

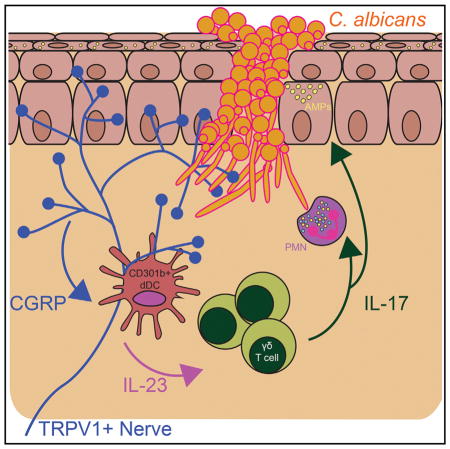
SUMMARY
Innate resistance to Candida albicans in mucosal tissues requires the production of interleukin-17A (IL-17A) by tissue-resident cells early during infection, but the mechanism of cytokine production has not been precisely defined. In the skin, we found that dermal γδ T cells were the dominant source of IL-17A during C. albicans infection and were required for pathogen resistance. Induction of IL-17A from dermal γδ T cells and resistance to C. albicans required IL-23 production from CD301b+ dermal den-dritic cells (dDCs). In addition, we found that sensory neurons were directly activated by C. albicans. Ablation of sensory neurons increased susceptibility to C. albicans infection, which could be rescued by exogenous addition of the neuropeptide CGRP. These data define a model in which nociceptive pathways in the skin drive production of IL-23 by CD301b+ dDCs resulting in IL-17A production from γδ T cells and resistance to cutaneous candidiasis.
Graphical Abstract
INTRODUCTION
Candida albicans is a common, highly morbid pathogen at barrier sites such as the skin and oral cavity in the setting of immunosuppression. For example, patients with defective T helper 17 (Th17) cell immunity such as AIDS or hyper-IgE syndrome are prone to develop chronic mucocutaneous candidiasis (CMC) (McDonald, 2012). Mice with defective Th17 cell type responses are also more susceptible to both oral and cutaneous candidiasis (Hernández-Santos et al., 2013; Kashem et al., 2015). Patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) disorder have circulating neutralizing antibodies against IL-17 and develop CMC (Kisand et al., 2010; Puel et al., 2010). Similarly, patients with IL-17 receptor A (IL-17RA) or IL-17F deficiencies also suffer from CMC (Puel et al., 2011; Smeekens et al., 2011). These patients have global defects in the IL-17 pathway that affects both IL-17 produced by Th17 cells as well as IL-17 derived from cells of the innate immune system.
It is becoming increasingly clear that innate sources of IL-17 are crucial for host defense against C. albicans at barrier sites (Conti et al., 2014; Gladiator et al., 2013). In the skin, dendritic epidermal T cells (DETCs), dermal γδ T cells, CD4+ and CD8+ αβ T cells, and innate lymphoid cells (ILCs) all can produce IL-17 (Gray et al., 2011; MacLeod et al., 2013; Naik et al., 2015, 2012). IL-17 from dermal γδ T cells is required for immunity against BCG, Staphylococcus aureus, and vaccinia virus and for autoimmune inflammation (Cho et al., 2010; Gray et al., 2011, 2013; Sumaria et al., 2011; Woodward Davis et al., 2015). CD4+ and CD8+ T cells generate IL-17 responses to commensal organisms such as Staphylococcus epidermitis and protect against opportunistic pathogens (Naik et al., 2012, 2015). In mouse models of oropharyngeal candidiasis, IL-17 from αβ and γδ T cells as well as ILC3s mediate protection, though the contribution of each cell type remains controversial (Conti et al., 2014; Gladiator et al., 2013).
Production of IL-17 by tissue-resident lymphocytes is mediated by pro-inflammatory cytokines, notably IL-23 (Sherlock et al., 2012; Sutton et al., 2009). IL-23 signaling plays a critical role against mucocutaneous candidiasis as shown by the fact that humans with IL-23R signaling defects are prone to CMC and mice with IL-23 deficiencies are susceptible to oral and cutaneous candidiasis (Gaffen et al., 2014; Kagami et al., 2010; Smeekens et al., 2011). There are at least three well-defined subsets of cutaneous dendritic cells (DCs): epidermal Langerhans cells (LCs), CD103+ dermal DCs (dDCs), and CD11b+ DCs (Kaplan, 2010). LCs and CD11b+ dDCs both secrete IL-23 and con-flicting reports suggest that each is an obligate source of IL-23 during imiquimod-induced dermatitis (Wohn et al., 2013; Yoshiki et al., 2014). Recently, the role of nociceptors has been appreciated in IL-23-mediated imiquimod-induced skin inflammation (Riol-Blanco et al., 2014). In addition, pathogens such as S. aureus can directly activate neurons isolated from the dorsal root ganglion (DRG) and induce pain hyper-responsiveness (Chiu et al., 2013). The relationship between nociception and innate immunity, however, remains poorly characterized.
