from 0.70 ± 0.03 (rest) to 0.84 ± 0.03 (GSK101; n = 4, P < 0.05); however [Ca2+]i did not increase further during hyperpolarizing current injection (for −3 nA: ∆F 340/F 380: 0.01 ± 0.01, ∆V m:−23 ± 5 mV). Thus, in the presence of ACh, TRPV4 activation in wild‐type endothelium contributes to Ca2+ influx during hyperpolarization. While genetic deletion of TRPV4 or its pharmacological inhibition with GSK219 impaired the effect of hyperpolarization on promoting Ca2+ entry during ACh stimulation, pharmacological activation of TRPV4 with GSK101 did not increase [Ca2+]i during hyperpolarization.
Discussion
The relationship between V m and EC [Ca2+]i is central to the regulation of tissue blood flow with Ca2+ serving as an integral second messenger during endothelium‐dependent vasodilatation (Busse et al. 2002; Ledoux et al. 2006; Bagher & Segal, 2011; Garland et al. 2011). Whether hyperpolarization promotes Ca2+ entry in vascular ECs has remained controversial for more than 25 years. The driving force for Ca2+ into the cell is manifested by a ∼20,000‐fold concentration gradient from the extracellular to the intracellular compartments complemented by electronegativity of the cell interior (Clapham, 2007). However, cell membranes must be permeable to Ca2+ for influx to occur. In this study, we questioned whether changes in V m alone or in conjunction with activation of muscarinic receptors could alter [Ca2+]i of intact endothelium freshly isolated from resistance arteries of mouse skeletal muscle. We reasoned that if Ca2+‐permeable channels were inactive at rest, then changing V m should be ineffective in altering resting [Ca2+]i. Complementary approaches altered V m under resting conditions and while Ca2+‐permeable channels were opened submaximally with ACh, i.e. during the plateau phase of signalling dominated by Ca2+ influx. Our principal findings demonstrate that, under resting conditions, [Ca2+]i was unaffected when V m was shifted through a range of ∼90 mV (i.e. from −80 mV to +10 mV). In striking contrast, in the presence of 100 nm ACh, [Ca2+]i increased in response to hyperpolarization and decreased in response to depolarization from a resting V m of ∼−40 mV. This effect of changing V m on [Ca2+]i was lost following removal of extracellular Ca2+ and was attenuated by half in endothelium of TRPV4−/− mice. Thus, V m modulates Ca2+ entry during submaximal activation of Ca2+‐permeable ion channels in the plasma membrane of native microvascular endothelium.
Experimental considerations
Complexity of Ca2+ signalling
A potentially confounding variable in studies of intact vessels is that ECs can be coupled directly (via myoendothelial gap junctions) to SMCs, which do express voltage activated ion channels (Jackson, 2005; Ledoux et al. 2006) and may affect EC [Ca2+]i indirectly via myoendothelial coupling (Sonkusare et al. 2012; Tran et al. 2012; Dora & Garland, 2013). Thus, to resolve the relationship between V m and Ca2+ entry strictly in accord with electrochemical driving force requires that (1) only the endothelium is present; (2) the experimental approach does not activate voltage‐gated ion channels; (3) V m is altered independent of [Ca2+]i while both variables are evaluated simultaneously; and (4) experimental manipulations are of sufficient intensity and duration to evoke definitive responses without saturating (and thereby masking) the variables under investigation. In light of previous efforts to resolve the effect of V m on [Ca2+]i (see citations in Dora & Garland, 2013), the present experiments endeavoured to ensure that respective criteria were satisfied to the extent possible.
The physiological integration between Ca2+ release from the ER and its influx through the plasma membrane can complicate experimental approaches attempting to resolve endothelial Ca2+ dynamics following muscarinic receptor stimulation. For example, preventing release of Ca2+ from the ER precludes the key physiological stimulus for Ca2+ influx (Bishara et al. 2002). Thus, our approach for investigating how Ca2+ influx may be governed by V m during submaximal stimulation of muscarinic receptors included removal of extracellular Ca2+, raising [Ca2+]o to 10 mm and determining the effect of eliminating an integral Ca2+‐permeable membrane channel by studying endothelial tubes of TRPV4−/− mice as well as pharmacological inhibition of TRPV4 in endothelial tubes of wild‐type mice.
