

Abstract
Purpose.
The association of single nucleotide polymorphisms of components of the complement alternative pathway with the risk of age-related macular degeneration (AMD) indicates that complement signaling plays an important role in retinal physiology. How genetic variation leads to retinal degeneration is unknown. It has been assumed that complement activation augments immune responses, which in turn initiate AMD pathogenesis. To better understand the relationship between complement and the outer retina, we examined mice lacking the main complement component C3 and the receptors for complement activation fragments C3a (C3aR) and/or C5a (C5aR).
Methods.
Complement mutant mice were studied along with wild-type (WT) littermates from 6 weeks to 14 months of age. Strobe flash electroretinography (ERG) was used to examine outer retinal function and a dc-ERG technique was used to measure ERG components generated by the retinal pigment epithelium. Retinas were examined by histology, immunohistochemistry, and biochemistry.
Results.
Mice lacking C3aR and/or C5aR developed early onset and progressive retinal degeneration, accompanied by cleaved caspase-3 upregulation. Genetic deletion of C3aR and/or C5aR led to cell-specific defects that matched the cellular localization of these receptors in the WT retina. Compared to WT, C3aR −/− and C3aR −/−C5aR −/− mice showed increased retinal dysfunction upon light exposure. C3aR −/−C5aR −/− mice immunized with 4-hydroxynonenal-adducted protein developed severe retinal impairment unrelated to immune response.
Conclusions.
C3aR- and C5aR-mediated signaling was necessary to maintain normal retinal function and structure. These receptors may be important biomarkers for predicting retinal degeneration including AMD.
Complement activation fragment receptors C3aR and C5aR as well as the central complement component C3 prevent retinal degeneration.
Introduction
Degeneration of photoreceptors underlies vision loss in early onset disorders such as retinitis pigmentosa (RP), cone-rod dystrophy, and Lebers congenital amaurosis, as well as in late onset conditions such as age-related macular degeneration (AMD). These retinal disorders affect more than 200,000 US children, 2 million US elderly, and more than 50 million people worldwide.1,2 A breakthrough finding in understanding AMD pathophysiology was the observation that single nucleotide polymorphisms (SNPs) of the complement alternative pathway components factor H, factor B, and complement C3 strongly associate with the risk of AMD development.3–9 Factor H is an inhibitor of complement activation, while factor B and C3 are essential components of complement activation. How variation in these genes is involved in the pathology of AMD remains unknown, although a current working hypothesis is that complement activation leads to AMD through an inflammatory mechanism.10–12
There are three pathways of complement activation, the classical, alternative, and lectin pathways. The main components of the alternative pathway include factor D, factor B, factor H, C3, and C5. The alternative pathway is initiated by spontaneous C3 hydrolysis C3(H2O), whose function is equal to that of C3b. C3(H2O) binds factor B so factor B is cleaved by factor D to form a C3 convertase, C3bBb. C3 convertases cleave C3 into C3a and C3b. A C3 convertase also binds C3b to form a C5 convertase (C3bBbC3b), which cleaves C5 to C5a and C5b. The coagulation/fibrinolysis proteases may also act as C3 and C5 convertases to produce C3a and C5a.13 Fragments C3a and C5a activate complement signaling by binding the C3a receptor (C3aR) or the C5a receptor (C5aR), respectively.14,15 C3 is a central component of all complement activation pathways. Outside of the retina, increased activation of C3aR or C5aR has been implicated in autoimmune diseases, transplant rejection, and inflammatory diseases.16–19 Presuming an analogous role in AMD pathogenesis, multiple groups are exploring the possibility that complement inhibition may be effective for preventing or slowing AMD progression.12,20–22 Several findings, however, argue against this strategy. For example, mice lacking C3a and/or C5a signaling have abnormal susceptibility to neuroexcitotoxicity,23 abnormal neurogenesis,24 abnormal differentiation and migration of neural progenitor cells,25 abnormal liver cell survival/regeneration,26 and abnormal remyelination.27 Furthermore, the pathology associated with a model of retinopathy of prematurity was exacerbated in mice lacking either C3 or C5aR.28 To date, the retinas of mice lacking C3aR or C5aR have not been examined. Here we report that C3aR −/− and/or C5aR −/− mice, as well as C3 −/− mice, developed progressive retinal degeneration and retinal dysfunction that matched the cellular distribution of these proteins in the retina. These results established a requirement for the alternative complement pathway in normal maintenance of the outer retina.
Materials and Methods
Animals
Mice were obtained from the Jackson Laboratory (Bar Harbor, ME). C3aR −/− mice at BALB/cJ background (stock number: 005712) and C5aR −/− (stock number: 005676; at unknown generations of C57BL/6J background) were backcrossed to C57BL/6J wild-type (WT) mice (stock number: 000664) for eight generations at the Cleveland Clinic Foundation. Back-crossed eight-generation C3aR −/− and C5aR −/− mice were then intercrossed to generate C3aR −/−C5aR −/− double knockouts. C3 −/− mice (stock number: 003641) were obtained on a C57BL/6J background. All of the mice in this study were on C57BL/6J background unless indicated specifically. The discovery29 that many mouse lines carry Crb1rd8 caused us to evaluate the mice studied here. We screened the available samples (14 C3aR −/−, nine C5aR −/−, 15 C3aR −/−C5aR −/−, two C3 −/−) for the Crb1rd8 allele. All mice were WT except for a single mutant that was Crb1+/rd8 heterozygous. All animal studies were approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic Foundation. The animals were treated in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the ARVO Statement on the Use of Animals in Ophthalmic and Vision Research.
