

Abstract
Thyroid hormones (THs) exert a number of physiological effects on the cardiovascular system. Some of the nongenomic actions of T3 are achieved by cross coupling the TH receptor (TR) with the phosphatidylinositol 3-kinase (PI3K)/protein kinase Akt (Akt) pathway. We observed that both T3 and T4 rapidly stimulated Akt phosphorylation and Ras-related C3 botulinum toxin substrate 1 (Rac1) activation, which resulted in cell migration, in a PI3K-dependent manner in human umbilical vein endothelial cells (HUVECs). We identified the expression of type 2 iodothyronine deiodinase (D2), which converts T4 to T3, and TRα1 in HUVECs. D2 activity was significantly stimulated by (Bu)2cAMP in HUVECs. The blockade of D2 activity through transfection of small interfering RNA (siRNA) specific to D2 as well as by addition of iopanoic acid, a potent D2 inhibitor, abolished Akt phosphorylation, Rac activation, and cell migration induced by T4 but not by T3. The inhibition of TRα1 expression by the transfection of siRNA for TRα1 canceled Akt phosphorylation, Rac activation, and cell migration induced by T3 and T4. These findings suggest that conversion of T4 to T3 by D2 is required for TRα1/PI3K-mediated nongenomic actions of T4 in HUVECs, including stimulation of Akt phosphorylation and Rac activation, which result in cell migration.
Thyroid hormones (THs) exert a number of physiological effects on the cardiovascular system given that they decrease systemic vascular resistance and arterial blood pressure, and increase renal sodium reabsorption and blood volume (1). THs are known as vasodilators that act directly on vascular smooth muscle cells to cause a relaxation of the coronary arteries (2) and aorta (3).
The classical genomic actions of THs are thought to be exerted by binding of T3 to high-affinity nuclear TH receptors (TRs) to regulate gene expression. TRs recognize specific TH-response elements (TREs) of target genes and activate or repress transcription in response to T3. The actions that are independent of these genomic or TRE-mediated actions are called nongenomic actions of THs (4). T4 and T3 binding sites have been shown to initiate short- and long-term effects via a plasma membrane receptor site located on integrin αvβ3 (5–7). THs bind to the integrin αvβ3 plasma membrane receptor without entering the cells. According to this model, the nongenomic actions of THs are mostly extranuclear and independent of TRs. In contrast, previous reports have described different mechanisms of TH action in which T3 binds to cytoplasmic TRs and interacts with the regulatory subunit of phosphatidylinositol 3-kinase (PI3K), p85α, which leads to activation of PI3K and its downstream-signaling cascade of protein kinase Akt (Akt) (8–13). Recently, T3 binding to membrane-localized TRs was demonstrated to result in activation of extracellular signal-regulated kinase (Erk) and Akt signaling (14).
TRs initiate rapid and nongenomic effects on the cardiovascular system through cross coupling with the PI3K/Akt-signaling pathway in the cytoplasm (9, 10). TH-induced rapid activation of PI3K/Akt/endothelial nitric oxide synthase (eNOS) has been demonstrated in endothelial cells (10). In addition, PI3K/Akt-signaling cascade (15) and Ras-related C3 botulinum toxin substrate 1 (Rac1) activation (16) have been reported to mediate the migration of endothelial cells, which is important for vessel repair and angiogenesis. Migration of vascular smooth muscle cells were mediated by Rac and inhibited by Ras homolog gene family member A (RhoA) (17, 18), and RhoA was stimulated by T4 (19). However, it remains unclear whether THs mediate Rac1 activation or migration of endothelial cells.
To bind to TRs and exert its biological activity, T4, which is the major secretory product of the thyroid gland, must be converted to T3 by selenocysteine-containing oxidoreductases, namely iodothyronine deiodinases (20). There are three types of iodothyronine deiodinase: type 1 (D1), type 2 (D2), and type 3 (D3). D1 and D2 remove iodine from the outer ring of T4 to form T3. D1 and D3 remove iodine from the inner rings of T4 and T3 to form the inactive thyroid hormones 3,3′,5′-triiodothyronine (rT3) and 3,3′-diiodothyronine, respectively. D1 is present in the thyroid gland, liver, kidney, and many other tissues, whereas D2 is present in a limited number of tissues, including the central nervous system, anterior pituitary, and brown fat in the rat. D3 is present in the placenta and central nervous system (20). Although both D1 and D2 catalyze conversion of T4 to T3, the properties of these two enzymes are remarkably different. The Km value of D2 is approximately 2nM for T4, which is 100-fold lower than that of D1 (20). D1 but not D2 is highly sensitive to inhibition by the antithyroid drug, 6-n-propylthiouracil (PTU). D1 activity is known to decrease in a hypothyroid state and is believed to have a primary role in maintaining circulating T3 levels (20). D2 activity, in contrast, is elevated in a hypothyroid state and is considered to play a pivotal role in providing local T3 to regulate intracellular T3 concentration (20). In humans, D2 mRNA has also been detected in the thyroid gland, skeletal muscle, and other tissues, suggesting physiological roles for D2 in those tissues (21–24).
We previously identified the expression of D2 mRNA, D2 activity and TRs in cultured human coronary artery smooth muscle cells (hCASMCs) and human aortic smooth muscle cells (hASMCs) (25). Because both T3 and T4 have been reported to regulate peripheral vascular function (26–28), D2 may contribute to the pathophysiology of the cardiovascular system by providing intracellular T3 from T4. However, the roles of D2 in the function of the cardiovascular system, including the rapid nongenomic actions of THs, remain to be elucidated.
In this study, we investigated the physiological roles of THs and the expression of iodothyronine deiodinases in human umbilical vein endothelial cells (HUVECs). We report that conversion of T4 to T3 by D2 is required for TRα1/PI3K-mediated nongenomic actions of T4, including stimulation of Akt phosphorylation and Rac activation, which result in migration of endothelial cells.
Materials and Methods
Materials
T4, T3, rT3, dibutyryl cyclic adenosine 3′,5′-monophosphate [(Bu)2cAMP], PTU, iopanoic acid (IOP), dithiothreitol (DTT) and fatty acid-free BSA were obtained from Sigma-Aldrich, Inc.; wortmannin from Calbiochem-Novabiochem Corp.; antibodies for Akt, phospho-Ser-473 Akt, p44/42MAPK (Erk1/2), phospho-p44/42MAPK (Erk1/2) (Thr202/Thr204), phospho-Src (Tyr416), and Src from Cell Signaling Technology, Inc. (Table 1); [125I]T4 from NEN Life Science Products Corp.; LH-20 from GE Healthcare Bio-Sciences Corp.; AG 50W-X2 resin and a protein assay kit from Bio-Rad Laboratories, Inc.; and sphingosine 1-phosphate (S1P) from Enzo Life Sciences.
Table 1.