In our previous work, we developed an epicutaneous C. albicans infection model that does not require immunosuppression or dysbiosis for productive infection (Igyártó et al., 2011). In this model, LCs are not required for pathogen clearance in naive mice. LCs were necessary and sufficient, however, for the development of antigen-specific Th17 cells that provided protection from a secondary infection in immune mice (Kashem et al., 2015). The cell types and cytokine networks responsible for resistance to C. albicans in the skin, however, have not been precisely defined. In this report, we examined the cellular sources of IL-17 and the inflammatory cascade that mediated resistance to cutaneous C. albicans infection in naive mice. We found that IL-23 from CD301b+ dDCs that includes the CD11b+ dDC subset drove expansion and IL-17 production by dermal γδ T cells, resulting in protection from C. albicans. In addition, transient receptor potential cation channel subfamily V member 1 (TRPV1) nociceptive neurons and the neuropeptide calcitonin gene-related peptide (CGRP) acted upstream of CD301b+ dDCs to drive IL-23 expression. Thus, we have defined a sequence of coordinated events occurring early during C. albicans skin infection that link nociceptive neurons, dendritic cells, and γδ T cells in establishing host defense against a fungal infection in the skin.
RESULTS
IL-17A from γδ T Cells Is Required for Resistance to Cutaneous Candidiasis
In the oral mucosa, IL-17 from αβ T cells, γδ T cells, and ILCs has been reported to be required for resistance to C. albicans infection (Conti et al., 2014, 2009; Gladiator et al., 2013; Gladiator and LeibundGut-Landmann, 2013). We have previously described a model of cutaneous candidiasis in which C. albicans yeast is applied onto the shaved back skin of immunocompetent mice. Infection, as measured by CFUs in the skin, peaks 2–3 days after inoculation but gradually declines over the course of 5 days (Igyártó et al., 2011). To determine whether IL-17 was required to control infection, we skin-infected mice deficient in IL-17A and IL-17F production (Il17af−/− mice). As expected, mice lacking IL-17A were highly susceptible to infection and had higher C. albicans burden 3 days after infection (Figure 1A). To identify the cellular source of IL-17A, we first analyzed the composition of the cellular infiltrate found in the skin of infected mice. Lymphoid cells in the skin can be identified by flow cytometric analysis of collagenase-digested skin gated on CD90.2+lin− (B220, CD11c, CD11b, F4/80) cells as TCRγδhi TCRβlo DETCs, TCRγδmidTCRβlo dermal γδ T cells, TCRγδlo TCRβhi αβ T cells (including CD4+ and CD8+ T cells), and a TCRlo population that includes NK and innate lymphoid cells (Figure 1B; Riol-Blanco et al., 2014). During infection, the numbers of dermal γδ T cells and CD4+ αβ T cells were increased compared with naive mice (Figure 1C). Intracellular flow cytometry revealed that approximately 3% of the CD90.2+ cells in infected skin expressed IL-17A (Figure 1D). When we subsetted these cells, we observed that dermal γδ T cells were the dominant source of IL-17A with a small amount contributed by CD4+TCRβ+ cells (Figure 1E). Individual dermal γδ T cells appeared to produce more IL-17A than CD4+TCRβ+ cells based on MFI. DETCs, CD8+ TCRβ+ cells, and CD90.2+TCR− innate lymphoid subsets did not produce appreciable amounts of IL-17.
Figure 1. Dermal γδ T Cells, but Not αβ T Cells, Are Required for IL-17-Mediated Protection against C. albicans.

Cohorts of mice were infected on their shaved dorsum with 2 × 108 CFU C. albicans.
(A) Day 3 whole skin homogenates from wild-type or Il17af−/− infected mice were plated onto YPAD agar plates and incubated at 30°C for 48 hr. The number of colony-forming units (CFU) per cm2 of skin is shown.
(B) Single-cell suspensions from whole skin was obtained by enzymatic digestion and analyzed by flow cytometry gated as singlets, live/dead excluded, CD90.2+, and Lineage− (B220−CD11c−CD11b−F4/80−). Expression of TCRβ and TCRγδ is used to identify lymphoid subsets in the skin.
(C) The numbers in each lymphocyte subset as in (B) in naive (white) and C. albicans-infected (black) mice are shown.
(D) Cells isolated from skin were re-stimulated with PMA and ionomycin in the presence of monensin and gated as in (B). Expression of IL-17A by CD90.2+ lymphocytes is shown.
(E) Representative expression of IL-17A and interferon-γ (IFN-γ) by individual subsets from (D) are shown.
(F) C. albicans CFUs isolated from the skin of WT, Rag1−/−, and Tcra−/− mice 3 days after infection is shown.
(G) Lethally irradiated B6.SJL-Ly5.2 were reconstituted with bone marrow from WT C57BL/6 or Tcrd−/− mice and rested for 12 weeks. C. albicans CFUs 3 days after infection is shown.
Error bars represent mean ± SEM. Student’s unpaired t test representative of *p < 0.05, **p < 0.01, ***p < 0.001. Data points in (A), (F), and (G) represent individual animals. Data are representative of at least three mice per group and at least three independent experiments.
To determine whether dermal γδ T cells and αβ T cells were required for resistance to C. albicans, we skin-infected control, Rag1−/− (lacking both T cell subsets), and Tcra−/− (lacking αβ T cells) mice (Figure 1F). Fungal burden was increased in Rag1−/− but not Tcra−/− mice, indicating a requirement for γδ T cells. To confirm this finding and exclude any role for DETCs, we generated mixed bone marrow chimeras in which Tcrd−/− bone marrow was transferred into irradiated B6.SJL-Ly5.2 (referred to as CD45.1 WT) hosts. Because DETCs are radio resistant, these mice have a selective absence of dermal γδ T cells (unpublished observations) (Honjo et al., 1990). As expected, Tcrd−/− → CD45.1 WT mice had a significantly higher fungal burden than the WT →CD45.1 WT controls (Figure 1G). These results indicate that resistance to primary C. albicans skin infection requires IL-17 produced by dermal γδ T cells.