Materials and Reagents
4-Hydroxynonenal (4-HNE) was from Alpha Diagnosis International Inc. (San Antonio, TX). Mouse serum albumin (MSA) was from Sigma (St. Louis, MO). Cleaved caspase 3 detection kits were obtained from Cell Signaling Technology (Danvers, MA). Antibodies: NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), p65: polyclonal rabbit (Cell Signaling Technology), phospho-IKKα/β, IkBα, and phospho-p65: polyclonal rabbit (Cell Signaling Technology), C3aR: monoclonal mouse (Santa Cruz Biotechnology, Santa Cruz, CA), C5aR: monoclonal mouse (Santa Cruz Biotechnology), Interleukin-17 (IL-17): monoclonal mouse (R&D Systems, Minneapolis, MN), PKCα: monoclonal mouse (Santa Cruz Biotechnology), Calbindin D-28: polyclonal rabbit (Millipore, Billerica, MA). PKCα was used to label rod bipolar cells30 and Calbindin D-28 was used to label horizontal and amacrine cells.31
Electroretinography
After overnight dark adaptation and pupil dilation (1% tropicamine; 2.5% phenylephrine HCl), electroretinograms (ERGs) were recorded from the corneal surface of anesthetized mice (ketamine 80 mg/kg; xylazine 16 mg/kg) using published procedures32–34 to examine function of the outer retina and retinal pigment epithelium (RPE).
Anatomical Analysis
Immediately after sacrifice, one eye was embedded in ornithine carbamoyltransferase freezing medium, flash frozen on powdered dry ice, and immediately transferred to a −80°C freezer. The other eye was fixed in 4% paraformaldehyde for 4 hours after the cornea and lens were removed. The eye cup was immersed through a graded series of sucrose solutions: 10% for 1 hour, 20% for 1 hour, and 30% overnight. 5-μm sections were cut on a cryostat. For immunofluorescence staining, retinal sections were incubated with primary antibodies (1:100) overnight at 4°C, in FITC-labeled secondary antibodies (1:1000) for 1 hour, and were then washed and mounted using mounting medium (VECTASHIELD; Vector Laboratories, Burlingame, CA). For electron microscopy sample preparation, ultrathin sections (85 nm) and electron micrographs were performed by the image core in the Cleveland Clinic Foundation. All images were taken along the horizontal meridian, approximately 200 μm from the edge of the optic nerve head.
Light Exposure
After pupil dilation (2.5% phenylephrine HCl; 1% cyclopentolate HCl; 1% mydriacyl), mice were exposed for 8 hours to white light of 7500 lux. ERGs were recorded on day 6 after light exposure, after which eyes were extracted and prepared for anatomical analysis.
HNE-MSA Immunization
We made 4-HNE-conjugated MSA (HNE-MSA) following a published method.35 4-HNE modification of MSA was confirmed by the Mass Spectrometry Core II of the Cleveland Clinic. C3aR −/−C5aR −/− and WT littermates were immunized subcutaneously with 200 μg of HNE-MSA, or MSA as control, in complete Freund's adjuvant at day 1, and in incomplete Freund's adjuvant at days 10 and 50. Two months later after first immunization, ERGs were recorded for those immunized mice, after which eyes were extracted and prepared for anatomic analysis.
Retinal Cell Isolation and Analysis
Retinal cells were isolated using a published procedure.36 Briefly, each mouse eyeball was enucleated and placed in sterile culture plates with sterile 1X PBS. All subsequent procedures were conducted in a sterile tissue culture hood. Each eyeball was cut from the middle of the cornea. After the lens was removed, the retina and RPE were isolated. To release cells, retinas were incubated in Ringer buffer containing 10 to 20 U/mL papain and 0.1 mg/mL cysteine for 30 minutes at 30°C; 11-cis retinal was not added. After rinsing with Ringer buffer, the retinas were triturated by a glass pipette. The retinal cells were counted by trypan blue, and few dead cells were seen. Neural retinal cells were incubated in Dulbecco's modified Eagle's medium (DMEM) (0% FCS) with 5 μg/mL HNE-MSA for 4 hours to detect apoptotic cells by Annexin V antibody staining/flow-cytometry. Another portion of retinal cells was grown on sterile cover slips overnight to seed cells and the next day exposed under light (FLUOREX 27W; Lights of America, Orange, CA) (fluorescent linear bulb at 10 cm distance) for 12 hours for p65 staining.
ELISPOT Assay
Splenocytes (6x105) from HNE-MSA immunized mice were incubated for 24 hours in IL-17 monoclonal antibody (R&D Systems)-coated and PBS-1% BSA-blocked 96-well MultiScreen HTS filter plates for the enzyme-linked immunosorbent spot (ELISPOT) (Whatman, Piscataway, NJ) with 0.5 μg/mL HNE-MSA. Spots were developed and quantitated with a CTL ImmunoSpot Analyzer (Cellular Technology, Shaker Heights, OH).
Mass Spectrometry
Mouse plasma was collected and frozen immediately. Mass spectrometric analyses for phospholipids were performed on-line using electrospray ionization tandem mass spectrometry in the positive ion mode with multiple reaction monitoring using the molecular cation [MH]+ and the m/z 184 daughter phosphocholine ion. For free fatty acid analysis, negative-ion mode with multiple reaction monitoring of parent and individual daughter ions of oxidized and unoxidized free fatty acids used the m/z transitions.
Statistics
ERG measures were evaluated with a two-way ANOVA. The power analysis was conducted by the F-test of one-way ANOVA, where we considered numbers as outcome and groups as the factor. P < 0.05 was considered statistically significant. All other comparisons between WT and mutant data were made using two-tailed, unpaired Student's t-tests.
