

Abstract
Hypoxic-ischemic (HI) encephalopathy is an important neurological problem of the perinatal period. Little is known of the long-term progression of HI insults or the maladaptive changes that lead to epilepsy. Using rats with unilateral carotid occlusion followed by hypoxia at postnatal day 7, this study provides an initial analysis of the epilepsy caused by a perinatal HI insult with chronic and continuous behavioral monitoring. The histopathology was investigated at postnatal day 30 and later at ≥6 months of age using cresyl violet, Timm, and rapid Golgi staining and immunocytochemistry. The resultant epilepsy showed an increase in seizure frequency over time, with a preponderance for seizure clusters and behavioral features of an ipsilateral cerebral syndrome. In addition to parasagittal infarcts and porencephalic cysts in severe lesions, columnar neuronal death was found with cytomegaly in isolated groups of dysmorphic cortical neurons. Cortical dysgenesis was seen in the form of deep laminar cell loss, microgyri, white matter hypercellularity, and blurring of the white and gray matter junction. Mossy fiber sprouting was not only detected in the atrophied ipsilateral dorsal hippocampus of HI rats with chronic epilepsy, but was also found in comparable grades in spared ipsi- and contralateral ventral hippocampi. The cortical lesions in this animal model show histological similarities with those found in humans after perinatal HI. The occurrence of cortical abnormalities that are associated with epilepsy in humans correlates with the consequent detection of spontaneous recurrent seizures.
Indexing terms: animal model, seizure clustering, cortical dysplasia, microgyri, porencephaly, dual pathology
Hypoxic-ischemic (HI)-induced perinatal encephalopathies are the major cause of acute mortality and chronic neurologic morbidity, such as in epilepsy (Bergamasco et al., 1984; Aso et al., 1990; Lanska et al., 1991; Volpe, 2001; Ramaswamy et al., 2004). The multifactorial causative factors range from prematurity, low birth weight, traumatic/prolonged labor, asphyxia, coagulation abnormalities, severe systemic cardiovascular hypotension, respiratory disorders, and maternal and fetal infections in the perinatal period (Courville, 1959; Golomb et al., 2001; Scher et al., 2002; Cowan et al., 2003; Nelson and Lynch, 2004). Refractory epilepsy is a common sequela (≈30%) in children who survive such insults (Marin-Padilla, 2000a). Autopsy and surgically resected neocortical tissues have revealed dysplasias and abnormal neuronal morphologies, which are thought to underlie the abnormal electrophysiological properties recorded during intraoperative and in vitro slice studies (Palmini et al., 1994; Avoli et al., 1999; Burneo et al., 2004; Najm et al., 2004; Calcagnotto et al., 2005; Matsumoto et al., 2005). A strong association has been found between disorders of cortical development and medically refractory epilepsy in children (Mischel et al., 1995; Spreafico et al., 1998). Animal models of HI provide a means to study the underlying mechanisms of HI-related pathogenesis (Kelly, 2002). Understanding how HI-induced plasticity can lead to chronic recurrent seizures after a latent period is an important question in epilepsy research. Clinically, perinatal insults leading to parasagittal infarcts, ulegyri, polymicrogyri, or massive porencephalic cysts are associated with progressive alterations that have been proposed to be the underlying pathognomic reason for the ensuing epilepsy (Marin-Padilla, 2000b). Epileptiform activity in the tissue adjacent to the lesion following massive loss of neuronal inputs to and from the damaged cortex is associated with hyperinnervation of spared cortical neurons (Neumann-Haefelin and Witte, 2000; Jacobs and Prince, 2005) and may represent at least one form of the plasticity of cortical structures. These clinical and experimental findings suggest that an appropriate animal model with a comparable perinatal insult would show development of seizures and a progressive form of epilepsy with characteristic histopathology in the cortex and hippocampi.
The present study examined this histopathological alteration in relation to the observation of behavioral seizures in rats with an HI insult at postnatal day (P)7. The presence of spontaneous behavioral seizures and the associated seizure semiology were determined by chronic behavioral monitoring. We first sought to determine the apparent latency-to-onset and temporal progression of the epilepsy. We also examined the possible epileptogenic histopathological features of the cortex and hippocampus (i.e., dual pathology). Histological, immunocytochemical, and stereological reconstructive techniques were used to determine the extent and nature of the injury and maladaptive reorganization that had occurred over time, at P30 and at ≥6 months of age. The combined behavioral seizure data and histopathological observations indicate that this is a good model to help understand the process of epileptogenesis following perinatal HI insults in term neonates. Some of these data have been presented in abstract form (Kadam et al., 2003).
MATERIALS AND METHODS
Surgical procedure for HI model
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Colorado State University and the University of Utah School of Medicine. Sprague-Dawley dams with dated P4 litters were obtained from Harlan Laboratories (Houston, TX), single-cage housed, and allowed to acclimate for 2 days before carotid-ligation surgeries were conducted at P7. Cerebral hypoxia was induced using the procedure described by Levine (1960), and modified by Rice et al. (1981). Sprague-Dawley male and female littermates were anesthetized using a 2% isoflurane/oxygen mixture. The ventral midline of the neck was surgically prepared and infused with bupivicane (0.5%, 0.2 mL). A 0.5-cm ventral midline incision was made and the right common carotid artery was exposed and double-ligated unilaterally with 4-0 surgisilk (Vicryl-polyglactin braided). The skin was closed with 4-0 surgisilk. Age-matched sham controls had the carotid artery exposed under the same anesthesia but not ligated. After a 2-hour recovery period the rat pups with the ligated carotid artery were placed in a hypoxia chamber (i.e., a chicken incubator from Lyon’s Electronics, National City, CA) where the temperature and humidity were maintained at 37°C and 90%, respectively. The hypoxic insult was achieved by a high flow rate (6 L/min) of 8%-oxygen balanced with nitrogen through the chamber, adjusted with a flow meter. An oxymeter placed in the chamber recorded consistent 8% oxygen levels in the chamber, which dropped to a minimum of 7.7% in the late phase of the hypoxia when most of the rat pups were hyperventilating. The rat pups were exposed to 8%-oxygen for 2 hours, then allowed to recover in room air and placed with their mother and littermates. Rectal temperatures taken before the pups were placed in the chamber averaged 32–33°C and just after hypoxia averaged 33–34°C. These temperatures were recorded to rule out hyperthermia as a variable during the acute insult.
Histological studies
Histological studies were done on the brains of HI-treated and control rats at P30 (n = 36, 20 HI and 16 control; this group of rats was not monitored) and at ≥6 months of age. Histological studies done in animals that were included in the ≥6 months of age group came either from the present behavioral monitoring study (described below: n = 12, 10 HI and 2 control; n/n = 3/10 HI were epileptic) or a chronic video-electroencephalogram (EEG) study (n = 28, 18 HI and 10 control; n/n = 10/18 HI were epileptic). The video-EEG data investigating the quantitative changes in seizure frequency and pattern, which essentially represents the natural history of the epileptogenesis, will be published separately. Therefore, whether the rats became epileptic (i.e., chronic spontaneous motor seizures were observed) or were categorized as nonepileptic (i.e., no spontaneous motor seizures were detected) following the perinatal HI treatment was only known for the ≥6 month group of rats. After pooling rats that were ≥6 months of age when histology was done (from the two studies mentioned above) the total sample sizes were: HI epileptic rats, n = 11 (n/n = 3/3 from behavioral study; n/n = 8/10 from video-EEG study [2 epileptic rats from the behavioral study were later introduced into the video-EEG study]); HI nonepileptic rats, n = 14 (n/n = 7/7 from behavioral study; n/n = 7/8 from video-EEG study [brain tissue of one HI nonepileptic rat that was found dead in the cage was not processed for histology]); Control, n = 12 (n/n = 2/2 from behavioral study, n/n = 10/10 from video-EEG study). Animals were anesthetized with halothane and then decapitated.
Cresyl violet at P30
The whole brain was rapidly removed and stored in ice-cold physiological solution (composition in mM: 124 NaCl, 3 KCl, 1.3 CaCl2, 26 NaHCO3, 1.3 MgSO4, 1.25 NaH2PO4, 10 glucose equilibrated with 95% O2, 5% CO2, pH 7.2–7.4) before being rapidly transferred into a fixative (4% paraformaldehyde). The fixed brain was then quick-frozen to cut 40-μm coronal sections on a sliding microtome in serial order starting at the genu of the corpus callosum anteriorly to the caudal end of the dorsal hippocampus posteriorly and every third section was mounted serially (20 HI-treated and 16 control). All sections were examined and images obtained with a Zeiss microscope and Microfire software in Neurolucida (Micro-BrightField, Colchester, VT).
Cresyl violet and Timm staining at ≥6 months of age
A modified Timm histological procedure was used to label the zinc-containing axons of the granule cells. The cerebrum was dissected out and prefixed in phosphate-buffered 0.37% Na2S to precipitate the zinc in the mossy fibers. This was followed by immersion fixation in phosphate-buffered 4% paraformaldehyde (pH 7.2). The tissue was then saturated with 30% sucrose and 40-μm coronal sections were cut on a sliding microtome in serial order starting at the genu of the corpus callosum anteriorly to the brain stem caudally. Every third section was serially mounted and processed for cresyl violet staining. An adjacent series of sections starting from the rostral tip of the dorsal hippocampus to the caudal end of the ventral hippocampus was serially mounted and processed for Timm and cresyl violet counterstaining (behavioral study: 10 HI treated and 2 control; chronic video-EEG study: 18 HI treated and 10 control). All slides with the relevant sections were coded and the intensity of the Timm stain in the dentate inner molecular layer was graded. True blinding of slides with respect to treatment (i.e., HI-treated versus control) was not possible due to the obvious lesion, but blinding achieved removal of bias when grading epileptic (n = 11) rats with variable seizure severities versus nonepileptic HI rats (n = 14). Timm stain was graded according to the rating scale of Tauck and Nadler (1985). The data were then grouped according to treatment, ipsilateral versus contralateral, and dorsal versus ventral hippocampus; then average grades with standard errors were obtained. A multiple comparisons (i.e., Bonferroni’s) test was used to assess differences between groups and anatomical locations. Serial reconstruction and solid modeling of lesioned brains was done with Virtual Slice software (MicroBrightField) to create three-dimensional closed and open contours of the ipsilateral lesion and nonlesioned contralateral hemisphere.
Immunocytochemistry
Antisera used for the immunohistochemical experiment were obtained from commercial sources. Neurons were identified using a mouse monoclonal antibody that recognizes the neuron-specific nuclear protein NeuN (Neuronal Nuclei, MAB377, Chemicon International, Temecula, CA; diluted 1:1,000). This antibody was raised against purified cell nuclei from mouse brain and has been shown to recognize 2–3 bands at 46–48 kDa and possibly one at 66 kDa by Western blot (Chemicon International product datasheet; Mullen and Buck, 1992). It reacts with most neuronal cell types throughout the nervous system of mice, rats, and humans and is primarily localized in the nucleus of the neurons with lighter staining in the cytoplasm. In this study the NeuN antibody provided specific labeling of neurons throughout the neocortex, and the staining pattern was very similar to that seen in other studies in which this antibody was used (e.g., Trotter et al., 2005). NeuN immunocytochemistry was done at P30. Coronal sections (40 μm) were cut on a cryostat and stored in cryoprotectant solution. Sections were then incubated in NeuN mouse monoclonal antibody (Chemicon) as primary in blocking solution overnight, followed by secondary incubation with goat antimouse IgG (A-11032, Molecular Probes, Eugene, OR, diluted 1:1,000) conjugated with red-fluorescent Alexa Fluor 594 dye. Sections were mounted serially and coverslipped with Vectashield (Vector Laboratories, La Jolla, CA).
Rapid Golgi staining
HI-treated and control rats (n = 10; ≥6 months of age; 8 HI-treated, 2 control) were anesthetized with halothane and then decapitated. The whole brain was rapidly removed and rinsed in ice-cold phosphate-buffered saline and then immediately submerged into rapid Golgi stain kit solutions (FD Neuro-Technologies, Ellicott City, MD). After 2 weeks brains were quick-frozen and 200-μm coronal sections were cut on a cryostat at −22°C. Sections were mounted on slides and coverslipped with mounting medium from the kit. Photomicrographs of Golgi-stained sections were taken as stacked images 1 μm apart on the Z axis and then fused with ImageJ (National Institutes of Health, Bethesda, MD, http://rsb.info.nih.gov/ij/, 1997–2006) to allow clear visualization of dendritic spines.
All photomicrographs were assembled using Adobe Photoshop (San Jose, CA). Only brightness and contrast were adjusted.
Behavioral monitoring for seizures in post-HI rats
The rat pups that underwent the HI insult at P7 were videotaped 24 hours per day for 1 week every month (i.e., 25% of the time) using a Panasonic WV-BP330 B/W camera connected to three Panasonic videocassette recorders, each with 8-hour videotapes. The rats were housed on a 12-h light/dark cycle from 6 AM to 6 PM. Night recordings were performed with a Kodak 1A filter over a safelight and day recordings were accomplished with diffuse fluorescent light. The recording started at P30 (1 month of age) after the rats were separated from their littermates at P28 and housed two rats per cage. Before this age they were housed in a single cage and tended to sleep and feed huddled together at most times under the mother (dam), which made it difficult if not impossible to detect any possible seizures with a videocamera, especially with litter sizes around 10–15. Once they were weaned the rats were same-sex housed 2 per cage. Seizures were scored using the Racine scale (Class 1–5) to determine the severity of motor seizure activity (Racine, 1972; Ben Ari, 1985): Class 1: Frozen (immobility with open eyes) and or “wet dog shakes” associated with facial convulsions (vibrissae twitching, jaw clonus, blepharospasms); Class 2: Same behaviors as Class 1 with head bobbing; Class 3: Forelimb clonus with a lordotic posture; Class 4: Forelimb clonus continued along with rearing; Class 5: Same behaviors as Class 3 and 4 seizure, but the rats also fall over. Some rats fell to one side first, then showed evidence of forelimb clonus. Ictal events lasted from 20 seconds to 3 minutes. The video analysis was conducted initially in fast-forward mode to detect behavioral postures (i.e., lordosis, straight tail, jumping/running, forelimb clonus, and/or rearing). Once a behavioral posture was observed the tape was reexamined at real-time speed and the seizure scored. After an ictal event was confirmed on real-time video, rat behaviors for a few minutes preceding and following the event were noted. The animal was scored as ‘inactive’ if there was 30 seconds of little to no volitional movement (i.e., animals were considered inactive even if they displayed slight head movement), or following 30 seconds of apparent sleep (i.e., lying still with head down and eyes apparently closed). Spontaneous motor seizures that occurred within 30 seconds after arousal were also considered to be seizures during inactivity. These behaviors were combined into one group based on similar methods described by Hellier and Dudek (1999). Motor seizures were considered to be during activity when the animal had spent more than 30 seconds in apparent volitional movement (e.g., grooming, walking, and eating). Although this definition required that the animal show a specific behavior for more than 30 seconds preceding a seizure, post-hoc analysis revealed that most rats were in a particular behavioral state (i.e., inactive or active) for several minutes prior to the onset of a seizure. This group of rats was monitored for a total of 7 months. With the 1-week-per-month protocol this amounted to 49 days of 24-hour video-monitoring.
