

Abstract
Background
Noonan syndrome (NS), a heterogeneous developmental disorder associated with variable clinical expression including short stature, congenital heart defect, unusual pectus deformity and typical facial features, is caused by activating mutations in genes involved in the RAS-MAPK signaling pathway.
Case presentation
Here, we present a clinical and molecular characterization of a small family with Noonan syndrome. Comprehensive mutation analysis of NF1, PTPN11, SOS1, CBL, BRAF, RAF1, SHOC2, MAP2K2, MAP2K1, SPRED1, NRAS, HRAS and KRAS was performed using targeted next-generation sequencing. The result revealed a recurrent mutation in NRAS, c.179G > A (p.G60E), in the index patient. This mutation was inherited from the index patient’s father, who also showed signs of NS.
Conclusions
We describe clinical features in this family and review the literature for genotype-phenotype correlations for NS patients with mutations in NRAS. Neither of affected individuals in this family presented with juvenile myelomonocytic leukemia (JMML), which together with previously published results suggest that the risk for NS individuals with a germline NRAS mutation developing JMML is not different from the proportion seen in other NS cases. Interestingly, 50 % of NS individuals with an NRAS mutation (including our family) present with lentigines and/or Café-au-lait spots. This demonstrates a predisposition to hyperpigmented lesions in NRAS-positive NS individuals. In addition, the affected father in our family presented with a hearing deficit since birth, which together with lentigines are two characteristics of NS with multiple lentigines (previously LEOPARD syndrome), supporting the difficulties in diagnosing individuals with RASopathies correctly. The clinical and genetic heterogeneity observed in RASopathies is a challenge for genetic testing. However, next-generation sequencing technology, which allows screening of a large number of genes simultaneously, will facilitate an early and accurate diagnosis of patients with RASopathies.
Keywords: NRAS, Noonan syndrome, Mutation, RAS-MAPK pathway, RASopathies
Background
Noonan syndrome (NS, OMIM 163950) is a relatively common developmental disorder belonging to the RASopathies, a group of clinically and genetically related syndromes [1, 2]. The molecular cause underlying RASopathies is dysregulation of the RAS-MAPK pathway and 15 different genes affecting this pathway have been associated to RASopathies. Of these 15 genes, eleven have been found to be involved in NS or NS-like conditions, where mutations in PTPN11 are the cause of ~50 % of the cases. The other genes are SOS1 [3, 4], CBL [5–7], BRAF [8], RAF1 [9, 10], SHOC2 [11], MAP2K1 [12], RIT1 [13], NRAS [14], KRAS [15, 16] and RRAS [17].
The main characteristics of NS are short stature, congenital heart defect, unusual pectus deformity and typical facial features, such as hypertelorism, ptosis, down-slanting palpebral fissures, low-set posteriorly rotated ears and a broad forehead. However, NS is a clinically variable disorder and additional associated features often present include neonatal failure to thrive, mild mental retardation, various skin manifestations, bleeding abnormalities and multiple skeletal defects [18, 19].
NRAS is a four-exon gene, encoding the widely expressed small GTPase NRAS, which act as a membrane-associated molecular switch in the RAS-MAPK pathway [14]. To date, only eight unrelated individuals with NS and three NS families have been identified with mutations in NRAS [14, 20–23].
Here, we performed a comprehensive molecular analysis of 13 RASopathy-associated genes; NF1, PTPN11, SOS1, CBL, BRAF, RAF1, SHOC2, MAP2K2, MAP2K1, SPRED1, NRAS, HRAS and KRAS, in a family with NS, which revealed a previously reported mutation in NRAS, c.179G > A (p.G60E). We describe clinical features in this family and review the literature for genotype-phenotype correlations for NS patients with mutations in NRAS.
Case presentation
Clinical investigations and genetic analyses were performed according to the guidelines in the Declaration of Helsinki and approved by the ethical committee of Uppsala University and Gothenburg University, Sweden. Informed consent was obtained from all patients and specific permission was given for photographs.