Results
Outer Retinal Function Is Impaired in Mice Lacking C3aR and/or C5aR
We used ERGs to compare outer retinal function of WT and complement mutant mice at ages ranging from 6 weeks to 12 months. Strobe flash ERGs were used to evaluate function of photoreceptors (a-wave) and depolarizing bipolar cells (b-wave). At 6 weeks of age, there was no significant difference between ERG a- and b-wave amplitudes of WT and C3aR−/− or C3aR−/−C5aR−/− mice (Figs. 1A, 1B). However, by 14 weeks of age, ERG a- and b-waves of C3aR−/− and C3aR−/−C5aR−/− mice were significantly reduced, and this reduction progressed with increasing age (Figs. 1A, 1B). In comparison, there was no difference between the a- or b-waves of WT and C5aR−/− mice, even at 12 months of age (Fig. 1C). ERG b-waves of C3−/− mice were also significantly decreased at 12 months old (Fig. 1D), but not at younger ages.
Figure 1.
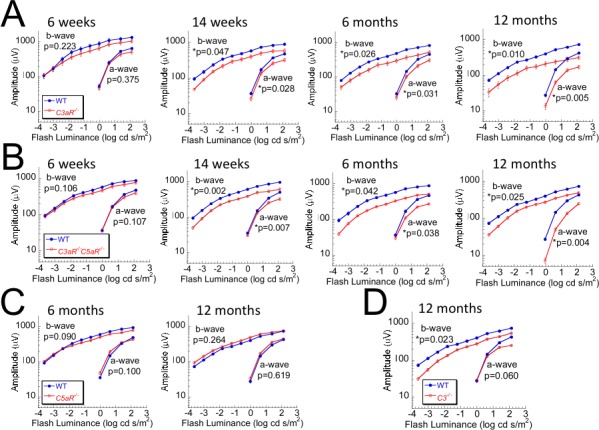
ERG measures of outer retinal function in complement mutant mice. ERGs were recorded in (A) C3aR −/−, (B) C3aR −/−C5aR −/−, (C) C5aR −/−, (D) C3 −/−, and their WT littermates at 6 weeks to 12 months of age. Data points indicate average (±SEM) of up to 14 mice. *P < 0.05.
We used dc-ERGs to document abnormal function of the RPE, whose activity is reflected in four major components (c-wave, fast oscillation, light peak, and off-response). At 8 weeks of age, we saw no significant difference in ERG potentials generated by the RPE between WT and complement mutant mice (Figs. 2A, 2B). At 12 months of age, however, the amplitude of the c-wave was significantly reduced in C3aR−/−, C5aR−/−, and C3−/− mice (Figs. 2A, 2C, 2D). No other component of the dc-ERG was significantly different between WT and C3aR−/−, C5aR−/−, or C3−/− mice. The amplitudes of the c-wave and the fast oscillation were reduced at an earlier time point (6 months) in C3aR−/−C5aR−/− mice (Figs. 2B, 2D).
Figure 2.

ERG measures of RPE function in complement mutant mice. DC ERG obtained from (A) C3aR −/−, C5aR −/−, (B) C3aR −/−C5aR −/−, (C) C3 −/−, and their WT littermates at 8 weeks to 12 months of age. Waveforms represent the grand average response of 6 to 10 mice. (D) Average (±SEM) amplitude of major components of DC-ERG (c-wave, fast-oscillation [FO], light peak [LP], off-response) in mutant mice and WT littermates. *P < 0.05.
Progressive Retinal Cell Loss in Mice Lacking C3aR and/or C5aR
Retinal structure was examined at ages where ERG studies were conducted. No significant changes in outer nuclear layer (ONL) thickness were noted at 3 months of age, but the ONL was disorganized at this age in C3aR−/− and C3aR−/−C5aR−/− mice (Figs. 3A, 3B). At older ages, we noted ONL thinning and disorganization in mice lacking C3aR (either C3aR−/− or C3aR−/−C5aR−/−) but not C5aR. The inner nuclear layer (INL) of C3aR−/− or C3aR−/−C5aR−/− mice was significantly thinner than WT littermates at 3 and 14 months of age (Figs. 3A, 3B). Because rod bipolar cells are a main component of the INL, we used PKCα immunolabeling to identify this cell type. In comparison to WT (60 ± 12; n = 5), the average (±SD)/200-μm field density of rod bipolar cells was significantly decreased at 3 months and declined further at 14 months in C3aR−/− (23 ± 7; n = 8) and C3aR−/−C5aR−/− (20 ± 5; n = 5) retinas (Fig. 4A). The number of rod bipolar cells was also decreased in 14 month old C3−/− mice (31 ± 10; n = 5) (Fig. 4A), but not in mice lacking only C5aR (65 ± 11; n = 5). We used calbindin to immunolabel horizontal cells. In comparison to WT (11 ± 3; n = 5), the average (±SD)/300-μm field density of horizontal cells was decreased in C3aR−/− (3 ± 2; n = 8), C3aR−/−C5aR−/−(3 ± 2; n = 5) and C3−/− (4 ± 3; n = 5) mice (Fig. 4A), but not in mice lacking only C5aR (12 ± 4; n = 5). Electron microscopy showed that RPE cell nuclei were shrunken or apoptotic and the normal structure of outer segment discs was lost in C3aR−/−C5aR−/− mice at 6 months (Fig. 4B).
Figure 3.
