

Abstract
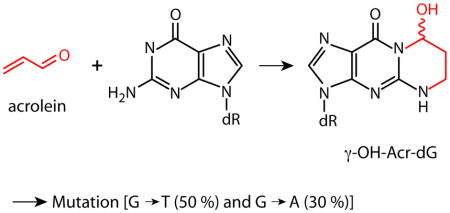
Acrolein (Acr) is a major toxicant in cigarette smoke (CS); it can interact with DNA forming two major adduct isomers: α-OH-Acr-dG and γ-OH-Acr-dG. Previously, we found that the Acr-DNA binding pattern in the human p53 gene coincides with the p53 mutational pattern in CS-related lung cancer; hence we proposed that Acr is a major lung cancer etiological agent (1). This hypothesis has been brought into question with recent work that failed to detect Acr-induced mutations in the pSP189 system (2). To resolve this controversy, we determined the level and the type of Acr-dG formation, and the mutagenicity of Acr-dG adducts in the same pSP189 system. We also mapped the Acr-dG adduct distribution at the nucleotide level, and the Acr-dG induced mutational spectrum in this system. We found that 1) γ-OH-Acr-dG is the major adduct formed in Acr-modified DNA based on the LC-ESI-MS/MS analysis; 2) the mutation frequency is proportional to the extent of Acr-modifications and the majority of which are G:C to T:A and G:C to A:T mutations; and 3) sequences with a run of G’s are the mutational hotspots. Using the UvrABC nuclease incision method to map the Acr-dG distribution in the supF gene sequence, we confirmed that Acr-DNA adducts preferentially form in guanine-rich sequences that are also mutational hotspots. These results reaffirm that Acr-dG adducts are mutagenic, and support our hypothesis that Acr is a major etiological agent for CS and cooking fume-related lung cancer.
Keywords: acrolein, acrolein-DNA adduct, mutation, lung cancer
Introduction
Acrolein (Acr), a ubiquitous environmental contaminant, is abundant in cigarette smoke (CS) and cooking fumes (3). Acr also can be produced endogenously in cells as a byproduct of lipid peroxidation (LPO) (3,4). Acr is one of the major toxic metabolites of the chemotherapeutic agents, cyclophosphamide and ifosfamide (5,6). Accordingly, it has been proposed that Acr is the major factor for inducing secondary human bladder tumors in patients treated with these agents; indeed, Acr has been demonstrated to induce urinary bladder papillomas in rats through intraperitoneal injection (5–8).
Acr is abundant in ambient air and its content in CS is more than 1000 fold greater than that of polycyclic aromatic hydrocarbons (PAHs) (9,10). It has been shown that Acr is mutagenic to human cells (11) and can cause lung injury, however, the role of Acr in lung cancer has not been established (5,7,12).
Acr can induce α- and γ-hydroxy-1, N2-propanodeoxyguanosine (α-OH-Acr-dG and γ-OH-Acr-dG) DNA adducts in cultured mammalian cells (13–15). Using an UvrABC incision procedure in combination with ligation-mediated PCR (LMPCR) to fingerprint the Acr-induced DNA damage, we recently found that the distribution of Acr-DNA damage in the p53 gene in normal human bronchial epithelial cells coincides with the p53 mutational spectrum in CS related lung cancer (1). Furthermore, we also found that Acr treatment induces an inhibitory effect on nucleotide excision repair (NER) (1). Hence, we have proposed that Acr is a major etiological agent for CS related lung cancer (1).
While the mutagenicity of α-OH-Acr-dG is well established, the mutagenicity of γ-OH-Acr-dG has been controversial (16–24). For example, using shuttle vector DNA modified with Acr in vitro, Kawanishi et al.,(24) have presented evidence to demonstrate that Acr-DNA adducts are mutagenic, inducing mainly G to T transversions in human cells. However, using a shuttle vector containing a site specific γ-OH-Acr-dG, Yang et al. (19) have found that γ-OH-Acr-dG is not mutagenic in human cells. Thus, the mutagenicity of γ-OH-Acr-dG in human cells remains to be clarified.
Recently, Pfeifer’s group (2) has reported that they were unable to detect any significant increase of mutations in an Acr-modified pSP189 shuttle vector DNA replicated in human cells, or in the cII transgene in Acr-treated mouse fibroblasts. They therefore concluded that Acr-dG adducts are not mutagenic and are not etiological agents for CS related lung cancer. It should be noted however, the results from Pfeifer’s group (2) as well as the Kawanishi et al.’s (24) were not based on the determination of either the amount or the type of Acr-dG adducts formed in the Acr-modified plasmid DNA and in the target gene used for mutation detection.
Because of the abundance of Acr in CS and environment, its extreme cytotoxicity, and the similarity of Acr-DNA binding spectrum to the p53 mutational spectrum in CS related lung cancer, the question of whether or not Acr-DNA adducts are mutagenic and carcinogenic is extremely important for risk assessment and for environmental regulation policy making; it therefore merits a careful re-evaluation. In this study, we addressed these issues by determining: 1) the types of Acr-DNA adducts formed in Acr-modified pSP189 shuttle vectors by the liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) analysis, and 2) the supF mutations in Acr-modified pSP189 shuttle vectors after replication in cultured human lung cells by DNA sequencing. We also mapped the Acr-dG distribution at the nucleotide level in the supF gene in the Acr-modified pSP189 vectors and determined the mutational spectrum in this supF gene induced by Acr-modifications. Our results affirm that Acr-DNA adducts are mutagenic and support our hypothesis that Acr is a major etiological agent for lung cancer.