Case 1
This is a 28-year-old woman, who got the clinical diagnosis of Noonan syndrome (NS) at the age of 4 years because of growth retardation, cardiomyopathy and facial features. She is the only child of non-related parents. The father (Case 2) has facial features of NS, but few additional clinical symptoms. She was born to a mother with diabetes during pregnancy with a birth weight of 4.7 kg (+3 SDS), a length of 52 cm (+1 SDS) and a head circumference of +2 SDS. She also had a large left ventricle, and a systolic murmur, but this disappeared at the age of six years. Postnatally, her growth decelerated and she had feeding difficulties. At 6.5 years of age, her height was 104 cm (−2 SDS) and her weight 18.5 kg (−2 SDS). She had low endogenous growth hormone (GH) secretion defined as “partial GH deficiency”, and started GH therapy within a formal clinical trial (NovoNordisk) from 6.5 years of age. She was treated with GH (dose of 66 μg/kg/day) and responded exceptionally well and treatment was discontinued after two years. However, at 10 years of age, she had her first pubertal signs and GH-treatment was started again using a standard dose of 33 μg/kg/day. At 12.3 years of age, she had menarche. The GH-treatment continued until final height (FH) was reached at the age of 14 years. Her FH is 164.5 cm (−0.45 SDS) and weight of 60 kg (+0.3 SDS). Her psychomotor development is normal, but she has slight problems of attention deficit. She attended regular school and works as an assistant nurse. At the age of 24 years, she has the following features of NS (Fig. 1a): a large skull (62 cm) with a broad forehead, hypertelorism, down slanted palpebral fissures, bilateral ptosis (especially of her left eye), short and broad neck with a low hairline, and low-set ears with broad helices. Her hair is normal. She has two large Café-au-lait spots on her back and >50 freckles (lentigines) all over her body, especially on her back (Fig. 1b) and arms (Fig. 1c).
Fig. 1.

Photograph of the index patient affected by NS with a mutation in NRAS, p.G60E. a Facial features. b The back with multiple lentigines. c The left arm with multiple lentigines
Case 2
This is the 62-year-old father of Case 1. He was clinically diagnosed after Case 1 was diagnosed. He has facial features of NS, but few additional clinical symptoms. Sensorineural hearing impairment was present at birth. His growth pattern was normal, but he had a delayed puberty. His FH is 175.0 cm (−0.4 SDS) and weight 75 kg (±0 SDS). The intellectual development was normal. He followed normal school and university education and worked as a librarian until the age of 55 years, when he had to retire because of tinnitus. At the age of 62 years, he has the following features of NS (Fig. 2a–c): slight macrocephaly (61 cm, +2 SDS), bilateral ptosis, hypertelorism and down-slanting palpebral fissures. He also has curly hair and lentigines on his back. He had a cardiac murmur in childhood that disappeared spontaneously.
Fig. 2.
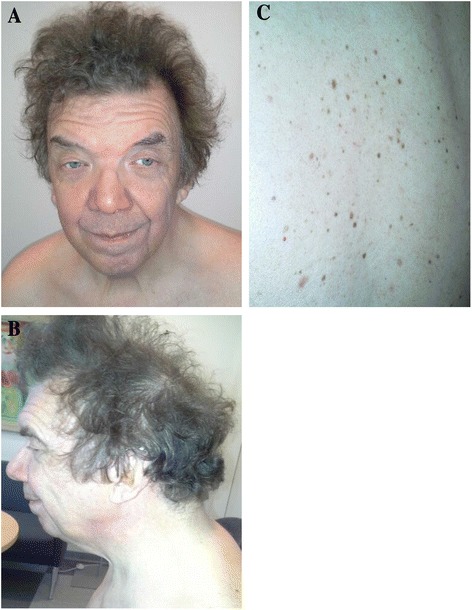
Photograph of the affected father with the same NRAS mutation, p.G60E. a Frontal facial features. b Additional facial features. c The back with multiple lentigines
Methods
Genomic DNA from the index patient and her father was extracted from peripheral blood leukocytes according to standard procedures.