Calcium photometry
Our evaluation of [Ca2+]i relied on photometry and reflects ‘global’ levels as recorded simultaneously from ∼50 ECs contained within the window used for detecting intracellular fluorescence (Fig. 1). This approach is consistent with methods employed by previous investigators evaluating the relationship between V m and [Ca2+]i (Busse et al. 1988; Cohen & Jackson, 2005; McSherry et al. 2005). In contrast, confocal imaging has been used to detect Ca2+ signalling events (i.e. ‘sparklets’) localized to single Ca2+ channels (e.g. TRPV4) in the plasma membrane of individual ECs within myoendothelial projections that contact SMCs (Bagher et al. 2012; Sonkusare et al. 2012; Tran et al. 2012; Sonkusare et al. 2014). However, during the isolation of endothelial tubes, removal of SMCs obviates myoendothelial projections. An advantage of Ca2+ photometry is the ability to acquire data from the same cells throughout protocols lasting 10–15 min (e.g. Figs 3, 5 and 7, 8, 9). Such prolonged recording is not possible with confocal imaging due to photobleaching of dye indicators. Thus, Fura‐2 photometry concomitant with intracellular recording enables resolution of whether, and under what conditions, ‘global’ [Ca2+]i of native microvascular endothelium can be governed in response to altering V m. Our use of 100 nm ACh in the present experiments was to approximate half‐maximal (i.e. EC50) levels of Ca2+ signalling and to limit the loss of charge during current injection to be able to control V m (Behringer & Segal, 2012 b). Using confocal imaging of the same preparations studied here, 100 nm ACh elicited spatially restricted Ca2+ events in single cells (i.e. Ca2+ ‘puffs’), whereas 1 μm ACh evoked the propagation of Ca2+ waves within and between neighbouring ECs (Socha et al. 2012). A question for imaging studies in the future is whether imposing hyperpolarization during submaximal stimulation with ACh can elevate Ca2+ signalling from discrete intracellular events to the generation and propagation of Ca2+ waves.
Intracellular recording
The V m of ECs was clamped for defined intervals (e.g. 20 s) by injecting negative or positive current through an intracellular microelectrode. As these cells are coupled electrically through gap junctions, the change in V m is conducted from cell to cell, decaying with distance from the site of current injection (Behringer & Segal, 2012 b). Monitoring V m adjacent to the site of Ca2+ photometry (Fig. 1) enabled the relationship between endothelial V m and [Ca2+]i to be evaluated remotely from the site of current injection while confirming the key physiological property of electrical conduction. Thus V m was controlled through a physiological range in accord with the magnitude and polarity of injected current. Nevertheless, current can ‘leak’ out of cells during ion channel activation as shown for SKCa/IKCa (Behringer & Segal, 2012 b). Thus the ability to control V m using current injection and thereby determine the effect of V m on [Ca2+]i is compromised with interventions that activate ion channels to a sufficient extent, e.g. during maximal stimulation with 3 μm ACh (Behringer & Segal, 2012 b). We therefore used an EC50 ACh stimulus to evaluate how V m affected Ca2+ entry while avoiding saturation of this signalling pathway.
Present and previous approaches
In the present experiments, multiple approaches were used to investigate the effect of V m on [Ca2+]i with ACh used as a tool to activate Ca2+‐permeable channels half‐maximally. Near‐maximal hyperpolarization was achieved by opening SKCa/IKCa directly with 10 μm NS309 (Behringer & Segal, 2012 b). Alternatively, maximal depolarization was imposed using 145 mm [K+]o to shift the Nernst potential for K+ to zero versus ∼−90 mV with 5 mm [K+]o. To explore the relationship between V m and [Ca2+]i with greater resolution, incremental changes in V m were imposed using graded levels of current injected through an intracellular microelectrode.