Antibodies Used in the Study
| Peptide/Protein Target | Antigen Sequence (if Known) | Name of Antibody | Manufacturer | Catalog #, and/or Name of Individual Providing the Antibody | Species Raised in; Monoclonal or Polyclonal | Dilution Used |
|---|---|---|---|---|---|---|
| Akt | Not known | Akt antibody | Cell Signaling | No. 9272S | Rabbit | 1000× |
| Phospho-Akt (Ser473) | Not known | Phospho-Akt (Ser473) antibody | Cell Signaling | No. 9271S | Rabbit | 1000× |
| Nonphospho-Src (Thr416) | Not known | Nonphospho-Src (Tyr416) (7G9) Mouse mAb | Cell Signaling | No. 2102S | Mouse | 1000× |
| Phospho-Src Family (Thr416) | Not known | Phospho-Src Family (Tyr416) Antibody | Cell Signaling | No. 2101S | Rabbit | 1000× |
| p44/42MAPK (Erk1/2) | Not known | p44/42 MAPK (Erk1/2) (137F5) Rabbit mAb | Cell Signaling | No. 4695S | Rabbit | 1000× |
| Phospho-p44/42MAPK (Erk1/2) (Thr202/Thr204) | Not known | Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) Antibody | Cell Signaling | No. 9101S | Rabbit | 1000× |
| Rac1 | Not known | Anti-Rac1 antibody | ThermoFisher Scientific, Inc. | No. 16118X | Mouse | 1000× |
| RhoA | Not known | Anti-RhoA monoclonal antibody | Cytoskeleton, Inc. | No. ARH03 | Mouse | 1000× |
Cell culture
HUVECs (passage 3) were purchased from Kurabo Industries, Ltd. Kurabo imported HUVECs from Lifeline Cell Technology, LLC. HUVECs were confirmed as positive for von Willebrand factor and negative for smooth muscle α-actin by Lifeline Cell Technology, LLC. In accordance with the manufacturer's instructions, cells were cultured in HuMedia-EG2 supplemented with 2% (v/v) fetal bovine serum (FBS), 10 μg/mL of heparin, 10 ng/mL of epidermal growth factor, 5 ng/mL of basic fibroblast growth factor, and 1.34 μg/mL of hydrocortisone in a humidified air/CO2 (19:1) atmosphere. We used cells passaged 5–8 times and confirmed the cobblestone-like cell shape. Before each experiment, cells were washed with fresh Roswell Park Memorial Institute (RPMI) 1640 medium containing 0.1% fatty acid–free BSA and incubated for 4 hours at 37°C with fresh RPMI 1640 medium containing 0.1% fatty acid–free BSA to avoid the effect of serum and THs.
Transfection of small interfering RNA
For the transfection of small interfering RNAs (siRNAs) specific to D2 and TRα1, and nonsilencing siRNA, the HUVECs were seeded at a density of 5.0 × 104 cells/cm2 in each well of six-well plates for Western blotting and qRT-PCR analyses, on a 10-cm plate for D2 activity measurement, and on a 6-cm plate for migration assay. After 16 hours, siRNA (100nM) was introduced into the cells using RNAiFect transfection reagent (QIAGEN) according to the manufacturer's instructions. The cells were further cultured for 48 hours in HuMedia-EG2 containing 2% FBS and growth factors as described above. The nonsilencing siRNA (D-001206-13) and siRNAs targeted for D2 mRNA (L-011171-01) and TRα1 mRNA (L-003446-00) were obtained from Dharmacon, Inc (Table 2).
Table 2.
siRNA Target Sequences
| siRNA Target | Catalog No. | Source | Target Sequence |
|---|---|---|---|
| Non target | d-001206-13 | Dharmacon, Inc | UAGCGACUAAACACAUCAA |
| UAAGGCUAUGAAGAGAUAC | |||
| AUGUAUUGGCCUGUAUUAG | |||
| AUGAACGUGAAUUGCUCAA | |||
| Human TRα1 | l-003446-00 | Dharmacon, Inc | CGGCCAAUGUUCCCUGAAA |
| GAACUGGGCAAGUCACUCU | |||
| GUAUAUCCCUAGUUACCUG | |||
| GAACCUCCAUCCCACCUAU | |||
| Human D2 | l-011171-01 | Dharmacon, Inc | CAACAUAGCUUACGGGGUA |
| ACAUGGAGCUAUCGGUUUA | |||
| GAUGAUAACUACUGACGAA | |||
| GAGUUUAUCUAUCGGAAGA |
qRT-PCR analysis
HUVECs were cultured with HuMedia-EG2 containing 2% FBS and growth factors as described above. Before the experiment, cells were washed twice with RPMI 1640 medium containing 0.1% fatty acid–free BSA and incubated for 4 hours at 37°C with RPMI 1640 medium containing 0.1% fatty acid–free BSA. The cells were collected after RPMI 1640 treatment, and total RNA was isolated using TRI RNA Isolation Reagent (Sigma-Aldrich) according to the manufacturer's instructions. After DNase I (Promega Corp.) treatment to remove possible traces of genomic DNA contaminating the RNA preparations, 5 μg of the total RNA was reverse transcribed using a High-Capacity cDNA Archive kit (Applied Biosystems) according to the manufacturer's instructions. To evaluate the mRNA expression level for D1 (Hs 00174944), D2 (Hs 00255341), D3 (Hs 00704811), TRα1 (Hs 00268470), and TRβ1 (Hs 00230861), qRT-PCR was performed using real-time TaqMan technology with the Sequence Detection System (model 7700; Applied Biosystems). The expression level of the target mRNA was normalized to the relative ratio of the expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA. Each qRT-PCR assay was performed at least three times, and the results are expressed as mean ± SEM.
Measurement of D2 activity
Iodothyronine deiodinase activity was measured as previously described (29), with minor modifications (30). Before the sample collection, HUVECs were washed twice with RPMI 1640 medium containing 0.1% fatty acid–free BSA and then incubated for 4 hours at 37°C with fresh RPMI 1640 medium containing 0.1% fatty acid–free BSA. Where indicated, cells were treated with 1mM (Bu)2cAMP or vehicle (PBS) for 6 hours before the sample collection. After siRNA transfection, cells were incubated for 4 hours at 37°C with fresh RPMI 1640 medium containing 0.1% fatty acid–free BSA with or without 1mM (Bu)2cAMP. Cells in each well were washed twice with PBS, scraped off, and transferred into tubes containing 1.5 mL ice-cold assay buffer [100mM potassium phosphate (pH 7.0), 1mM ethylenediaminetetraacetic acid (EDTA), and 20mM DTT]. After centrifugation at 3000 rpm for 10 minutes at 4°C, the resultant precipitates were sonicated in 100 μL of the assay buffer per tube and then incubated in a total volume of 50 μL with 2nM or the indicated amount of [125I]T4, which was purified using LH-20 column chromatography on the day of the experiment, in the presence or absence of 1mM PTU or 1mM IOP, for 1 hour at the indicated temperatures, in duplicate. After the characterization of deiodination activity of the HUVECs, the sonicates were routinely incubated with 2nM [125I]T4 in the presence of 1mM PTU at 37°C for 1 hour. The reaction was terminated by adding 100 μL of ice-cold 2% fatty acid–free BSA and 800 μL of ice-cold 10% trichloroacetic acid. After centrifugation at 3000 rpm for 10 minutes at 4°C, the supernatant was placed in a small column packed with AG 50W-X2 resin (bed volume = 1 mL) and then eluted with 2 mL of 10% glacial acetic acid. Separated 125I− was counted with a γ-counter. Nonenzymatic deiodination was corrected by subtracting readings of 125I− released in control tubes without cell sonicates. The protein concentration was determined by Bradford's method using BSA as a standard (31). The deiodination activity was calculated as femtomoles of I− released/mg protein/h, after multiplication by a factor of two to correct random labeling at the equivalent 3′ and 5′ positions. In some experiments, reaction products were analyzed by HPLC (Hitachi) (32, 33). In brief, the incubation mixtures were extracted with two volumes of absolute ethanol, evaporated, dissolved in acetonitrile/water (32:68), placed in a C18 column (Shimazu Co.), and eluted with acetonitrile/water/phosphoric acid (32:68:0.1). The flow rate was 1 mL/min, and each 0.5-minute fraction was collected and counted for radioactivity.
Construction of adenoviral vector and infection of recombinant adenovirus
A dominant-negative form of human Rac1 (T17NRac1; Upstate Biotechnology) was used for the construction of recombinant adenovirus and infected. In brief, 80% confluent HUVECs were infected with the recombinant at a multiplicity of infection of 30 for 2 hours at 37°C in RPMI 1640 medium containing 2% FBS. Cells were cultured for an additional 48 hours at 37°C in HuMedia-EG2 supplemented with 2% FBS and 10 μg/mL of heparin, 10 ng/mL epidermal growth factor, 5 ng/mL of basic fibroblast growth factor, and 1.34 μg/mL of hydrocortisone. Under these conditions, infection with adenovirus-coding green fluorescent protein (GFP) resulted in virtually all cells being GFP positive.