All of the “n” in each group for histological findings refer to the number of animals. All statistical values are presented as mean ± SEM. Statistical differences were measured using the paired Student’s t-test, unless indicated otherwise.
RESULTS
Animal behavior
Routine observation and handling of the HI-treated rats did not elicit any abnormal behaviors or disturbances in eating and grooming that differentiated them from their sham-control littermates. Continuous video-behavioral monitoring demonstrated these rats to be equally active as their control counterparts. Although specific tests to examine contralateral limb power were not done, there was no apparent contralateral loss of power associated with the unilateral infarct. Ipsilateral torticollis was noted for two rats that may be attributed to the ipsilateral trauma to neck muscles during the carotid ligation surgery (also reported by Levine, 1960).
Behavioral monitoring for motor seizures in post-HI rats
No behavioral seizures were detected in the first month of video monitoring. Out of 10 HI rats, three were detected to manifest spontaneous behavioral seizures in the second month of monitoring (Fig. 1A), which was substantially shorter than the previously reported latent period to epilepsy after HI insult at P7 (Williams et al., 2004; i.e., 194 ± 43 days compared to 68.3 ± 0.3 days). Given the intermittent nature of the monitoring (i.e., 1 week per month) and the lack of early monitoring the latent period could be shorter than measured here. Overall total counts of seizures (Racine scale; Class 1–5, n = 97; Class 1 and 2 = 14, Class 3 = 18, Class 4 = 7, Class 5 = 58) in the 3 out of 10 HI-treated rats that were detected to be epileptic showed an increase in seizure frequency of behavioral convulsive seizures over time (i.e., data with ≥Class 3 seizures on the Racine scale (Racine, 1972), n = 83, Fig. 1A). Individual counts, although variable, also showed the same trend. The mean seizure rate for the three HI rats with spontaneous recurrent seizures increased from 0.43 ± 0.08 seizures per day at 2 months of age to 1.62 ± 1.09 seizures per day at 7 months of age. The mean seizure rate for the total number of seizures observed over the 7 months of monitoring with the 1-week-per-month monitoring protocol was 0.66 ± 0.35 seizures per day. The 7-day continuous monitoring period detected clusters of seizures. As seen in Figure 1B (1 and 2), the ictal events were detected to cluster within 24-hour periods in spite of low seizure frequencies. Evidence for the preponderance for seizures to occur in clusters was that 83.5% of the seizures occurred in groups of ≥2 and 62.9% occurred in groups of ≥3 seizures within 24-hour time bins (Fig. 1C). The total number of clusters detected per month showed a gradual increase as a function of time (Fig. 1D) with an associated increase in mean number of seizures within each cluster (Fig. 1E). In addition to the three epileptic rats, episodes of Class 1 and 2 behaviors (Racine, 1972) were noted in three more HI-treated rats (i.e., total number of episodes detected = 5). On histological analysis, their ipsilateral cerebral hemispheres were found to have lesions and Timm staining comparable to the HI-treated epileptic rats. All Class 1 and 2 behaviors detected were not included in data analysis because of lack of concurrent EEG data for verification of these episodes as ictal events. Since this protocol did monitor the rats for 25% of the time, and given the propensity of the HI-treated rats to have seizures in clusters separated by days of seizure-free periods (Fig. 1B), it is possible that the 1-week-per-month protocol failed to detect seizures in some of the epileptic rats with a low seizure frequency.
Fig. 1.
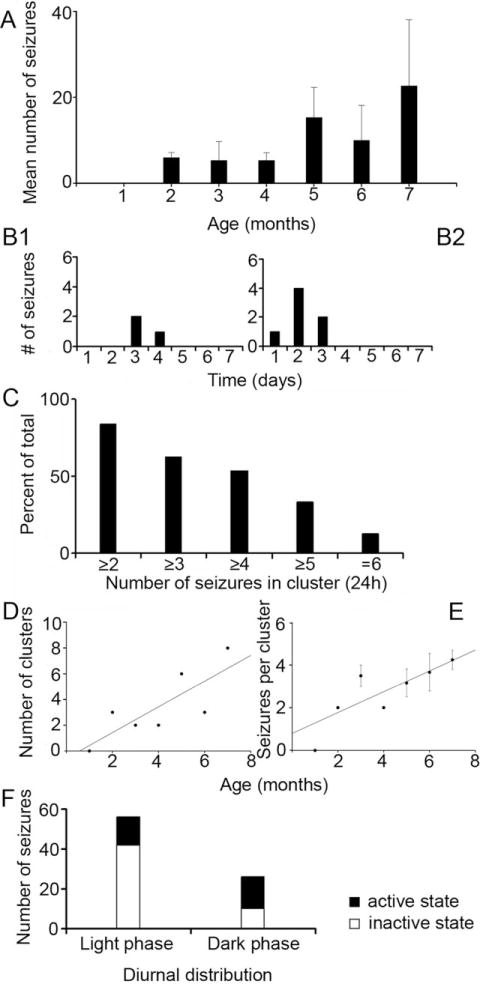
Progressive increases in post-HI seizure frequencies and occurrence of seizure clusters in rats that were detected to be epileptic. A: Mean number of Class 3, 4, and 5 (Racine scale) seizures detected with the 1-week-per-month video-monitoring protocol for rats that were found to have recurrent spontaneous seizures (n/n = 3/10). Histogram shows means counts for three HI-treated rats, which showed an increase in recurrent spontaneous seizures over the 7 consecutive months of video-monitoring. Data from the second and third month (B1 and B2, respectively) of monitoring for an individual epileptic HI-treated rat showed clustering of seizures within 24-hour periods in the 7-day monitoring protocol. The data in B2 show an increase in the number of seizures within the detected cluster compared to the cluster detected in B1. The histogram in C shows the percent of seizures occurring as clusters (within a 24-hour period) as a function of the minimum number of seizures in the cluster over the 7-month period. Thus, about 84% of the seizures were associated with at least one other seizure in a 24-hour period and 54% of the seizures were part of a cluster of four or more seizures. A gradual increase in the number of clusters (≥2 seizures in 24 hours; D) and in the number of seizures within a cluster (normalized to number of clusters, E) was observed as a function of time. F: Diurnal distribution of seizure occurrence and dependence on activity state. A stacked histogram shows the diurnal variation of total ictal events (≥Class 3) detected, presented as seizures occurring during the light phase (n = 52) versus the dark phase (n = 30) that was not statistically significant (P > 0.1). The data are further differentiated by seizures that occurred when the animal was active or inactive within the respective phase of the diurnal cycle. Activity dependence was significant during the light phase (P = 0.001).
Diurnal variation of seizures
The time-of-day of ictal events (≥Class 3, Racine scale seizures) was noted for the HI epileptic rats over the 7-month period of monitoring. The average ratio of the light-phase seizures (6 AM to 6 PM) to the dark-phase seizures was 1.7:1 (Fig. 1F, 63.4% vs. 36.6%). Although overall more seizures occurred during the light phase versus the dark phase, the difference was not significant in a pairwise comparison for individual animals (P > 0.1). Fifty-five of the total 83 seizures (67.1%) occurred while the rats were in an inactive state (see Materials and Methods). A significant number of light-phase seizures (80.8%) occurred at arousal from inactive states (P = 0.001) compared to 46.7% of the dark-phase seizures (Fig. 1F). Similar observations have been noted in the kainate model of temporal lobe epilepsy (Hellier and Dudek, 1999).
Seizure semiology of resultant epilepsy in post HI-treated rats
Seizure semiology consisted of a cerebral syndrome wherein most seizure onsets were associated with preictal versive head movements, predominantly ipsiversive and/or start of tonic-clonic activity first in the left forelimb and post-ictal circular swaying movements. Among other observed behaviors were pre-ictal arousals from sleep, followed by counterclockwise turning (i.e., toward side of lesion). Most ictal events were initiated by a subtle contraversion of the head followed by an exaggerated ipsiversion of the head and torso and a tonic-clonic seizure. Most seizures that were classified as Class 3 (Racine, 1972, i.e., bilateral forelimb clonus) were actually unilateral left-forelimb tonic-clonic seizures (n/n = 16/18). Many Class 5 seizures started as partial seizures (i.e., left forelimb clonus) and then became secondarily generalized (n/n = 11/58). Early seizures in the 2-month-old rats were partial seizures. These motor ictal events had an average duration of 71.7 ± 2.9 seconds. Seizure durations for the motor seizures ranged from 10 seconds to 2 minutes. Every episode was followed by variable periods (seconds to minutes) of a group of behaviors that included volitional walking around the cage with head bobbing, freeze posturing with eyes open, and inactivity with eyes closed. These periods were not included in the quantification of the duration involved in the actual convulsive motor seizure itself. Thus, there was evidence of progression in the post-HI epilepsy by the secondary generalization of partial seizures. Behaviorally, the partial seizures indicated the ipsilateral cortex as the origin.
Cortical histopathology
The HI insult at P7 produced either a distinct ipsilateral infarct lesion or no visibly discernable lesion. The apparent lack of a lesion was noted at both timepoints (i.e., 1 month and ≥6 months) and probably results from an insufficient drop in perfusion pressure during hypoxia due to a sufficiently patent carotid or collateral circulation via the posterior communicating arteries. All of the HI-treated rats that were found to be epileptic had cystic infarcts (n/n = 3/10 from behavioral study and n/n = 10/18 from video-EEG study; total n = 11 [2 epileptic rats from the behavioral study were later introduced into the video-EEG study]). Out of the 14 HI-treated rats that were categorized nonepileptic (n/n = 7/10 from behavioral study and n/n = 7/18 from video-EEG study), 11 had no discernable HI infarcts and were similar to control. The remaining three rats had HI infarcts and were the same three rats (referred to previously in the behavioral monitoring section) that had a few Class 1 and 2 (Racine scale) behaviors but no observed motor (≥Class 3) seizures. As mentioned previously, the 25% monitoring protocol used on these rats may have failed to detect epilepsy in these lesioned rats; however, since no spontaneous motor seizures were detected they were included in the nonepileptic group. This study found similarities between the histopathological features of the neocortical lesions in all of the animals with the infarct lesions and the corresponding clinical findings of the human condition which include porencephalic cysts, microgyri, dislamination of cortical cytoarchitecture, and neuronal dysmorphisms (see Table 1). Specifically, the modified Levine’s method (1960) in postnatal-day-7 rat pups mimicked the infarct defined by the region lying between perfusion territories of the major cerebral arteries (“watershed zone”) seen in human neonates after HI encephalopathy (Fig. 2, 1-month group). The cores of the infarcts were parasagittal in location with variability in its dimensions of middle cerebral artery territory involvement between animals. The cingulate and paracingulate cortex supplied by the anterior cerebral artery was spared (Fig. 2A), possibly because it retained collateral circulation during the unilateral ischemic insult through the anterior communicating artery. This presented the paracingulate cortex as a consistent parainfarct zone to the core of the infarct for both moderate as well as severe lesions. In Figure 2A–C the representative coronal slices from an HI-treated rat with a cystic infarct (1-month group) are shown according to their rostral-to-caudal position in relation to bregma (Paxinos and Watson, 1998). Note that the border between the unaffected cortex of the paracingulate cortex and the core of the infarct formed a distinct border marking the perfusion territories of the anterior and middle cerebral arteries. In contrast, at the ventral border of the infarct broad-spectrum involvement of the middle cerebral artery and possibly poor collateral circulation of the posterior cerebral artery blurred the margin of their respective perfusion territories. Marked atrophy of the ipsilateral hemisphere was seen (Fig. 2E) and the lateral ventricles were enlarged ipsilaterally, at the more caudal coordinates (compare B and C to A in Fig. 2). Enlarged ipsilateral ventricles eventually fused with the infarct cavity to form the porencephalic cyst in severely lesioned rats. The dilatation represented lost brain tissue compensated by cerebrospinal fluid filling rather than a nonobstructive hydrocephaly, because the contralateral ventricle remained unchanged and the aqueducts and the third and fourth ventricles were undilated and similar to controls. The 3D reconstruction of the HI lesion (Fig. 2D,E) was performed with “Virtual Slice” software (Neurolucida) and revealed shrinkage of the ipsilateral hippocampus such that its anterior septal anatomical coordinates were pushed back to a more caudal location (Fig. 2D, green contours) and replaced by the porencephalic cyst (infarct cavitation coalescing with the lateral ventricle to form a large cystic lesion lined by ependymal layer and inclusive of the choroid plexus). This was evident by comparing the rostrocaudal location and dimensions of the ipsi- and contralateral hippocampal tracings in the 3D wireframe and solid-model reconstructions (green, Fig. 2D,E). The porencephalic cyst involved the posterior cerebrum predominantly, representing the watershed territory of all of the three major cerebral arteries. Sparing of the more rostral and temporal border zones may be explained by perfusion of those regions by larger proximal cerebral vessels. The neocortical neuronal cell death was predominantly columnar when viewed at 1 month of age. This occurs due to immaturity of penetrating cerebral vasculature at the age of P7 (Rorke, 1992), which was seen as bands of scar tissue (tissue between end arteriolar branches) separating columns of surviving neurons (Fig. 3A,B). NeuN immunocytochemistry allowed better visualization of these columns of surviving neurons in the core of the infarct (Fig. 3C,D) and revealed that the scar tissue was devoid of NeuN staining. However, the columnar pattern of neuronal loss became less apparent in the ≥6-month group of HI-treated rats. Apparent morphogenesis of the scar tissue over time had occurred such that spared cortex appeared as patches of isolated neurons in contrast to the columns seen in the 1-month group. As the infarct increased in severity it affected the neocortex supplied by the middle cerebral artery in addition to the watershed zone between the middle and anterior cerebral artery territories, thus extending over the parietal cortical convexities. Sometimes the ipsilateral gray matter was reduced to a thin membrane comprised of infarcted tissue and the piamater, coalesced to form the outer wall of the porencephalic cyst. The deep gray matter showed moderate-to-severe striatal and thalamic atrophy in the ipsilateral hemisphere in the HI-treated epileptic rats. Mineralization was commonly seen within the deep gray matter as chalky spots in the posterior lateral thalamic nuclei in unprocessed sections from HI-treated epileptic rats (1-month and ≥6-month groups). After cresyl violet staining these spots became a dark violet hue (asterisk in C in Fig. 2). Calcifications of ischemic lesions in brains of children are well documented (Ansari et al., 1990) and similar mineralization noted in the HI-treated brains with infarct lesions represents one more similarity this animal model was found to have to the clinical condition. Histology done with cresyl violet ≥6 months after the insult elicited a loss of the columnar pattern of neuronal loss in the neocortex seen at ≈1 month of age. This indicates that the HI-induced infarct underwent morphological changes over time that at 1 month was representative of microvascular perfusion failures during the perinatal insult, but at 6 months that distinction was lost.