Under resting conditions, neither chemical nor electrical interventions altered [Ca2+]i despite V m ranging from −80 to +10 mV (Figs 2, 3, 4, 5). Endothelium of the mouse SEA lacks voltage‐gated ion channel activity as demonstrated by strictly linear voltage responses through this range in V m during current injection (Fig. 3; Behringer & Segal, 2012 b; Behringer et al. 2012). Such behaviour is consistent with findings from native endothelium in several vascular beds including skeletal muscle, and mesenteric and cerebral arteries of mice, rats and hamsters (Cohen & Jackson, 2005; Jackson, 2005; Ledoux et al. 2008). In contrast, studies of cultured ECs have detected a progressive decrease in resting [Ca2+]i from a V m of −100 mV to +60 mV (Cannell & Sage, 1989; Laskey et al. 1990; Sharma & Davis, 1995), suggesting the presence of voltage‐sensitive ion channels. The expression of membrane proteins can be altered when ECs are grown in culture. Thus, the acquisition of large‐conductance Ca2+ activated K+ channels (Sandow & Grayson, 2009) may impart voltage sensitivity while the loss of muscarinic receptors (Tracey & Peach, 1992) has necessitated the use of alternative agonists (e.g. bradykinin) to initiate Ca2+ signalling, thereby complicating comparisons to experiments performed on native endothelium stimulated with ACh.
Near‐maximal hyperpolarization with NS309 amplified the [Ca2+]i response to ACh (Fig. 2 A, B and D) while maximal depolarization with 145 mm [K+]o reduced the plateau phase of the [Ca2+]i response to ACh (Fig. 2 C and E). In the presence of 100 nm ACh, −5 nA hyperpolarized the endothelium and enhanced plateau [Ca2+]i similar to what was achieved using 10 μm NS309 (compare Fig. 2 D with Figs 4 C and 5 B). Conversely, injection of +3 nA depolarized the endothelium and decreased the plateau [Ca2+]i response to ACh similar to the action of 145 mm [K+]o (compare Fig. 2 E with Figs 4 C and 5 B). In contrast, studies of endothelium freshly isolated from arterioles of the hamster cremaster muscle found that the plateau [Ca2+]i responses to methacholine (1 μm) was unaffected by depolarization with 145 mm [K+]o (Cohen & Jackson, 2005) while experiments performed on intact rat mesenteric arteries found that the [Ca2+]i response to 0.3 μm ACh was unaffected by depolarization with 35 mm KCl (McSherry et al. 2005). These latter findings are consistent with studies of native endothelium from several laboratories indicating that changes in endothelial [Ca2+]i were independent of changes in V m (Marrelli et al. 2003; Takano et al. 2004; Cohen & Jackson, 2005; McSherry et al. 2005; Qian et al. 2014). It is possible that the level of Ca2+ influx stimulated by muscarinic receptor activation (and Ca2+ depletion from the ER) superseded any effect of hyperpolarization on Ca2+ influx. Nevertheless, several studies of cultured ECs have supported a role for V m in modulating Ca2+ influx during stimulation of G protein‐coupled receptors. For example, hyperpolarization and depolarization using voltage clamp, manipulation of [K+]o and/or altering K+ channel activity were found to increase and decrease [Ca2+]i, respectively during stimulation with ACh, bradykinin or histamine (Cannell & Sage, 1989; Laskey et al. 1990; Luckhoff & Busse, 1990; Sharma & Davis, 1995; Li et al. 1999).
An unexpected yet consistent finding was a transient secondary rise of F 340/F 380 values during depolarization with 145 mm [K+]o in the presence of 100 nm ACh. The amplitude of this secondary response was not different from the plateau F 340/F 380 values of paired controls with 100 nm ACh alone (Table 1; compare Fig. 2 C bottom trace at arrow to Fig. 2 A bottom trace during plateau]. While the mechanism underlying this secondary rise in [Ca2+]i is unknown, it may entail a delayed decrease in the function of the Na+/Ca2+ exchanger as a consequence of a low [Na+]o due to equimolar replacement with K+ during exposure to 145 mm [K+]o. Thus the time window used for data acquisition during the plateau phase of Ca2+ signalling may be critical for resolving the effects of depolarization using elevated [K+]o.