Estimation of Rac1 activation by the pull-down reaction
Before the experiment, HUVECs were incubated for 4 hours at 37°C with fresh RPMI 1640 medium containing 0.1% fatty acid–free BSA. These cells were treated with or without 1nM T3, or 1nM T4 for 10 minutes at 37°C in RPMI 1640 medium containing 0.1% fatty acid–free BSA. The incubation with T3 or T4 was terminated by washing twice with ice-cold PBS and adding 0.8 mL of a lysis buffer according to the instruction manual of the Active GTPase Pull-down and Detection Kits (ThermoFisher Scientific, Inc.). Rac1 activity was then assessed by the pull-down assay of the active form of Rac1 with glutathione S-transferase (GST)-RalGDS Rho-binding domain (RBD) and GST-Rhotekin RBD fusion proteins according to the manufacturer's instructions. Transfection with nonsilencing siRNA, siRNA against D2 or TRα1, or treatment with wortmannin or IOP was performed before experiments. The intensity of signal was analyzed by densitometry using Bio-1D version 15.04 (Vilber Lourmat). The activation ratio of Rac was calculated as activated Rac/total protein for Rac, and normalized by control experiments. The results are presented as the mean ± SEM of three independent experiments.
Estimation of RhoA activation by the pull-down reaction
Before the experiment, HUVECs were incubated for 4 hours at 37°C with fresh RPMI 1640 medium containing 0.1% fatty acid–free BSA. These cells were treated with or without, 1nM T, 1nM T4, 1nM rT3, or 1μM S1P for 10 minutes at 37°C in RPMI 1640 medium containing 0.1% fatty acid–free BSA. Incubation with the indicated agents was terminated by washing twice with ice-cold PBS and adding 0.45 mL of lysis buffer according to the instruction manual of the Rho Activation Assay Biochem Kit (Cytoskeleton, Inc.). RhoA activity was then assessed by the pull-down assay of the active form of RhoA with Rhotekin RBD with or without bound RhoA according to the manufacturer's instructions.
Western blotting
Before the experiment, HUVECs were incubated for 4 hours at 37°C with fresh RPMI 1640 medium containing 0.1% fatty acid–free BSA. The cells were pretreated for 20 minutes at 37°C with or without 100nM wortmannin or 1μM of IOP. These cells were stimulated for 10 minutes at 37°C in RPMI 1640 medium containing 0.1% fatty acid–free BSA and indicated concentrations of T3, T4, rT3, S1P, wortmannin, or IOP. To detect phosphorylation of Akt, Erk, or Src, the reaction was terminated by washing twice with ice-cold PBS and adding 0.1 mL of lysis buffer containing 1% Triton X-100, 50mM Tris-HCl (pH 7.5), 150mM NaCl, 2mM EDTA, 8mM ethylene glycol tetracetic acid, 25mM NaF, 10mM Na4P2O7, 1mM Na3VO4, 5 μg/mL leupeptin, 5 μg/mL pepstatin, 5 μg/mL aprotinin, and 0.5mM phenylmethanesulfonylfluoride. The lysate was separated by 10% SDS-PAGE and analyzed by Western blotting with antibodies specific for Akt, phospho-Akt, pErk42/44, phospho-pErk42/44, Src, or phospho-Src. The intensity of signal was analyzed by densitometry using Bio-1D version 15.04 (Vilber Lourmat). The phosphorylation intensity of Akt was calculated as phosphorylated Akt/total protein for Akt and normalized by control experiments. The results are presented as the mean ± SEM of three independent experiments. For the detection of Rac1 and RhoA, the procedures were essentially the same as those for Akt phosphorylation except for the primary antibody.
Migration assay
Transwell migration of HUVECs was determined in a modified Boyden chamber (Neuro Probe, Inc.) using polycarbonate filters with 8-μm pores as described previously (17, 18, 34). Before the experiment, HUVECs were washed twice with RPMI 1640 medium containing 0.1% fatty acid–free BSA and then incubated with RPMI 1640 medium containing 0.1% fatty acid–free BSA for 4 hours at 37°C. Next, the cells were suspended in RPMI 1640 medium containing 0.1% fatty acid–free BSA and pretreated for 20 minutes at 37°C with or without 100nM wortmannin or 1μM IOP. These cells were loaded into the upper wells with or without 100nM wortmannin or 1μM IOP, whereas the lower wells were filled with the same medium containing indicated concentrations of PBS (none), T3, T4, rT3, or S1P. The cells were allowed to migrate across the porous filter for 4 hours at 37°C in a tissue culture incubator. After staining the migrated cells with Diff-Quick staining solution (Sysmex) and scraping the upper surface of the filter, the number of migrated cells that was attached to the lower surface of the filters was counted under a microscope at 400× magnification. The results are presented as the mean ± SEM of three independent experiments.
Data analysis
All experiments were performed in duplicate or triplicate. The results of multiple observations are presented as the mean ± SEM or as a representative result of more than two different separate experiments, unless otherwise stated. Statistical significance was assessed by ANOVA or the Student t test. Values were considered significant at P < .05 (*).
Results
THs rapidly stimulate Akt phosphorylation in HUVECs
Previous studies have suggested that THs initiated rapid nongenomic actions through TR cross coupling to the PI3K/Akt signaling pathway in the cytoplasm of endothelial cells (10). However, it has not been revealed whether T4 is able to activate Akt phosphorylation. Therefore, we tested whether T4 as well as T3 could activate Akt phosphorylation in HUVECs. A 10-minute treatment of both T3 and T4 stimulated Akt phosphorylation in HUVECs in concentration-dependent manners (Figure 1, A and B). T3 (1nM) stimulated Akt phosphorylation within 5 minutes, whereas T4 (1nM) stimulated the reaction within 10 minutes (Figure 1, C and D).
Figure 1.