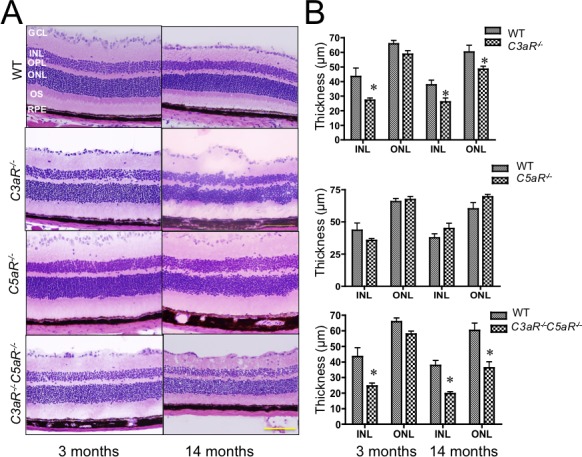
Anatomical analysis of complement mutant mice. (A) H&E staining of retinal cross-sections obtained from C3aR −/−, C5aR −/−, C3aR −/−C5aR −/−, and WT littermates. Images are representative of at least 10 mice from each group. All sections of photos were taken at the central areas (200 μm from the optic nerve head). Magnification: ×40; scale bar indicates 100 μm. (B) Thickness of ONL and INL. Bars indicate average (±SEM) of at least 10 mice per group. *P < 0.05.
Figure 4.
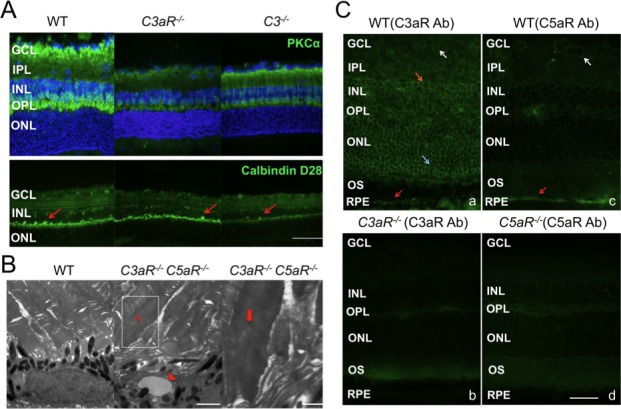
Immunohistochemical analysis. (A) Staining of retinal sections of 14-month-old WT, C3aR−/−, and C3 −/− mice for rod bipolar cells (PKCα antibody; upper panels; magnification: ×60) or for horizontal and amacrine cells (Calbindin D-28 antibody; lower panels; magnification: ×40; scale bar indicates 100 μm). Images are representative of at least five mice from each group. All sections of photos were taken at the central areas (200 μm from the optic nerve head). (B) Electron microscopy of RPE-outer segment interface in 6-month-old WT (left) and C3aR −/−C5aR −/− mice (middle and right). Triangle indicates the apoptotic RPE nucleus (middle), magnification: ×7,100; scale bar indicates 2 μm for left and middle images. Arrows indicate swollen outer segment (right; magnification: ×19,500; scale bar indicates 1 μm). (C) Distribution of C3aR and C5aR in retina. C3aR antibody labeling of WT (a) showed C3aR expression in INL (orange arrow), ONL (light blue arrow), RPE (red arrow), and ganglion cell layer (white arrow), but was missing in the C3aR −/− retina (b). C5aR antibody labeling of WT retina (c) showed C5aR expression in RPE (red arrow) and ganglion cells (white arrow), which was missing in C5aR −/− retina (d). Magnification: ×60; scale bar indicates 50 μm.
We used antibodies to define the location of C3aR and C5aR. Consistent with a recent report,37 C3aR is expressed in multiple layers of the WT retina (INL, ONL, RPE, ganglion cell layer) and is missing in the C3aR−/− retina (Fig. 4C). C5aR is expressed in RPE and ganglion cells of the WT retina, but not in the C5aR−/− retina (Fig. 4C). These distributions match the functional and anatomical changes in these mutant mice, indicating that C3aR and C5aR play critical and cell-specific roles for the maintenance of neural retina and RPE cells.
Increased Activated Caspase-3 and Decreased NF-κB Activation in the Absence of C3aR or C3aR/C5aR
We studied cellular apoptosis to examine the mechanism underlying cell loss and dysfunction in C3aR−/− and C3aR−/−C5aR−/− retinas. Levels of apoptotic cleaved caspase-3 were elevated in C3aR−/− and C3aR−/−C5aR−/− retinas at 3 months and increased further at 14 months (Fig. 5A).
Figure 5.
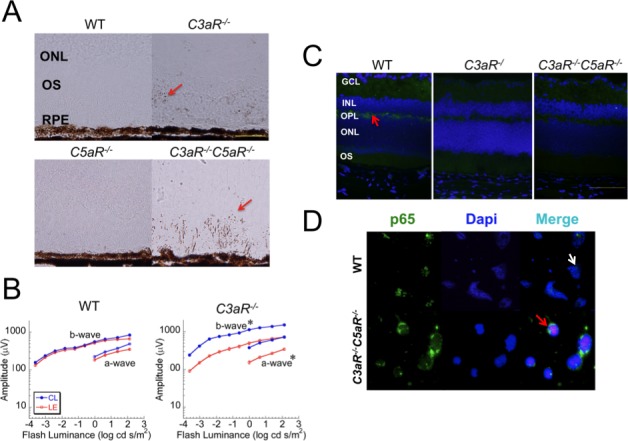
Light exposure. (A) Retinal sections of 14-month-old WT, C3aR−/−, C5aR−/−, and C3aR−/−C5aR−/− mice probed for cleaved casepase-3, which appears as brown spots (pointed by arrows). Scale bar indicates 50 μm. (B) ERG studies 6 days following 8-hour light exposure of 6-week-old C3aR−/−C5aR−/− and WT mice (at Balb/c background). Data points indicate the average (±SEM) of five mice per group at each condition. *P < 0.05. (C) Retinal cross-sections made following ERG analysis and reacted with phospho-IKKα/β. Bright green staining (pointed by an arrow) was detected in the OPL of WT littermates but not C3aR−/− or C3aR−/−C5aR−/− mice. Pictures represent two independent experiments with the same results, magnification: ×60; scale bar indicates 500 μm. (D) Antibody staining for p65 nuclear translocation of isolated neural retinal cells obtained from WT (top) or C3aR−/−C5aR−/− (bottom) mice after 12-hour light exposure. The white arrow points to p65 (green) in the nucleus of WT retinal cells and the red arrow points to the p65 (green) in the cytoplasm of C3aR−/−C5aR−/− retinal cells.