New insights into V m and Ca2+ signalling in endothelium of resistance vessels
Using dual intracellular microelectrodes to investigate the effect of controlling V m on the regulation of [Ca2+]i, the present findings illustrate that the relationship between V m and [Ca2+]i during stimulation with 100 nm ACh is sigmoidal in nature (Fig. 5 B) and requires extracellular Ca2+ (Fig. 7). Values for F 340/F 380 effectively plateaued with V m > ∼−60 mV or <∼−25 mV (Fig. 5 B) and the effect of V m on F 340/F 380 when [Ca2+]o was raised to 10 mm was significantly different from control ([Ca2+]o = 2 mm) only with V m > −80 mV) (Fig. 8). Thus, with a resting V m of ∼−30 to −40 mV (Welsh & Segal, 1998; Emerson & Segal, 2000; Wolfle et al. 2011; Behringer & Segal, 2012 b), the V m range of −25 to −60 mV appears to have the greatest physiological relevance to the regulation of [Ca2+]i by V m during submaximal activation of Ca2+‐permeable channels in the plasma membrane of ECs. Indeed, maximal dilatation of feed arteries and arterioles can be evoked with 10–15 mV of hyperpolarization (Emerson & Segal, 2000; Wolfle et al. 2011). Thus, a sigmoidal relationship between endothelial V m and [Ca2+]i signalling that is centred near resting V m is consistent with the physiological regulation of blood flow via endothelium‐dependent hyperpolarization. In turn, finding that the plateau elevation in [Ca2+]i can be reversed by depolarizing the cell interior to < −10 mV (Figs. 4 C and 5 B) suggests that sufficient hyperpolarization is required for increases in [Ca2+]i to be governed by V m. An unexpected finding is that, while the ability to increase [Ca2+]i during incremental levels of hyperpolarization did not correlate with the magnitude of the [Ca2+]i response to ACh (Fig. 6 A), decreases in [Ca2+]i during incremental depolarization were correlated with the magnitude of the plateau [Ca2+]i response to ACh (Fig. 6 B). These results suggest that the ability of depolarization to restore [Ca2+]i to resting levels reflects the marked ability of ECs to sequester or extrude cytosolic Ca2+ during a reduced electrical gradient for Ca2+ influx.
Role of TRPV4
Recent studies have demonstrated an integral role of TRPV4 for promoting endothelial Ca2+ influx and dilatation of resistance arteries via activation of SKCa/IKCa (i.e. endothelium‐dependent hyperpolarization) (Zhang et al. 2009; Sonkusare et al. 2012; Qian et al. 2014). In intact resistance arteries, pharmacological blockade and/or genetic deletion of TRPV4 were shown to reduce Ca2+ signalling and vasodilatation in response to ACh by ≥70% (Zhang et al. 2009; Sonkusare et al. 2012; Qian et al. 2014). Our findings using endothelial tubes from TRPV4−/− mice indicate that TRPV4 may account for nearly half of the capacity to increase [Ca2+]i in response to V m ≥ −60 mV during half‐maximal stimulation with ACh (Fig. 9) and that this effect can be mimicked in the endothelium of wild‐type mice using pharmacological inhibition with GSK219. The remaining [Ca2+]i response under these conditions may reflect Ca2+‐permeable channels comprising other TRP channels (e.g. TRPC1,3,4,5,6/TRPV1,3/TRPA1; Yue et al. 2015) that assemble in a homomeric or heteromeric fashion (Cheng et al. 2007; Earley et al. 2007; Ma et al. 2010). The contribution of other TRP channels may also explain why the activation of TRPV4 alone with GSK101 increased baseline [Ca2+]i yet had no further effect during current injection. Taken together, previous and current findings indicate that TRPV4, at least in part, contributes to both Ca2+‐induced hyperpolarization and hyperpolarization‐induced Ca2+ entry underlying endothelium‐dependent vasodilatation in the regulation of tissue blood flow.