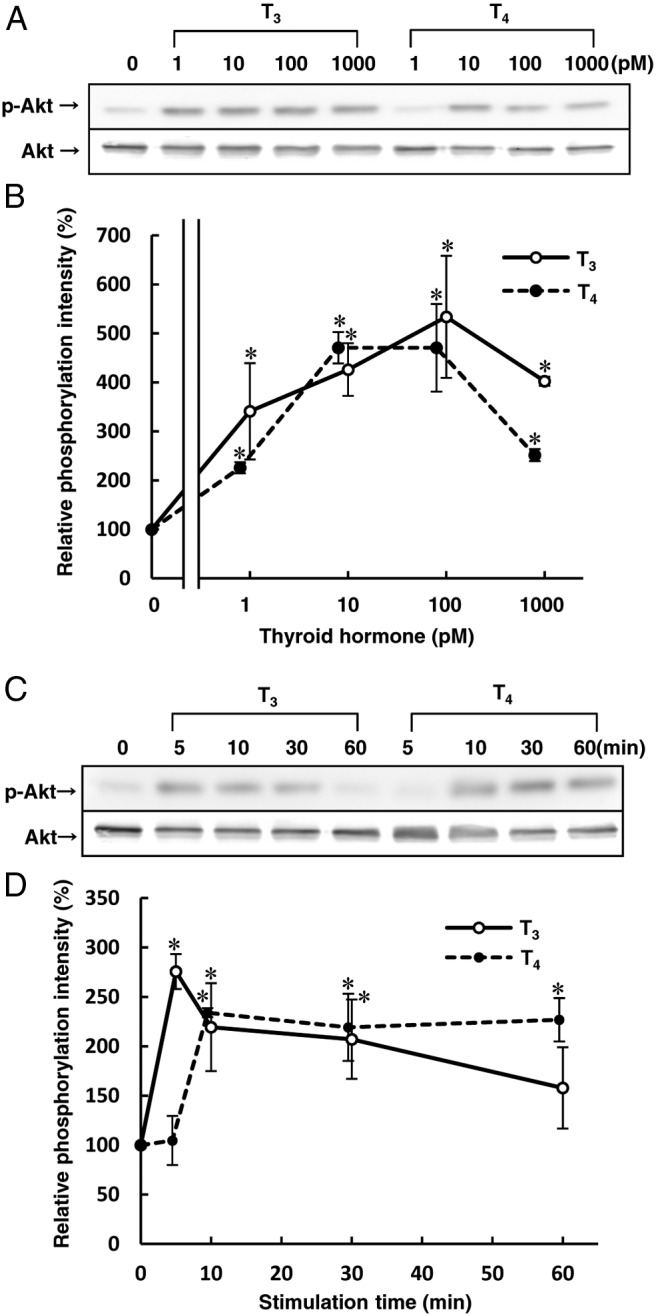
THs promote Akt phosphorylation in HUVECs. Before performing the experiment described below, HUVECs were incubated for 4 h at 37°C with RPMI 1640 medium containing 0.1% fatty acid–free BSA to avoid the effect of serum and THs. A, HUVECs were incubated for 10 min at 37°C with the indicated concentrations of T3 or T4 to measure Akt phosphorylation activity by Western blotting analysis. B, The relative amount of Akt phosphorylation was calculated from three independent experiment shown in panel A. C, HUVECs were incubated for the indicated times with T3 (1nM) or T4 (1nM) to measure Akt phosphorylation activity by Western blotting analysis. D, The relative amount of Akt phosphorylation was calculated from three independent experiments shown in C. Representative Western blots of three separate experiments are shown in panels A and C. Results are means ± SEM of three separate experiments in panels B and D. *, P < .05 vs 0. Zero is none in panel B, and 0 minutes (baseline) in panel D.
T3 and T4, but not rT3, stimulate migration of HUVECs
Because Akt phosphorylation has been reported to mediate the migration of endothelial cells (15), we investigated the migration-promoting activity of THs in HUVECs and found that both T3 and T4 clearly stimulated migration of HUVECs in dose dependent manners (Figure 2A). We tested whether rT3 could stimulate Akt phosphorylation and migration in HUVECs using S1P as a positive control. Although S1P significantly stimulated both Akt phosphorylation and cell migration, rT3 did not stimulate Akt phosphorylation or migration in HUVECs (Figure 2, B–D).
Figure 2.
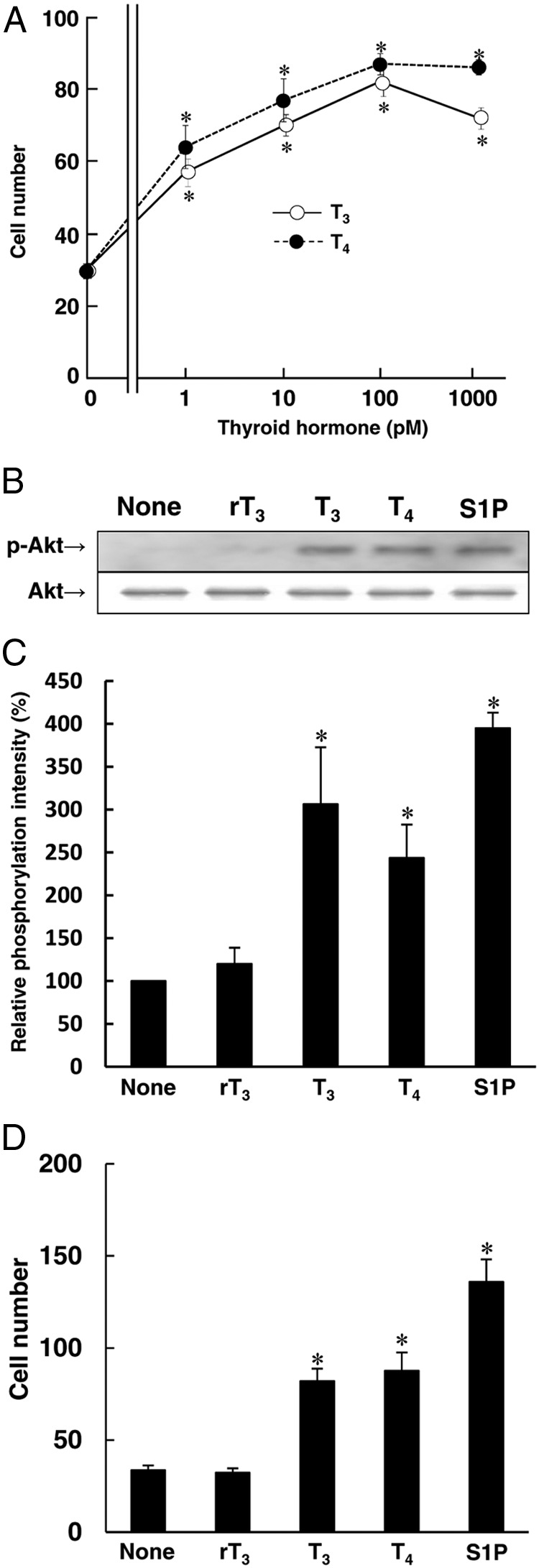
T3 and T4 but not rT3 induce Akt phosphorylation and migration in HUVECs. Before performing the experiment described below, HUVECs were incubated for 4 h at 37°C with RPMI 1640 medium containing 0.1% fatty acid–free BSA. A, HUVECs were incubated for 4 h at 37°C with the indicated concentrations of T3 or T4 to investigate migration. TH-induced migration assay was performed using a blind Boyden chamber apparatus. Migrated cells attached to the lower surface of the filters were stained with Diff-Quick staining solution and the number of migrated cells attached to the lower surface of the filters was counted under a microscope at 400× magnification. B and C, HUVECs were incubated for 10 min at 37°C with PBS (none), rT3 (1nM), T3 (1nM), T4 (1nM), or S1P (1μM) to analyze Akt phosphorylation by Western blotting in panel B, the relative amount of Akt phosphorylation from these three independent experiment is shown in panel C. D, HUVECs were incubated for 4 h at 37°C with PBS (none), rT3 (1nM), T3 (1nM), T4 (1nM), or S1P (1μM) to investigate the migration of HUVECs using a blind Boyden chamber apparatus. Representative results of three separate experiments are shown in panel B. Results are presented as the means ± SEM of three separate experiments in panels A, C, and D. *, P < .05 vs none.
THs promote Akt phosphorylation and cell migration through a PI3K-dependent pathway
To study the role of PI3K, we used wortmannin to inhibit PI3K. Wortmannin inhibited Akt phosphorylation and cell migration induced by T3 and T4 (Figure 3, A–C). In these experiments, we used S1P as a positive control to stimulate phosphorylation of Akt, Erk, and Src in HUVECs. S1P but not THs stimulated Erk and Src phosphorylation in HUVECs. Wortmannin inhibited Akt phosphorylation induced by S1P (Figure 3A), whereas S1P-induced phosphorylation of Erk and Src was not blocked by wortmannin in HUVECs, indicating the viability of HUVECs after treatment with wortmannin. These results indicated that THs promoted endothelial cell migration through the PI3K/Akt-signaling pathway. Although several reports showed rapid activation of Erk and Src as well as Akt by THs in the neuron-like cells and glioma cells (35, 36), THs stimulated phosphorylation of Akt but not Erk and Src in HUVECs under our experimental conditions.
Figure 3.