TABLE 1.
Summary of Histopathological Features of Post-perinatal Hypoxia-ischemia (HI) in the Rodent Model Compared to the Reported Clinical Conditions
| Neuropathology | Model of perinatal HI in Sprague-Dawley rats | Clinical condition |
|---|---|---|
| Porencephalic cyst | Ipsilateral to ligation | HIE, Neonatal stroke |
| Parasagittal cerebral injury | Ipsilateral to ligation | HIE |
| Microgyri | Ipsilateral to ligation | HIE, Polymicrogyria |
| Deep cortical neuronal loss | Ipsilateral to ligation | HIE |
| Dyslamination of cortical cytoarchitecture | Ipsilateral to ligation | HIE |
| Dysplastic neurons | Bilateral | HIE |
| White matter hypercellularity | Bilateral | Focal cortical dysplasia |
| Blurring of white-gray matter junction | Bilateral | HIE, Focal cortical dysplasia |
| Calcification of ischemic lesions | Ipsilateral to ligation | HIE |
| Spastic plegia | Absent | Common |
| Dual pathology (hippocampus and neocortex) | Bilateral | HIE, TLE |
| Epilepsy | Post-lesion epilepsy | HIE,* Neonatal stroke,** Cortical dysplasia*** |
HIE, hypoxic-ischemic encephalopathy; TLE, temporal lobe epilepsy.
~30% (Marin-Padilla, 2000a);
~67% (Koelfen et al., 1995);
very common (Alonso-Nanclares et al., 2005).
Fig. 2.
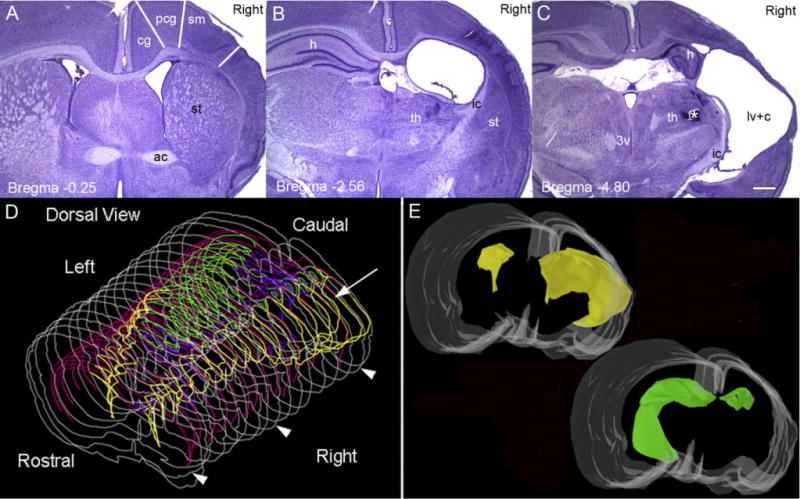
Location of cystic infarct. A–C: Cresyl-violet-stained coronal sections from a P30 rat following the HI insult at P7. A1, 2, and 3 are images of 40-μm coronal sections shown in reference to bregma and demonstrate the core of the infarct in the parasagittal zone of the right hemisphere. In the section through the anterior commissure (ac) at −0.25 bregma (A), the lateral ventricles (lv) are seen bilaterally and are comparable in size. Striatal (st) atrophy is evident in right (ipsilateral) hemisphere. Columnar bands of cell death are seen predominantly in the sensorimotor cortex (sm) with a relatively preserved cingulate (cg) and paracingulate cortex (pcg). The sm and pcg cortices are both parasagittal in location with the pcg cortex being more medial in location (Paxinos and Watson, 1998). In the section through dorsal hippocampus (h) at −2.56 bregma (B), right thalamic (th) atrophy was especially marked in the dorsolateral nuclei. The right dorsal hippocampus is conspicuously absent in this section, and replaced with an enlarged lateral ventricle lined with an ependymal layer and a visible choroid plexus. The section through the posterior thalamus at −4.80 bregma (C) shows mineralized lesions in the region of the ventral posteromedial thalamic nucleus (asterisk); the cystic infarct (c) has coalesced with the lateral ventricle to form a porencephalic cyst and the third ventricle is undilated. D,E: Serial reconstruction using Virtual Slice software in Neurolucida representing the rostrocaudal dimensions of the cystic infarct. The 3D dorsal view of the wireframe generated by serial reconstruction is shown in D and anatomical structures are color coordinated with 3D reconstructions in E to assist visualization within the wireframe. The cystic infarct fused with the lateral ventricle (yellow) is seen in the right cerebral hemisphere predominantly in the middle and posterior cerebral artery vascular territory (arrow). Arrowheads represent the sections shown in A, B, and C along the rostrocaudal axis, respectively. A solid model of the reconstructed lesioned brain in the same orientation as in D is shown in E. A basofrontal view allows better visualization of the asymmetry of the lateral ventricles (yellow), with the ipsilateral lateral ventricle coalescing with the infarct cyst to form the porencephalic cyst (as seen in D). The severely atrophied dorsal hippocampus (green, also seen in wireframes in D) in the ipsilateral hemisphere is seen in comparison to the contralateral hippocampus. Color code for D and E: white, section contour / cerebral surface; pink, corpus callosum; yellow, porencephalic cyst merged with lateral ventricle; green, hippocampus; purple, third and fourth ventricles. Scale bar = 500 μm in C (applies to A,B).
Fig. 3.
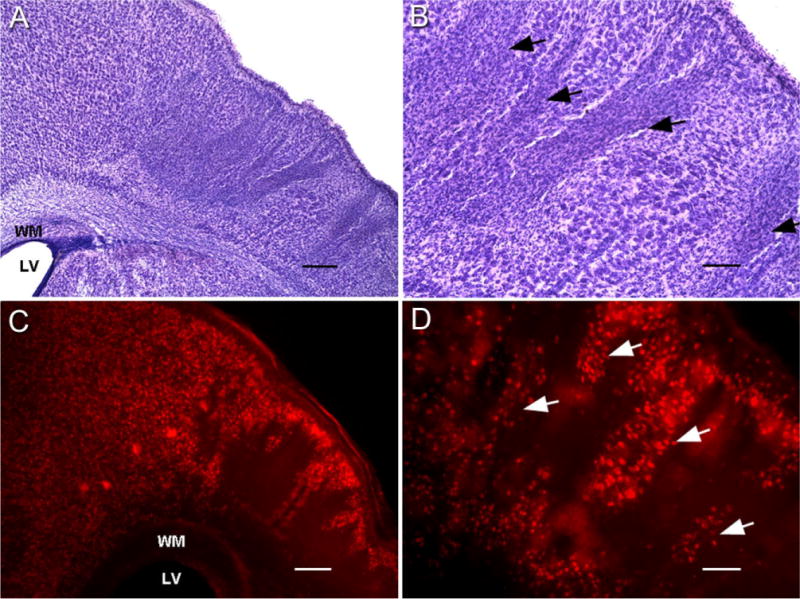
Columnar cell death visualized with cresyl violet and NeuN immunocytochemistry at P30. Coronal section of cresyl-violet-stained sensorimotor cortex shows the core of the infarct with columnar cell death at lower magnification (A). At higher magnification, columns of surviving gray matter separate bands of scarred neocortex (arrows, B). NeuN-stained 40-μm coronal sections demonstrate the marked dorsal border of the infarct that lies between the spared paracingulate cortex (anterior cerebral artery perfusion) and the “watershed zone” delineated by a massive loss of neurons in the parasagittal neocortex (C). Magnified view (D) shows columns of NeuN-positive surviving neurons (arrows) separated by bands of tissue devoid of any stained cells (corresponding to the bands of scar tissue in the cresyl-violet-stained sections in A,B). WM, white matter; LV, lateral ventricle. Scale bars = 250 μm in A,B; 100 μm in C,D.
Acquired microgyri
Cortical dysplasias occurred in the form of deep laminar cell loss (Fig. 4A,B; seen at 1 month of age), microgyri (Fig. 4C,D, at ≥6 months of age; n/n = 5/11 epileptic rats; 0/14 HI-treated nonepileptic rats), and cortical layering abnormalities in the affected neocortex of all HI-treated rats with lesions (Fig. 4F, seen at 1 and ≥6 months). They were detected in the spared frontal and sensorimotor neocortices of the HI-treated rats with infarcts. In the more severe forms of infarcts, these cortical malformations overlaid the porencephalic cysts. These findings correlate with the concomitant human neuropathogy (Courville, 1958; Levine et al., 1974) where porencephaly and microgyri were both found after in utero perfusion failures. These observations were summarized as microgyri not only being manifestations of the same pathogenic event, but also a milder form of the abnormality that produced the porencephaly in the severe form. Histology performed at 1 month of age in HI-treated rats showed the presence of deep-laminar loss of cortical neurons with possible signs of a microgyrus in its early stages of formation (Fig. 4A,B, arrows show indentation at cortical surface above the laminar cell loss, but a gyrus has not yet formed). Acquired microgyri were detected in 5 out of 11 of the epileptic HI-treated rats (≥6-month group) in the ipsilateral neocortex (Fig. 4C,D) and in none of the 1-month group of HI-treated rats with infarct lesions. These microgyri were characterized by marked disruption of cortical cytoarchitecture and deep-laminar cell loss, flanked by clusters of misaligned hypertrophic neurons representing areas of surviving gray matter that seemed isolated within scar tissue (Fig. 4E). Comparing the histological sections from the early and late timepoints in HI-treated rats suggested that there was progressive postinjury evolution of the microgyric formation in this model, which along with the increasing dysplastic features may also involve increased epileptogenesis. In addition to the presence of the microgyri, other forms of dysplasia included cortical warts (Fig. 4F) and obliteration or marked thinning of molecular layer I overlying the affected neocortex seen at both time-points. All of the rats that were detected to be epileptic had a HI-induced infarct lesion. This indicates that the epilepsy that ensues after the perinatal insult in the model is a postlesion epilepsy. It is likely that massive laminar and focal cell loss within the infarcted tissue may induce maladaptive reorganization in the surviving neurons to compensate for the loss of intra- and interlaminar connections that is epileptogenic.
Fig. 4.
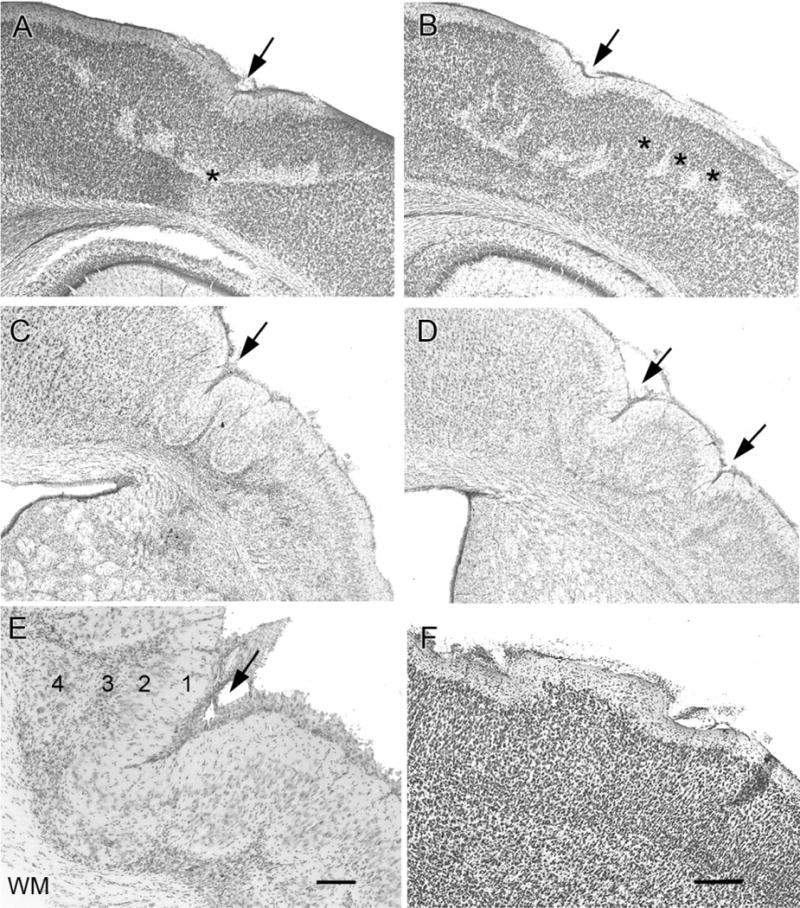
Dislamination of parasagittal cortex. Coronal cresyl-violet-stained sections showing deep laminar cell loss at P30 (A,B) and microgyri in the chronic (≥6 month age) group of HI-treated rats (C–E). A: Early stages of the possible progressive collapse (arrow) of the neocortex to a microgyrus at the cortical surface overlying the deep laminar cell loss (asterisk). The pattern of patchy deep laminar cell loss coincides with the columnar predilection to cell death that was commonly observed (B), which is likely linked to immaturity of the cerebral vasculature (three asterisks) at the time of the HI. C: The presence of acquired microgyral-like structures (arrows) in tissue fixed from a chronically epileptic rat (age >6 months). D: Two such microgyri are seen, compared to the one microgyrus seen in C. The rat that generated the section in D also had twice as many seizures compared to the rat with the section in C, as determined with a continuous and chronic recording of electroencephalograms. E: A microgyrus from an epileptic rat is shown at higher magnification to demonstrate the collapse of the neocortex into a four-layered structure. Layer 1 is contiguous with the molecular layer of the undamaged cortex and fuses to form a microsulcus (arrow). Layer 2 is contiguous with layers II and III of undamaged cortex, but is unlaminated. Layer 3 is the scar that is the remnant of the original injury and hence also called lamina dissecans. Layer 4 is contiguous with layer 5 of intact cortex. F: Cortical surface irregularities were accompanied by underlying cell death, and could be compared to cortical warts where layer II/III neurons protrude along with the molecular layer above a normally lissencephalic brain. WM, white matter. Scale bars = 250 μm in F (applies to A–D); 100 μm in E.