IL-17-Producing γδ T Cells Require IL-23
We next sought to identify the factors required for production of IL-17 by dermal γδ T cells. In vitro, γδ T cell activation has been shown to require IL-1R and/or IL-23R signaling (Cai et al., 2014; Gray et al., 2011). Because patients with deficiencies in IL-23 signaling but not IL-1 signaling suffer from chronic mucocutaneous candidiasis, we focused on the role of IL-23 in response to C. albicans infection (Huppler et al., 2012; Smeekens et al., 2011). Skin infection of IL-23p19-deficient mice (referred to as Il23a−/−) resulted in a large reduction of dermal γδ T cells compared with naive controls (Figure 2A). In addition to fewer cells, there was also a reduced frequency of cells expressing IL-17, resulting in a dramatic decrease in the number of dermal γδ T cells producing IL-17 (Figures 2B and 2C). Skin infection with C. albicans was associated with a strong induction of dermal γδ T cell proliferation as evidenced by the large number of Ki-67-positive cells after infection (Figure 2D). In the absence of IL-23, proliferation was greatly curtailed, consistent with observations that IL-23 drives proliferation of γδ T cells in vitro (Cai et al., 2014). Consistent with the importance of IL-17 from dermal γδ T cells, we found that the fungal burden was greatly increased in Il23a−/− mice (Figure 2E). Exogenous administration of IL-17A restored fungal burdens to the level observed in WT mice, indicating that IL-23 acts upstream of IL-17 (Figure 2E). These results show that IL-23 is required for proliferation and production of IL-17A by dermal γδ T cells during C. albicans infection.
Figure 2. IL-23 Is Required for IL-17 Secretion and Proliferation of Dermal γδ T Cells.

Mice were infected on their dorsum with 2 × 108 CFUs C. albicans. Three days later, single-cell suspensions from whole skin was obtained by enzymatic digestion and subjected to flow cytometry.
(A) Total number of dermal γδ T cells in WT or Il23a−/− mice is shown in naive (white) or infected (black) mice.
(B) As in (A), cells were re-stimulated with PMA and ionomycin in the presence of monensin and analyzed for IL-17 expression.
(C) The number of IL-17A-expressing dermal γδ T cells in (B) is shown in naive (white) or infected (black) mice.
(D) Dermal γδ T cells in WT or Il23a−/− mice were analyzed for proliferation by nuclear Ki-67 staining compared to naive mice.
(E) C. albicans CFU from day 3 skin homogenates from WT, Il23a−/−, or Il23a−/− mice administered 1 μg intradermal recombinant IL-17A (rIL-17A) 1 day prior to and 1 day after infection.
Error bars represent mean ± SEM. Student’s unpaired t test representative of *p < 0.05, **p < 0.01, ***p < 0.001. Data points in (E) represent individual animals. Data are representative of at least three independent experiments with cohorts of at least six mice.
IL-23 from CD301b+ Dermal DCs Is Critical for IL-17 Production by Dermal γδ T Cells and Pathogen Resistance
In other models, DCs have been shown to be an important source of IL-23 (Satpathy et al., 2013; Wohn et al., 2013). To determine whether IL-23 from DCs was required for cutaneous resistance to C. albicans, we skin-infected control and CD11c-DTR mice 1 day after treatment with diphtheria toxin (DT) to deplete all dendritic cells. DC-depleted mice expressed lower amounts of Il23a mRNA in the skin 16 hr after infection (Figure 3A). In addition, DC-deficient mice were unable to clear C. albicans as efficiently as controls (Figure 3B). These findings indicate that dendritic cells are a non-redundant source of IL-23 that is required for clearance of C. albicans skin infection in vivo.
Figure 3. CD301b+ Dermal DCs Are Required for IL-23-Mediated Protection against C. albicans.

CD11c-DTR mice were treated with 1 μg of diphtheria toxin (DT) or vehicle 1 day before infection with 2 × 108 CFU C. albicans.
(A) Il23a expression in whole skin tissue 16 hr after infection as analyzed by qRT-PCR is shown.
(B) C. albicans CFUs in whole skin from control and DC-depleted mice 3 days after infection is shown.
(C–F) Mice lacking LCs and CD103+ dDCs (LCΔBatf3Δ) were infected with C. albicans.
(C) Il23a expression in skin was analyzed 16 hr after infection by qRT-PCR.
(D and E) Number (D) and percent (E) of IL-17A-expressing dermal γδ T cells in mice kept naive (white) or infected with C. albicans (black) is shown.
(F) Skin CFUs 3 days after C. albicans infection in LCΔBatf3Δ depleted mice is shown.
(G–J) Mgl2-DTR mice were treated with 1 μg of DT or vehicle 1 day before infection with C. albicans.
(G) Il23a expression in skin was assessed by qRT-PCR 16 hr after infection.
(H and I) Number (H) and percent (I) of IL-17A-expressing dermal γδ T cells in mice kept naive (white) or infected with C. albicans (black) is shown.
(J) CFUs in skin 3 days after infection is shown.
Error bars represent mean ± SEM. Student’s unpaired t test representative of *p < 0.05, **p < 0.01, ***p < 0.001. Data points in (B), (F), and (J) represent individual animals. Data are representative of at least three independent experiments with cohorts of at least three animals.