Physiological significance of hyperpolarization‐induced Ca2+ entry
The use of hyperpolarizing agents (e.g. K+ channel activators) may help to alleviate decrements in vasodilatation and thereby restore tissue perfusion during cardiovascular disease (Wulff & Kohler, 2013). Indeed, pharmacological activation of SKCa/IKCa with SKA‐31 has been found to produce EC hyperpolarization and a reduction in arterial blood pressure (Damkjaer et al. 2012). Such actions have been found to ameliorate complications of hypertension (Radtke et al. 2013) and to promote coronary blood flow in diabetic hearts (Mishra et al. 2014). The mechanism underlying such benefits of SKCa/IKCa activation may reflect hyperpolarization‐induced Ca2+ entry to amplify SKCa/IKCa activity and/or NO production and can be evoked via submaximal stimulation of muscarinic receptors with ACh. However, there is a balance between the strength of initiating hyperpolarization at sites of stimulation and the ability to conduct the electrical signal along the vessel wall through gap junctions coupling ECs to each other and to surrounding SMCs. Activating SKCa/IKCa to the extent where V m exceeds −60 mV results in current leakage that restricts the spread of hyperpolarization by more than half (Behringer & Segal, 2012 b; Behringer et al. 2013). Under such conditions, charge loss through electrically leaky membranes impairs the ability of conducted vasodilatation to coordinate blood flow regulation among branches of vascular resistance networks (Behringer & Segal, 2012 a). Thus, invoking hyperpolarization‐induced Ca2+ entry (and activation of SKCa/IKCa in general) should be viewed with caution, as increasing SKCa/IKCa activation may be beneficial up to the point at which vasodilator signals fail to initiate and/or spread effectively. In light of current understanding with respect to how hyperpolarization is initiated and conducted to promote vasodilatation (Emerson & Segal, 2000; Wolfle et al. 2011; Behringer & Segal, 2012 b; Behringer et al. 2013), we suggest that excessive activation of SKCa/IKCa may supersede effective physiological regulation of tissue perfusion.
Summary and perspective
An integral component of endothelial function in the resistance vasculature entails Ca2+ and electrical signalling to govern vessel diameter and tissue perfusion. The increase in EC [Ca2+]i leads to SMC relaxation and vasodilatation through production of vasodilator autacoids (e.g. NO) and the initiation of hyperpolarization (via SKCa/IKCa activation) that can spread from cell to cell along and among the branches of resistance networks to coordinate blood flow distribution and magnitude according to local and global metabolic demand. The present findings illustrate that V m can modulate Ca2+ entry into the ECs (e.g. through TRPV4) during submaximal stimulation of muscarinic receptors. However this signalling pathway is effectively closed under resting conditions and Ca2+ entry can be saturated during maximal receptor stimulation, likely to be a consequence of depleting Ca2+ stores within the endoplasmic reticulum. Nevertheless, when Ca2+ entry is submaximal, EC [Ca2+]i increases with hyperpolarization and decreases with depolarization through a physiological range when initiated from resting V m. The interplay between V m and [Ca2+]i signalling revealed here in native microvascular endothelium helps to resolve earlier controversy regarding the role of V m in governing [Ca2+]i and provides new insight into the physiological regulation of vascular resistance. The ability to modulate [Ca2+]i by changing V m (e.g. through activation or inhibition of specific ion channels) may be employed judiciously to adjust irregularities in the behaviour of resistance vessels that control blood pressure and tissue perfusion during cardiovascular disease.
Additional information
Competing interests
The authors have declared no competing interests.
Author contributions
E.J.B. designed and performed experiments in the laboratory of S.S.S., analysed and interpreted the data, drafted the manuscript and prepared the figures. S.S.S. contributed to experimental design and the interpretation and presentation of data, and edited the manuscript. Both authors approved the final version of the manuscript for publication.
Funding
This research was supported by National Institutes of Health grant R37‐HL041026 to S.S.S. E.J.B. was supported by NIH grants 5T32‐AR048523 and 1K99‐AG047198. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Acknowledgements
Dr William F. Jackson provided helpful comments in the early stages of these experiments.
Author's present address
E. J. Behringer: Loma Linda University, School of Medicine, Basic Sciences: Division of Pharmacology, Risley Hall, 11041 Campus Street, Loma Linda, CA 92354, USA.