The spared ipsilateral sensorimotor cortex in HI-treated epileptic rats showed cortical dysplastic features (Fig. 5B,E compared to control in 5A,D; n/n = 11/11, ≥6-month group) in the form of cytomegalic neurons. Cytomegaly was seen in two groups of cresyl-violet-stained cells, one group was darkly stained cells with larger cell bodies that retained their pyramidal structure (Fig. 5E,F), and the other group contained lightly stained cells with large rounded or misshapen cell bodies (Fig. 5, see insets in 5E,F). Although the hypertrophied pyramidal cells were predominantly seen in the deep laminar layers of the motor cortices, they were larger in size compared to those in control brains (Fig. 5D). Similar dysplastic cells were also seen in the contralateral sensorimotor cortex (Fig. 5C,F). Comparable findings have been described in cortical tissue resected from patients diagnosed with intractable epilepsy secondary to cortical dysplasia (Ferrer et al., 1992; Alonso-Nanclares et al., 2004). Cytomegalic neurons and large cells with atypical morphology have been suggested to play an important role in the generation of epileptic activity due to their abnormal electrophysiological properties (Kerfoot et al., 1999; Cepeda et al., 2003).
Fig. 5.
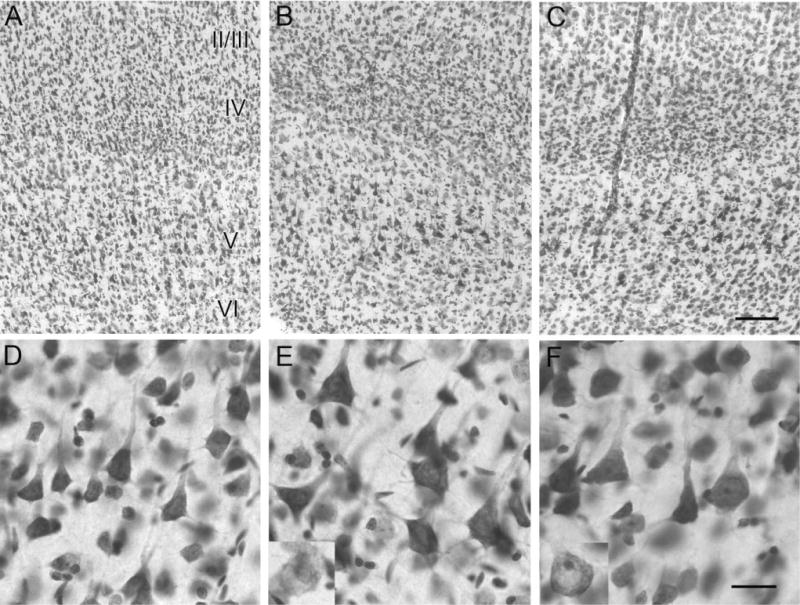
Cresyl-violet-stained coronal sections showing cytomegalic neurons in deep laminar layers of the spared paracingulate neocortex. All panels show paracingulate neocortex with layer II/III at the top of the panel and layer VI at the base (A–C; ≈3.3 mm caudal to bregma [Paxinos and Watson, 1998]) with the lower panels showing the respective deeper layers at higher magnification (D–F). Compared to control (A), ipsi- (B), and contralateral (C) neocortex showed the presence of cortical dysplasia in the form of hypertrophic, strongly Nissl-stained pyramidal neurons and lightly stained misshapen and/or cytomegalic neurons (insets in E and F) in the deep laminar layers (compare D [control] to E and F [ipsi- and contralateral, respectively]). Scale bars =100 μm in C (applies to top row); 50 μm in F (applies to bottom row).
White matter/layer VI junction
In the control sections the corpus callosal white matter tract formed a distinct border with layer VI of the sensorimotor cortex (Fig. 6A). This junction of white matter and layer VI underlying the lesion in the HI-treated epileptic rat was blurred. Hypercellularity in the form of an increased number of Nissl-stained cells were seen in the white matter tract of the corpus callosum underlying the injured cortex in the ipsi- and also the contralateral cortex (compare Fig. 6C,E to 6A; n/n =11/11, ≥6-month group) of epileptic rats. Future studies will be needed to investigate the cell lineage of the hypercellularity seen in these epileptic animals. Although postinjury glial proliferation originating from the neural progenitor cells of the subventricular zone is a distinct possibility, it is only speculative at this juncture. Studies conducted in Wistar rats using a similar model of perinatal HI (Hayashi et al., 2005) have shown an increase in endogenous stem cell proliferation in the subventricular zone after HI. The proliferating stem cells identified by bromodeoxyuridine incorporation migrated toward the injured area and immunohistochemically colabeled for astroglial, oligodendroglial progenitor, and microglial cell markers. White matter hypercellularity has been noted in resected human samples of cortical dysplasia (Meencke and Janz, 1985; Armstrong and Mizrahi, 1998; Andres et al., 2005) but remains poorly understood. In this study closer microscopic examination showed blurring of the white matter and layer VI junction to result from patchy neuronal loss at the junction causing the loss of definition of the overlying neocortex from the hypercellular white matter (compare Fig. 6D,F to 6A). Pathogenesis of the cellularity of white matter tissue and the relationship to the pathogenesis of epilepsy requires study both in humans and animal models.
Fig. 6.
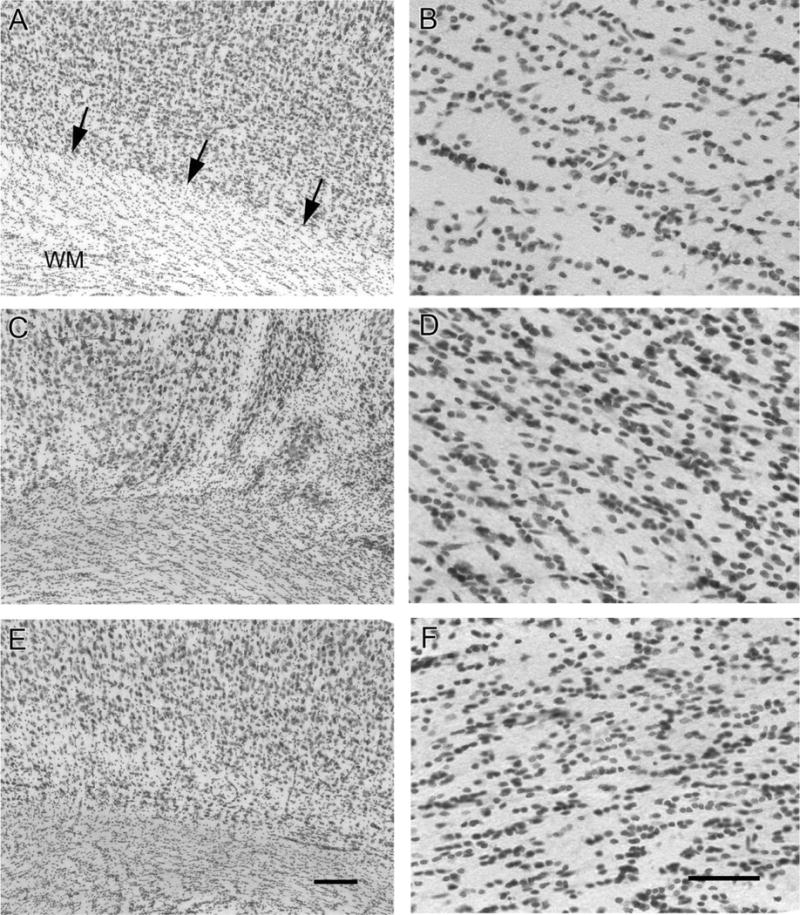
Layer VI /white matter (WM) junction dysplasia and WM hypercellularity. The left panels show the layer VI/WM junction of the parasagittal sensorimotor neocortices and the right panels are magnified views of the WM underlying the sensorimotor cortices at the corresponding locations of the left panels. The normal distinct layer VI/WM junction (arrows) is seen in a control rat (A), while blurring of this junction was noted in the ipsilateral (C) and contralateral (E) sensorimotor neocortex of an HI-treated epileptic rat. Note the hypercellularity of the WM underlying the HI-treated ipsi- and contralateral cortex (D,F, respectively) compared to control (B). Scale bars = 100 μm in E (applies to A,C); 25 μm in F (applies to B,D).
Rapid Golgi
Golgi staining of HI-treated rat brains with infarct-lesions (n = 6; n/n = 6/8), using the Rapid-Golgi technique revealed marked loss of laminarity leading to neuronal disorganization in the sensorimotor cortices. Dysmorphic features dominated these sections (Figs. 7, 8). The Golgi technique stains few neurons randomly, which is why it was possible to study individual neuron morphology in its entirety. In spite of the random staining pattern the laminar cytoarchitecture of the neocortex was clearly visible in the control cortex (Fig. 7A). The spared paracingulate cortex that appeared laminar in cresyl-violet-stained sections (Fig. 5A–C) revealed dysmorphisms in the form of maloriented clumps of neurons forming dense dendritic networks (n = 6; Fig. 7B). Similar loss of laminarity and neuronal clumping has been reported by Marin-Padilla et al. (2003) in the irradiation-induced cortical dysplasia model with Golgi staining. For the initial analysis conducted in this study the morphology of neurons in the ipsilateral spared sensorimotor cortices was studied because of their proximity to the core of the infarct. Previous studies with cortically induced lesions have shown that neurons in seemingly unaffected columns next to the lesion, rather than neurons within lesions, demonstrated epileptiform plasticity (see Introduction). Cytomegalic (25–30 μm) pyramidal neurons in the ipsilateral spared parasagittal cortex were noted to have dysmorphic dendritic branching and a multitude of pedunculate and mushroom spines on their dendrites (compare Fig. 7D to C). These morphological changes were noted in the spared ipsilateral parasagittal cortices of all HI-treated brains with infarct lesions (n/n = 6/8). Neurolucida tracings of stained sections revealed the 1) ipsilateral hemispherical atrophy (Fig. 8, compare B to A); 2) consistent parasagittal location of microgyri for the model (Fig. 8D); and 3) pyramidal neurons with transformed morphologies (Fig. 8, compare F to E). These changes included altered locations, increase in cell body size, distorted and sometimes increased branching of apical and basal dendrites. These findings demonstrate that spared ipsilateral parasagittal cortices forming the medial border of the ischemic infarct undergo postinjury plasticity in the form of morphological changes in the structure of the spared neurons. These initial observations in Golgi-stained sections will require further quantification and functional studies to investigate the significance of these changes consistently seen in the spared cortices of the brains with infarct lesions for this model.
Fig. 7.
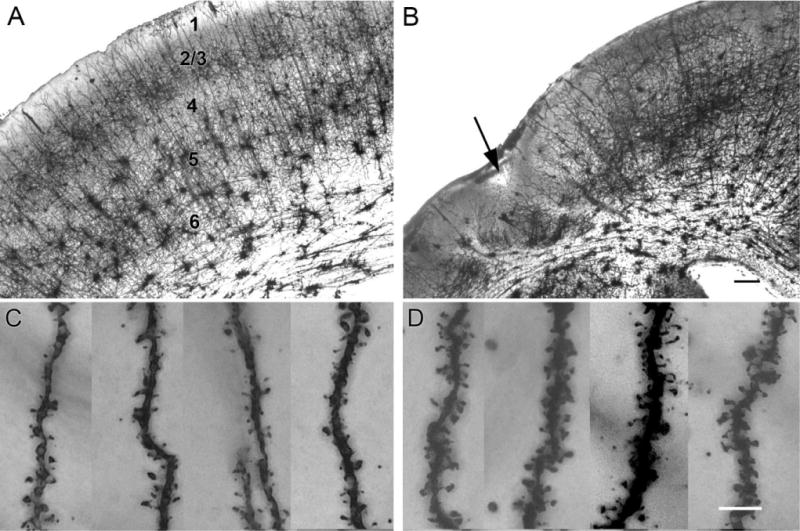
Dislamination of spared paracingulate cortex as revealed by the rapid-Golgi stain. The upper panels show low-magnification images of control (A) and HI-treated neocortex (B), and the lower panels are high-magnification images showing the corresponding apical dendrites and spines. A: Golgi-stained paracingulate neocortex from a control rat shows isocortical cytoarchitecture, starting outside with the molecular layer I, followed by the outer granular and pyramidal layer (II/III), the inner granular layer IV, the inner pyramidal layer V, and mutiforme layer VI followed by corpus callosal white matter tract. B: The ipsilateral Golgi-stained section from an HI-treated brain with a parasagittal microgyrus (arrow). Note the lack of Golgi staining in the bands of scar tissue around the microgyrus and the loss of laminar cytoarchitecture in the spared paracingulate cortex, which appeared comparatively laminar in cresyl-violet-stained sections. Pedunculate spines were seen on apical dendrites reaching the molecular layer from pyramidal neurons in layer II in control (C) and lesioned (D) sensorimotor neocortices. The distal apical dendrites from HI-treated rats showed an abundance of pedunculate and mushroom spines compared to control (counts done on 2D images of fused Z stack images in ImageJ [see Materials and Methods] were 42.3 ± 0.9 and 23.8 ± 1.9 per 25 μm distal dendrite, respectively, sample size =4 each) Scale bars = 250 μm in B (applies to A); 5 μm in D (applies to C).
Fig. 8.