Materials and Methods
Shuttle Vector pSP189 DNA Preparation and Acrolein Modifications
The shuttle vector DNA pSP189 was prepared as previously described (25). The DNA was modified with Acr as described prior (1). Briefly, DNA was reacted with different concentrations of Acr in 0.1x TE buffer (10 mM Tris, pH 8.0, and 1 mM EDTA) at 37 °C for 16 h, the unreacted Acr was removed by phenol/diethyl ether extractions and the modified DNA was precipitated by ethanol and then dissolved in 1x TE buffer. The amount of Acr-dG formed in the pSP189 DNA was determined by UvrABC nuclease incision and by LC-ESI-MS/MS method as described below.
UvrABC Nuclease Incision on Acr-Modified DNA and Determination of Acr-DNA Adduct Distribution in the supF Gene
UvrA, UvrB and UvrC proteins are gene products that can function in concert (termed UvrABC nuclease) in incising bulky DNA damage in Escherichia coli cells (26). Methods for the UvrA, UvrB and UvrC protein preparations and the UvrABC nuclease incision conditions were the same as previously described (26). The number of UvrABC incisions on pSP189 supercoiled DNA was calculated based on Poisson distribution equation P(0) = e−n, where P(0) represents the fraction of supercoiled DNA and n represents the number of UvrABC incisions (26). Since we have found that UvrABC nuclease can incise Acr-dG quantitatively and specifically (1), we used this incision method to identify and quantify the Acr-dG distribution in the supF sequence and in the pSP189 plasmid DNA, respectively. To map the Acr-dG distribution in the supF sequence, pSP189 DNA was first digested with restriction enzyme EcoR I, 5′-end-labeled with 32P-γ-ATP and further digested with restriction enzyme BamH I to generate a single 5′-end-32P labeled 131 bp DNA fragment containing supF sequence. To generate a single 5′-end-32P labeled 131 bp DNA fragment containing supF sequence in the opposite strand, pSP189 DNA was first digested with restriction enzyme BamH I, 5′-end-labeled with 32P-γ-ATP and further digested with restriction enzyme EcoR I. These DNA fragments were modified with Acr and then reacted with UvrABC nuclease. The resultant DNAs were separated by electrophoresis in 8% denaturing polyacrylamide gels in parallel with Maxam and Gilbert sequencing reaction products as previously described (26,27).
Acr-DNA Adduct Analysis by LC-ESI-MS/MS Method
The method for determination of Acr-dG adducts by LC-ESI-MS/MS method was the same as previously described (28). Briefly, DNA (0.1 mg) was dissolved in sodium succinate/CaCl2 (10/5 mM) buffer, pH 7.0, with 50 fmol of [13C10,15N5]Acr-dG (α-OH-dG and γ-OH-dG) as internal standards, heated at 100 °C, 30 min and then quenched in an ice bath. The denatured DNA was digested with micrococcal nuclease (75 U) and phosphodiesterase II (0.5 U) at 37 °C, 6 h, and then alkaline phosphatase (150 U) was added and the mixtures were further incubated at 37 °C overnight. The hydrolysate was further purified using solid-phase extraction (SPE) cartridge, the hydrolysate was loaded first, washed with 1 mL H2O, 1 mL 5% CH3OH, and then eluted with 1 mL 70% CH3OH. The eluant was dried and dissolved in 20 μL H2O for LC-ESI-MS/MS analysis.
Determination of Acr-DNA Induced Mutational Frequency and Mutational Spectrum in the supF Gene
Acr mutagenicity was determined as previously described (29). Briefly, lung fibroblasts CCL-202 cells (American Type Culture Collection (ATCC), Manassas, VA), were grown to 70% confluence in 150 mm tissue-culture dishes and transfected with pSP189 DNA modified by different concentrations of Acr as described previously (1). After transfection, medium containing the transfection mixture was removed and cells were cultured in fresh medium for another 72 h. The transfected plasmids were then rescued from the transfected cells by the alkaline lysis method (1,30). DNA was extracted with phenol and chloroform, precipitated with ethanol, dissolved in TE buffer and then treated with the DpnI restriction enzyme (New England Biolabs, Beverly, MA) to remove the unreplicated plasmids, which bear the bacterial adenine methylation pattern. A known quantity of the replicated plasmids was then electroporated into indicator MBM7070 bacteria, which carry a lacZ gene with an amber mutation. The transformed bacteria were plated on LB plates containing ampicillin (50 μg/ml), isopropyl β-D-thiogalactoside (IPTG) (190 μg/ml), and 5-bromo-4-chloro-3-indolyl β-D-galactoside (X-Gal) (0.8 mg/ml). After overnight incubation at 37 °C, white and light blue mutant colonies were picked from the background of blue wild-type colonies and re-streaked. Plasmids were then extracted and purified using the QIApre-spin plasmid kit (Qiagen, Valencia, CA). The sequences of the supF gene of mutant plasmids were determined with the primer 5′-GGC GAC ACG GAA ATG TTG AA-3′.