Excessive light will induce retinal degeneration.38 If C3aR and C5aR normally function to ameliorate light-induced retinal damage, the effect should be more pronounced in mice lacking these proteins. In comparison to WT, after an 8-hour light exposure C3aR−/− mice showed a significantly greater loss in retinal function in both C57BL/6J and BALC/cJ backgrounds. In comparison to WT, C3aR−/− mice in BALB/cJ background showed a greater decrease in retinal function (Fig. 5B). Histologic studies showed the same findings in both C57BL/6J and BALB/cJ mice. We used four NF-κB pathway antibodies that work for immunohistochemistry to examine cellular stimulation in retinas removed seven days after 8-hour light exposure. We found that level of phospho-IKKα/β (not IkBα, p65, and phospho-p65) was significantly increased in the WT OPL but not in the OPL of C3aR−/− or C3aR−/−C5aR−/− mice (Fig. 5C).
In isolated retinal cells, light exposure of WT and C3aR−/− C5aR−/− retinal cells was carried out in parallel in six-well plates. We observed p65-nuclear translocation in almost all WT but not in C3aR−/−C5aR−/− cells after the 12 hour-light stimulation (Fig. 5D). These results indicate that C3aR could play an important role in protecting retinal cells from light-induced retinal degeneration through the NF-κB pathway.
Plasma Lysophosphatidylcholine or Free Polyunsaturated Fatty Acids Are Significantly Decreased in the Absence of Complement Proteins
Light and aging produce reactive oxygen species, which attack phospholipids, polyunsaturated fatty acids (PUFAs), proteins, and DNA to cause lipid peroxidation, fatty acid oxidation, protein modification, and DNA breakage, respectively. Phospholipids and PUFAs are essential components of cellular membranes and are also the primary targets of oxidative attack. In comparison to WT littermates, the levels of lysophosphatidylcholine (LPC) were significantly reduced in plasma of C3aR−/− and C3aR−/−C5aR−/− mice at 3 and 14 months of age (Fig. 6A). LPC may be important not only for ganglion cell survival39 but also for INL and ONL cell survival and regeneration. We found that the free PUFAs linoleic acid (LA) and arachidonic acid (AA) were significantly decreased in all complement mutant mice at 14 months of age (Fig. 6B), but not in mice aged 2 to 3 months, perhaps reflecting an age-related increase in oxidative stress in the receptor mutant retina.
Figure 6.
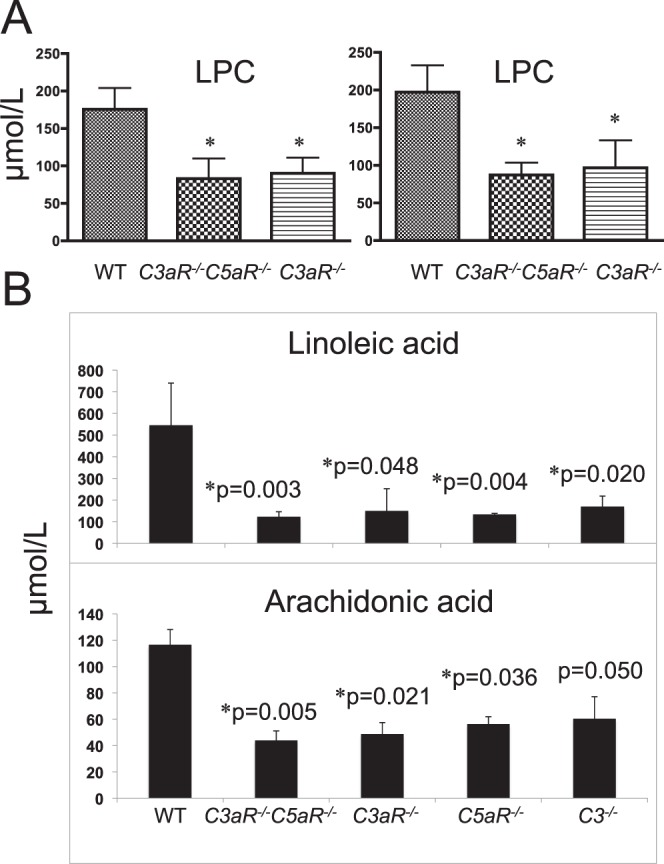
Mass spectrometric analysis of mouse plasma for phospholipids and free fatty acids. (A) LPC levels in mice at 2 to 3 months (left) or 14 months (right) of age. (B) Levels of LA and AA in 14-month-old WT and complement mutant mice. Bars indicate average (±SEM) of 8 to 10 mice *P < 0.05. Figures are a representative of three independent experiments with equal numbers of males and females.
Retinal Dysfunction in C3aR/C5aR Deficiency Is Independent of Immune Responses
In comparison to WT, after immunization with HNE-MSA, C3aR−/−C5aR−/− mice developed a more severe reduction in ERG amplitude (Fig. 7A). These changes are unlikely to reflect an immune-mediated response because this pathway is truncated in C3aR−/−C5aR−/− mice. Annexin V, indicative of cell apoptosis, was elevated in HNE-MSA immunized C3aR−/− C5aR−/− retina (Fig. 7B). The impaired retinal function was independent of T-cell activation as IL17-producing cells were markedly diminished in the HNE-MSA immunized C3aR−/− C5aR−/− mice (Fig. 7C, left panel). Consistent with this observation, incubation of retinal cells with 5 μg/mL of HNE-MSA showed greater cell death in C3aR−/−C5aR−/− than in WT retinas (Fig. 7C, right panel). In summary, when mice were immunized with 4-HNE-adducted protein, C3aR−/−C5aR−/− showed a more severe phenotype than WT and this phenotype was not related to immune responses. The heightened sensitivity of C3aR−/−C5aR−/− retinal cells to the fatty acid oxidation 4-HNE fragment was observed in vivo and in vitro, indicating that a decreased capacity to ameliorate oxidative damage may underlie the retinal changes observed in these animal models.