The effects of wortmannin and IOP on TH-induced Akt phosphorylation and migration in HUVECs. Before performing the experiment described below, HUVECs were treated for 4 h at 37°C with RPMI 1640 medium containing 0.1% fatty acid–free BSA. A, HUVECs were pretreated with or without 100nM wortmannin or 1μM IOP for 20 min at 37°C in RPMI 1640 medium containing 0.1% fatty acid–free BSA. The cells were then stimulated for 10 min at 37°C with T3 (1nM), T4 (1nM), S1P (1μM), PBS (none), wortmannin, or IOP to measure the phosphorylated form and total Akt, pErk42/44 and Src by Western blotting. B, The relative amount of Akt phosphorylation was calculated from three independent experiments shown in panel A. C, HUVECs were pretreated with or without 100nM wortmannin or 1μM IOP for 20 min at 37°C in RPMI 1640 medium containing 0.1% fatty acid–free BSA. The cells were then stimulated for 4 h at 37°C with T3 (1nM), T4 (1nM), S1P (1μM), PBS (none), wortmannin, or IOP in RPMI 1640 medium containing 0.1% fatty acid–free BSA to investigate the migration of HUVECs using a blind Boyden chamber apparatus. The number of migrated cells attached to the lower surface of the filters was counted under a microscope at 400× magnification. Representative results of three separate experiments are shown in panel A. Results are presented as means ± SEM of three separate experiments in panels B and C. *, P < .05 vs none.
THs stimulate cell migration through Rac activation
Because Rac-dependent endothelial-cell spreading and migration as well as angiogenesis have been previously demonstrated (16), we tested whether THs-induced migration was mediated by Rac1 in endothelial cells. As shown in Figure 4, A and B, both T3 and T4 promoted Akt phosphorylation, Rac activation, and cell migration. Furthermore, infection of adenovirus containing the dominant-negative form of human Rac1 inhibited THs-induced migration in HUVECs (Figure 4B). Both T3- and T4-induced Rac activation was inhibited by the addition of wortmannin in HUVECs. These results suggest that THs stimulate cell migration through PI3K/Rac activation. Given that Rho as well as Rac was reported to play a critical role in the regulation of cell migration (17, 18), we also tested whether THs could stimulate Rho activation using S1P as a positive control. As shown in Figure 4C, T3, T4, or rT3 did not stimulate Rho activation in HUVECs.
Figure 4.

THs stimulate migration through PI3K/Rac activation in HUVECs. Before performing the experiment described below, HUVECs were incubated for 4 h at 37°C with RPMI 1640 medium containing 0.1% fatty acid–free BSA. A, HUVECs were pretreated with or without 100nM wortmannin or 1μM IOP for 20 min at 37°C. The cells were then stimulated for 10 min at 37°C with T3 (1nM), T4 (1nM), PBS (none), wortmannin, or IOP to measure the phosphorylated form and total Akt, activated Rac, and total Rac by Western blotting. B, HUVECs were infected with adenovirus carrying GFP or a dominant negative form of Rac1 before the experiment. The cells were then stimulated for 4 h at 37°C with T3 (1nM), T4 (1nM), or PBS (none) to investigate the migration of HUVECs using a blind Boyden chamber apparatus. The number of migrated cells attached to the lower surface of the filters was counted under a microscope at 400× magnification. C, HUVECs were incubated for 10 min at 37°C with PBS (none), rT3 (1nM), T3 (1nM), T4 (1nM), or S1P (1μM) to analyze RhoA activation by Western blotting. Representative results of three separate experiments are shown in panels A and C. Results are presented as means ± SEM of three separate experiments in panel B. *, P < .05 vs none.
IOP inhibits T4-induced Akt phosphorylation, Rac activation, and cell migration
T4 required a longer time than T3 to stimulate Akt phosphorylation in the present study (Figure 1, C and D). Therefore, we hypothesized that T4 might stimulate Akt phosphorylation, Rac activation, and cell migration in HUVECs after conversion to T3. We employed IOP, a potent iodothyronine deiodinase inhibitor, to investigate the possible role of T4 activation in Akt phosphorylation, Rac activation, and cell migration induced by THs in HUVECs. IOP blocked Akt phosphorylation, Rac activation, and cell migration induced by T4, but not by T3 or S1P (Figure 3, A–C, and Figure 4A). S1P stimulated phosphorylation of Akt, Erk, and Src with or without IOP (Figure 3A), indicating the viability of HUVECs after treatment with IOP. These results suggest that deiodination of T4 is a key step in T4-induced Akt phosphorylation, Rac activation, and cell migration of HUVECs.
Expression of D2 and TR in HUVECs
In this study, we have tested whether T4-deiodination activity is present in endothelial cells. As shown in Figure 5A, qRT-PCR analysis revealed the presence of mRNA of D2 but not D1 or D3 in HUVECs. A double-reciprocal plot of T4-deiodination activity demonstrated that the Km for T4 was 3.48nM, and the maximum velocity (Vmax) was 5.93 fmol/mg protein/h (Figure 5B). Incubation at 4°C or preheating the cell sonicate at 56°C for 30 minutes abolished the deiodination (data not shown). T4-deiodination activity was not influenced by the addition of 1mM PTU, but was completely inhibited by 1mM IOP (data not shown). When sonicates of HUVECs were incubated with 2nM [125I]T4 in the presence of 20mM DTT and 1mM PTU, subsequent HPLC analysis showed that there were only three definable peaks corresponding to I−, T4, and T3, and radioactivity at the I− peak was comparable with that at the T3 peak (data not shown). These results suggest that the characteristics of T4-deiodination activity in HUVECs are compatible with those of D2. A 6-hour treatment with 1mM (Bu)2cAMP significantly increased D2 activity in HUVECs (Figure 5C), indicating the regulation of its activity through a cAMP-mediated pathway. We also tested whether the expression of TR mRNA was observed in endothelial cells, and found the predominant expression of TRα1, but not TRβ1 in HUVECs by qRT-PCR analysis (Figure 5D).
Figure 5.

Expression of D2 and TR in HUVECs. Before performing the experiment described below, HUVECs were incubated for 4 h at 37°C with RPMI 1640 medium containing 0.1% fatty acid–free BSA. A, Expression of iodothyronine deiodinase mRNA in HUVECs was measured by qRT-PCR using real-time TaqMan technology. The results for D1, D2, and D3 are shown. B, A double-reciprocal plot of iodothyronine deiodinase activity in HUVECs. Cell sonicates were incubated with various concentrations of [125I]T4 and T4 in assay buffer [100mM potassium phosphate (pH 7.0), 1mM EDTA, and 20mM DTT] at 37°C for 1 h. A double-reciprocal plot of deiodination activity demonstrated that the Km for T4 was 3.48nM. The maximum velocity (Vmax) was 5.93 fmol/mg protein/h. C, Cells were treated with or without 1mM (Bu)2cAMP for 6 h. Cell sonicates were incubated with 2nM [125I]T4 in the presence of 1mM PTU at 37°C for 1 h to measure D2 activity. D, Expression of TRα1 and TRβ1 mRNA was measured by a qRT-PCR using real-time TaqMan technology. Results are presented as means ± SEM of three separate experiments in panels A, C, and D. *, P < .05 vs control. n.d., not detected.
D2 plays a critical role in T4-induced Akt phosphorylation, Rac activation, and cell migration
To study the role of D2 in HUVECs, we employed siRNA against D2 mRNA. The transfection of siRNA specific for D2 mRNA inhibited D2 mRNA expression, and basal and (Bu)2cAMP-stimulated D2 activity (Figure 6,A and B). T4-induced but not T3-induced Akt phosphorylation, Rac activation, and cell migration were blocked by the knockdown of D2 mRNA in HUVECs (Figure 6, C–F). As described above, IOP also blocked T4-induced but not T3-induced Akt phosphorylation, Rac activation, and cell migration (Figure 3, A–C; Figure 4A). Furthermore, transfection of siRNA against TRα1 mRNA decreased TRα1 mRNA expression (Figure 6A), and blocked both T3- and T4-induced Akt phosphorylation, Rac activation, and cell migration in HUVECs (Figure 6, C–F). S1P-induced cell migration was not blocked by transfection of siRNA against D2 and TRα1 (Figure 6F), indicating viability of HUVECs after transfection of siRNA against D2 and TRα1. These results suggest that conversion of T4 to T3 by D2 is required for TRα1/PI3K-mediated nongenomic actions of T4 in HUVECs, including stimulation of Akt phosphorylation and Rac activation, which result in cell migration.