There are at least three major subsets of DCs in the skin. To evaluate whether LCs or CD103+ dDCs are an obligate source of IL-23, we crossed huLangerin-DTA mice that lack LCs with Batf3−/− mice that lack CD103+ dDCs to generate human Langerin-DTA×Batf3−/− mice (referred to as LCΔBatf3Δ) that lack both DC subsets (Edelson et al., 2010; Kashem et al., 2015; Welty et al., 2013). Production of IL-23, numbers of dermal γδ T cells, expression of IL-17 by dermal γδ T cells, and C. albicans CFUs were all unaffected by the absence of these DC subsets (Figures 3C–3F). This indicates that CD103+ dDCs and LCs are not required for innate responses to C. albicans and that another source of IL-23 is sufficient.
The C-type lectin macrophage galactose lectin type 2 receptor (Mgl2; CD301b) is expressed by the bulk of CD103− dDCs in the dermis and can be ablated with Mgl2-DTR mice (Kashem et al., 2015; Kumamoto et al., 2013). CD301b+ dDCs are heterogeneously comprised of CD11b+CD64− conventional DCs and the recently defined CD11b+CD64+ cells that are now ontogenically defined as tissue macrophages (McGovern et al., 2014). C. albicans infection of Mgl2-DTR mice depleted of CD301b+ DCs by injection of DT showed greatly decreased IL-23 expression in the skin compared to PBS-treated mice (Figure 3G). In addition, the numbers of dermal γδ T cells and IL-17 production by γδ T cells was reduced (Figures 3H and 3I). Fungal burdens were also significantly increased in these mice (Figure 3J).
To determine whether IL-23 from the CD301b+ dDCs was required for dermal γδ T cell production of IL-17, we generated a series of mixed bone marrow chimeras. A 1:1 ratio of bone marrow isolated from Mgl2-DTR mice and bone marrow from either WT or Il23a−/− mice was transferred into irradiated CD45.1 mice. After 12 weeks of reconstitution, mice were treated with either vehicle or DT to generate cohorts in which CD301b+ dDCs are either IL-23 sufficient or deficient. Mgl2-DTR+Il23a−/− → WT mice in which CD301b+ dDCs are IL-23 deficient demonstrated reduced expression of IL-17 by dermal γδ T cells compared with controls (Figure 4A). The numbers and proliferation of dermal γδ T cells was also reduced (Figures 4A–4D). Finally, the CFUs of C. albicans were increased in mice that lacked IL-23 production from CD301b+ dDCs (Figure 4E). Thus, CD301b+ dDCs are an obligate source of IL-23 that is required for activation and expansion of dermal γδ T cells during C. albicans infection.
Figure 4. IL-23 from CD301b+ Dermal DCs Is Necessary for IL-17 Secretion and Proliferation of Dermal γδ T Cells.

Lethally irradiated B6.SJL-Ly5.2 (CD45.1) mice were re-constituted with 1:1 ratio of bone marrow from Mgl2-DTR mice and either WT or Il23a−/− mice. After 12 weeks, mice were treated with 1 μg of diphtheria toxin (DT) or vehicle 1 day before infection with 2 × 108 CFU C. albicans.
(A) IL-17A expression of dermal γδ T cells in mice 3 days after C. albicans infection is shown.
(B) The number of IL-17-expressing dermal γδ T cells in the skin from (A) is shown.
(C and D) As in (A), Ki-67 staining of dermal γδ T cells (C) and quantification of Ki-67+ dermal γδ T cells (D) 3 days after infection is shown.
(E) C. albicans skin CFUs from mixed bone marrow chimeric mice 3 days after infection.
Student’s unpaired t test representative of *p < 0.05, **p < 0.01, ***p < 0.001. Error bars represent mean ± SEM. Data points in (E) represent individual animals. Data are representative of at least three independent experiments with cohorts of at least three mice.
Nociceptive Neurons Augment Immunity to C. albicans
Cutaneous sensation of temperature and pain is transmitted by sensory fibers that express the cation channel TRPV1. The role of nociceptors has been recently appreciated for IL-23-mediated imiquimod-induced inflammation (Riol-Blanco et al., 2014). In addition, pathogens such as S. aureus can directly activate neurons isolated from the dorsal root ganglion (DRG) and induce pain hyper-responsiveness (Chiu et al., 2013). To determine whether C. albicans could directly affect sensory neurons, we measured changes in intracellular calcium via intracellular Fura2-AM fluorescence in primary cultures of mouse DRG neurons exposed to heat-killed C. albicans (HKCA). Cells were stimulated with 30 mM potassium chloride (KCl) initially to determine viability before being exposed to HKCA (Figure 5A). After a 10 min wash-out period, cells were re-stimulated with 500 mM of the TRPV1-specific agonist capsaicin (Cap) to assess neuronal specificity. Out of 53 total DRG neurons examined, Cap induced calcium transients in 24 neurons, identifying them as TRPV1+ nociceptors (Figure 5B). Within the group of Cap-sensitive neurons, 17 also responded to HKCA. Furthermore, zymosan, a yeast cell wall derivative, also directly activated sensory neurons from the DRG (Figure 5C). Thus, the majority of TRPV1+ neurons are directly activated by C. albicans as measured by calcium mobilization.
Figure 5. Nociception and Sensory Nerves Are Crucial Mediators of Anti-fungal Immunity.