Mutation analysis
The index patient was analyzed for variants in all coding exons and exon-intron boundaries of NF1 (NM_000267.3), PTPN11 (NM_002834), SOS1 (NM_005633), CBL (NM_005188), BRAF (NM_004333), RAF1 (NM_002880), SHOC2 (NM_007373), MAP2K2 (NM_030662), MAP2K1 (NM_002755), SPRED1 (NM_152594), NRAS (NM_002524), HRAS (NM_005343) and KRAS (NM_004985) using Agilent HaloPlex Target Enrichment (Agilent Technologies, Inc., Santa Clara, CA, USA), followed by next-generation sequencing on MiSeq, Illumina (Illumina, Inc., San Diego, CA, USA). Data analysis was performed by NextGENe software v2.3.1 (SoftGenetics, LLC., State College, PA, USA) and BENCHlab NGS (Cartagenia, Inc., Cambridge, MA, USA) [Ekvall et al. Manuscript in preparation].
Variants observed in NRAS exon 2 were verified by bi-directional Sanger sequencing in the index patient and her father. Primer sequences and PCR conditions are available upon request.
Results
DNA sequencing analysis of the index patient (Fig. 1) was performed on 13 RASopathy-associated genes using HaloPlex target enrichment (Agilent) and next-generation sequencing on MiSeq (Illumina). Coverage and read depth of the RASopathy genes in the index patient is shown in Table 1. Targeted bases in region of interest (ROI) with >30X read depth was 100 % for all genes, except NF1 (99.8 %) and SOS1 (99.9 %). No complementary Sanger sequencing was performed. A heterozygous missense mutation, c.179G > A; p.G60E, in exon 2 of NRAS was identified and verified using Sanger sequencing. This mutation was inherited from the father (Fig. 2), who also shows signs of NS. No additional variants of significance were identified in the index patient.
Table 1.
Average read depth and coverage of RASopathy-associated genes in this study
| Gene | Reference sequence | ROIa bases | Exons | Average read depth | Coverage >30X (%) |
|---|---|---|---|---|---|
| BRAF | NM_004333 | 3021 | 18 | 12 637 | 100. 0 |
| CBL | NM_005188 | 3361 | 16 | 12 016 | 100.0 |
| HRAS | NM_005343 | 812 | 6 | 14 291 | 100.0 |
| KRAS | NM_004985 | 768 | 5 | 10 811 | 100.0 |
| MAP2K1 | NM_002755 | 1622 | 11 | 12 525 | 100.0 |
| MAP2K2 | NM_030662 | 1643 | 11 | 11 127 | 100.0 |
| NF1 | NM_000267 | 10737 | 58 | 12 728 | 99.8 |
| NRAS | NM_002524 | 861 | 7 | 16 378 | 100.0 |
| PTPN11 | NM_002834 | 6521 | 16 | 16 262 | 100.0 |
| RAF1 | NM_002880 | 2587 | 16 | 15 985 | 100.0 |
| SHOC2 | NM_007373 | 2069 | 8 | 17 812 | 100.0 |
| SOS1 | NM_005633 | 4922 | 23 | 13 439 | 99.9 |
| SPRED1 | NM_152594 | 1615 | 7 | 10 509 | 100.0 |
a ROI Region of interest
Conclusions
To date, only eight unrelated patients with NS and three NS families have been reported positive for NRAS mutations (p.G13D, p.I24N, p.P24L, p.T50I and p.G60E) [14, 20–23]. In this study, we describe an additional family with NS, where the index patient and her father have c.179G > A (p.G60E). This mutation has been identified in both sporadic and familial NS patients of European origin [14, 23] and is the most common germline mutation in NRAS.
NS patients with NRAS mutations often show a relatively mild phenotype of typical Noonan facial features. A comparison of previously reported NRAS-associated NS cases shows that all of the patients present with typical Noonan facial features (14/14) and 11/13 have macrocephaly or relative macrocephaly, but only half of them display congenital heart defects (7/14). All previously reported patients also show short stature. However, in the family reported here the father’s height was normal, while the daughter had short stature successfully treated with GH. The majority has pterygium or webbing of the neck (10/12). Thorax deformity (pectus excavatum) occurs in 5/14 patients, while easy bruising is less common (3/14). Half of the males show cryptorchidism (6/10) and ophthalmological problems appear in 4/14 patients. Motor delay is common (9/14 patients) and as previously reported, intellectual development is often mildly delayed (6 patients normal and 8 mildly delayed). Keratosis pilaris/hyperkeratosis is less common (4/12 patients) and hair abnormalities occur in about half of the patients. Of note, lentigines are observed in six patients, but leukemia/cancer are rarely seen (1 patient with JMML) (Table 2) [14, 20–23]. Somatic mutations affecting genes in the RAS-MAPK pathway are associated with cancer, and NS and related disorders are known to cause a predisposition to cancer [24]. Somatic mutations in NRAS are involved in the development of hematological malignancies and in a variety of solid tumors (COSMIC database; http://cancer.sanger.ac.uk/). However, germline NRAS mutations differ from most common somatic NRAS mutations associated with malignancies and are less activating in dysregulating intracellular signaling [18].