Figure 6.

D2 plays a critical role in T4-induced migration mediated by Akt/Rac activation in HUVECs. Before the experiment, HUVECs were transfected with nonsilencing siRNA (control), siD2, or siTRα1. After transfection of siRNA, HUVECs were treated for 4 h at 37°C with RPMI 1640 medium containing 0.1% fatty acid–free BSA. Next, the experiments were performed as described below. A, Expression of D2 and TRα1 mRNA were measured by qRT-PCR using real-time TaqMan technology. The expression of target mRNA normalized to the relative ratio of the expression in those transfected with nontarget siRNA. B, After transfection of siRNA, HUVECs were treated for 4 h at 37°C with RPMI 1640 medium containing 0.1% fatty acid–free BSA with or without 1mM (Bu)2cAMP. Cell sonicates were incubated with 2nM [125I]T4 in the presence of 1mM PTU at 37°C for 1 h to measure D2 activity. C, Cells were incubated for 10 min at 37°C with T3 (1nM), T4 (1nM), or PBS (none) to analyze Akt phosphorylation and Rac activation by Western blotting. D, The relative amount of Akt phosphorylation amount was calculated from three independent experiments described in panel C. E, The relative degree of Rac activation was calculated from three independent experiments shown in panel C. F, Cells were stimulated for 4 h at 37°C with T3 (1nM), T4 (1nM), S1P (1μM), or PBS (none) to analyze migration using a blind Boyden chamber apparatus. The number of migrated cells attached to the lower surface of the filters was counted under a microscope at 400× magnification. A representative (C) or mean ± SEM (A, B, D–F) of three separate experiments is shown. *, P < .05 vs control in panels A and B. *, P < .05 vs none in panels D–F. n.d., not detected.
Discussion
We demonstrated that both T3 and T4 stimulated rapid Akt phosphorylation and cell migration in a PI3K-dependent manner in HUVECs. The inhibition of TRα1 expression by siRNA against TRα1 mRNA blocked Akt phosphorylation and cell migration induced by T3 and T4. A number of rapid effects induced by THs in the cytoplasm and at the plasma membrane have recently been identified (4–14), which are known as the nongenomic or TRE-independent actions of THs. T3 stimulates Akt through cross coupling of T3/TR with PI3K in the cytoplasm (8–13) or by T3 binding to membrane-localized TRs (14). T4, as well as T3, also stimulates MAPK by TH binding to integrin αVβ3, which is located at the plasma membrane (5–7). In fibroblasts, T3 binds to TRβ1, and then this complex stimulates Akt phosphorylation (11). In endothelial cells, T3 increases the association of TRα1 with the regulatory subunit of PI3K, p85α, in a ligand-dependent manner (10). T3/TRα1-mediated PI3K/Akt activation promote cytoprotection in cardiomyocytes (37) and nitric oxide synthase activation in endothelial cells (9, 10). The present results clearly demonstrated that both T3 and T4 stimulated cell migration of HUVECs. Although endothelial cell migration may involve both nongenomic and genomic signaling, nongenomic stimulation through TRα1/PI3K/Akt pathway is required for TH-induced cell migration of HUVECs. Endothelial cell migration activity plays a critical role in preventing atherosclerosis and neovascularization that is associated with occlusive vascular diseases (15). Taken together, TH-regulated endothelial cell migration is considered to be involved in the actions of THs in cardiovascular system. We tested the effect of tetrac, which was shown to inhibit association of T4 and integrin αVβ3 (6). T4- or T3-induced Akt phosphorylation was not attenuated by the addition of tetrac in HUVECs (data not shown), indicating the role of integrin αVβ3 is not involved in the nongenomic actions of THs in HUVECs. Additional studies will be required to elucidate whether THs stimulation of PI3K observed in HUVECs requires direct association of TRα1 with PI3K (10), or whether membrane-localized TRs are involved in the nongenomic actions of THs in HUVECs (14).
Second, we showed for the first time that THs provoked direct Rac activation and stimulated cell migration through the T3/TRα1/PI3K/Rac-signaling pathway in endothelial cells. TH-induced rapid activation of PI3K/Akt/eNOS has been demonstrated in endothelial cells (10), and PI3K/Akt-signaling cascade (15) and Rac1 activation (16) have been reported to mediate the migration of endothelial cells. Under our experimental conditions, neither T3 nor T4 activated Src in endothelial cells, and PP2, a Src kinase family inhibitor, did not attenuate TH-induced Akt phosphorylation in HUVECs (data not shown). These results suggest that THs are able to activate the TRα1/PI3K/Akt-signaling pathway independent of Src in endothelial cells. Previous observations suggested that the Rho family signaling pathway mediated several TH-induced actions (38). Src-mediated T3/TRα1/PI3K activation was recently demonstrated in neuronal cells (35). T3 activated PI3K through binding to integrin at the plasma membrane in a Src-mediated manner (36). Although direct Rho activation by T4 through binding to integrin was demonstrated in previous observations (19), TH-induced Rac activation has not been shown. In this study, we showed that Rac mediated the T3/TRα1/PI3K/Akt signaling pathway in the cytoplasm and not via Src activation in HUVECs. These results suggest the contribution of the Rho family GTPases in TH-induced nongenomic actions and that the TH-induced activation of Rho family GTPases is mediated by at least two pathways, T3/TRα1/PI3K in the cytoplasm and T4/integrin at the plasma membrane.
Third, we demonstrated D2 mRNA expression and functional D2 activity in HUVECs. Furthermore, D2 activity was significantly increased by (Bu)2cAMP treatment. These characteristics of D2 activity in HUVECs are compatible with those in hCASMCs and hASMCs (25). We have also demonstrated that beraprost sodium increased D2 activity in hCASMCs (39). The expression of D2 in endothelial cells and vascular smooth muscle cells suggests that the regulation of D2 activity through a cAMP-mediated pathway could be a therapeutic target for cardiovascular disease. D2 mRNA and its splicing variants, but not D2 activity, were previously demonstrated in ECV304 cells that were thought to be HUVECs (40). However, ECV304 cells have been reported to be cross contaminated with T24 bladder carcinoma cells (41). Although immunohistochemistry of term umbilical cord demonstrated D3 staining in endothelial cells of the umbilical arteries and vein (42), we were not able to detect D3 mRNA in HUVECs under our culture conditions.
Finally, we showed for the first time that conversion of T4 to T3 by D2 is a critical step in T4-induced rapid nongenomic, TRE-independent actions in endothelial cells. So far, the possible role of iodothyronine deiodinase in rapid nongenomic actions of THs has not been suggested. In the present study, we observed that T4 stimulated the PI3K/Akt/Rac-signaling pathway by binding to TRα1 after conversion to T3 by D2. The inhibition of D2 expression by siRNA against D2 mRNA or IOP, a potent D2 inhibitor, blocked T4-promoted but not T3-promoted actions in HUVECs. These findings suggest that conversion of T4 to T3 by D2 is a critical step in T4-induced nongenomic, TRE-independent actions in endothelial cells. T4-induced Akt phosphorylation, Rac activation, and cell migration were blocked by wortmannin treatment or the transfection of siRNA against TRα1 mRNA. The present results suggest that T4-promoted activation of the Akt phosphorylation, Rac activation, and cell-migration pathway is mediated by the D2-catalyzed conversion of T4 to T3, which binds to TRα1 in HUVECs (Figure 7). In the present study, T4 required a longer time than T3 to stimulate Akt phosphorylation, which could be explained by the requirement of deiodination for T4 action.