Figure 7.

4-HNE-MSA immunization. C3aR−/−C5aR−/− and WT littermates were immunized at 8 weeks of age with 4-HNE-MSA or MSA. (A) The ERG results indicate no significant impact of immunization on WT mice (left), but a significant reduction in C3aR−/−C5aR−/− animals. Data points indicate average (±SEM) of 5 WT or 7-8 C3aR−/−C5aR−/− mice per group. *P < 0.05. (B) Retinal cross-sections of immunized C3aR−/−C5aR−/− mice exposed to Annexin V antibody. Annexin V expression is increased in the ONL of HNE-MSA immunized C3aR−/−C5aR−/− mice (right panel arrow) but not in MSA immunized C3aR−/−C5aR−/− mice (left). Magnification: ×60; scale bar indicates 100 μm. (C) Left: Spleen cells were isolated from HNE-MSA immunized C3aR−/−C5aR−/− or HNE-MSA immunized WT littermates and incubated with 0.5 μg of HNE-MSA for 24 hours. IL-17 producing cells were analyzed by ELISPOT assays (left). *P < 0.05. Right: Retinal cells isolated from C3aR−/−C5aR−/− mice or WT littermates were incubated with 5 μg/mL of HNE-MSA for 4 hours, stained by Annexin V antibody, and then analyzed by flow cytometry (right). The black peak depicts the WT flow histogram and the white peak shows the flow histogram of C3aR−/−C5aR−/− retinal cells. Data are representative of three independent experiments.
Discussion
A critical role for the alternative pathway of the complement system has been implicated in AMD pathogenesis based on the identification of complement components in the drusen of AMD patient eyes40,41 and genetic studies in which AMD risk is related to a series of SNPs of factor H, factor B, and C3.1–7 Complement is an essential component of the innate immune system, and is ubiquitously located on cell surfaces and in the circulation. How variation in these genes is involved in the pathology of AMD is unknown. The current interpretation holds that complement activation leads to AMD through an inflammatory mechanism, and has led to the development of treatment strategies based on complement inhibition.42–44
In this project, we examined mice with truncated complement activation due to mutations in the central complement component C3 or the receptors for one or both complement activation fragments. We report a progressive loss of photoreceptor, bipolar cells and horizontal cells in mice lacking C3aR, and RPE dysfunction in mice lacking C5aR or C3aR. These changes match the tissue distribution of these proteins.37,45,46 The retinal dysfunction in C3−/− mice was variable at young ages, but became consistent and statistically significant in older mice. Overall, C3−/− and C3aR−/− mice displayed a similar phenotype, but the onset was later in the C3−/− mutant. Increased intrinsic apoptosis may play an important role in retinal cell loss in these models. The ERG reductions seen in C3aR−/− and C3−/− mice were consistent with a panretinal loss of retinal cells, which we confirmed by anatomic evaluation. Although this phenotype is most similar to that of human disorders like RP, no disease loci have been linked to the region near C3 or C3aR (https://sph.uth.tmc.edu/Retnet/). However, low serum C3 levels were reported in RP patients.47 Whether C3 or C3aR mutations underlie some forms of RP remains to be determined.
Our results indicated that signaling through C3aR and C5aR was important for the normal retina to maintain their structure and their function. Both of these proteins are members of the class A subfamily 8 of G protein-coupled receptors (GPCRs). In general, GPCR signaling is important for cell growth, activity, and survival. GPCR signaling can vary widely across different cell types, and the role of C3aR and C5aR signaling in the retina remains to be clarified. In the brain, macrophages, dendritic cells, and neutrophils, C5a•C5aR activates MAPK, PI3K-Akt, NF-κB, and cAMP response element-binding signaling.48–52 C3a•C3aR activates NF-κB in tubular epithelial cells,53 nerve growth factor-induced nuclear factor of activated T-cells (NFAT) activation in human mast cells,54 and transcription factors activator protein-1 in glial cells.55 C3a•C3aR and C5a•C5aR both activate PI3K-Akt pathway in T cells.18 Our results suggested that the NF-κB pathway may be involved in retinal repair and regeneration.
Oxidation of PUFA generates the chemically reactive 4-HNE fatty acid fragment, and light exposure increases 4-HNE-protein modification in the retina prior to cell apoptosis.56 4-HNE triggers mitochondrial apoptosis. Mice immunized against the protein adduct carboxyethylpyrrole developed focal RPE pathology and photoreceptor degeneration due to increased immune responses.57 Because 4-HNE is found in the mouse retina, we expected C3aR−/−C5aR−/− mice to have less retinal damage from suppressed immune responses following HNE-MSA immunization. Surprisingly, the impact was much greater in C3aR−/−C5aR−/− than in WT mice. This result suggested that the retinal dysfunction did not result from complement-mediated immune responses because the immunized C3aR−/−C5aR−/− had significantly suppressed immune responses compared to the immunized WT mice.
Our findings indicate a novel role of complement signaling in retinal maintenance. This result will have particular relevance to current therapeutic approaches and clinical trials based upon inhibition of complement activation.11,42–44 Our observations indicate that retinal cells require a certain level of complement activity and raise questions concerning the long-term use of complement inhibition as a treatment for human disease.