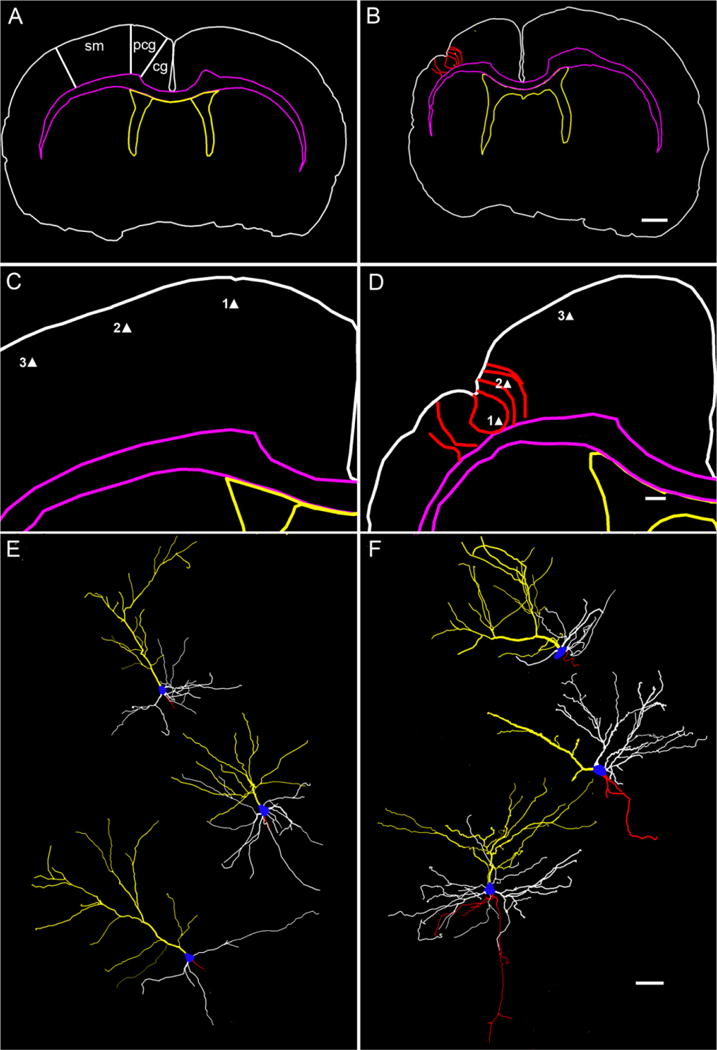
Cortical neuronal morphologies revealed by neuronal reconstructions from rapid-Golgi-stained sections. Top panels show low-magnification traces of sections from control (A) and HI-treated rats (B). Regions of interest in the parasagittal zone (Paxinos and Watson, 1998) are marked in A: sensorimotor cortex (sm), cingulate (cg), and paracingulate cortex (pcg). Middle panels show higher magnification of the traces in the top panels from the corresponding right parasagittal neocortices. Bottom panel shows the dysmorphic features of reconstructed layer II/III pyramidal neurons from HI-treated sections compared to corresponding neurons from control sections. The tracing of a Golgi-stained 200-μm coronal section from control and HI-treated rats (color code same as in Fig. 2D) shows bilateral symmetry in control (A) and atrophied ipsilateral hemisphere with a microgyri in the sensorimotor cortex (red) in a lesioned rat (B). In contrast to control (C), the presence of a parasagittal microgyrus and the surrounding gliotic bands are traced (red contours) in D to give reference to the location of the reconstructed pyramidal neurons (i.e., solid triangles 1, 2, and 3). The reconstructed pyramidal neurons are depicted in the exact dorsoventral orientation they were found in the coronal sections and are shown in a numerically ascending order from top to bottom (control [E] and HI-treated [F], respectively). Neuron 1 from HI-treated rat (F) is a displaced layer II/III neuron showing a dominant apical dendritic tree (yellow) with basal dendrites (white) running along the curvature formed by the pit of the microgyrus. Neuron 2 is a layer II/III neuron adjacent to the microgyrus bordering a gliotic band. It shows a sparsely branched apical dendrite running along the gliotic band toward the molecular layer and a unilateral tree of basal dendrites directed away from the gliotic band and toward the parainfarct cortex. Neuron 3 is a layer II/III layer pyramidal neuron in the parainfarct sensorimotor cortex with a well-formed apical and basal dendritic tree and axon collaterals (red); cell body, blue; apical dendrite, yellow; basal dendrite, white; axon, red. Scale bars = 1 mm in B (applies to A); 250 μm in D (applies to C); 50 μm in F (applies to E).
Hippocampal lesion and mossy fiber sprouting in the ≥6-month group
The hippocampus showed massive loss of neurons in the ipsilateral CA3 region (Fig. 9A,B) of all HI-treated epileptic rats where the atrophied dorsal hippocampus was clearly visible (n/n = 7/11). The subjective lesion grades for ipsilateral dorsal hippocampi were as follows: grade 0: no apparent loss of neurons in CA3, CA1, or the granule cell layer (i.e., equal to control tissue [epileptic rats, n/n = 0/11; HI nonepileptic rats, n/n = 11/14]); grade 1: mild loss of neurons in CA3, or CA1, or the granule cell layer (epileptic rats, n/n = 0/11; HI nonepileptic rats, n/n = 1/14); grade 2: severe loss of neurons in CA3, CA1, or the dentate gyrus with atrophy (epileptic rats, n/n = 7/11; HI nonepileptic rats, n/n = 2/14); and grade 3: no hippocampus present or very little tissue remaining that could be histologically identified as hippocampus (epileptic rats, n/n = 4/11; HI nonepileptic rats, n/n = 0/14). Examination of the entorhinal cortex revealed marked laminar neuronal cell death ipsilaterally (Fig. 9C and D; n/n = 11/11 of the epileptic rats); similar findings have also been reported in the pilocarpine-treated rat model of epilepsy (Kobayashi et al., 2003). Timm stain was seen bilaterally in the inner molecular layer of the dentate gyrus in the HI-treated epileptic rat (Figs. 10, 11), both in the dorsal (Fig. 10E,H) and ventral (Fig. 11C,E) hippocampus. The average grades of Timm stain product were compared between sides (ipsi- and contralateral) and the three groups of monitored animals (HI-treated epileptic rats, n = 11; HI-treated nonepileptic rats, n = 14; and sham control rats, n = 12; Fig. 12) from the ≥6-month group. In HI-treated epileptic rats, significantly higher average grades were found in the ipsilateral dorsal (septal) hippocampus compared to the contralateral dorsal hippocampus (P < 0.0001). In comparison, a similar difference was not found to be significant in HI-treated nonepileptic rats (P = 0.02). Both of the above-mentioned grades in HI-treated epileptic rats were significantly different from average grades for controls (P < 0.0001 and P < 0.0001, respectively). The average staining grades for the HI-treated nonepileptic rats were only significantly different from control ipsilaterally but not contralaterally (P < 0.0001 and P = 0.07, respectively). On the other hand, the ventral hippocampi of HI-treated epileptic rats showed significantly higher grades of stain product compared to both the HI-treated nonepileptic and control rats (P = 0.001 and P = 0.0001, respectively) contralaterally, but ipsilaterally they were only significantly different from control (P < 0.0001). Average grades for HI-treated nonepileptic rats as a group were significantly different from average grades for control rats on both counts. These data suggest that HI-treated epileptic rats not only have significantly greater mossy fiber sprouting ipsilaterally both in the severely atrophied dorsal and relatively spared ventral hippocampus, but also in the contralateral hippocampus, with significantly greater grades in the ventral hippocampus than the dorsal. This contralateral sprouting in the ventral hippocampus of the HI-treated epileptic and nonepileptic rats, which anatomically is representative of the anterior head of the human hippocampus, suggests chronic interhemispheric diaschisis associated with this perinatal insult that was significantly different for rats with seizures versus rats without detectable seizures. Similar significance was not found for the contralateral dorsal hippocampus in this study. The presence of hippocampal sclerosis with neuroplasticity in the form of mossy fiber sprouting seen in this animal model was similar to findings in tissue derived from human lesional epilepsies after term perinatal insults as described by Cendes et al. (1995), where dual pathologies (i.e., histopathology involving both cortex and hippocampus) have been shown to be prevalent.
Fig. 9.
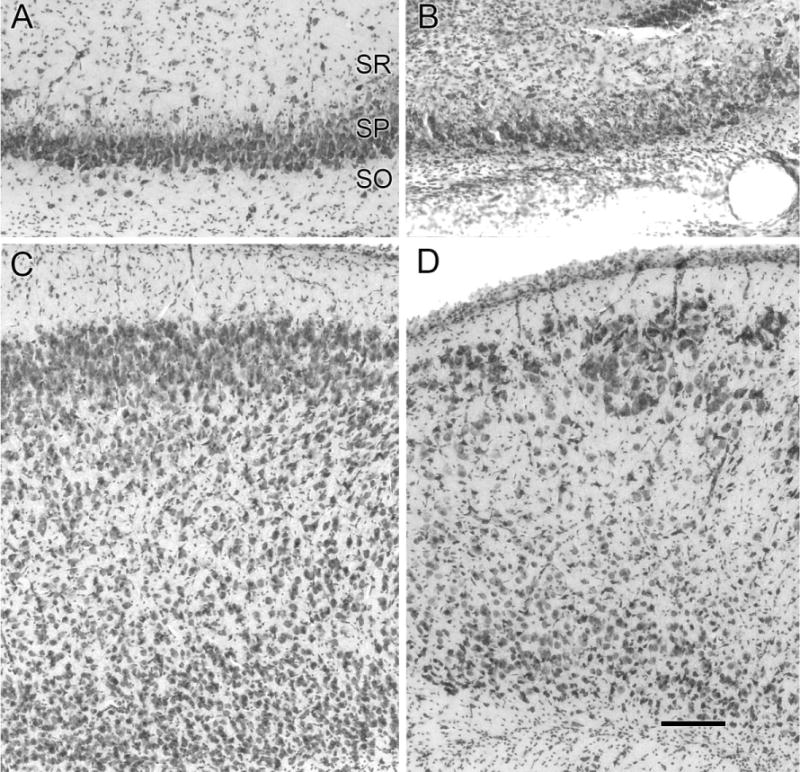
Cell loss in the dorsal hippocampus and the entorhinal cortex. Cresyl-violet-stained coronal sections of the ipsilateral dorsal hippocampus (A,B) and entorhinal cortex (C,D) with the molecular layer I, at the top of the panels are shown. Neurons in the stratum pyramidale (SP) are seen populating the CA3 region of the right hippocampus in a control rat (A). Massive loss of neurons is shown in the corresponding CA3 region in an HI-treated rat (B). Note the proliferation of cells in the stratum oriens (SO) and stratum radiatum (SR) in the HI-lesioned section that may be due to reactive gliosis. Compared to control (C) the entorhinal cortex shows laminar loss of neurons marked by the superficial presence of large neurons normally located in the deeper layers in the section from an HI-treated epileptic rat (D). Note the thinning/obliteration of the molecular layer I in D. Scale bars = 50 μm in D (applies to A–C).
Fig. 10.
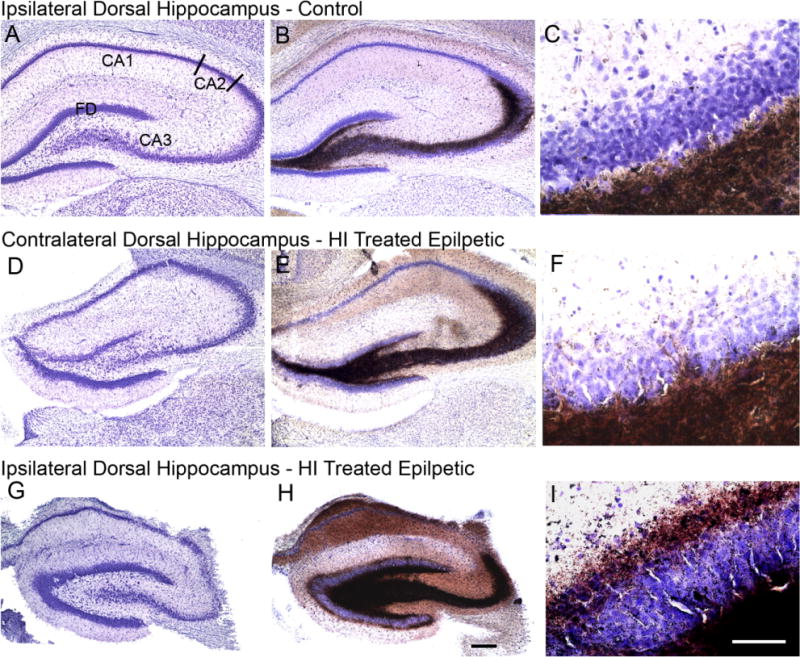
Representative coronal cresyl-violet and Timm-stained-sections focusing on the ipsi- and contralateral dorsal hippocampi. Top and bottom panels show ipsilateral cresyl-violet (A,G), Timm-stained sections (B,H) and magnified views of the inner molecular layer of the dentate gyrus (C,I) from a control and HI-treated epileptic rat, respectively. Middle panel shows the corresponding contralateral sections from the same HI-treated epileptic rat as shown in panel C (D–F). The dorsal hippocampus from a control rat (A) shows fascia dentata (FD) and the Ammon’s horn (CA3, CA2, and CA1). The Timm stained section shows dark brown precipitate in the region of mossy fiber innervation (B). Grade 0 Timm stain product is seen in dentate inner molecular layer (C). The contralateral dorsal hippocampus (D) shows no apparent cell loss of CA3 neurons. Timm stain shows a more robust distribution compared to control (E) and minimal (grade 1) stain product is seen in dentate inner molecular layer (F). Massive loss of lateral CA3 neurons causing a distinct demarcation between the CA3 and CA2 (relatively spared) junction is seen in the ipsilateral region of the epileptic rat (G); note the relative preservation of the fascia dentata (FD) medial CA3 and hilar neurons. Robust innervation of the CA3 region till the CA3/CA2 junction by mossy fibers masks presence of the few surviving neurons in the region by cresyl violet counterstain (H). The dentate inner molecular layer shows grade 3 stain product (I); note atrophy of ipsilateral dorsal hippocampus in ipsilateral compared to the contralateral and control sections. Scale bars = 250 μm in H (applies to A,B,D,E,G); 50 μm in I (applies to C,F).
Fig. 11.
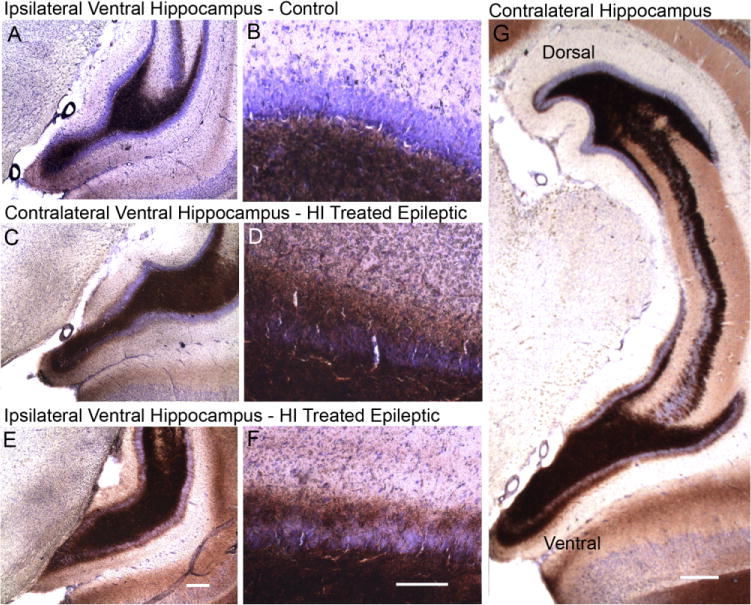
Representative coronal Timm-stained sections focusing on the ipsi- and contralateral ventral hippocampi. Top and bottom panels show ipsilateral Timm stained sections (A,E) and magnified views of the inner molecular layer of the dentate gyrus (B,F) from a control and HI-treated epileptic rat, respectively. Middle panel shows the corresponding contralateral sections from the same HI-treated epileptic rat as shown in E (C,D). The ventral hippocampus from a control rat (A) shows Timm staining in the dentate hilus and grade 0-1 stain product in the dentate inner molecular layer (B). A more robust stain product is seen in both C and E with grade 3 staining (D,F) in the dentate inner molecular layer bilaterally. Contralateral Timm stain showed a differential grade of stain in the dorsal versus ventral hippocampus of epileptic rats. A representative coronal section from an epileptic rat (G) showing grade 1 compared to grade 3 of Timm stain in the dentate inner molecular of the dorsal hippocampus at the top of the panel and the ventral hippocampus in the bottom of the panel, respectively. Scale bars = 250 μm in E (applies to A,C); 100 μm in F (applies to B,D); 500 μm in G.
Fig. 12.