UV-irradiation Induced Mutation Detection in the supF Gene
For comparison purposes, ultraviolet (UV) light (254 nm)-irradiation induced mutations in pSP189 plasmid DNA were also determined. The mutagenicity of the photoproducts induced by UV irradiation in this shuttle system is well established (31–33). The method of detection is the same as described above except that DNA was irradiated with germicidal lamp (>95% emission at 254 nm) for different time periods and the relative amount of DNA lesion formed in the plasmid DNA was determined by the UvrABC incision assay (26).
Results
The Major Type of Adduct Formed in Acr-Modified DNA Is γ-OH-Acr-dG
Although Acr can interact with DNA to form covalent DNA adducts, this process is slow at neutral pH and ambient temperature. In order to obtain reproducible Acr-DNA modifications, we incubated DNA with different concentrations of Acr in 0.1x TE buffer (pH 8.0) at 37 °C for 16 h. To identify and quantify Acr-DNA adducts formed under these conditions, the modified and mock-modified DNAs were first heat denatured, then digested with micrococcal nucleases, phosphodiesterases and alkaline phosphatases, and the adducted nucleosides in the hydrolysates were further purified by using an SPE cartridge for LC-ESI-MS/MS analysis. It should be noted that Zhang et al. (28) has shown that this method is able to achieve >90 % recovery for both α-OH-Acr-dG and γ-OH-Acr-dG adducts. The results in Table 1 and Figure 2 show that 95% of Acr-dG adducts formed under in vitro modification conditions are γ-OH-Acr-dG and 5% are α-OH-Acr-dG. The levels of both γ-OH-Acr-dG and α-OH-Acr-dG adduct formation are proportional to the Acr concentrations used for DNA modifications.
Table 1.
LC-MS/MS Analysis of Acrolein-Treated pSP189.
| Acrolein concentration (mM) | Acr-dG concentration (μ mol/mol dG)
|
||
|---|---|---|---|
| α-OH-Acr-dG | γ-OH-Acr-dG | Total | |
| 0 | 0.1 | 0.5 | 0.6 |
| 0.2 | 7.6 | 148 | 155 |
| 0.4 | 15.8 | 307 | 323 |
| 1.0 | 41.6 | 848 | 890 |
Figure 2.

Typical chromatograms obtained from LC-ESI-MS/MS analysis of Acr-modified pSP189 DNA. Double-stranded pSP189 DNA modified with different concentrations of Acr were denatured, digested with micrococcal nuclease, phosphodiesterase II, and alkaline phosphatase; the Acr-dG adducts were purified by SPE, and analyzed. Acr-dG adducts (upper trace) and standards of purified α-OH-Acr-dG and γ-OH-Acr-dG adducts (lower trace) was shown in. Note: two (+/−) stereoisomers of α-OH-Acr-dG were well separated while two (+/−) stereoisomers of γ-OH-Acr-dG cochromatographed at the same position (28).
In earlier published work, we demonstrated that the E. coli NER enzyme UvrABC nuclease is able to incise Acr-modified DNAs quantitatively and specifically (1). To determine the amount of Acr-dG adduct formed in the Acr-modified supercoiled pSP189 shuttle vector DNAs were reacted with UvrABC nuclease and the resultant DNAs separated electrophoretically on 1% agarose gels. The number of UvrABC incisions was calculated based on Poisson equation P(0) = e−n assuming the distribution of Acr-dG adducts in the plasmid DNA is random (26). The results in Figure 3A show that the number of UvrABC incisions on Acr-modified pSP189 DNA is proportional to the amount of Acr used for DNA modifications. The number of UvrABC incisions is slightly lower than the amount of Acr-dG adduct per molecule of pSP189 plasmid DNA calculated based on LC-ESI-MS/MS analysis (Figure 3B). These results indicate that the LC-ESI-MS/MS method is more sensitive than the UvrABC incision method for quantifying Acr-dG adducts. Since 95% Acr-dG adducts in the pSP189 DNA are γ-OH-Acr-dG, and more than 50% of these Acr-dG adducts can be detected by UvrABC incision method, these results indicate that γ-OH-Acr-dG adduct is a substrate for the UvrABC nuclease.
Figure 3.

UvrABC incisions on Acr-modified supercoiled pSP189 DNA. The same Acr-modified supercoiled pSP189 DNAs described in Figure 2 were reacted with UvrABC nuclease and the resultant DNAs were separated by electrophoresis in a 1 % agarose and stained with ethidium bromide. A) Photograph of a typical gel. Symbols: OC, open circle and CCC, closed covalent circle. B) Quantitative determination of Acr-dG formation in supercoiled pSP189 DNA. The number of Acr-dG adducts in the pSP189 plasmid DNA modified with different concentrations of Acr was calculated from the number of UvrABC nuclease incisions based on Poisson distribution equation (26) (—▲—). The numbers represent the average of two independent experiment results. For comparison the number of Acr-dG adduct per plasmid was also calculated from results of LC-ESI-MS/MS analysis (—○—).