(A) Sensory neurons were dissociated from dorsal root ganglion of WT mice and cultured for 18–24 hr before being loaded with the ratiometric intracellular calcium indicator Fura2-AM. Cultured neurons were stimulated sequentially with 30 mM KCl (10 s), heat-killed 1 × 107 CFU C. albicans (HKCA; 15 s), and 500 mM capsaicin (Cap; 10 s). Traces showing calcium flux as indicated by the 340/380 ratio visualized by microscopy from three individual cells are shown. Dark blue line represents cell that responded to HKCA and Cap, dotted blue line represents cell that responded to Cap but not HKCA, and red dashed line represents cell that responded to HKCA but not Cap. Data are representative of 53 cells tested.
(B) Scaled venn diagram representation of the 53 DRG neuron responsiveness to different stimuli.
(C) Cultured neurons were stimulated sequentially with 30 mM KCl (10 s), 1 × 107 CFU zymosan (15 s), and 30 mM KCl (10 s). Traces showing calcium flux as indicated by the 340/380 ratio visualized by microscopy from a single neuronal cell representative of ten cells.
(D–J) Cohorts of WT mice were injected s.c. with resineferatoxin (RTX) or vehicle (EtOh).
(D) Mice were checked for their latency to tail-flick after submersion in a 52°C waterbath.
(E and F) Nocifensive behavior (E) was assessed for 10 min after injection of vehicle or 5 μg of capsaicin into the hindpaws of EtOh- or RTX-treated mice. Neg is defined as EtOH mice treated with vehicle. Mice were infected with C. albicans and (F) Il23a expression was quantified by qRT-PCR in skin 16 hr later.
(G) IL-17A expression by dermal γδ T cells 3 days after infection is shown.
(H and I) The number (H) and percentage (I) of IL-17-expressing dermal γδ T cells is shown in naive (white) and infected (black) mice.
(J) C. albicans CFU in vehicle- or RTX-treated mice 3 days after infection is shown.
(K–M) Mice were surgically denervated on one lateral side of their dorsum and infected 7 days later with equal CFUs of C. albicans.
(K and L) Il23a (K) and Il17a (L) expression in skin was quantified by qRT-PCR 16 hr and 60 hr later, respectively.
(M) CFU from mechanically denervated side of infected mice and mock denervated side are shown. Data from individual mice are connected by a line.
Error bars represent mean ± SEM. Student’s unpaired (D–F; H–J) or paired (K–M) t test representative of *p < 0.05, **p < 0.01, ***p < 0.001. Data are representative of at least three independent experiments with cohorts of at least five mice.
We next examined whether sensory nociceptive fibers participated in anti-fungal responses. To ablate TRPV1+ sensory neurons, cohorts of mice were treated subcutaneously with escalating amounts of the capsaicin analog resineferatoxin (RTX) and rested for 4 weeks (Riol-Blanco et al., 2014). Treated mice demonstrated increased tail flick latency to heat and absent nocifensive response to capsaicin administration, confirming effective ablation of TRPV1+ nociceptors (Figures 5D and 5E). Mice were then skin infected with C. albicans. RTX-treated mice demonstrated significantly decreased levels of IL-23 compared with vehicle-treated controls (Figure 5F). In addition, the numbers IL-17+ dermal γδ T cells were reduced and the fungal burden was increased (Figures 5G–5J).
Recently, TRPV1 has been shown to be expressed and functional on T cells and keratinocytes (Bertin et al., 2014; Graham et al., 2013). To insure that RTX treatment was not affecting these cells in addition to nociception, we mechanically disrupted the cutaneous nerves on one lateral half of the mice dorsum (Ostrowski et al., 2011). Infection on the denervated side demonstrated less Il23a and Il17a mRNA expression as well as higher fungal burden than the mock denervated side (Figures 5K–5M). These data indicate that sensory nerves and TRPV1+ nociceptors are crucial for the generation of cutaneous IL-23 and IL-17 responses and protection against C. albicans.
CGRPα Drives IL-23 Expression in CD301b+ dDCs
TRPV1+ neurons release numerous neuropeptides in the periphery (Engel et al., 2011; Meng et al., 2009). Given the importance of CD301b+ dDCs in our system, we explored the expression pattern of neuropeptide receptors on cutaneous DC subsets from data generated by Immunological Genome Consortium (Miller et al., 2012). Notably, CD11b+ DCs express high levels of the CGRP receptors Calrcl and Ramp3 (Figure 6A). We confirmed the microarray findings by analyzing cell surface CALCRL expression on CD11b+ dDCs by flow cytometry (Figure 6B). This is consistent with other reports showing expression of CGRP receptors by CD11b+ DCs in other tissues (Li et al., 2014).
Figure 6. Neuropeptide CGRPα Drives IL-23 Response and Cutaneous Fungal Resistance.

(A) Gene expression patterns of CGRP, substance P, neuropeptide Y, somatostatin, and vaso-active intestinal peptide receptors on the indicated skin dendritic cell subsets. Raw data were obtained from the Immunological Genome Consortium.
(B) Expression of CGRP receptor CALCRL (red) with isotype control (blue) on skin CD11b+ DC subsets.
(C) Cultured DRG neurons were incubated for 10 min in HEPES buffer to measure basal release (Neg) and then incubated for 10 min in HEPES buffer (release, open bars) or HKCA (release, black bars). CGRP levels in supernatants and cell lysates (lysate) were determined by ELISA.
(D–I) Cohorts of WT mice were treated intradermally with either PBS or 0.5 μg of peptide CGRPα or inhibitor CGRP32-37 on days −1, 0, 1, and 2 of C. albicans infection.
(D) IL-17A expression of PMA and ionomycin-stimulated dermal γδ T cells in mice 3 days after infection is shown.