Figure 7.
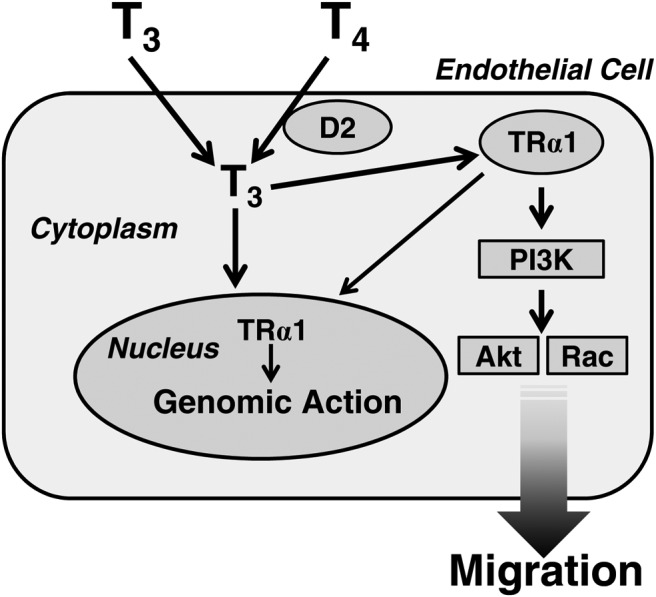
Postulated mechanisms for TH actions in HUVECs. T4 to T3 conversion by D2 is a critical step for T4-induced rapid nongenomic action in HUVECs. T3, converted from T4, activates Akt/Rac through the TRα1/PI3K pathway. As a result, cell migration occurs.
The nongenomic actions of THs have been demonstrated in several cell types other than endothelial cells. THs were demonstrated to stimulate ether-a-go-go-related potassium channel in rat pituitary GH4C1 cells (8), the expression of an endogenous calcineurin inhibitor in human fibroblasts (11), and nitric oxide and cyclic guanosine monophosphate in human osteoblasts (14) through the nongenomic actions. In preliminary studies, we found that TH rapidly stimulated Akt phosphorylation in hCASMCs, which were demonstrated to express D2 (25). Because D2 expression was also shown in GH4C1 cells (43), human fibroblasts (44), and human osteoblasts (33), the possible role of D2 in the nongenomic actions of T4 in those cells needs to be elucidated in additional studies.
In the animal model, T3 did not stimulate Akt kinase activity in brain tissues from TRα1−/−β−/− mice, indicating that T3 activates the PI3K/Akt pathway through TR (10). Recently, introduction of whole-body knockout of TRα loci in a background of ApoE−/− was demonstrated to accelerate atherosclerosis development, which suggested the protective role of TRα against atherosclerosis (45).
In summary, T3 and T4 stimulate Akt phosphorylation and Rac activation through TRα1/PI3K-mediated mechanisms in cytoplasm of HUVECs, which results in cell migration. The conversion of T4 to T3 by D2 is a critical step for T4-induced nongenomic actions in those cells, which may open a novel perspective on understanding of the actions of THs.
Acknowledgments
This work was supported in part by Grants-in-Aid 23390146 and 26293125 (to M.M.) for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. We are indebted to Prof Fumikazu Okajima for helpful discussion and encouragement.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- Akt
- protein kinase Akt
- (Bu)2cAMP
- dibutyryl cyclic adenosine 3′,5′-monophosphate
- D1
- type 1 iodothyronine deiodinase
- D2
- type 2 iodothyronine deiodinase
- D3
- type 3 iodothyronine deiodinase
- DTT
- dithiothreitol
- Erk
- extracellular signal-regulated kinase
- eNOS
- endothelial nitric oxide synthase
- FBS
- fetal bovine serum
- GFP
- green fluorescent protein
- GST
- glutathione S-transferase
- hASMC
- human aortic smooth muscle cell
- hCASMC
- human coronary artery smooth muscle cell
- HUVECs
- human umbilical vein endothelial cells
- IOP
- iopanoic acid
- PTU
- 6-n-propylthiouracil
- Rac1
- Ras-related C3 botulinum toxin substrate 1
- RBD
- Rho-binding domain
- RhoA
- Ras homolog gene family member A
- RPMI
- Roswell Park Memorial Institute
- S1P
- sphingosine 1-phosphate
- siRNA
- small interfering RNA
- TH
- thyroid hormone
- TR
- thyroid hormone receptor
- TRE
- thyroid hormone response element.
References
- 1. Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. N Engl J Med. 2001;344:501–509. [DOI] [PubMed] [Google Scholar]
- 2. Ojamaa K, Klemperer JD, Klein I. Acute effects of thyroid hormone on vascular smooth muscle. Thyroid. 1996;6:505–512. [DOI] [PubMed] [Google Scholar]
- 3. Carrillo-Sepúlveda MA, Ceravolo GS, Fortes ZB, et al. Thyroid hormone stimulates NO production via activation of the PI3K/Akt pathway in vascular myocytes. Cardiovasc Res. 2010;85:560–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31:139–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Botta JA, de Mendoza D, Morero RD, Farías RN. High affinity L-triiodothyronine binding sites on washed rat erythrocyte membranes. J Biol Chem. 1983;258:6690–6692. [PubMed] [Google Scholar]
- 6. Bergh JJ, Lin HY, Lansing L, et al. Integrin alphaVbeta3 contains a cell surface receptor site for thyroid hormone that is linked to activation of mitogen-activated protein kinase and induction of angiogenesis. Endocrinology. 2005;146:2864–2871. [DOI] [PubMed] [Google Scholar]
- 7. Davis PJ, Davis FB, Mousa SA, Luidens MK, Lin HY. Membrane receptor for thyroid hormone: physiologic and pharmacologic implications. Annu Rev Pharmacol Toxicol. 2011;51:99–115. [DOI] [PubMed] [Google Scholar]
- 8. Storey NM, O'Bryan JP, Armstrong DL. Rac and Rho mediate opposing hormonal regulation of the ether-a-go-go-related potassium channel. Curr Biol. 2002;12:27–33. [DOI] [PubMed] [Google Scholar]
- 9. Vicinanza R, Coppotelli G, Malacrino C, Nardo T, Buchetti B, Lenti L, Celi FS, Scarpa S. Oxidized low-density lipoproteins impair endothelial function by inhibiting non-genomic action of thyroid hormone-mediated nitric oxide production in human endothelial cells. Thyroid. 2013;23:231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hiroi Y, Kim HH, Ying H, Furuya F, Huang Z, Simoncini T, Noma K, Ueki K, Nguyen NH, Scanlan TS, Moskowitz MA, Cheng SY, Liao JK. Rapid nongenomic actions of thyroid hormone. Proc Natl Acad Sci U S A. 2006;103:14104–14109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cao X, Kambe F, Moeller LC, Refetoff S, Seo H. Thyroid hormone induces rapid activation of Akt/protein kinase B-mammalian target of rapamycin-p70S6K cascade through phosphatidylinositol 3-kinase in human fibroblasts. Mol Endocrinol. 2005;19:102–112. [DOI] [PubMed] [Google Scholar]
- 12. Gauthier KF. Nongenomic, TRβ-dependent, thyroid hormone response gets genetic support. Endocrinology. 2014;155:3206–3209. [DOI] [PubMed] [Google Scholar]
- 13. Martin NP, Marron Fernandez de Velasco E, Mizuno F, et al. A rapid cytoplasmic mechanism for PI3 kinase regulation by the nuclear thyroid hormone receptor, TRβ, and genetic evidence for its role in the maturation of mouse hippocampal synapses in vivo. Endocrinology. 2014;155:3713–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kalyanaraman H, Schwappacher R, Joshua J, et al. Nongenomic thyroid hormone signaling occurs through a plasma membrane-localized receptor. Sci Signal. 2014;7:ra48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nagata D, Mogi M, Walsh K. AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J Biol Chem. 2003;278:31000–31006. [DOI] [PubMed] [Google Scholar]
- 16. Dormond O, Foletti A, Paroz C, Rüegg C. NSAIDs inhibit alpha V beta 3 integrin-mediated and Cdc42/Rac-dependent endothelial-cell spreading, migration and angiogenesis. Nat Med. 2001;7:1041–1047. [DOI] [PubMed] [Google Scholar]