Determination of the Mutagenicity of the Acr-dG DNA Adducts
Having establishing that γ-OH-Acr-dG is the major DNA adduct formed in the Acr modified pSP189 shuttle vectors, we then determined the mutagenicity of these DNA adducts. Cultured primary human lung fibroblasts (CCL-202) were transfected with a supercoiled form of pSP189 DNAs modified with different concentrations of Acr; the transfected cells were then incubated for 72 h to allow the transfected shuttle vector to replicate. The replicated DNAs were subsequently isolated by Hirt’s method (30) and the nonreplicated pSP189 DNAs were removed by restriction enzyme DpnI digestion. The mutagenicity of Acr-dG adducts was determined by transforming a known quantity of the replicated pSP189 DNA recovered from transfected human cells into E. coli MB7070 indicator cells and the transformed cells were plated onto growth medium containing ampicillin, IPTG and X-Gal. Only the cells carrying pSP189 will form colonies in this growth medium. MB7070 cells carry an amber codon in the lac Z gene; if the cells are transformed with pSP189 shuttle vectors then supF will be able to suppress the nonsense mutation and the phenotype of the transformed cells will be Lac Z+. On the other hand, if the cells are transformed with a mutated supF gene that is no longer able to suppress the nonsense mutation, then the transformed cells will remain phenotypically LacZ−. A total of ~10,000 colonies that resulted from transfection of the pSP189 plasmid DNA modified with each concentration of Acr were counted and the white colonies were picked and confirmed by re-streak onto indicator plates. The mutation frequency was calculated as the ratio of white colonies (which carry mutated supF) to total colonies including blue colonies (which carry functional supF). Results in Figure 4 and Table 2 show that Acr-dG adducts induce mutations and that the mutation level is proportional to the amount of Acr used for DNA modifications; the increase of mutations in pSP189 plasmid DNA modified with 0.1, 0.5, 1 and 2.5 mM Acr is 3.5, 6, 13, and 23 fold, respectively.
Figure 4.

The effect of Acr-modifications and UV-irradiation on the mutation induction in the supF gene. A) The same Acr-modified supercoiled pSP189 DNAs as described in Figures 2 and 3 were used for transfection in human lung fibroblasts CCL-202. The methods of transfection and the supF gene mutation detection are described in Materials and Methods. The mutant plasmids were purified and the mutations in supF gene in these plasmids were confirmed by DNA sequencing. Mutation frequency was calculated based the ratio of the number of white colonies over the number of total colonies. Results represent three independent experiments. In B) mutagenicity of Acr-dG was compared with mutagenicity of UV-irradiation induced DNA lesions. The amount of Acr-dG formed in the pSP189 DNA was calculated based on the UvrABC incision assay (26). For detection of UV-induced mutations, pSP189 DNA was irradiated with different fluences of UV and the mutation detection was the same as for Acr-modification-induced mutation detection. The amount of DNA lesions produced by UV-irradiation was also based on the UvrABC incision assay (26).
Table 2.
Types of Mutations in the supF Gene in Acrolein-Treated pSP189 Plasmids Replicated in Human Normal Lung Fibroblasts CCL-202.
| CCL-202
|
||
|---|---|---|
| Control | Treated* | |
| Single base substition | ||
| G•C to T•A | 6 (43%) | 44 (53%) |
| G•C to A•T | 7 (50%) | 25 (30%) |
| G•C to C•G | 1 (7%) | 10 (12%) |
| A•T to T•A | 0 (0%) | 1 (1%) |
| A•T to C•G | 0 (0%) | 1 (1%) |
| A•T to G•C | 0 (0%) | 2 (2%) |
| Total | 14 | 83 |
| Base substition** | ||
| G•C to T•A | 6 (43%) | 45 (48%) |
| G•C to A•T | 7 (50%) | 27 (29%) |
| G•C to C•G | 1 (7%) | 17 (18%) |
| A•T to T•A | 0 (0%) | 1 (1%) |
| A•T to C•G | 0 (0%) | 1 (1%) |
| A•T to G•C | 0 (0%) | 2 (2%) |
| Total | 14 | 93 |
| Single base substition | 14 (78%) | 83 (92%) |
| Single base deletion | 0 (0%) | 1 (1%) |
| Fragment deletion | 4 (22%) | 2 (2%) |
| Multiple muation*** | 0 (0%) | 3 (3%) |
| Single base insertion | 0 (0%) | 1 (1%) |
| Total | 18 | 90 |
Plasmids were treated with 1 or 2.5 mM Acr.
Including single base substitution, and multiple mutations.
Two or more than two mutations occur in the same plasmid.
The relative mutagenicity of Acr-dG was compared with UV-induced DNA lesions [cyclobutane pyrimidine dimer (CPD) and <6-4>photoproduct], a well-known mutagenic DNA damage. Plasmid pSP189 was irradiated with different doses of UV and the mutations induced were determined by the same method as for Acr modifications. The number of Acr-dG adducts and UV-induced DNA lesions in the pSP189 DNA was determined by UvrABC incision. Results in Figure 4B show that the mutagenicity of Acr-dG adducts, defined as mutant/DNA adduct, is significantly greater than that of UV-induced DNA lesion, defined as mutant/UV-induced DNA lesion (18.3×10−4 mutant/Acr-dG vs. 8.7×10−4 mutant/UV-induced DNA lesion).
Comparison of the Acr-dG Adduct Distribution and Acr-dG Induced Mutation Distribution in the supF gene
The mutations induced by Acr-dG adduct in the pSP189 vectors were sequenced and the types of mutation induced by Acr-dG and the distribution of mutations in the supF gene are presented in Figure 5 and Table 2. From these results, we conclude: 1) base substitutions at G:C positions are the major type of mutations induced by Acr-modifications indicating Acr induces mainly targeted mutations; 2) Acr-dG adducts induce mainly G:C to T:A and G:C to A:T mutations; and 3) 4 out 5 mutation hotspots induced by Acr occur in sequence with runs of 2–5 G’s.