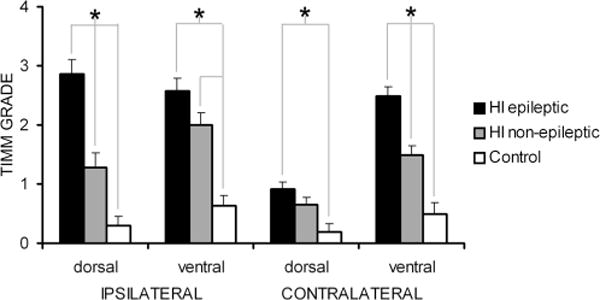
Comparison of Timm stain grades in the dentate inner molecular layer of ipsi- and contralateral dorsal and ventral hippocampi of i) HI-treated rats that were detected to be epileptic (n = 11); ii) HI-treated rats that were not detected to be epileptic (n = 14); iii) sham control rats (n = 12). Significant differences between groups for each anatomical location are indicated by asterisks.
DISCUSSION
The objectives of this study were to investigate the progression and temporal distribution of spontaneous motor seizures occurring in this rodent model for perinatal HI damage and to assess the histopathological features of the resultant lesion in the neocortex and hippocampus. The perinatal HI insult resulted in a progressive type of epilepsy with an apparent latent period of ≈2 months. The increase in the frequency of spontaneous seizures as a function of time was associated with a predilection for clustering. The histopathological features studied at a late timepoint (i.e., ≥6 months of age) after the perinatal HI insult revealed cystic infarcts in epileptic rats associated with multiple areas of cortical dysplasia. The presence of porencephalic cysts, microgyri, neocortical dislamination, and deep laminar cell loss and dysmorphic neurons in the ipsilateral neocortices of the epileptic rats (≥6 month group) suggests that this animal model may provide a useful tool to help understand the human condition with similar histopathological properties. Dysphasic features were not only observed in the spared ipsilateral cortex, but also in the contralateral hemisphere. These contralateral features occurred in the form of cortical dysplasia, white matter hypercellularity, and mossy fiber sprouting without the concomitant massive cell loss and atrophy seen on the ipsilateral side. This suggests diachisis due to the unilateral lesion or unilateral epileptogenic zone (or both). Thus, the animal model demonstrates “dual pathology” by having histopathological features in both the neocortex and hippocampus, which have been linked to epileptogenic lesions in humans and multiple animal models of epilepsy.
In this study the HI lesion was performed at P7, because the developmental stage of the rat brain has been compared to that of a 32–36-week gestational human fetus (see review, Vannucci and Vannucci, 2005), thus representing a periterm injury for the animal model. A preterm insult has been shown to have distinctive histopathology in the form of periventricular leucomalacia, highlighting the role of developmental stages in the vulnerability of specific brain areas to similar insults (Back et al., 2002; du Plessis and Volpe, 2002; Sizonenko et al., 2003). In this model, ligation of the unilateral carotid artery alone does not decrease cerebral perfusion below critical levels, and the addition of hypoxia is required to cause the pathogenic ischemia (Vannucci and Vannucci, 2005). Thus, once the rat pups are removed from the hypoxia chamber, cerebral blood flow returns to prehypoxia levels, which represents the reperfusion stage (Silverstein et al., 1984; Lynch et al., 2002). The return of oxygenated blood to previously ischemic tissue causes further damage (i.e., reperfusion injury), which is a major contributor to the pathogenesis of the ischemic lesions (Palmer, 1995). This particular animal model, therefore, seemed highly appropriate for chronic analysis of the histopathology and development of seizures in reference to an equivalent human insult.
Spontaneous motor seizures
Focal onset
The postnatal-day-7 model of perinatal HI in the rat with the modified Levine’s method develops spontaneous motor seizures that behaviorally reflect a right-sided lesion. All three epileptic rats (i.e., 100%; from the behavioral study reported here) in the video-monitoring study showed left forelimb clonus as their first detected behavioral seizure; this may indicate a focal ipsilateral brain involvement, at least initially. Even in the generalized forms of the motor seizures a left forelimb onset was noted in animals with gross infarcts in the perfusion territory of the middle cerebral artery with no obvious left-sided motor disability. This likely represents secondary generalization of partial seizures. A study conducted in children with perinatally acquired unilateral porencephalic cysts and refractory epilepsy (Guzzetta et al., 2006) found that these patients consistently presented with partial epilepsy.
Latent period and progression
The 25% monitoring protocol used in this study led to the detection of a shorter latent period to the first motor seizure (68.3 ± 0.3 days) than the previously reported latent period of 194 ± 43 days (Williams et al., 2004), which used a random 6-hour per week monitoring protocol (i.e., animals were monitored ≈4% of the time). If seizure occurrence was random, the mean seizure rate (0.66 ± 0.35 seizures per day) suggests that these rats had at least one seizure every 2 days. However, we found that only 16% of the total detected seizures occurred as isolated seizures at the rate of ≤1 seizure per 24 hours, whereas 63% of the seizures occurred at the rate of ≥3 seizures per 24 hours. The continuous video-monitoring over 1-week periods revealed that the seizures occurred in close temporal proximity to each other, forming clusters. Seizure clustering is commonly seen in human epilepsies (Haut, 2006) and is defined as a deviation from random distribution. Continuous monitoring for 1-week periods every 4 weeks over 7 consecutive months revealed a gradual progression of increasing seizure frequency as a function of time, due largely to an increase in the number of seizure clusters and seizures within each cluster. The low seizure rate and the lack of a continuous monitoring protocol suggest that we may have failed to detect all of the epileptic rats with this protocol. The seizure rate of the epileptic rats at 7 months after HI (maximum time studied after HI) compared to kainate-treated rats (Hellier et al., 1998) at 6 months after treatment was ≈10-fold lower (1.62 seizures per day to 16.3 seizures per day, respectively). Thus, the apparent latent period was about 2 months with 25% monitoring (i.e., the actual latent period is presumably shorter) and the mean behavioral seizure frequency was about 0.66 seizures per day; however, because most seizures occurred in clusters, many seizures could occur in 1 day and most days had no motor seizures. These findings suggest that the seizure rate in this model is much lower than the rate in models based on status epilepticus (see Williams et al., 2004) and that the seizures primarily occur in clusters, which emphasizes the value of long-term continuous monitoring in animal models of epilepsy.
Diurnal distribution
Seizure frequencies for the light and dark phase of the diurnal cycle were not significantly different, but significantly more seizures occurred during inactive states of the light phase. These data suggest that the activity state rather than the light-dark cycle influences seizure occurrence. Activity state did not appear to affect seizure occurrence in the dark phase, possibly because the rats had fewer episodes of nonactive states during the dark phase. These data are comparable to results obtained in rats with kainate-induced epilepsy (Hellier et al., 1999), where inactivity more than the phase of the light/dark cycle was associated with an increased occurrence of motor seizures.
Neocortical lesion and its implications for progressive epilepsy
Every HI-treated rat detected to have spontaneous recurrent seizures had ipsilateral cystic infarcts. Surviving frontal, paracingulate (anterior cerebral artery perfusion), and entorhinal cortices (posterior cerebral artery perfusion) that were spared from the liquifactive necrosis involved in the formation of a porencephalic cyst showed multiple forms of cortical dysplasias. Analogous human HI insults show different combinations of similar postlesion histopathology, although the neurological loss of function in the form of prominent motor disabilities, a common clinical feature of the human HI syndrome, was not obvious in these rats. The histological findings at the early timepoint (P30) were similar in their “watershed zone” distribution and cortical and subcortical involvement to those of human neonatal encephalopathies due to HI. At the later timepoint (≥6 months of age), histopathological data suggest shrinking of the infarct-induced scar tissue in the neocortex. This caused the collapse of the columnar and laminar bands of scar tissue, distinctly seen at 1 month of age, to form microgyri and islands of isolated neurons with altered orientation and morphology. This transformation over time may be associated with additional chronic neuroplasticity and progressive epilepsy. Microgyri associated with encephaloclastic porencephaly have been reported for neonatal lesions resulting from perinatal brain damage (Levine et al., 1974; Norman, 1981) and therefore represent the similarity the model has to the human condition. Similarly, neuropathology of spared cortical regions in the human fetal brain (Norman, 1981; Marin-Padilla, 1999, 2000a,b) after perinatal encephalopathies found postinjury development that was characterized by progressive alterations compatible with acquired cortical dysplasia. Neurons underwent postinjury structural and functional hypertrophy by acquiring new morphologic and functional features. These human studies have proposed that, in acquired encephalopathies, the progressive postinjury reorganization of the undamaged cortex and its consequences (acquired cortical dysplasia), rather than the original lesion, may represent the main underlying mechanism of pathogenesis. The ensuing neurological sequelae, such as epilepsy, cerebral palsy, dyslexia, cognitive impairment, and/or poor school performance are the result of such pathogenesis.
Hippocampal histopathology has been a major focus of studies of epileptogenesis. The field of epilepsy is not settled on whether the hippocampus generates or propagates seizures or both. Clinical data suggest that nonhippocampal sources of epileptiform discharges are common even in patients diagnosed with temporal lobe epilepsy (i.e., dual pathology; Mathern et al., 1994; Porter et al., 2003). Dual pathology has been commonly reported in human cases with porencephalic cysts and reactive gliosis (Cendes et al., 1995; Ho et al., 1998). Cortical dysplasias are commonly detected in resected temporal lobe tissues associated with refractory temporal lobe epilepsy (Kasper et al., 1999; Porter et al., 2003). Disrupted cortical lamination and dystrophic and maloriented neurons are characteristic findings of the cortical dysplasia found in the temporal neocortex of these patients. Similar cortical dysplasias detected in conjunction with the robust mossy fiber sprouting in the dentate gyrus emphasize the need to investigate the functional alterations associated with the dysmorphisms in this model. Microgyral malformations in the parasagittal cortex of the HI-treated epileptic rats from the late group are analogous with numerous human resective and autopsy studies of ulegyric/polymicrogyric tissue. Similarly located in watershed zones in neonates and children, they are thought to have a common pathogenic mechanism related to perfusion failure in that area (Villani et al., 2003). To our knowledge, this finding has never been reported for this model. Conditions like polymicrogyri are also thought to result from ischemic insults in the perinatal period (Dvorak et al., 1993). Golgi preparations from resected human tissue demonstrate that neurons of the mid-cortical layers are replaced by a tangential band of fibrous astrocytes, variably layers II–V, in the microgyric cortex. These findings confirm impressions from general cell and fiber stains that acquired microgyria are the result of a destructive process predominantly affecting mid-cortical regions. This must occur after migration is complete but before the development of secondary and tertiary gyri (Williams et al., 1976). With the HI insult at P7, we saw similar mid-cortical loss of neurons (seen at 1 month of age) that eventually collapsed into the four-layered acquired microgyral structure (seen at 6 months of age). Since neuronal migration in rat neocortex is thought to be complete at P7 (Hagberg et al., 2002), microgyral formations in the model represent an acquired form of the dysplasia. Microgyral-like structures were noted for their absence in a previous study in this model by Towfighi et al. (1991). Since these dysmorphisms are commonly found in the human fetal brain, it was agreed that the animal model was different from the human condition in this matter. However, all of the histological data presented in that publication were from P30 rats. The present study also failed to detect classical microgyral structures in the P30 sections, but they were found in sections from older HI-treated rats (≥6 months of age) documented to be epileptic (45%). The gray and white matter anomalies detected in this study months after initial injury suggest that it would not be advisable to use the contralateral hemisphere as “control cortex” in this model, as previously reported with histological studies done at early (P30) timepoints (Towfighi et al., 1994). Although all of the examples of acquired neocortical dysplasia reported here were only observed in the epileptic rats, it must emphasized that with the current experimental design it remains unclear whether these features are causes or consequences (or both) of the epileptogenesis.
Mossy fiber sprouting
Detection of Timm stain product in the dentate inner molecular layer is thought to represent sprouting of mossy fiber axons, which has been detected in children with extrahippocampal seizures (Mathern et al., 1994). Although more Timm stain has been seen after more evoked seizures (i.e., kindled seizures; Cavazos et al., 1991), no correlation has been found between density of sprouting and severity of the chronic epilepsy (i.e., recurrent spontaneous motor seizures; Buckmaster and Dudek, 1997; Pitkanen, 2005). Similarly, this study found no clear association. The animals with spontaneous motor seizures (≥6-month group) demonstrated mossy fiber sprouting in both the ipsi- and contralateral dorsal and ventral hippocampi. The main points from the Timm-stain data for epileptic rats were, first, HI lesions were consistently associated with bilateral Timm stain in the inner molecular layer. Second, average scores for Timm stain product in the inner molecular layer were comparable ipsilaterally in the dorsal and ventral hippocampi (2.86 ± 0.14 and 2.57 ± 0.20, respectively), but significantly different in the contralateral hippocampi at those two anatomic locations (0.91 ± 0.16 and 2.5 ± 0.22, respectively). Third, in every instance the grade for Timm stain in the contralateral ventral hippocampus was higher than the contralateral dorsal hippocampus. Similar differences in the extent of Timm staining have been reported in rats that were chronically epileptic after kainate-induced status epilepticus (Buckmaster and Dudek, 1997), where higher grades of Timm-stain product were found in the temporal hippocampi. The septo-temporal difference in mossy fiber sprouting after HI was also similar to observations reported for kindled rats (Cavazos et al., 1992). The ventral hippocampus in the rodent is thought to represent the anterior pole in the human hippocampus, which is often surgically resected in patients with refractory temporal lobe epilepsy. Therefore, this robust sprouting in the ipsi- and contralateral ventral hippocampi is potentially of considerable interest, and is evidence of distant neuroplasticity of apparently noninjured cortical structures. It represents one of the possible effects of trans-hemispherical diaschisis in this model. Lastly, the HI-treated animals that were not observed to have spontaneous motor seizures were always found to have lower average grades of mossy fiber sprouting in the ipsi- and contralateral hippocampi compared to the average grades found bilaterally in the epileptic rats. However, they were significantly different only in the ipsilateral dorsal and contralateral ventral hippocampi. These data indicate that the HI lesions in the rats with spontaneous seizures in this model were consistently associated with significantly more Timm stain in the inner molecular layer compared to the rats without observed seizures and control rats. This increase was statistically significant in the ipsilateral dorsal hippocampus, which may be attributed to the HI-insult-induced neuron loss. However, it was also statistically significant in the contralateral temporal hippocampus, which may indicate trans-hemispheric diaschisis induced neuroplasticity. Timm stain was not done in the early (P30) group of animals because at that timepoint we had no behavioral data to determine the epileptic status of that group of HI-treated animals. Understanding the significance of the robust mossy fiber sprouting in transhemispheric ventral hippocampus of HI-treated epileptic rats may help shed light on the mechanisms of epileptogenesis for this model.