Table 2.
Clinical features of patients with Noonan syndrome caused by NRAS mutations
| # | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 9 M | 10 | 11 | 12 | 12 F |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | De Filippi et al. [20] | Runtuwene et al. [21] | Denayer et al. [22] | Denayer et al. [22] | Denayer et al. [22] | Cirstea et al. [14] | Cirstea et al. [14] | Cirstea et al. [14] | Cirstea et al. [14] | Cirstea et al. [14] | Kraoua et al. [23] | Kraoua et al. [23] | Present study | Present study |
| NRAS mutation | p.G13D | p.I24N | p.I24N | p.P24L | p.T50I | p.T50I | p.T50I | p.G60E | p.G60E | p.G60E | p.G60E | p.G60E | p.G60E | p.G60E |
| Origin of mutation | de novo | de novo | de novo | Inherited | ND | de novo | de novo | de novo | Inherited | ND | ND (probably inherited) | de novo | Inherited | ND (probably inherited) |
| Paternal age at conception | ND | 26 years | ND | ND | ND | 50 years | 34 years | 31 years | 47 years | 44 years | 45 years | 47 years | 34 years | ND |
| Age at last examination | 3 years | 30 years | 13 years | 19 years | 2.5 years | 14 years | 7 years | 3.3 years | 20 years | 50 years | 24 years | 3 months | 28 years | 62 years |
| Gender | Male | Male | Male | Male | Male | Male | Male | Female | Male | Female | Male | Female | Female | Male |
| Prenatal findings | ND | Polyhydramnios | ND | ND | ND | Nuchal edema, Polyhydramnios | Polyhydramnios | Single umbilical artery | - | - | Polyhydramnios | Pyelectasis | - | - |
| Congenital heart defect | - | - | - | ND | Coarctation aortae, Patent foramen ovale | HCM | PS | Mild HCM, Mitral valve dysplasia, PS | - | - | - | PS | ASD, HCM | Cardiac murmur |
| Rythm disturbance | ND | - | ND | ND | - | SVES | - | - | - | - | - | - | - | - |
| Typical facial features | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Stature | 5–10th centile | Mild short | <3rd centile | 10th–25th centile | 10th–25th centile | 10th centilea | <3rd centile | <3rd centile | >10th centile | 10th centile | 3rd centile | 3rd–10th centile | 50th centilea | 50th centile |
| Macrocephaly | Relative | >90th centile | >97th centile | ND | 25th–50th centile | + | Relative | - | + | + | Relative | Relative | + | Relative |
| Pterygium colli/Webbed neck | - | + | ND | ND | + | + | - | + | + | + | + | + | + | + |
| Thorax deformity | - | Pectus excavatum | Pectus excavatum | ND | Pectus excavatum | + | - | Pectus excavatum | + | + | Mildly depressed thorax | Pectus excavatum | - | - |
| Easy bruising | - | - | ND | + | ND | - | - | - | - | - | + | ND | + | - |
| Cryptorchidism | - | + | + | ND | + | + | + | NA | + | NA | - | NA | NA | - |
| Ophthalmological problems | ND | - | Strabismus, Bilateral keratoconus of the cornea | ND | ND | Myopia | - | - | - | Myopia | - | - | Astigmatis, Myopia, Strabismus | - |
| Motor delay/Muscular hypotonia | - | Motor delay | Mild | ND | ND | Mild | + | + | + | + | Mild | + | - | - |
| Mental development | Normal | Mild learning difficulties | Normal | Learning difficulties | Normal | Normal | Borderline | Speech delay | Normal-borderline | Normal | Speech delay, dyscalculy | NA | ADHD, normal IQ | Normal |
| Keratosis pilaris/Hyperkeratosis | ND | - | ND | ND | ND | Severe | - | + | + | + | ND | - | - | - |
| Hair abnormalities | - | - | ND | ND | ND | Curly hair | Curly hair | Sparse thin hair | Curly hair | - | - | Curly hair | - | Curly hair |
| Lentigines/Café-au-lait spots | + | Some lentigines | - | - | - | - | - | - | - | - | + | + | + | + |