Figure 5.

Acr-modification induced mutational spectrum in the supF gene in pSP189 plasmids replicated in human lung fibroblasts CCL-202. Mutations induced by Acr-modifications are shown in the lower panel and spontaneous mutations are shown in the upper panel. Methods for transfection and supF gene mutation detection are described in Materials and Methods. The mutant plasmids were purified (90 mutant plasmids resulted from plasmid modified with 1 and 2.5 mM Acr and 18 mutants plasmids resulted from unmodified plasmids) and the supF gene in these plasmids was sequenced. The symbols denoted are as follows: X, single base deletions; # ^, double mutations (two mutations occurring in the same plasmid); *, multiple mutation (more than two mutations occurring in the same plasmid); +, single base insertion; —X— fragment deletion.
Two possible mechanisms may account for the mutation hotspots in the supF gene: one, Acr-dG adducts are preferentially formed at these hotspot sequences, and two, that error-prone translesion synthesis occurs more often at these hotspots. Previously we have shown that UvrABC nuclease is able to incise Acr-dG DNA adducts specifically and quantitatively (1). Hence, to test the first possibility, we determined the Acr-dG formation in the supF gene sequence using UvrABC incision method. Acr-modified pSP189 DNAs were first cut with restriction enzyme EcoRI, followed by 5′-32P labeling of the supF gene nontranscribed strand, and then digested with a second restriction enzyme (BamH I). Alternatively, the sample was cut with BamH I first, 5′-32P labeled end of the transcribed strand of the supF gene, and then digested with the second restriction enzyme (EcoR I). In both cases, the resulting fragments were then incubated with UvrABC nucleases and subsequently separated by electrophoresis in a sequencing gel in parallel with Maxam and Gilbert sequencing reaction products (27). The results in Figure 6 show that almost all UvrABC incision bands can be attributed to G residues indicating that Acr-DNA adducts formed mainly at G residues; the results are consistent with the results shown in Figure 2 that Acr-modifications produce Acr-dG adducts. The results in Figure 6 also show that the intensities of UvrABC incision bands are great for sequence runs of G’s. Since UvrABC is able to incise Acr-dG adducts quantitatively, these results indicate that Acr-dG adducts are preferentially formed in sequence runs of G’s.
Figure 6.

The Acr-dG adduct distribution detected by UvrABC incision method in the supF gene sequence. The supF DNA fragments were prepared from pSP189 plasmid DNA modified with different concentrations of Acr, single 5′-end labeled with 32P at the nontranscribed stand (NT strand) or at the transcribed strand (T strand), reacted with UvrABC, and the resultant DNAs were separated by 8 % denatured polyacrylamide gel electrophoresis as described in Materials and Methods. A) represents a typical radiogram resulting from single 5′-end labeled with 32P at the transcribed strand, and B) represents a typical radiogram resulting from single 5′-end labeled with 32P at the nontranscribed strand. Symbols: GA and TC represent Maxam and Gilbert sequencing reaction products, and +/− represents reaction with Acr or UvrABC nuclease. Guanine residue numbers in the sequence are labeled at the left and the corresponding UvrABC incision bands are labeled at the right. Note: we were unable to assign the UvrABC incision bands within the (?) area to corresponding guanine residues in the supF sequence.
In order to determine the role of the sequence context and extent of Acr-dG formation in mutation induction, the relative Acr-dG formation at both strands of the supF gene was plotted against the mutational spectrum in this gene (Figure 7). The results show that mutational hotspots occurred at these preferential sites for Acr-dG formation, leading us to conclude that preferential Acr-dG formation contributes to Acr-induced mutational hotspots in the supF sequence. However, while the formations of Acr-dG at positions 4 and 35 are modest, mutation frequencies at these sites are relatively high, suggesting that either Acr-dG formed at these sites are relatively resistant to repair mechanism and/or adducts formed at these sites allow high frequency of translesion synthesis with a compromised fidelity.
Figure 7.

Comparison of the Acr-dG adduct distribution detected by UvrABC incision method with the relative mutation frequency induced by Acr modification in the supF gene sequence. The relative Acr-dG adduct formation at different sequences of the nontranscribed strands (NT strands) and the transcribed strands (T strands) of the supF gene were determined by the UvrABC incision method as shown in Figure 6. The Acr-modification induced relative mutation frequency at different sequences of the supF gene was calculated from Figure 5.
Discussion
In this study we address three major questions regarding Acr mutagenicity: first and foremost, are the Acr-dG DNA adducts mutagenic? Second, which isomer of Acr-dG is the major adduct formed in Acr-modified DNA? Third, what role does DNA sequence context play in determining Acr-dG adduct formation and mutations?
Based on LC-ESI-MS/MS analysis, we conclude that γ-OH-Acr-dG is the major adduct isomer formed when Acr reacts with DNA at 37 °C in TE buffer at pH 8.0. Our results are thus consistent with those reported by Chung et al (13) for Acr-modified calf thymus DNA.
We found that Acr-modifications induce mutations in the supF gene, and that the level of mutations is proportional to the extent of Acr-DNA adduct formation in the pSP189 plasmid DNA. Furthermore, we found that the mutagenicity of the Acr-dG is higher than the mutagenicityof the UV-induced DNA lesion. Our results are consistent with prior observations by Kawanishi et al (24) that Acr-dG adducts are mutagenic. Since more than 95% of Acr-induced DNA adducts in the pSP189 plasmids are γ-OH-Acr-dG adducts, our results indicate that these isomers are mutagenic.