- 17. Takashima S, Sugimoto N, Takuwa N, et al. G12/13 and Gq mediate S1P2-induced inhibition of Rac and migration in vascular smooth muscle in a manner dependent on Rho but not Rho kinase. Cardiovasc Res. 2008;79:689–697. [DOI] [PubMed] [Google Scholar]
- 18. Okamoto H, Takuwa N, Yokomizo T, et al. Inhibitory regulation of Rac activation, membrane ruffling, and cell migration by the G protein-coupled sphingosine-1-phosphate receptor EDG5 but not EDG1 or EDG3. Mol Cell Biol. 2000;20:9247–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zvibel I, Atias D, Phillips A, Halpern Z, Oren R. Thyroid hormones induce activation of rat hepatic stellate cells through increased expression of p75 neurotrophin receptor and direct activation of Rho. Lab Invest. 2010;90:674–684. [DOI] [PubMed] [Google Scholar]
- 20. Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev. 2002;23:38–89. [DOI] [PubMed] [Google Scholar]
- 21. Davey JC, Becker KB, Schneider MJ, St Germain DL, Galton VA. Cloning of a cDNA for the type II iodothyronine deiodinase. J Biol Chem. 1995;270:26786–26789. [DOI] [PubMed] [Google Scholar]
- 22. Croteau W, Davey JC, Galton VA, St Germain DL. Cloning of the mammalian type II iodothyronine deiodinase. A selenoprotein differentially expressed and regulated in human and rat brain and other tissues. J Clin Invest. 1996;98:405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salvatore D, Bartha T, Harney JW, Larsen PR. Molecular biological and biochemical characterization of the human type 2 selenodeiodinase. Endocrinology. 1996;137:3308–3315. [DOI] [PubMed] [Google Scholar]
- 24. Salvatore D, Tu H, Harney JW, Larsen PR. Type 2 iodothyronine deiodinase is highly expressed in human thyroid. J Clin Invest. 1996;98:962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mizuma H, Murakami M, Mori M. Thyroid hormone activation in human vascular smooth muscle cells: Expression of type II iodothyronine deiodinase. Circ Res. 2001;88:313–318. [DOI] [PubMed] [Google Scholar]
- 26. Ishikawa T, Chijiwa T, Hagiwara M, Mamiya S, Hidaka H. Thyroid hormones directly interact with vascular smooth muscle strips. Mol Pharmacol. 1989;35:760–765. [PubMed] [Google Scholar]
- 27. Zwaveling J, Pfaffendorf M, van Zwieten PA. The direct effects of thyroid hormones on rat mesenteric resistance arteries. Fundam Clin Pharmacol. 1997;11:41–46. [DOI] [PubMed] [Google Scholar]
- 28. Park KW, Dai HB, Ojamaa K, Lowenstein E, Klein I, Sellke FW. The direct vasomotor effect of thyroid hormones on rat skeletal muscle resistance arteries. Anesth Analg. 1997;85:734–738. [DOI] [PubMed] [Google Scholar]
- 29. Leonard JL, Rosenberg IN. Iodothyronine 5′-deiodinase from rat kidney: Substrate specificity and the 5′-deiodination of reverse triiodothyronine. Endocrinology. 1980;107:1376–1383. [DOI] [PubMed] [Google Scholar]
- 30. Murakami M, Tanaka K, Greer MA, Mori M. Anterior pituitary type II thyroxine 5′-deiodinase activity is not affected by lesions of the hypothalamic paraventricular nucleus which profoundly depress pituitary thyrotropin secretion. Endocrinology. 1988;123:1676–1681. [DOI] [PubMed] [Google Scholar]
- 31. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. [DOI] [PubMed] [Google Scholar]
- 32. Richard K, Hume R, Kaptein E, et al. Ontogeny of iodothyronine deiodinases in human liver. J Clin Endocrinol Metab. 1998;83:2868–2874. [DOI] [PubMed] [Google Scholar]
- 33. Morimura T, Tsunekawa K, Kasahara T, et al. Expression of type 2 iodothyronine deiodinase in human osteoblast is stimulated by thyrotropin. Endocrinology. 2005;146:2077–2084. [DOI] [PubMed] [Google Scholar]
- 34. Bornfeldt KE, Graves LM, Raines EW, et al. Sphingosine-1-phosphate inhibits PDGF-induced chemotaxis of human arterial smooth muscle cells: Spatial and temporal modulation of PDGF chemotactic signal transduction. J Cell Biol. 1995;130:193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cao X, Kambe F, Yamauchi M, Seo H. Thyroid-hormone-dependent activation of the phosphoinositide 3-kinase/Akt cascade requires Src and enhances neuronal survival. Biochem J. 2009;424:201–209. [DOI] [PubMed] [Google Scholar]
- 36. Lin HY, Sun M, Tang HY, et al. L-Thyroxine vs. 3,5,3′-triiodo-L-thyronine and cell proliferation: Activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. Am J Physiol Cell Physiol. 2009;296:C980–C991. [DOI] [PubMed] [Google Scholar]
- 37. Ojamaa K. Signaling mechanisms in thyroid hormone-induced cardiac hypertrophy. Vascul Pharmacol. 2010;52:113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zamoner A, Funchal C, Jacques-Silva MC, Gottfried C, Barreto Silva FR, Pessoa-Pureur R. Thyroid hormones reorganize the cytoskeleton of glial cells through Gfap phosphorylation and Rhoa-dependent mechanisms. Cell Mol Neurobiol. 2007;27:845–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kasahara T, Tsunekawa K, Seki K, Mori M, Murakami M. Regulation of iodothyronine deiodinase and roles of thyroid hormones in human coronary artery smooth muscle cells. Atherosclerosis. 2006;186:207–214. [DOI] [PubMed] [Google Scholar]
- 40. Ohba K, Yoshioka T, Muraki T. Identification of two novel splicing variants of human type II iodothyronine deiodinase mRNA. Mol Cell Endocrinol. 2001;172:169–175. [DOI] [PubMed] [Google Scholar]
- 41. Dirks WG, Drexler HG, MacLeod RA. ECV304 (endothelial) is really T24 (bladder carcinoma): Cell line cross-contamination at source. In Vitro Cell Dev Biol. 1999;35:558–559. [DOI] [PubMed] [Google Scholar]
- 42. Huang SA, Dorfman DM, Genest DR, Salvatore D, Larsen PR. Type 3 iodothyronine deiodinase is highly expressed in the human uteroplacental unit and in fetal epithelium. J Clin Endocrinol Metab. 2003;88:1384–1388. [DOI] [PubMed] [Google Scholar]
- 43. Koenig RJ. Regulation of thyroxine 5′-deiodinase by thyroid hormones and activators of protein kinase C in GH4C1 cells. Endocrinology. 1986;118:1491–1497. [DOI] [PubMed] [Google Scholar]
- 44. Dumitrescu AM, Liao XH, Abdullah MS, et al. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat Genet. 2005;37:1247–1252. [DOI] [PubMed] [Google Scholar]
- 45. Billon C, Canaple L, Fleury S, et al. TRα protects against atherosclerosis in male mice: Identification of a novel anti-inflammatory property for TRα in mice. Endocrinology. 2014;155:2735–2745. [DOI] [PubMed] [Google Scholar]