References
- Bagher P, Beleznai T, Kansui Y, Mitchell R, Garland CJ & Dora KA (2012). Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca2+ events, and IKCa channels, reducing arteriolar tone. Proc Natl Acad Sci USA 109, 18174–18179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagher P & Segal SS (2011). Regulation of blood flow in the microcirculation: role of conducted vasodilation. Acta Physiol (Oxf) 202, 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ & Segal SS (2012. a). Spreading the signal for vasodilatation: implications for skeletal muscle blood flow control and the effects of ageing. J Physiol 590, 6277–6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ & Segal SS (2012. b). Tuning electrical conduction along endothelial tubes of resistance arteries through Ca2+‐activated K+ channels. Circ Res 110, 1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ, Shaw RL, Westcott EB, Socha MJ & Segal SS (2013). Aging impairs electrical conduction along endothelium of resistance arteries through enhanced Ca2+‐activated K+ channel activation. Arterioscler Thromb Vasc Biol 33, 1892–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ, Socha MJ, Polo‐Parada L & Segal SS (2012). Electrical conduction along endothelial cell tubes from mouse feed arteries: Confounding actions of glycyrrhetinic acid derivatives. Br J Pharmacol 166, 774–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishara NB, Murphy TV & Hill MA (2002). Capacitative Ca2+ entry in vascular endothelial cells is mediated via pathways sensitive to 2 aminoethoxydiphenyl borate and xestospongin C. Br J Pharmacol 135, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM & Weston AH (2002). EDHF: bringing the concepts together. Trends Pharmacol Sci 23, 374–380. [DOI] [PubMed] [Google Scholar]
- Busse R, Fichtner H, Luckhoff A & Kohlhardt M (1988). Hyperpolarization and increased free calcium in acetylcholine‐stimulated endothelial cells. Am J Physiol Heart Circ Physiol 255, H965–H969. [DOI] [PubMed] [Google Scholar]
- Busse R & Mulsch A (1990). Calcium‐dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS Lett 265, 133–136. [DOI] [PubMed] [Google Scholar]
- Cannell MB & Sage SO (1989). Bradykinin‐evoked changes in cytosolic calcium and membrane currents in cultured bovine pulmonary artery endothelial cells. J Physiol 419, 555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W, Yang F, Takanishi CL & Zheng J (2007). Thermosensitive TRPV channel subunits coassemble into heteromeric channels with intermediate conductance and gating properties. J Gen Physiol 129, 191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE (2007). Calcium signaling. Cell 131, 1047–1058. [DOI] [PubMed] [Google Scholar]
- Cohen KD & Jackson WF (2005). Membrane hyperpolarization is not required for sustained muscarinic agonist‐induced increases in intracellular Ca2+ in arteriolar endothelial cells. Microcirculation 12, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damkjaer M, Nielsen G, Bodendiek S, Staehr M, Gramsbergen JB, de Wit C, Jensen BL, Simonsen U, Bie P, Wulff H & Kohler R (2012). Pharmacological activation of KCa3.1/KCa2.3 channels produces endothelial hyperpolarization and lowers blood pressure in conscious dogs. Br J Pharmacol 165, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dora KA & Garland CJ (2013). Linking hyperpolarization to endothelial cell calcium events in arterioles. Microcirculation 20, 248–256. [DOI] [PubMed] [Google Scholar]
- Earley S, Reading S & Brayden JE (2007). Functional significance of transient receptor potential channels in vascular function In TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades, ed. Liedtke WB. & Heller S, pp. 361–376. CRC Press, Boca Raton. [PubMed] [Google Scholar]
- Emerson GG & Segal SS (2000). Endothelial cell pathway for conduction of hyperpolarization and vasodilation along hamster feed artery. Circ Res 86, 94–100. [DOI] [PubMed] [Google Scholar]
- Garland CJ, Hiley CR & Dora KA (2011). EDHF: spreading the influence of the endothelium. Br J Pharmacol 164, 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M & Tsien RY (1985). A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260, 3440–3450. [PubMed] [Google Scholar]