(E) Skin CFUs 3 days after infection from mice treated with PBS, CGRPα, or inhibitor CGRP32-37 is shown.
(F and G) 4-week-old WT mice were injected with RTX or vehicle (EtOH) subcutaneously in flank. 4 to 6 weeks later, mice were treated with either PBS, CGRPα, or inhibitor CGRP32-37.
(F) Expression of Il23a in skin 16 hr after infection was determined by qRT-PCR.
(G) Skin CFUs 3 days after infection is shown.
(H and I) Mgl2-DTR mice were treated with 1 μg of diphtheria toxin (DT) or vehicle 1 day before infection with C. albicans. Mice were given intradermal PBS or CGRPα on days −1, 0, 1, and 2 of C. albicans infection.
(H) Expression of Il23a in skin 16 hr after infection was assessed by qRT-PCR.
(I) Skin CFU 3 days after infection is shown.
Student’s unpaired t test representative of *p < 0.05, **p < 0.01, ***p < 0.001. Data are representative of at least three independent experiments with cohorts of at least three mice.
To determine whether sensory neurons released CGRP in response to C. albicans, we measured CGRP levels after stimulation of cultured DRG neurons with HKCA. HKCA stimulation resulted in increased concentrations of CGRP in the supernatant of DRG cultures (Figure 6C) relative to control treatment. Lysates of HKCA- or control-treated cells had similar concentrations of CGRP, indicating that the CGRP increase in the supernatant of HKCA-stimulated cells was not due to HKCA-induced neuronal damage. Thus, C. albicans directly induce CGRP release from DRG neurons.
Because ablation of nociception by RTX has been shown to reduce the expression of CGRP in the skin and because DRGs exposed to S. aureus supernatant or C. albicans have been shown to increase CGRP expression in vivo, we next tested whether CGRP participated in the anti-C. albicans immune response (Chiu et al., 2013; Hsieh et al., 2012). We found that daily intradermal administration of CGRPα into the site of C. albicans infection increased the number of IL-17+ dermal γδ T cells in the skin and reduced the fungal burden. Conversely, injection of the CGRP antagonist, CGRP32-37, reduced levels of IL-23, reduced the number of IL-17+ dermal γδ T cells in the skin, and increased the fungal burden (Figures 6D–6F). Moreover, injection of CGRPα in RTX-treated mice was able to restore IL-23 expression and fungal burden to levels seen in control mice (Figures 6F and 6G).
CGRP, as well as other neuropeptides, have been previously determined to be directly anti-microbial against C. albicans (Augustyniak et al., 2012; El Karim et al., 2008). To determine whether CGRP could suppress C. albicans infection independently of IL-23-mediated inflammation, we treated CD301b+ depleted Mgl2-DTR mice with CGRPα or vehicle. As shown previously, DT-treated Mgl2-DTR mice showed reduced IL-23 and an exaggerated fungal burden (Figures 6H and 6I). Unlike RTX-treated mice, administration of CGRPα to Mgl2-DTR mice did not rescue the phenotype. Thus, these data indicate that CGRPα, produced by sensory nerves in response to C. albicans, acts upstream of CD301b+ dDCs to induce IL-23 expression that induces IL-17 production from dermal γδ T cells, resulting in an effective innate immune response to cutaneous C. albicans infection.
DISCUSSION
Our work shows that resistance to C. albicans skin infection requires IL-17A and identifies dermal γδ T cells as the dominant and primary source of this cytokine. IL-23 derived from CD301+ dDCs was required for proliferation and IL-17A production by dermal γδ T cells. Ablation of dermal γδ T cells, CD301b+ dDCs, or selective deletion of IL-23 in CD301b+ dDCs greatly increased susceptibility to C. albicans skin infection. In addition, we found that sensory neurons were capable of directly responding to C. albicans. Ablation of nociceptors or cutaneous nerves reduced production of IL-23 from CD301b+ dDCs and resulted in inefficient clearance of C. albicans through a CGRP-dependent mechanism. Thus, we have defined a series of coordinated events linking nociceptive sensory neurons, dendritic cells, and γδ T cells that together provide critical host defense to a fungal pathogen early during skin infection.
Our observation that Il17af−/− mice were highly susceptible to skin infection with C. albicans is consistent with the well-documented requirement for this cytokine in protection against C. albicans at barrier surfaces in both humans and mice (Bär et al., 2014; Conti et al., 2009, 2011, 2014; Conti and Gaffen, 2010; Gladiator et al., 2013; Gladiator and LeibundGut-Landmann, 2013; Smeekens et al., 2013; Trautwein-Weidner et al., 2015). In mice, Th17 cells provide protection from secondary infection with C. albicans (Hernández-Santos et al., 2013; Kashem et al., 2015). C. albicans is a commensal microorganism in humans, but not in mice. Thus, protection in naive mice relies on IL-17 from cells of the innate immune system that can provide immediate protection rather than Th17 cells that arise only several days after infection. In the well-studied oropharyngeal model of mucosal candidiasis, γδ T cells, natural IL-17+ αβ T cells, and ILC3s have been demonstrated to be important sources of IL-17 (Conti et al., 2009, 2014; Gladiator et al., 2013). In the skin, C. albicans infection induced production of IL-17 primarily from dermal γδ T cells. These cells are also the dominant source of IL-17 in the response to S. aureus skin infection or imiquimod application (Cho et al., 2010; Gray et al., 2011, 2013). Ablation of dermal γδ T cells increased fungal burden, confirming the importance of this cell type. We also observed a small subset of IL-17-producing CD4+ T cells that might represent natural Th17 cells, but they were not an obligate source of IL-17 in the skin. Notably, DETCs, ILC3s, and CD8+ T cells that produce IL-17 in other models were not important sources during C. albicans infection.