| Leukemia/Cancer | JMML | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Other | - | Oligospermia | - | Inadequate visio-spatial orientation skills, Inguinal hernia, Delayed pubertal development | - | Pes equinovarus | - | Palpebral ptosis | Ichtyosiform eczema, Acanthosis nigricans, Scoliosis | Mother of patient 9 | Palpebral ptosis, Inguinal hernia, Scoliosis | Palpebral ptosis, Unilateral pyelectasis | - | Sensory-neural hearing deficit, Father of patient 12 |
ASD atrial septal defect, HCM hypertrophic cardiomyopathy, JMML juvenile myelomonocytic leukemia, NA not applicable, ND not determined, PS pulmonic stenosis, SVES supraventricular extrasystole
aReceived growth hormone treatment from the age of 8 years, when partial growth hormone deficiency had been noted
In summary, we report an NS family with a p.G60E in NRAS. Neither of affected individuals presented with JMML. Thus, the proportion of JMML observed in NRAS patients (1/12) is comparable with the observed proportion of JMML in NS in general [25].
Interestingly, half of the patients (including affected individuals in our family) presented with lentigines and/or Café-au-lait spots, which is high compared to the prevalence of 3 % for lentigines and 10 % for Café-au-lait spots in the general NS population [26]. Multiple nevi, lentigines and/or Café-au-lait spots are also detectable in one-third of NS individuals with a RAF1 mutation and previous studies have demonstrated a higher prevalence of these features in BRAF-positive NS individuals as well. This suggests that NS individuals with a mutation in NRAS, RAF1 or BRAF have a predisposition to hyperpigmented cutaneous lesions [8, 27]. Of note, the father in our family presented with congenital sensorineural hearing impairment, which together with lentigines are two common features in Noonan syndrome with multiple lentigines (NS-ML, previously LEOPARD syndrome). This demonstrates the wide spectrum of phenotypes within each syndrome as well as the clinical overlap between RASopathies, which makes diagnosis of NS and related disorders challenging.
However, by using the advent of next-generation sequencing technology, which allow for screening of a large number of genes simultaneously, an early and accurate genetic diagnosis of patients with RASopathies will be facilitated.
Consent
We have obtained written informed consent from the patients for publication of this case report and accompanying images. A copy of the written consent is available from the Editor of this journal.
Acknowledgements
This study was supported by grants from the Swedish Research Council, the Borgström foundation, the Sävstaholm foundation and foundations at the Medical Faculty of Uppsala University, Sweden. The authors wish to thank the family for participating in this study.
Abbreviations
- NS
Noonan syndrome
- MAPK
Mitogen-activated protein kinase
- JMML
Juvenile myelomonocytic leukemia
- GH
Growth hormone
- ROI
Region of interest
- COSMIC
Catalogue of somatic mutations in cancer
- ASD
Atrial septal defect
- HCM
Hypertrophic cardiomyopathy
- NA
Not applicable
- ND
Not determined
- PS
Pulmonic stenosis
- SVES
Supraventricular extrasystole
Footnotes
Competing interests
The authors declare that there are no conflicts of interest in connection with this article.
Authors’ contributions
MLB, GA and SE conceived the study and participated in its design. GA, JD, OW, EL and AVH performed clinical examination of the patients. SE, MLB and MW carried out the molecular genetic studies and interpreted the data. SE drafted the manuscript with input from the other co-authors. All authors read and approved the final manuscript.
Contributor Information
Sara Ekvall, Email: sara.ekvall@igp.uu.se.
Maria Wilbe, Email: maria.wilbe@igp.uu.se.
Jovanna Dahlgren, Email: jovanna.dahlgren@gu.se.
Eric Legius, Email: Eric.Legius@uzleuven.be.