Acknowledgments
We thank Bruce Levison for helping to conjugate HNE-MSA, Belinda Willard for analysis of MSA with HNE modification, Jingwei Huang for technical assistance in retinal cell isolation, Renliang Zhang for technical support in mass spectrometry, Gwen Sturgill-Short for Crb1rd8 genotyping, Patsy Nishina for sharing her Crb1rd8 PCR protocol, and Daniel I. Sessler for critical comments in manuscript writing.
Footnotes
Supported by American Health Assistance Foundation Award M2008-063, Foundation Fighting Blindness Center grant; Research to Prevent Blindness; VA Medical Research Service.
Disclosure: M. Yu, None; W. Zou, None; N.S. Peachey, None; T.M. McIntyre, None; J. Liu, None
References
- 1. Friedman DS, O'Colmain BJ, Munoz B, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004; 122: 564– 572. [DOI] [PubMed] [Google Scholar]
- 2. Klein R, Chou CF, Klein BE, Zhang X, Meuer SM, Saaddine JB. Prevalence of age-related macular degeneration in the US population. Arch Ophthalmol. 2011; 129: 75– 80. [DOI] [PubMed] [Google Scholar]
- 3. Edwards AO, Ritter R III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005; 308: 421– 424. [DOI] [PubMed] [Google Scholar]
- 4. Francis PJ, Hamon SC, Ott J, Weleber RG, Klein ML. Polymorphisms in C2, CFB and C3 are associated with progression to advanced age related macular degeneration associated with visual loss. J Med Genet. 2009; 46: 300– 307. [DOI] [PubMed] [Google Scholar]
- 5. Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005; 102: 7227– 7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005; 308: 419– 421. [DOI] [PubMed] [Google Scholar]
- 7. Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005; 308: 385– 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006; 38: 458– 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yates JR, Sepp T, Matharu BK, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007; 357: 553– 561. [DOI] [PubMed] [Google Scholar]
- 10. Gehrs KM, Jackson JR, Brown EN, Allikmets R, Hageman GS. Complement, age-related macular degeneration and a vision of the future. Arch Ophthalmol. 2010; 128: 349– 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khandhadia S, Cipriani V, Yates JR, Lotery AJ. Age-related macular degeneration and the complement system. Immunobiology. 2012; 217: 127– 146. [DOI] [PubMed] [Google Scholar]
- 12. Zipfel PF, Lauer N, Skerka C. The role of complement in AMD. Adv Exp Med Biol. 2010; 703: 9– 24. [DOI] [PubMed] [Google Scholar]
- 13. Amara U, Flierl MA, Rittirsch D, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. 2010; 185: 5628– 5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tornetta MA, Foley JJ, Sarau HM, Ames RS. The mouse anaphylatoxin C3a receptor: molecular cloning, genomic organization, and functional expression. J Immunol. 1997; 158: 5277– 5282. [PubMed] [Google Scholar]
- 15. Gerard NP, Gerard C. The chemotactic receptor for human C5a anaphylatoxin. Nature. 1991; 349: 614– 617. [DOI] [PubMed] [Google Scholar]
- 16. Asgari E, Zhou W, Sacks S. Complement in organ transplantation. Curr Opin Organ Transplant. 2010; 15: 486– 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu J, Lin F, Strainic MG, et al. IFN-gamma and IL-17 production in experimental autoimmune encephalomyelitis depends on local APC-T cell complement production. J Immunol. 2008; 180: 5882– 5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Strainic MG, Liu J, Huang D, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008; 28: 425– 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010; 11: 785– 797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clark SJ, Perveen R, Hakobyan S, et al. Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch's membrane in human retina. J Biol Chem. 2010; 285: 30192– 30202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hecker LA, Edwards AO. Genetic control of complement activation in humans and age related macular degeneration. Adv Exp Med Biol. 2010; 703: 49– 62. [DOI] [PubMed] [Google Scholar]
- 22. Karagianni N, Adamis AP. The case for complement and inflammation in AMD: open questions. Adv Exp Med Biol. 2010; 703: 1– 7. [DOI] [PubMed] [Google Scholar]
- 23. Mukherjee P, Pasinetti GM. The role of complement anaphylatoxin C5a in neurodegeneration: implications in Alzheimer's disease. J Neuroimmunol. 2000; 105: 124– 130. [DOI] [PubMed] [Google Scholar]
- 24. Rahpeymai Y, Hietala MA, Wilhelmsson U, et al. Complement: a novel factor in basal and ischemia-induced neurogenesis. Embo J. 2006; 25: 1364– 1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shinjyo N, Stahlberg A, Dragunow M, Pekny M, Pekna M. Complement-derived anaphylatoxin C3a regulates in vitro differentiation and migration of neural progenitor cells. Stem Cells. 2009; 27: 2824– 2832. [DOI] [PubMed] [Google Scholar]
- 26. Markiewski MM, DeAngelis RA, Strey CW, et al. The regulation of liver cell survival by complement. J Immunol. 2009; 182: 5412– 5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ingersoll SA, Martin CB, Barnum SR, Martin BK. CNS-specific expression of C3a and C5a exacerbate demyelination severity in the cuprizone model. Mol Immunol. 2010; 48: 219– 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langer HF, Chung KJ, Orlova VV, et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood. 2010; 116: 4395– 4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mattapallil MJ, Wawrousek EF, Chan CC, et al. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci. 2012; 53: 2921– 2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Greferath U, Grunert U, Wassle H. Rod bipolar cells in the mammalian retina show protein kinase C-like immunoreactivity. J Comp Neurol. 1990; 301: 433– 442. [DOI] [PubMed] [Google Scholar]