- Himmel HM, Whorton AR & Strauss HC (1993). Intracellular calcium, currents, and stimulus‐response coupling in endothelial cells. Hypertension 21, 112–127. [DOI] [PubMed] [Google Scholar]
- Jackson WF (2005). Potassium channels in the peripheral microcirculation. Microcirculation 12, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knot HJ & Nelson MT (1998). Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol 508, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskey RE, Adams DJ, Johns A, Rubanyi GM & van Breemen C (1990). Membrane potential and Na+‐K+ pump activity modulate resting and bradykinin‐stimulated changes in cytosolic free calcium in cultured endothelial cells from bovine atria. J Biol Chem 265, 2613–2619. [PubMed] [Google Scholar]
- Ledoux J, Bonev AD & Nelson MT (2008). Ca2+‐activated K+ channels in murine endothelial cells: block by intracellular calcium and magnesium. J Gen Physiol 131, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledoux J, Werner ME, Brayden JE & Nelson MT (2006). Calcium‐activated potassium channels and the regulation of vascular tone. Physiology (Bethesda) 21, 69–78. [DOI] [PubMed] [Google Scholar]
- Li L, Bressler B, Prameya R, Dorovini‐Zis K & Van Breemen C (1999). Agonist‐stimulated calcium entry in primary cultures of human cerebral microvascular endothelial cells. Microvasc Res 57, 211–226. [DOI] [PubMed] [Google Scholar]
- Li W, Halling DB, Hall AW & Aldrich RW (2009). EF hands at the N‐lobe of calmodulin are required for both SK channel gating and stable SK‐calmodulin interaction. J Gen Physiol 134, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckhoff A & Busse R (1990). Activators of potassium channels enhance calcium influx into endothelial cells as a consequence of potassium currents. Naunyn Schmiedebergs Arch Pharmacol 342, 94–99. [DOI] [PubMed] [Google Scholar]
- Ma X, Cao J, Luo J, Nilius B, Huang Y, Ambudkar IS & Yao X (2010). Depletion of intracellular Ca2+ stores stimulates the translocation of vanilloid transient receptor potential 4‐c1 heteromeric channels to the plasma membrane. Arterioscler Thromb Vasc Biol 30, 2249–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrelli SP, Eckmann MS & Hunte MS (2003). Role of endothelial intermediate conductance KCa channels in cerebral EDHF‐mediated dilations. Am J Physiol Heart Circ Physiol 285, H1590–H1599. [DOI] [PubMed] [Google Scholar]
- McSherry IN, Spitaler MM, Takano H & Dora KA (2005). Endothelial cell Ca2+ increases are independent of membrane potential in pressurized rat mesenteric arteries. Cell Calcium 38, 23–33. [DOI] [PubMed] [Google Scholar]
- Mishra RC, Wulff H, Cole WC & Braun AP (2014). A pharmacologic activator of endothelial KCa channels enhances coronary flow in the hearts of type 2 diabetic rats. J Mol Cell Cardiol 72, 364–373. [DOI] [PubMed] [Google Scholar]
- Nelson MT & Quayle JM (1995). Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol Cell Physiol 268, C799–C822. [DOI] [PubMed] [Google Scholar]
- Qian X, Francis M, Kohler R, Solodushko V, Lin M & Taylor MS (2014). Positive feedback regulation of agonist‐stimulated endothelial Ca2+ dynamics by KCa3.1 channels in mouse mesenteric arteries. Arterioscler Thromb Vasc Biol 34, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke J, Schmidt K, Wulff H, Kohler R & de Wit C (2013). Activation of KCa3.1 by SKA‐31 induces arteriolar dilatation and lowers blood pressure in normo‐ and hypertensive connexin40‐deficient mice. Br J Pharmacol 170, 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhle B & Trebak M (2013). Emerging roles for native Orai Ca2+ channels in cardiovascular disease. Curr Top Membr 71, 209–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandow SL & Grayson TH (2009). Limits of isolation and culture: intact vascular endothelium and BKCa . Am J Physiol Heart Circ Physiol 297, H1–H7. [DOI] [PubMed] [Google Scholar]