Dermal γδ T cells constitutively express both subunits of the receptor for IL-23 and proliferate in response to IL-23 in vitro (Cai et al., 2014; Gray et al., 2011). In vivo, both C. albicans and imiquimod application induce IL-17 production by dermal γδ T cells (Awasthi et al., 2009; Gray et al., 2013). We found that dermal γδ T cells required IL-23 for both induction of IL-17 and proliferation. Notably, although proliferation and IL-17 production was greatly diminished in Il23a−/− mice, it was not completely abolished. This might represent a varying requirement for IL-23 within heterogeneous populations of dermal γδ T cells. In addition, IL-1β is known to be induced during C. albicans infection and might be partially sufficient to activate dermal γδ T cells in the absence of IL-23 (Cai et al., 2014; Gray et al., 2011; Joly et al., 2009).
In both the skin and the small intestine, DCs are the primary source of IL-23 during inflammation (Igyártó et al., 2011; Kinnebrew et al., 2012; Wohn et al., 2013; Yoshiki et al., 2014). We have previously observed that LCs and CD11b+ dDCs produce much higher levels of IL-23 mRNA than CD103+ dDCs during C. albicans skin infection (Igyártó et al., 2011). Our current observation that mice lacking CD301b+ dDCs (Mgl2-DTR mice) had reduced levels of IL-23 and reduced resistance to C. albicans infection coupled with the absence of a phenotype in mice lacking both LCs and CD103+ dDCs (LCΔBatf3Δ mice) argues that CD301b+ dDCs are the source of IL-23. This was confirmed using mixed bone marrow chimeras in which CD301b+ dDCs were rendered IL-23 deficient. It is important to note that CD301b+ dDCs are heterogeneous and include all CD11b+ conventional DCs in the dermis but also include populations of CD64+ monocyte-derived DCs and resident macrophages (Kashem et al., 2015). The depletion of CD301b using DTR-depleter mice precluded a definitive identification of which subset within the CD301b+ population was required. In support of CD11b+ dDCs, others have found using Itgax-cre × Irf4fl mice that CD11b+ DCs induce IL-17 expression in skin CD8+ T cells after S. epidermidis colonization and in lung CD4+ T cells after Aspergillus fumigatus infection (Naik et al., 2015; Schlitzer et al., 2013). Human homologs of all these subsets, however, make IL-23 in similar amounts, raising the possibility that CD301b+ cells other than CD11b+ dDCs might also contribute IL-23 during C. albicans infection (Haniffa et al., 2012; Schlitzer et al., 2013).
The interaction of nociceptive neurons and dermal dendritic cells has recently been appreciated. TRPV1+ nociceptive nerve fibers in the skin are in close contact with dermal DCs and are required for their production of IL-23 in the context of imiquimod-induced skin inflammation (Riol-Blanco et al., 2014). Denervation also reduces disease severity in patients with the IL-17- and IL-23-mediated autoimmune diseases psoriasis and rheumatoid arthritis, as well as in mouse models of these diseases (Joseph et al., 2005; Thompson and Bywaters, 1962; Ostrowski et al., 2011; Riol-Blanco et al., 2014; Stangenberg et al., 2014). Patients with spinal cord injuries have decreased levels of IL-23 and IL-17 as well as frequent fungal skin infections below the site of neurological lesion (Jones and Jones, 2014; Nash, 2000; Rubin-Asher et al., 2005). In addition, the skin pathogen S. aureus directly activates sensory nerves resulting in hyper-responsiveness to pain, and the yeast cell wall product, zymosan, has been used extensively to experimentally induce pain (Chiu et al., 2013; Ren and Dubner, 1999).
Our data connect these observations and demonstrate that sensory neurons can directly sense a pathogen and then enhance host resistance via secretion of neuropeptides. We found that CD301b+ dDCs have reduced expression of IL-23 after RTX ablation of TRPV1+ neurons or physical denervation resulting in reduced γδ T cell activation and reduced resistance to C. albicans infection. Expression of IL-23 and host defense could be inhibited by administering the CGRP peptide antagonist, CGRP32-37, and augmented by the addition of the CGRP agonist, CGRPα. Moreover, CGRPα rescued IL-23 responses in RTX-treated WT mice but did not rescue mice lacking CD301b+ dDCs, indicating that CGRPα does not directly inhibit C. albicans proliferation or function indirectly through other cell types such as keratinocytes or fibroblasts (Roggenkamp et al., 2013). Thus, CGRPα acts downstream of TRPV1+ nociceptors but acts upstream of CD301b+ dDCs. Notably, cultured DRGs were able to flux calcium and release CRGP in response to exposure to C. albicans. Many of these DRG neurons were also capsaicin responsive, which is consistent with the observed reduction of anti-C. albicans responses in RTX-treated mice. Capsaicin-insensitive DRGs also responded to C. albicans, suggesting that other sensory neurons might also participate in the anti-C. albicans response. Although our data indicate that C. albicans can directly activate sensory neurons in DRG cultures, it does not negate the likely role of inflammation-induced pain response or indirect activation of nociceptors in vivo.