Arie van Haeringen, Email: A.van_Haeringen@lumc.nl.
Otto Westphal, Email: otto.westphal@gu.se.
Göran Annerén, Email: goran.anneren@igp.uu.se.
Marie-Louise Bondeson, Phone: +46-18-6115939, Email: marielouise.bondeson@igp.uu.se.
References
- 1.Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013;381(9863):333–342. doi: 10.1016/S0140-6736(12)61023-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tartaglia M, Gelb BD. Disorders of dysregulated signal traffic through the RAS-MAPK pathway: phenotypic spectrum and molecular mechanisms. Ann N Y Acad Sci. 2010;1214:99–121. doi: 10.1111/j.1749-6632.2010.05790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roberts AE, Araki T, Swanson KD, Montgomery KT, Schiripo TA, Joshi VA, Li L, Yassin Y, Tamburino AM, Neel BG, et al. Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat Genet. 2007;39(1):70–74. doi: 10.1038/ng1926. [DOI] [PubMed] [Google Scholar]
- 4.Tartaglia M, Pennacchio LA, Zhao C, Yadav KK, Fodale V, Sarkozy A, Pandit B, Oishi K, Martinelli S, Schackwitz W, et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat Genet. 2007;39(1):75–79. doi: 10.1038/ng1939. [DOI] [PubMed] [Google Scholar]
- 5.Martinelli S, Checquolo S, Consoli F, Stellacci E, Rossi C, Silvano M, Franciosa G, Flex E, Cossu C, De Luca A, et al. Loss of CBL E3-ligase activity in B-lineage childhood acute lymphoblastic leukaemia. Br J Haematol. 2012;159(1):115–119. doi: 10.1111/j.1365-2141.2012.09245.x. [DOI] [PubMed] [Google Scholar]
- 6.Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, Bunin NJ, Bunda S, Finklestein JZ, Sakamoto KM, Gorr TA, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet. 2010;42(9):794–800. doi: 10.1038/ng.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez B, Mechinaud F, Galambrun C, Ben Romdhane N, Isidor B, Philip N, Derain-Court J, Cassinat B, Lachenaud J, Kaltenbach S, et al. Germline mutations of the CBL gene define a new genetic syndrome with predisposition to juvenile myelomonocytic leukaemia. J Med Genet. 2010;47(10):686–691. doi: 10.1136/jmg.2010.076836. [DOI] [PubMed] [Google Scholar]
- 8.Sarkozy A, Carta C, Moretti S, Zampino G, Digilio MC, Pantaleoni F, Scioletti AP, Esposito G, Cordeddu V, Lepri F, et al. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: molecular diversity and associated phenotypic spectrum. Hum Mutat. 2009;30(4):695–702. doi: 10.1002/humu.20955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pandit B, Sarkozy A, Pennacchio LA, Carta C, Oishi K, Martinelli S, Pogna EA, Schackwitz W, Ustaszewska A, Landstrom A, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39(8):1007–1012. doi: 10.1038/ng2073. [DOI] [PubMed] [Google Scholar]
- 10.Razzaque MA, Nishizawa T, Komoike Y, Yagi H, Furutani M, Amo R, Kamisago M, Momma K, Katayama H, Nakagawa M, et al. Germline gain-of-function mutations in RAF1 cause Noonan syndrome. Nat Genet. 2007;39(8):1013–1017. doi: 10.1038/ng2078. [DOI] [PubMed] [Google Scholar]
- 11.Cordeddu V, Di Schiavi E, Pennacchio LA, Ma'ayan A, Sarkozy A, Fodale V, Cecchetti S, Cardinale A, Martin J, Schackwitz W, et al. Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet. 2009;41(9):1022–1026. doi: 10.1038/ng.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nava C, Hanna N, Michot C, Pereira S, Pouvreau N, Niihori T, Aoki Y, Matsubara Y, Arveiler B, Lacombe D, et al. Cardio-facio-cutaneous and Noonan syndromes due to mutations in the RAS/MAPK signalling pathway: genotype-phenotype relationships and overlap with Costello syndrome. J Med Genet. 