- Sharma NR & Davis MJ (1995). Substance P‐induced calcium entry in endothelial cells is secondary to depletion of intracellular stores. Am J Physiol Heart Circ Physiol 268, H962–H973. [DOI] [PubMed] [Google Scholar]
- Socha MJ, Domeier TL, Behringer EJ & Segal SS (2012). Coordination of intercellular Ca2+ signaling in endothelial cell tubes of mouse resistance arteries. Microcirculation 19, 757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socha MJ, Hakim CH, Jackson WF & Segal SS (2011). Temperature effects on morphological integrity and Ca2+ signaling in freshly isolated murine feed artery endothelial cell tubes. Am J Physiol Heart Circ Physiol 301, H773–H783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socha MJ & Segal SS (2013). Isolation of microvascular endothelial tubes from mouse resistance arteries. J Vis Exp 81, e50759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill‐Eubanks DC & Nelson MT (2012). Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336, 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Dalsgaard T, Bonev AD, Hill‐Eubanks DC, Kotlikoff MI, Scott JD, Santana LF & Nelson MT (2014). AKAP150‐dependent cooperative TRPV4 channel gating is central to endothelium‐dependent vasodilation and is disrupted in hypertension. Sci Signal 7, ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobaek D, Teuber L, Jorgensen TD, Ahring PK, Kjaer K, Hansen RS, Olesen SP, Christophersen P & Skaaning‐Jensen B (2004). Activation of human IK and SK Ca2+‐activated K+ channels by NS309 (6,7‐dichloro‐1H‐indole‐2,3‐dione 3‐oxime). Biochim Biophys Acta 1665, 1–5. [DOI] [PubMed] [Google Scholar]
- Takano H, Dora KA, Spitaler MM & Garland CJ (2004). Spreading dilatation in rat mesenteric arteries associated with calcium‐independent endothelial cell hyperpolarization. J Physiol 556, 887–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorneloe KS, Sulpizio AC, Lin Z, Figueroa DJ, Clouse AK, McCafferty GP, Chendrimada TP, Lashinger ES, Gordon E, Evans L, Misajet BA, Demarini DJ, Nation JH, Casillas LN, Marquis RW, Votta BJ, Sheardown SA, Xu X, Brooks DP, Laping NJ & Westfall TD (2008). N‐((1S)‐1‐{[4‐((2S)‐2‐{[(2,4‐Dichlorophenyl)sulfonyl]amino}‐3‐hydroxypropanoyl)‐1‐piperazinyl]carbonyl}‐3‐methylbutyl)‐1‐benzothiophene‐2‐carboxamide (GSK1016790A), a novel and potent transient receptor potential vanilloid 4 channel agonist induces urinary bladder contraction and hyperactivity: Part I. J Pharmacol Exp Ther 326, 432–442. [DOI] [PubMed] [Google Scholar]
- Tracey WR & Peach MJ (1992). Differential muscarinic receptor mRNA expression by freshly isolated and cultured bovine aortic endothelial cells. Circ Res 70, 234–240. [DOI] [PubMed] [Google Scholar]
- Tran CH, Taylor MS, Plane F, Nagaraja S, Tsoukias NM, Solodushko V, Vigmond EJ, Furstenhaupt T, Brigdan M & Welsh DG (2012). Endothelial Ca2+ wavelets and the induction of myoendothelial feedback. Am J Physiol Cell Physiol 302, C1226–C1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh DG & Segal SS (1998). Endothelial and smooth muscle cell conduction in arterioles controlling blood flow. Am J Physiol Heart Circ Physiol 274, H178–H186. [DOI] [PubMed] [Google Scholar]
- Wolfle SE, Chaston DJ, Goto K, Sandow SL, Edwards FR & Hill CE (2011). Non‐linear relationship between hyperpolarisation and relaxation enables long distance propagation of vasodilatation. J Physiol 589, 2607–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H & Kohler R (2013). Endothelial small‐conductance and intermediate‐conductance KCa channels: an update on their pharmacology and usefulness as cardiovascular targets. J Cardiovasc Pharmacol 61, 102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, Xie J, Yu AS, Stock J, Du J & Yue L (2015). Role of TRP channels in the cardiovascular system. Am J Physiol Heart Circ Physiol 308, H157–H182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DX, Mendoza SA, Bubolz AH, Mizuno A, Ge ZD, Li R, Warltier DC, Suzuki M & Gutterman DD (2009). Transient receptor potential vanilloid type 4‐deficient mice exhibit impaired endothelium‐dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension 53, 532–538. [DOI] [PMC free article] [PubMed] [Google Scholar]