Models of epilepsy from perinatal insults
Chronic models of pediatric epilepsy related to prenatal and perinatal insults have focused on cortical dysplasias, which are the hypothesized epileptogenic zones in affected brains. Microgyri-like malformations have been induced with the freeze-lesion model and cortical dysplasias by prenatal irradiation and the neuroteratogen, methylazoxymethanol (MAM) (Smith et al., 1999; Chevassus-Au-Louis et al., 1999; Kondo et al., 2001; Schwartzkroin et al., 2004). Animal models using perinatal rats have induced acute seizures using hyperthermia, hypoxia, and Flurothyl (Jensen, 2006) to mimic seizures during the neonatal period. Neonatal seizures, from poorly understood causes, are predictors of adverse neurological outcomes, one of which is chronic epilepsy (Lombroso, 1996). Until recently, the acute insults or the induced dysplasias have not been shown to result in spontaneous recurrent motor seizures in these models. Therefore, the presence of cortical dysplasias in the present model occurring after perinatal perfusion failure is more analogous to the human condition under study. There is no consensus on whether neonatal seizures simply reflect an existing underlying brain injury or are a de-novo causative factor for further damage causing chronic epilepsy (Scher, 2003). Animal models have been designed to address this issue by combining a hyperthermic insult to a postnatal rat with a perinatally induced cortical malformation created either by the freeze lesion or MAM exposure. The MAM-treated rat with hyperthermia-induced seizures showed a lower threshold to the hyperthermia-induced behavioral seizures (Germano et al., 1996), but the high mortality (≈90%) and low yield after the second insult made it difficult to test for recurrent spontaneous seizures in these animals. A rat model with the freeze-lesion-induced microgyral cortical malformations, when subjected to hyperthermia at a postnatal age of 10 days, has recently been shown to have abnormal EEG recordings in the amygdala associated with freeze behaviors (Scantlebury et al., 2004). This monitoring protocol was conducted ≈2 months after the second insult and did not detect any generalized motor seizures. Another animal model designed to mimic early-life prolonged seizures that result in chronic epilepsy is the lithium-pilocarpine model of status epilepticus (Raol et al., 2003). This model has shown a 67% incidence of chronic epilepsy in rats with the chemotoxin-induced status epilepticus at a postnatal age of 20 days, but it is not clear that an insult at that age can be classified as a model for developmental epileptogenesis. Immature rodents below the age of 3 weeks have been shown to be resistant to chemoconvulsant-induced status epilepticus in terms of neuronal death in the hippocampus and resultant spontaneous seizures (Priel et al., 1996; Haas et al., 2001). In light of these recent findings, the model of perinatal HI used in this study with histopathological lesions similar to the human condition and the evolution of postlesion epilepsy has advantages compared to the other rodent models. Additionally, the present study revealed that rats that did not develop infarct lesions after the perinatal insult of carotid ligation followed by 120 minutes of hypoxia (8% O2) did not develop epilepsy (i.e., HI nonepileptic rats, n/n = 17/28; HI nonepileptic rats with no lesion, n/n = 14/17). Thus, no spontaneous recurrent seizures (i.e., no evidence for epilepsy) were detected in any of the HI-treated rats without infarcts (n/n = 14/14).
CONCLUSION
This study reports the cortical and hippocampal histopathological features of a model of perinatal HI with spontaneous recurrent motor seizures. Similar histopathological alterations have been reported in a variety of multicausal human perinatal injuries and insults (Rorke, 1992; Villani et al., 2003). Evidence is presented for a postinjury transformation of the damaged and spared neocortex, with the possibility of surviving cortical neurons adapting by transforming their structure (hypertrophy) and connectivity (increase in dendritic spines) to compensate for the massive laminar and columnar neuronal loss seen at the early timepoint (P30). Postlesional plasticity of the developing brain has been proposed to occur due to reorganization of thalamocortical, callosal, and intracortical circuitry, and failure to prune immature connections that possibly are epileptogenic due to a hyperinnervated circuitry by previous studies (Luhmann et al., 1998; Jacobs et al., 2000). The postinjury structural plasticity in this model coincides with the progressive nature of the resultant epilepsy. Whether this structural plasticity also functions as the pathological mechanism contributing to the epileptogenesis in this model with an apparent latent period of 2 months requires further studies. The diaschisis seen in the form of histological alterations in the contralateral somatosensory cortex, underlying white matter, and hippocampus are relevant to alterations noted in human nonaffected cortices (Nakashima et al., 1985; Andrews, 1991; Kraemer et al., 2004). The presence of the dual pathology present in this model, in the form of neocortical and hippocampal histological aberrations, provides the means to study the interrelationship of these features with the porencephalic lesion morphology similar to the one found in humans; this approach may provide insights into the epileptogenesis in patients with congenital porencephaly (Cusmai et al., 1993; Ho et al., 1998). Finally, the epilepsy following perinatal HI in this model was found to be lesion-induced; HI-treated rats that did not develop any of the infarct-related histological lesions described above did not develop epilepsy. Future studies in the model need to investigate the role of the neuropathological features in the epileptogenesis; however, without the associated neuropathogy, perinatal HI did not result in epilepsy.
Acknowledgments
This work was performed in partial fulfillment of the Ph.D. degree requirements of Colorado State University graduate program of Molecular, Cellular and Integrative Neurosciences.
Grant sponsor: American Heart Association; Grant numbers: 0410026Z and 0610146Z (to S.D.K.); Grant sponsor: National Institutes of Health; Grant numbers: NS16683 and NS45144 (to F.E.D).
LITERATURE CITED
- Alonso-Nanclares L, Garbelli R, Sola RG, Pastor J, Tassi L, Spreafico R, DeFelipe J. Microanatomy of the dysplastic neocortex from epileptic patients. Brain. 2005;128:158–173. doi: 10.1093/brain/awh331. [DOI] [PubMed] [Google Scholar]
- Andres M, Andre VM, Nguyen S, Salamon N, Cepeda C, Levine MS, Leite JP, Neder L, Vinters HV, Mathern GW. Human cortical dysplasia and epilepsy: an ontogenetic hypothesis based on volumetric MRI and NeuN neuronal density and size measurements. Cereb Cortex. 2005;15:194–210. doi: 10.1093/cercor/bhh122. [DOI] [PubMed] [Google Scholar]
- Andrews RJ. Transhemispheric diaschisis. A review and comment. Stroke. 1991;22:943–949. doi: 10.1161/01.str.22.7.943. [DOI] [PubMed] [Google Scholar]
- Ansari MQ, Chincanchan CA, Armstrong DL. Brain calcification in hypoxic-ischemic lesions: an autopsy review. Pediatr Neurol. 1990;6:94–101. doi: 10.1016/0887-8994(90)90041-x. [DOI] [PubMed] [Google Scholar]
- Armstrong DD, Mizrahi EM. Pathologic basis of the symptomatic epilepsies in childhood. J Child Neurol. 1998;13:361–371. doi: 10.1177/088307389801300801. [DOI] [PubMed] [Google Scholar]
- Aso K, Scher MS, Barmada MA. Cerebral infarcts and seizures in the neonate. J Child Neurol. 1990;5:224–228. doi: 10.1177/088307389000500315. [DOI] [PubMed] [Google Scholar]
- Avoli M, Bernasconi A, Mattia D, Olivier A, Hwa GG. Epileptiform discharges in the human dysplastic neocortex: in vitro physiology and pharmacology. Ann Neurol. 1999;46:816–826. doi: 10.1002/1531-8249(199912)46:6<816::aid-ana3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Ari Y. Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience. 1985;14:375–403. doi: 10.1016/0306-4522(85)90299-4. [DOI] [PubMed] [Google Scholar]
- Bergamasco B, Benna P, Ferrero P, Gavinelli R. Neonatal hypoxia and epileptic risk: a clinical prospective study. Epilepsia. 1984;25:131–136. doi: 10.1111/j.1528-1157.1984.tb04168.x. [DOI] [PubMed] [Google Scholar]
- Buckmaster PS, Dudek FE. Neuron loss, granule cell axon reorganization, and functional changes in the dentate gyrus of epileptic kainate-treated rats. J Comp Neurol. 1997;385:385–404. [PubMed] [Google Scholar]
- Burneo JG, Bebin M, Kuzniecky RI, Knowlton RC. Electroclinical and magnetoencephalographic studies in epilepsy patients with polymicrogyria. Epilepsy Res. 2004;62:125–133. doi: 10.1016/j.eplepsyres.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Calcagnotto ME, Paredes MF, Tihan T, Barbaro NM, Baraban SC. Dysfunction of synaptic inhibition in epilepsy associated with focal cortical dysplasia. J Neurosci. 2005;25:9649–9657. doi: 10.1523/JNEUROSCI.2687-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazos JE, Golarai G, Sutula TP. Mossy fiber synaptic reorganization induced by kindling: time course of development, progression, and permanence. J Neurosci. 1991;11:2795–2803. doi: 10.1523/JNEUROSCI.11-09-02795.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazos JE, Golarai G, Sutula TP. Septotemporal variation of the supragranular projection of the mossy fiber pathway in the dentate gyrus of normal and kindled rats. Hippocampus. 1992;2:363–372. doi: 10.1002/hipo.450020404. [DOI] [PubMed] [Google Scholar]
- Cendes F, Cook MJ, Watson C, Andermann F, Fish DR, Shorvon SD, Bergin P, Free S, Dubeau F, Arnold DL. Frequency and characteristics of dual pathology in patients with lesional epilepsy. Neurology. 1995;45:2058–2064. doi: 10.1212/wnl.45.11.2058. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Flores-Hernandez J, Hernandez-Echeagaray E, Klapstein GJ, Boylan MK, Calvert CR, Jocoy EL, Nguyen OK, Andre VM, Vinters HV, Ariano MA, Levine MS, Mathern GW. Morphological and electrophysiological characterization of abnormal cell types in pediatric cortical dysplasia. J Neurosci Res. 2003;72:472–486. doi: 10.1002/jnr.10604. [DOI] [PubMed] [Google Scholar]
- Chevassus-Au-Louis N, Jorquera I, Ben Ari Y, Represa A. Abnormal connections in the malformed cortex of rats with prenatal treatment with methylazoxymethanol may support hyperexcitability. Dev Neurosci. 1999;21:385–392. doi: 10.1159/000017388. [DOI] [PubMed] [Google Scholar]
- Courville CB. Etiology and pathogenesis of laminar cortical necrosis; its significance in evaluation of uniform cortical atrophies of early life. AMA Arch Neurol Psychiatry. 1958;79:7–30. doi: 10.1001/archneurpsyc.1958.02340010025002. [DOI] [PubMed] [Google Scholar]
- Courville CB. Antenatal and paranatal circulatory disorders as a cause of cerebral damage in early life. J Neuropathol Exp Neurol. 1959;18:115–140. doi: 10.1097/00005072-195901000-00008. [DOI] [PubMed] [Google Scholar]
- Cowan F, Rutherford M, Groenendaal F, Eken P, Mercuri E, Bydder GM, Meiners LC, Dubowitz LM, de Vries LS. Origin and timing of brain lesions in term infants with neonatal encephalopathy. Lancet. 2003;361:736–742. doi: 10.1016/S0140-6736(03)12658-X. [DOI] [PubMed] [Google Scholar]
- Cusmai R, Ricci S, Pinard JM, Plouin P, Fariello G, Dulac O. West syndrome due to perinatal insults. Epilepsia. 1993;34:738–742. doi: 10.1111/j.1528-1157.1993.tb00455.x. [DOI] [PubMed] [Google Scholar]
- du Plessis AJ, Volpe JJ. Perinatal brain injury in the preterm and term newborn. Curr Opin Neurol. 2002;15:151–157. doi: 10.1097/00019052-200204000-00005. [DOI] [PubMed] [Google Scholar]
- Dvorak K, Feit J. Pathogenesis of micropolygyria. Cesk Patol. 1993;29:125–133. [PubMed] [Google Scholar]
- Ferrer I, Pineda M, Tallada M, Oliver B, Russi A, Oller L, Noboa R, Zujar MJ, Alcantara S. Abnormal local-circuit neurons in epilepsia partialis continua associated with focal cortical dysplasia. Acta Neuropathol (Berl) 1992;83:647–652. doi: 10.1007/BF00299415. [DOI] [PubMed] [Google Scholar]
- Germano IM, Zhang YF, Sperber EF, Moshe SL. Neuronal migration disorders increase susceptibility to hyperthermia-induced seizures in developing rats. Epilepsia. 1996;37:902–910. doi: 10.1111/j.1528-1157.1996.tb00044.x. [DOI] [PubMed] [Google Scholar]
- Golomb MR, MacGregor DL, Domi T, Armstrong DC, McCrindle BW, Mayank S, deVeber GA. Presumed pre- or perinatal arterial ischemic stroke: risk factors and outcomes. Ann Neurol. 2001;50:163–168. doi: 10.1002/ana.1078. [DOI] [PubMed] [Google Scholar]
- Guzzetta F, Battaglia D, Di Rocco C, Caldarelli M. Symptomatic epilepsy in children with poroencephalic cysts secondary to perinatal middle cerebral artery occlusion. Child’s Nervous System. 2006;22:922–930. doi: 10.1007/s00381-006-0150-3. [DOI] [PubMed] [Google Scholar]
- Haas KZ, Sperber EF, Opanashuk LA, Stanton PK, Moshe SL. Resistance of immature hippocampus to morphologic and physiologic alterations following status epilepticus or kindling. Hippocampus. 2001;11:615–625. doi: 10.1002/hipo.1076. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Ichord R, Palmer C, Yager JY, Vannucci SJ. Animal models of developmental brain injury: relevance to human disease. A summary of the panel discussion from the Third Hershey Conference on Developmental Cerebral Blood Flow and Metabolism. Dev Neurosci. 2002;24:364–366. doi: 10.1159/000069040. [DOI] [PubMed] [Google Scholar]