Finally, our results show that all base substitution mutations in the supF sequence induced by Acr modification are located at guanine residues, and that the Acr-induced mutational spectrum is distinctly different from the spontaneous mutational spectrum. These results strongly suggest that Acr modification induced mutations are due to Acr-dG adduct formation. Using the UvrABC incision method to map the Acr-DNA adduct distribution in the supF sequence we found that most, if not all, Acr-DNA adducts form at deoxyguanosine positions. Furthermore, we found that Acr-dG adducts preferentially form at runs of G sites in the supF sequences; these sites are also the mutational hotspots induced by Acr modifications.
Taken in sum, these results not only strengthen the conclusion that Acr-dG adduct is mutagenic but also suggest that Acr-induced mutations result from translesional synthesis. However, we also found that a relatively low level of Acr-dG formation at sequences, such as positions 4 and 35, nevertheless resulted in a relatively high level of mutations. We hypothesize that Acr-dG adducts formed at these sequences allow more erroneous translesional synthesis and/or are less susceptible to DNA repair mechanisms. It is worth noting that 50% of Acr modification induced mutations are G to T transversions and 30% are G to A transitions; this mutational signature is similar to that found in the p53 gene in the CS related lung cancer (1,34).
It is not clear why Pfeifer laboratory (2) was unable to detect a significant increase of mutation in pSP189 shuttle vectors modified with up to 1 mM Acr in human cells, except to note that these authors did not use direct adduct analysis method to quantify the Acr-dG formation in the pSP189 plasmid DNA used for transfection. However, based on the terminal transferase dependent (TD)-LMPCR signals presented by these authors, the extent of Acr-DNA modifications appears to be 10–20 times less than the number of UVB-DNA modifications that induced 4 to 20 fold increase in mutations compared to unmodified DNA in repair proficient and deficient human fibroblasts, respectively. If this indeed is the case, then this low level of Acr-dG in pSP189 would not be expected to induce mutations significantly above background, even though using the same detection system, our results show that the mutagenicity of the Acr-dG adduct is higher than UV-induced DNA lesions. Hence, it is likely that the extent of Acr-dG formations in the pSP189 DNA used by Pfeifer laboratory (2) was too low to generate significant increase of mutations.
Recently Zhang et al (28) have reported that almost equal amounts of α- and γ-OH-Acr-dG are found in human lung tissues, and that the amount of these adducts is comparable to the total DNA adduct burden detected in cigarette smokers with similar cigarette smoke history. Gupta laboratory (35) recently reported that the majority of diagonal radioactive zones (DRZs) detected in cigarette smokers using 32P post-labeling and 2 dimensional thin layer chromatograph (2D-TLC) method are not PAHs and aromatic amines. Interestingly, these workers also found that DNA modified with acetyl aldehydes and formaldehydes produced the same DRZs as DNA from lung tissues of cigarette smokers. It should be noted that the amount of Acr in CS is more than 1000 fold higher than PAHs (7,8). Among aldehydes found in CS, Acr is one of the most active and is readily taken up by cells (3,36).
We previously found that the Acr-dG distribution in the p53 gene coincides with the p53 mutational spectrum in CS related lung cancer, and that Acr can have an inhibitory effect on DNA nucleotide excision repair (1). Since the results presented here further confirm that Acr-dG adducts are mutagenic and induce G to T and G to A mutations that are similar to the mutational signature found in the p53 gene in the CS related lung cancer, together these results support the hypothesis that Acr is a major etiological agent for CS related lung cancer and cooking fume related lung cancer. It is possible that the susceptibility to Acr-induced adduct formation, and the repair capacity for Acr-dG adducts may play a major role in determining an individual’s susceptibility to CS-induced lung carcinogenesis.
Figure 1.
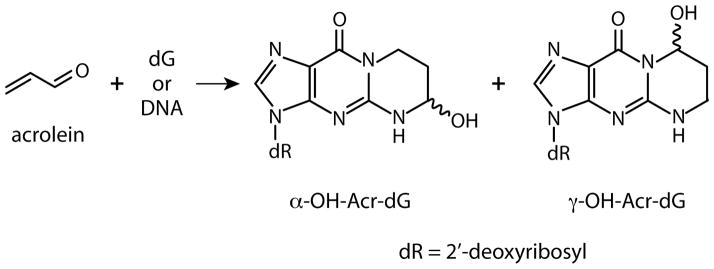
Structures of α-OH-Acr-dG and γ-OH-Acr-dG adducts resulting from reaction of Acr with deoxyguanosine (dG) residues.
Acknowledgments
We thank Drs. Michael Patrick and Cathy Klein for critical review. This study was supported by National Institutes of Health Grants CA114541, ES014641, CA99007 and ES00260.