- 31. Oguni M, Setogawa T, Shinohara H, Kato K. Calbindin-D 28 kD and parvalbumin in the horizontal cells of rat retina during development. Curr Eye Res. 1998; 17: 617– 622. [PubMed] [Google Scholar]
- 32. Yu M, Sturgill-Short G, Ganapathy P, Tawfik A, Peachey NS, Smith SB. Age-related changes in visual function in cystathionine-beta-synthase mutant mice, a model of hyperhomocysteinemia. Exp Eye Res. 2012; 96: 124– 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu M, Peachey NS. Attenuation of oscillatory potentials in nob2 mice. Doc Ophthalmol. 2007; 115: 173– 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu J, Peachey NS, Marmorstein AD. Light-evoked responses of the mouse retinal pigment epithelium. J Neurophysiol. 2004; 91: 1134– 1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Uchida K, Stadtman ER. Modification of histidine residues in proteins by reaction with 4-hydroxynonenal. Proc Natl Acad Sci U S A. 1992; 89: 4544– 4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Varela C, Igartua I, De la Rosa EJ, De la Villa P. Functional modifications in rod bipolar cells in a mouse model of retinitis pigmentosa. Vision Res. 2003; 43: 879– 885. [DOI] [PubMed] [Google Scholar]
- 37. Chen M, Muckersie E, Robertson M, Forrester JV, Xu H. Up-regulation of complement factor B in retinal pigment epithelial cells is accompanied by complement activation in the aged retina. Exp Eye Res. 2008; 87: 543– 550. [DOI] [PubMed] [Google Scholar]
- 38. Wenzel A, Grimm C, Samardzija M, Reme CE. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res. 2005; 24: 275– 306. [DOI] [PubMed] [Google Scholar]
- 39. da Silva Cunha KC, Fuly AL, de Araujo EG. A phospholipase A isolated from Lachesis muta snake venom increases the survival of retinal ganglion cells in vitro. Toxicon. 2011; 57: 580– 585. [DOI] [PubMed] [Google Scholar]
- 40. Crabb JW, Miyagi M, Gu X, et al. Drusen proteome analysis. An approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A. 2002; 99: 14682– 14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci U S A. 2006; 103: 2328– 2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Troutbeck R, Al-Qureshi S, Guymer RH. Therapeutic targeting of the complement system in age-related macular degeneration: a review. Clin Experiment Ophthalmol. 2012; 40: 18– 26. [DOI] [PubMed] [Google Scholar]
- 43. Yehoshua Z, Rosenfeld PJ, Albini TA. Current clinical trials in dry AMD and the definition of appropriate clinical outcome measures. Semin Ophthalmol. 2011; 26: 167– 180. [DOI] [PubMed] [Google Scholar]
- 44. Schmitz-Valckenberg S, Mossner A, Fleckenstein M, Wiedemann P, Holz FG. Therapy approaches for geographic atrophy. Ophthalmologe. 2010; 107: 1016– 1019. [DOI] [PubMed] [Google Scholar]
- 45. Wang YP, Hui YN, Wang YS, Wang HY. Detection of C5a receptor expressions on human epiretinal membranes and cultured RPE cells by immunohistochemical staining. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2003; 19: 221– 222. [PubMed] [Google Scholar]
- 46. Vogt SD, Barnum SR, Curcio CA, Read RW. Distribution of complement anaphylatoxin receptors and membrane-bound regulators in normal human retina. Exp Eye Res. 2006; 83: 834– 840. [DOI] [PubMed] [Google Scholar]
- 47. Heredia CD, Huguet J, Cols N, Engel P, Garcia-Calderon PA. Immune complexes in retinitis pigmentosa. Br J Ophthalmol. 1984; 68: 811– 814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bamberg CE, Mackay CR, Lee H, et al. The C5a receptor (C5aR) C5L2 is a modulator of C5aR-mediated signal transduction. J Biol Chem. 2010; 285: 7633– 7644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ward PA. Functions of C5a receptors. J Mol Med (Berl). 2009; 87: 375– 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. O'Barr SA, Caguioa J, Gruol D, et al. Neuronal expression of a functional receptor for the C5a complement activation fragment. J Immunol. 2001; 166: 4154– 4162. [DOI] [PubMed] [Google Scholar]
- 51. Kastl SP, Speidl WS, Kaun C, et al. The complement component C5a induces the expression of plasminogen activator inhibitor-1 in human macrophages via NF-kappaB activation. J Thromb Haemost. 2006; 4: 1790– 1797. [DOI] [PubMed] [Google Scholar]
- 52. Peng Q, Li K, Wang N, et al. Dendritic cell function in allostimulation is modulated by C5aR signaling. J Immunol. 2009; 183: 6058– 6068. [DOI] [PubMed] [Google Scholar]
- 53. Thurman JM, Lenderink AM, Royer PA, et al. C3a is required for the production of CXC chemokines by tubular epithelial cells after renal ischemia/reperfusion. J Immunol. 2007; 178: 1819– 1828. [DOI] [PubMed] [Google Scholar]
- 54. Ahamed J, Venkatesha RT, Thangam EB, Ali H. C3a enhances nerve growth factor-induced NFAT activation and chemokine production in a human mast cell line, HMC-1. J Immunol. 2004; 172: 6961– 6968. [DOI] [PubMed] [Google Scholar]
- 55. Martin CB, Ingersoll SA, Martin BK. Transcriptional control of the C3a receptor gene in glial cells: dependence upon AP-1 but not Ets. Mol Immunol. 2007; 44: 703– 712. [DOI] [PubMed] [Google Scholar]
- 56. Tanito M, Haniu H, Elliott MH, Singh AK, Matsumoto H, Anderson RE. Identification of 4-hydroxynonenal-modified retinal proteins induced by photooxidative stress prior to retinal degeneration. Free Radic Biol Med. 2006; 41: 1847– 1859. [DOI] [PubMed] [Google Scholar]
- 57. Hollyfield JG, Bonilha VL, Rayborn ME, et al. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med. 2008; 14: 194– 198. [DOI] [PMC free article] [PubMed] [Google Scholar]