- Haut SR. Seizure clustering. Epilepsy Behav. 2006;8:50–55. doi: 10.1016/j.yebeh.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Iwai M, Ikeda T, Jin G, Deguchi K, Nagotani S, Zhang H, Sehara Y, Nagano I, Shoji M, Ikenoue T, Abe K. Neural precursor cells division and migration in neonatal rat brain after ischemic/hypoxic injury. Brain Res. 2005;1038:41–49. doi: 10.1016/j.brainres.2004.12.048. [DOI] [PubMed] [Google Scholar]
- Hellier JL, Dudek FE. Spontaneous motor seizures of rats with kainate-induced epilepsy: effect of time of day and activity state. Epilepsy Res. 1999;35:47–57. doi: 10.1016/s0920-1211(98)00127-2. [DOI] [PubMed] [Google Scholar]
- Ho SS, Kuzniecky RI, Gilliam F, Faught E, Bebin M, Morawetz R. Congenital porencephaly: MR features and relationship to hippocampal sclerosis. AJNR Am J Neuroradiol. 1998;19:135–141. [PMC free article] [PubMed] [Google Scholar]
- Jacobs KM, Graber KD, Kharazia VN, Parada I, Prince DA. Postlesional epilepsy: the ultimate brain plasticity. Epilepsia. 2000;41(Suppl 6):S153–S161. doi: 10.1111/j.1528-1157.2000.tb01574.x. [DOI] [PubMed] [Google Scholar]
- Jacobs KM, Prince DA. Excitatory and inhibitory postsynaptic currents in a rat model of epileptogenic microgyria. J Neurophysiol. 2005;93:687–696. doi: 10.1152/jn.00288.2004. [DOI] [PubMed] [Google Scholar]
- Jensen FE. Pediatric epilepsy models. Epilepsy Res. 2006;68:28–31. doi: 10.1016/j.eplepsyres.2005.09.013. [DOI] [PubMed] [Google Scholar]
- Kadam SD, Williams PA, Dudek FE. Modified Levine’s method in the P7 rat pup causes a lesion similar to the hypoxic encephalopathy-induced parasagittal injury of human neonates. Soc Neurosci Abstr. 2003;29:211.5. [Google Scholar]
- Kasper BS, Stefan H, Buchfelder M, Paulus W. Temporal lobe microdysgenesis in epilepsy versus control brains. J Neuropathol Exp Neurol. 1999;58:22–28. doi: 10.1097/00005072-199901000-00003. [DOI] [PubMed] [Google Scholar]
- Kelly KM. Poststroke seizures and epilepsy: clinical studies and animal models. Epilepsy Currents. 2002;2:173–177. doi: 10.1046/j.1535-7597.2002.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerfoot C, Vinters HV, Mathern GW. Cerebral cortical dysplasia: giant neurons show potential for increased excitation and axonal plasticity. Dev Neurosci. 1999;21:260–270. doi: 10.1159/000017405. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Wen X, Buckmaster PS. Reduced inhibition and increased output of layer II neurons in the medial entorhinal cortex in a model of temporal lobe epilepsy. J Neurosci. 2003;23:8471–8479. doi: 10.1523/JNEUROSCI.23-24-08471.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelfen W, Freund M, Varnholt V. Neonatal stroke involving the middle cerebral artery in term infants: clinical presentation, EEG and imaging studies, and outcome. Dev Med Child Neurol. 1995;37:204–212. doi: 10.1111/j.1469-8749.1995.tb11993.x. [DOI] [PubMed] [Google Scholar]
- Kondo S, Najm I, Kunieda T, Perryman S, Yacubova K, Luders HO. Electroencephalographic characterization of an adult rat model of radiation-induced cortical dysplasia. Epilepsia. 2001;42:1221–1227. doi: 10.1046/j.1528-1157.2001.38300.x. [DOI] [PubMed] [Google Scholar]
- Kraemer M, Schormann T, Hagemann G, Qi B, Witte OW, Seitz RJ. Delayed shrinkage of the brain after ischemic stroke: preliminary observations with voxel-guided morphometry. J Neuroimaging. 2004;14:265–272. doi: 10.1177/1051228404264950. [DOI] [PubMed] [Google Scholar]
- Lanska MJ, Lanska DJ, Horwitz SJ, Aram DM. Presentation, clinical course, and outcome of childhood stroke. Pediatr Neurol. 1991;7:333–341. doi: 10.1016/0887-8994(91)90062-p. [DOI] [PubMed] [Google Scholar]
- Levine S. Anoxic-ischemic encephalopathy in rats. Am J Pathol. 1960;36:1–17. [PMC free article] [PubMed] [Google Scholar]
- Levine DN, Fisher MA, Caviness VS., Jr Porencephaly with microgyria: a pathologic study. Acta Neuropathol (Berl) 1974;29:99–113. doi: 10.1007/BF00684769. [DOI] [PubMed] [Google Scholar]
- Lombroso CT. Neonatal seizures: historic note and present controversies. Epilepsia. 1996;37(Suppl 3):5–13. doi: 10.1111/j.1528-1157.1996.tb01814.x. [DOI] [PubMed] [Google Scholar]
- Luhmann HJ, Raabe K, Qu M, Zilles K. Characterization of neuronal migration disorders in neocortical structures: extracellular in vitro recordings. Eur J Neurosci. 1998;10:3085–3094. doi: 10.1046/j.1460-9568.1998.00311.x. [DOI] [PubMed] [Google Scholar]
- Lynch JK, Hirtz DG, DeVeber G, Nelson KB. Report of the National Institute of Neurological Disorders and Stroke Workshop on Perinatal and Childhood Stroke. Pediatrics. 2002;109:116–123. doi: 10.1542/peds.109.1.116. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage. III: gray matter lesions of the neocortex. J Neuropathol Exp Neurol. 1999;58:407–429. doi: 10.1097/00005072-199905000-00001. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M. Neocortical epilepsies. 1st. Philadelphia: Lippincott Williams & Wilkins; 2000a. Perinatal brain damage, cortical reorganization (acquired cortical dysplasias), and epilepsy; pp. 153–172. [PubMed] [Google Scholar]
- Marin-Padilla M. Neuropathologic correlates of perinatal asphyxia. Int Pediatr. 2000b;15:221–228. [Google Scholar]
- Marin-Padilla M, Tsai RJ, King MA, Roper SN. Altered corticogenesis and neuronal morphology in irradiation-induced cortical dysplasia: a Golgi-Cox study. J Neuropathol Exp Neurol. 2003;62:1129–1143. doi: 10.1093/jnen/62.11.1129. [DOI] [PubMed] [Google Scholar]
- Mathern GW, Leite JP, Pretorius JK, Quinn B, Peacock WJ, Babb TL. Children with severe epilepsy: evidence of hippocampal neuron losses and aberrant mossy fiber sprouting during postnatal granule cell migration and differentiation. Brain Res Dev Brain Res. 1994;78:70–80. doi: 10.1016/0165-3806(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Matsumoto R, Kinoshita M, Taki J, Hitomi T, Mikuni N, Shibasaki H, Fukuyama H, Hashimoto N, Ikeda A. In vivo epileptogenicity of focal cortical dysplasia: a direct cortical paired stimulation study. Epilepsia. 2005;46:1744–1749. doi: 10.1111/j.1528-1167.2005.00284.x. [DOI] [PubMed] [Google Scholar]
- Meencke HJ, Janz D. The significance of microdysgenesia in primary generalized epilepsy: an answer to the considerations of Lyon and Gastaut. Epilepsia. 1985;26:368–371. doi: 10.1111/j.1528-1157.1985.tb05665.x. [DOI] [PubMed] [Google Scholar]
- Mischel PS, Nguyen LP, Vinters HV. Cerebral cortical dysplasia associated with pediatric epilepsy. Review of neuropathologic features and proposal for a grading system. J Neuropathol Exp Neurol. 1995;54:137–153. doi: 10.1097/00005072-199503000-00001. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Najm I, Ying Z, Babb T, Crino PB, Macdonald R, Mathern GW, Spreafico R. Mechanisms of epileptogenicity in cortical dysplasias. Neurology. 2004;62:S9–13. doi: 10.1212/01.wnl.0000114506.49267.bb. [DOI] [PubMed] [Google Scholar]
- Nakashima K, Kanba M, Fujimoto K, Sato T, Takahashi K. Somatosensory evoked potentials over the non-affected hemisphere in patients with unilateral cerebrovascular lesions. J Neurol Sci. 1985;70:117–127. doi: 10.1016/0022-510x(85)90081-4. [DOI] [PubMed] [Google Scholar]
- Nelson KB, Lynch JK. Stroke in newborn infants. Lancet Neurol. 2004;3:150–158. doi: 10.1016/S1474-4422(04)00679-9. [DOI] [PubMed] [Google Scholar]
- Neumann-Haefelin T, Witte OW. Periinfarct and remote excitability changes after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2000;20:45–52. doi: 10.1097/00004647-200001000-00008. [DOI] [PubMed] [Google Scholar]
- Norman MG. On the morphogenesis of ulegyria. Acta Neuropathol (Berl) 1981;53:331–332. doi: 10.1007/BF00690375. [DOI] [PubMed] [Google Scholar]
- Palmer C. Hypoxic-ischemic encephalopathy. Therapeutic approaches against microvascular injury, and role of neutrophils, PAF, and free radicals. Clin Perinatol. 1995;22:481–517. [PubMed] [Google Scholar]
- Palmini A, Gambardella A, Andermann F, Dubeau F, da Costa JC, Olivier A, Tampieri D, Robitaille Y, Paglioli E, Paglioli NE. Operative strategies for patients with cortical dysplastic lesions and intractable epilepsy. Epilepsia. 1994;35(Suppl 6):S57–S71. doi: 10.1111/j.1528-1157.1994.tb05989.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic Press; 1998. [DOI] [PubMed] [Google Scholar]
- Pitkanen A. Epilepsy and plasticity. Epilepsia. 2005;46:204–207. [Google Scholar]
- Porter BE, Judkins AR, Clancy RR, Duhaime A, Dlugos DJ, Golden JA. Dysplasia: a common finding in intractable pediatric temporal lobe epilepsy. Neurology. 2003;61:365–368. doi: 10.1212/01.wnl.0000076487.28227.6e. [DOI] [PubMed] [Google Scholar]
- Priel MR, dos Santos NF, Cavalheiro EA. Developmental aspects of the pilocarpine model of epilepsy. Epilepsy Res. 1996;26:115–121. doi: 10.1016/s0920-1211(96)00047-2. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Ramaswamy V, Miller SP, Barkovich AJ, Partridge JC, Ferriero DM. Perinatal stroke in term infants with neonatal encephalopathy. Neurology. 2004;62:2088–2091. doi: 10.1212/01.wnl.0000129909.77753.c4. [DOI] [PubMed] [Google Scholar]
- Raol YS, Budreck EC, Brooks-Kayal AR. Epilepsy after early-life seizures can be independent of hippocampal injury. Ann Neurol. 2003;53:503–511. doi: 10.1002/ana.10490. [DOI] [PubMed] [Google Scholar]
- Rice JE, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol. 1981;9:131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- Rorke LB. Anatomical features of the developing brain implicated in pathogenesis of hypoxic-ischemic injury. Brain Pathol. 1992;2:211–221. doi: 10.1111/j.1750-3639.1992.tb00694.x. [DOI] [PubMed] [Google Scholar]
- Scantlebury MH, Ouellet PL, Psarropoulou C, Carmant L. Freeze lesion-induced focal cortical dysplasia predisposes to atypical hyperthermic seizures in the immature rat. Epilepsia. 2004;45:592–600. doi: 10.1111/j.0013-9580.2004.51503.x. [DOI] [PubMed] [Google Scholar]
- Scher MS. Neonatal seizures and brain damage. Pediatr Neurol. 2003;29:381–390. doi: 10.1016/s0887-8994(03)00399-0. [DOI] [PubMed] [Google Scholar]
- Scher MS, Wiznitzer M, Bangert BA. Cerebral infarctions in the fetus and neonate: maternal-placental-fetal considerations. Clin Perinatol. 2002;29:693–724. doi: 10.1016/s0095-5108(02)00055-6. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Roper SN, Wenzel HJ. Cortical dysplasia and epilepsy: animal models. Adv Exp Med Biol. 2004;548:145–174. doi: 10.1007/978-1-4757-6376-8_12. [DOI] [PubMed] [Google Scholar]
- Silverstein F, Buchanan K, Johnston MV. Pathogenesis of hypoxic-ischemic brain injury in a perinatal rodent model. Neurosci Lett. 1984;49:271–277. doi: 10.1016/0304-3940(84)90301-x. [DOI] [PubMed] [Google Scholar]
- Sizonenko SV, Sirimanne E, Mayall Y, Gluckaman PD, Inder T, Williams C. Selective cortical alteration after hypoxic-ischemic injury in the very immature rat brain. Pediatr Res. 2003;54:263–269. doi: 10.1203/01.PDR.0000072517.01207.87. [DOI] [PubMed] [Google Scholar]
- Smith BN, Dudek FE, Roper SN. Synaptic responses of neurons in heterotopic gray matter in an animal model of cortical dysgenesis. Dev Neurosci. 1999;21:365–373. doi: 10.1159/000017386. [DOI] [PubMed] [Google Scholar]
- Spreafico R, Battaglia G, Arcelli P, Andermann F, Dubeau F, Palmini A, Olivier A, Villemure JG, Tampieri D, Avanzini G, Avoli M. Cortical dysplasia: an immunocytochemical study of three patients. Neurology. 1998;50:27–36. doi: 10.1212/wnl.50.1.27. [DOI] [PubMed] [Google Scholar]
- Tauck DL, Nadler JV. Evidence of functional mossy fiber sprouting in the hippocampal formation of kainic acid-treated rats. J Neurosci. 1985;5:1016–22. doi: 10.1523/JNEUROSCI.05-04-01016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towfighi J, Yager JY, Housman C, Vannucci RC. Neuropathology of remote hypoxic-ischemic damage in the immature rat. Acta Neuropathol (Berl) 1991;81:578–587. doi: 10.1007/BF00310141. [DOI] [PubMed] [Google Scholar]
- Towfighi J, Housman C, Vannucci RC, Heitjan DF. Effect of unilateral perinatal hypoxic-ischemic brain damage on the gross development of opposite cerebral hemisphere. Biol Neonate. 1994;65:108–118. doi: 10.1159/000244036. [DOI] [PubMed] [Google Scholar]
- Trotter SA, Kapur J, Anzivino MJ, Lee KS. GABAergic synaptic inhibition is reduced before seizure onset in a genetic model of cortical malformation. J Neurosci. 2006;26:10756–10767. doi: 10.1523/JNEUROSCI.2323-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannucci RC, Vannucci SJ. Perinatal hypoxic-ischemic brain damage: evolution of an animal model. Dev Neurosci. 2005;27:81–86. doi: 10.1159/000085978. [DOI] [PubMed] [Google Scholar]
- Villani F, D’Incerti L, Granata T, Battaglia G, Vitali P, Chiapparini L, Avanzini G. Epileptic and imaging findings in perinatal hypoxic-ischemic encephalopathy with ulegyria. Epilepsy Res. 2003;55:235–243. doi: 10.1016/s0920-1211(03)00121-9. [DOI] [PubMed] [Google Scholar]
- Volpe JJ. Neurology of the newborn. Philadelphia: WB Saunders; 2001. pp. 217–331. [Google Scholar]
- Williams RS, Ferrante RJ, Caviness VS., Jr The cellular pathology of microgyria. A Golgi analysis. Acta Neuropathol (Berl) 1976;36:269–283. doi: 10.1007/BF00685371. [DOI] [PubMed] [Google Scholar]
- Williams PA, Dou P, Dudek FE. Epilepsy and synaptic reorganization in a perinatal rat model of hypoxia-ischemia. Epilepsia. 2004;45:1210–1218. doi: 10.1111/j.0013-9580.2004.60403.x. [DOI] [PubMed] [Google Scholar]