Abbreviations
- CS
cigarette smoke
- Acr
acrolein
- dG
deoxyguanosine
- α-OH-Acr-dG
α-OH-propanodeoxyguanosine
- γ-OH-Acr-dG
γ-OH-propanodeoxyguanosine
- LC-ESI-MS/MS
liquid chromatography-electrospray ionization-tandem mass spectrometry
- LPO
lipid peroxidation
- PAHs
polycyclic aromatic hydrocarbons
- LMPCR
ligation-mediated polymerase chain reactions
- TD-LMPCR
terminal transferase-dependent LMPCR
- NER
nucleotide excision repair
- TE
tris-EDTA
- IPTG
isopropyl β-D-thiogalactoside
- X-Gal
5-bromo-4-chloro-3-indolyl β-D-galactoside
- CPD
cyclobutane pyrimidine dimer
- DRZ
diagonal radioactive zones
- TLC
thin layer chromatography
References
- 1.Feng Z, Hu W, Hu Y, Tang M-s. Acrolein is a major cigarette-related lung cancer agent: Preferential binding at p53 mutational hotspots and inhibition of DNA repair. Proc Natl Acad Sci USA. 2006;103:15404–15409. doi: 10.1073/pnas.0607031103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim SI, Pfeifer GP, Besaratinia A. Lack of mutagenicity of acrolein-induced DNA adducts in mouse and human cells. Cancer Res. 2007;67:11640–11647. doi: 10.1158/0008-5472.CAN-07-2528. [DOI] [PubMed] [Google Scholar]
- 3.Stevens JF, Maier CS. Acrolein: sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol Nutr Food Res. 2008;52:7–25. doi: 10.1002/mnfr.200700412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uchida K, Kanematsu M, Morimitsu Y, Osawa T, Noguchi N, Niki E. Acrolein is a product of lipid peroxidation reaction. J Biol Chem. 1998;273:16058–16066. doi: 10.1074/jbc.273.26.16058. [DOI] [PubMed] [Google Scholar]
- 5.Ghilarducci DP, Tjeerdema RS. Fate and effects of acrolein. Rev Environ Contam Toxicol. 1995;144:95–146. doi: 10.1007/978-1-4612-2550-8_2. [DOI] [PubMed] [Google Scholar]
- 6.Kehrer JP, Biswal SS. The molecular effects of acrolein. Toxicol Sci. 2000;57:6–15. doi: 10.1093/toxsci/57.1.6. [DOI] [PubMed] [Google Scholar]
- 7.Comes RMM, Eggleton M. Concise International Chemical Assessment Document. 43. World Health Organization; Geneva: 2002. Acrolein. [Google Scholar]
- 8.Cohen SM, Garland EM, John ST, Okamura T, Smith RA. Acrolein initiates rat urinary bladder carcinogenesis. Cancer Res. 1992;52:3577–3581. [PubMed] [Google Scholar]
- 9.Hoffman D, Hect SS. Advances in tobacco carcinogenesis. In: Cooper CS, Grover PL, editors. Handbook of experimental pharmacology. Heidelberg (Germany): Springer-Veriag; 1990. pp. 63–102. [Google Scholar]
- 10.Fujioka K, Shibamoto T. Determination of toxic carbonyl compounds in cigarette smoke. Environ Toxicol. 2006;21:47–54. doi: 10.1002/tox.20153. [DOI] [PubMed] [Google Scholar]
- 11.Curren RD, Yang LL, Conklin PM, Grafstrom RC, Harris CC. Mutagenesis of xeroderma pigmentosum fibroblasts by acrolein. Mutat Res. 1988;209:17–22. doi: 10.1016/0165-7992(88)90104-2. [DOI] [PubMed] [Google Scholar]
- 12.Tungeln LSV, Yi P, Bucci TJ, Samokyszyn VM, Chou MW, Kadlubar FF, Fu PP. Tumorigenicity of chloral hydrate, trichloroacetic acid, trichloroethanol, malondialdehyde, 4-hydroxy-2-nonenal, crotonaldehyde, and acrolein in the B6C3F1 neonatal mouse. Cancer Lett. 2002;185:13–19. doi: 10.1016/s0304-3835(02)00231-8. [DOI] [PubMed] [Google Scholar]
- 13.Chung FL, Young R, Hecht SS. Formation of cyclic 1,N2-propanodeoxyguanosine adducts in DNA upon reaction with acrolein or crotonaldehyde. Cancer Res. 1984;44:990–995. [PubMed] [Google Scholar]
- 14.Nath RG, Chung FL. Detection of exocyclic 1,N2-propanodeoxyguanosine adducts as common DNA lesions in rodents and humans. Proc Natl Acad Sci USA. 1994;91:7491–7495. doi: 10.1073/pnas.91.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nath RG, Ocando JE, Chung FL. Detection of 1,N2-propanodeoxyguanosine adducts as potential endogenous DNA lesions in rodent and human tissues. Cancer Res. 1996;56:452–456. [PubMed] [Google Scholar]
- 16.Yang IY, Johnson F, Grollman AP, Moriya M. Genotoxic mechanism for the major acrolein-derived deoxyguanosine adduct in human cells. Chem Res Toxicol. 2002;15:160–164. doi: 10.1021/tx010123c. [DOI] [PubMed] [Google Scholar]
- 17.Yang IY, Chan G, Miller H, Huang Y, Torres MC, Johnson F, Moriya M. Mutagenesis by acrolein-derived propanodeoxyguanosine adducts in human cells. Biochemistry. 2002;41:13826–13832. doi: 10.1021/bi0264723. [DOI] [PubMed] [Google Scholar]