2007;44(12):763–771. doi: 10.1136/jmg.2007.050450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aoki Y, Niihori T, Banjo T, Okamoto N, Mizuno S, Kurosawa K, Ogata T, Takada F, Yano M, Ando T, et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet. 2013;93(1):173–180. doi: 10.1016/j.ajhg.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cirstea IC, Kutsche K, Dvorsky R, Gremer L, Carta C, Horn D, Roberts AE, Lepri F, Merbitz-Zahradnik T, Konig R, et al. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet. 2010;42(1):27–29. doi: 10.1038/ng.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carta C, Pantaleoni F, Bocchinfuso G, Stella L, Vasta I, Sarkozy A, Digilio C, Palleschi A, Pizzuti A, Grammatico P, et al. Germline missense mutations affecting KRAS Isoform B are associated with a severe Noonan syndrome phenotype. Am J Hum Genet. 2006;79(1):129–135. doi: 10.1086/504394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schubbert S, Zenker M, Rowe SL, Boll S, Klein C, Bollag G, van der Burgt I, Musante L, Kalscheuer V, Wehner LE, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38(3):331–336. doi: 10.1038/ng1748. [DOI] [PubMed] [Google Scholar]
- 17.Flex E, Jaiswal M, Pantaleoni F, Martinelli S, Strullu M, Fansa EK, Caye A, De Luca A, Lepri F, Dvorsky R, et al. Activating mutations in RRAS underlie a phenotype within the RASopathy spectrum and contribute to leukaemogenesis. Hum Mol Genet. 2014;23(16):4315–4327. doi: 10.1093/hmg/ddu148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allanson JE. Noonan syndrome. J Med Genet. 1987;24(1):9–13. doi: 10.1136/jmg.24.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van der Burgt I. Noonan syndrome. Orphanet J Rare Dis. 2007;2:4. doi: 10.1186/1750-1172-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Filippi P, Zecca M, Lisini D, Rosti V, Cagioni C, Carlo-Stella C, Radi O, Veggiotti P, Mastronuzzi A, Acquaviva A, et al. Germ-line mutation of the NRAS gene may be responsible for the development of juvenile myelomonocytic leukaemia. Br J Haematol. 2009;147(5):706–709. doi: 10.1111/j.1365-2141.2009.07894.x. [DOI] [PubMed] [Google Scholar]
- 21.Runtuwene V, van Eekelen M, Overvoorde J, Rehmann H, Yntema HG, Nillesen WM, van Haeringen A, van der Burgt I, Burgering B, den Hertog J. Noonan syndrome gain-of-function mutations in NRAS cause zebrafish gastrulation defects. Dis Model Mech. 2011;4(3):393–399. doi: 10.1242/dmm.007112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denayer E, Peeters H, Sevenants L, Derbent M, Fryns JP, Legius E. NRAS Mutations in Noonan Syndrome. Mol Syndromol. 2012;3(1):34–38. doi: 10.1159/000338467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kraoua L, Journel H, Bonnet P, Amiel J, Pouvreau N, Baumann C, Verloes A, Cave H. Constitutional NRAS mutations are rare among patients with Noonan syndrome or juvenile myelomonocytic leukemia. Am J Med Genet A. 2012;158A(10):2407–2411. doi: 10.1002/ajmg.a.35513. [DOI] [PubMed] [Google Scholar]
- 24.Kratz CP, Franke L, Peters H, Kohlschmidt N, Kazmierczak B, Finckh U, Bier A, Eichhorn B, Blank C, Kraus C, et al. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer. 2015;112(8):1392–1397. doi: 10.1038/bjc.2015.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strullu M, Caye A, Lachenaud J, Cassinat B, Gazal S, Fenneteau O, Pouvreau N, Pereira S, Baumann C, Contet A, et al. Juvenile myelomonocytic leukaemia and Noonan syndrome. J Med Genet. 2014;51(10):689–697. doi: 10.1136/jmedgenet-2014-102611. [DOI] [PubMed] [Google Scholar]
- 26.Noonan Syndrome.[http://www.ncbi.nlm.nih.gov/books/NBK1124/]
- 27.Tartaglia M, Zampino G, Gelb BD. Noonan syndrome: clinical aspects and molecular pathogenesis. Mol Syndromol. 2010;1(1):2–26. doi: 10.1159/000276766. [DOI] [PMC free article] [PubMed] [Google Scholar]