- 18.Kanuri M, Minko IG, Nechev LV, Harris TM, Harris CM, Lloyd RS. Error prone translesion synthesis past gamma-hydroxypropano deoxyguanosine, the primary acrolein-derived adduct in mammalian cells. J Biol Chem. 2002;277:18257–18265. doi: 10.1074/jbc.M112419200. [DOI] [PubMed] [Google Scholar]
- 19.Yang IY, Hossain M, Miller H, Khullar S, Johnson F, Grollman A, Moriya M. Responses to the major acrolein-derived deoxyguanosine adduct in Escherichia coli. J Biol Chem. 2001;276:9071–9076. doi: 10.1074/jbc.M008918200. [DOI] [PubMed] [Google Scholar]
- 20.VanderVeen LA, Hashim MF, Nechev LV, Harris TM, Harris CM, Marnett LJ. Evaluation of the mutagenic potential of the principal DNA adduct of acrolein. J Biol Chem. 2001;276:9066–9070. doi: 10.1074/jbc.M008900200. [DOI] [PubMed] [Google Scholar]
- 21.Minko IG, Washington MT, Kanuri M, Prakash L, Prakash S, Lloyd RS. Translesion synthesis past acrolein-derived DNA adduct, gamma-hydroxypropanodeoxyguanosine, by yeast and human DNA polymerase eta. J Biol Chem. 2003;278:784–790. doi: 10.1074/jbc.M207774200. [DOI] [PubMed] [Google Scholar]
- 22.Yang IY, Miller H, Wang Z, Frank EG, Ohmori H, Hanaoka F, Moriya M. Mammalian translesion DNA synthesis across an acrolein-derived deoxyguanosine adduct: Participation of pol eta in error-prone synthesis in human cells. J Biol Chem. 2003;278:13989–13994. doi: 10.1074/jbc.M212535200. [DOI] [PubMed] [Google Scholar]
- 23.Sanchez AM, Minko IG, Kurtz AJ, Kanuri M, Moriya M, Lloyd RS. Comparative evaluation of the bioreactivity and mutagenic spectra of acrolein-derived alpha-HOPdG and gamma-HOPdG regioisomeric deoxyguanosine adducts. Chem Res Toxicol. 2003;16:1019–1028. doi: 10.1021/tx034066u. [DOI] [PubMed] [Google Scholar]
- 24.Kawanishi M, Matsuda T, Nakayama A, Takebe H, Matsui S, Yagi T. Molecular analysis of mutations induced by acrolein in human fibroblast cells using supF shuttle vector plasmids. Mutat Res. 1998;417:65–73. doi: 10.1016/s1383-5718(98)00093-x. [DOI] [PubMed] [Google Scholar]
- 25.Canella KA, Seidman MM. Mutation spectra in supF: approaches to elucidating sequence context effects. Mutat Res. 2000;450:61–73. doi: 10.1016/s0027-5107(00)00016-6. [DOI] [PubMed] [Google Scholar]
- 26.Tang M-s. Mapping and quantification of bulky chemicals-induced DNA damage using UvrABC nucleases. In: Pfeifer G, editor. Technologies for Detection DNA Damage and Mutation. Chapter 11. Plenum Press; 1996. pp. 139–152. [Google Scholar]
- 27.Maxam AM, Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Meth Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 28.Zhang S, Villalta PW, Wang M, Hecht SS. Detection and quantitation of acrolein-derived 1,N2-propanodeoxyguanosine adducts in human lung by liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem Res Toxicol. 2007;20:565–571. doi: 10.1021/tx700023z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng Z, Hu W, Amin S, Tang M-s. Mutational spectrum and genotoxicity of the major lipid peroxidation product, trans-4-hydroxy-2-nonenal, induced DNA adducts in nucleotide excision repair-proficient and -deficient human cells. Biochemistry. 2003;42:7848–7854. doi: 10.1021/bi034431g. [DOI] [PubMed] [Google Scholar]
- 30.Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- 31.Moriwaki S, Ray S, Tarone RE, Kraemer KH, Grossman L. The effect of donor age on the processing of UV-damaged DNA by cultured human cells: reduced DNA repair capacity and increased DNA mutability. Mutat Res. 1996;364:117–123. doi: 10.1016/0921-8777(96)00029-8. [DOI] [PubMed] [Google Scholar]
- 32.Moriwaki S, Tarone RE, Kraemer KH. A potential laboratory test for dysplastic nevus syndrome: ultraviolet hypermutability of a shuttle vector plasmid. J Invest Dermatol. 1994;103:7–12. doi: 10.1111/1523-1747.ep12388847. [DOI] [PubMed] [Google Scholar]
- 33.Parris CN, Seidman MM. A signature element distinguishes sibling and independent mutations in a shuttle vector plasmid. Gene. 1992;117:1–5. doi: 10.1016/0378-1119(92)90482-5. [DOI] [PubMed] [Google Scholar]
- 34.International Agency for Research on Cancer. TP53 Mutation Database. ( http://www-p53.iarc,fr)
- 35.Arif JM, Dresler C, Clapper ML, Gairola CG, Srinivasan C, Lubet RA, Gupta RC. Lung DNA adducts detected in human smokers are unrelated to typical polyaromatic carcinogens. Chem Res Toxicol. 2006;19:295–299. doi: 10.1021/tx0502443. [DOI] [PubMed] [Google Scholar]
- 36.Fujioka K, Shibamoto T. Determination of toxic carbonyl compounds in cigarette smoke. Environ Toxicol. 2006;21:47–54. doi: 10.1002/tox.20153. [DOI] [PubMed] [Google Scholar]