We focused on CGRP rather than other neuropeptides based on the high expression levels of CGRP receptors on CD11b+ dDCs and the ability of C. albicans to directly induce CGRP release by DRG neurons. In the lung, these DCs also express CGRP receptors and promote airway hyperresponsiveness (Li et al., 2014). In murine vulvovaginal candidiasis, pain was accompanied by increased expression of CGRP in peripheral nerves (Farmer et al., 2011). In addition, CGRP has also been implicated in the pathogenesis of psoriasis (Assas et al., 2014; Saraceno et al., 2006). It is important to note that although our data highlight the importance of CGRPα, we have not excluded a role for other neuropeptides in host defense against C. albicans.
In summary, we have elucidated a mechanism of innate host defense against C. albicans infection in the skin that involves pain-sensing neurons, dermal dendritic cells, and dermal γδ T cells as well as the soluble factors CGRPα, IL-23, and IL-17. This work provides insight into the complex interplay of cells of the innate immune system at a barrier tissue and offers potential avenues of therapeutic interventions to suppress IL-23- and/or IL-17-mediated autoimmune skin diseases and augment antimicrobial therapy.
EXPERIMENTAL PROCEDURES
Mice
Mgl2eGFP-DTR/wt (referred to as Mgl2-DTR), human Langerin-DTA×Batf3−/− (referred to as LCΔBatf3Δ), CD11c-DTR, Rag1−/−, Tcra−/−, Tcrd−/−, Il23a−/−, and Il17af−/− mice have been previously described (Edelson et al., 2010; Ghilardi et al., 2004; Haas et al., 2012; Itohara et al., 1993; Jung et al., 2002; Kaplan et al., 2005; Kumamoto et al., 2013; Mombaerts et al., 1992a, 1992b; Welty et al., 2013). See Supplemental Experimental Procedures for additional details.
Generation and Testing of Bone Marrow Chimeric Mice
Recipient mice received two split doses at 500 cGy each followed by 5 × 106 bone marrow donor cells 24 hr later. See Supplemental Experimental Procedures for additional details.
DC Depletion with Diphtheria Toxin
CD11c-DTR and Mgl2-DTR mice were i.p. injected with 1 μg of diphtheria toxin (List Biological Laboratories) 1 day before infection.
Flow Cytometry
Single-cell skin suspensions and flow cytometry were performed as previously described (Kashem et al., 2015). Antibodies used in study and detailed methods are further described in the Supplemental Experimental Procedures.
qPCR
Isolation of RNA and qPCR was performed as previously described (Kashem et al., 2015). Detailed methods are further described in the Supplemental Experimental Procedures.
Candida albicans Strains
Growth of C. albicans strain SC5314 occurred after inoculation of a colony at 30°C in YPAD overnight and, the next day, diluted 1:10 and cultured in 30°C in YPAD until OD600 reached 1.5 and then washed and re-suspended at 4 × 109 CFU/ml in PBS.
Infection Models
The skin infection was performed as described (Haley et al., 2012; Igyártó et al., 2011) and is fully described in the Supplemental Experimental Procedures. In some instances, CGRPα or CGRP32-37 (Genscript) was administered intradermally at days −1, 0, 1, and 2 during infection at total 0.5 μg per dose spread over 10 sites in the back. When specified, 1.0 μg recombinant IL-17A (R&D Biosystems) was administered on days −1 and 1 during infection.
Denervation
Ablation of nociception was obtained using resineferatoxin (RTX; LC Laboratories) as previously described (Riol-Blanco et al., 2014). Mechanical denervation was performed as previously described (Ostrowski et al., 2011; Siebenhaar et al., 2008). Both are further described in the Supplemental Experimental Procedures.
Behavioral Assays
Ethanol- or resineferatoxin-treated mice were rested 24 hr prior to experiment to allow for adjustment to the experimenter before behavioral assessment assays. Responsiveness to noxious heat stimuli was performed as previously described (Bannon and Malmberg, 2007). Assay is further described in the Supplemental Experimental Procedures.
DRG Cultures
Primary cultures of mouse DRG neurons were prepared using methods similar to those previously described (Khasabova et al., 2002). In brief, DRG were enzymatically dissociated in Collagenase-D (1.5 mg/ml, two 60 min incubations; Roche Diagnostics), filtered, and plated onto poly-l-lysine coverslips. See Supplemental Experimental Procedures for additional details.
Calcium Imaging
In brief, 18–24 hr after plating onto coverslips, cells were incubated in the ratiometric intracellular calcium indicator, Fura2-AM (Invitrogen Life Technologies) at 37°C for 1 hr. Coverslips were then transferred to a recording chamber and cells were stimulated with 30 mM KCl (10 s), heat-killed 1 × 107 Candida albicans (15 s), and 500 mM capsaicin (10 s) or 30 mM KCl (10 s). Excitation wavelengths (340- and 380-nm) were used to calculate calcium flux. See Supplemental Experimental Procedures for additional details.
CGRP Release Assay
After 48 hr in culture, DRG neurons (approx. 20,000 cells/well) were washed once with HEPES buffer and then incubated in HEPES buffer for 10 min (for measurement of basal release) followed by a 10 min incubation with HEPES or HKCA resuspended in HEPES buffer (for measurement of stimulated release). CGRP levels in the supernatant were measured by ELISA (Cayman Chemical). See Supplemental Experimental Procedures for additional details.
Microarray Gene Expression Data for Murine Skin DC Subsets
Microarray gene expression data for three murine skin DC subsets (with replicates) was obtained from the Immunological Genome Consortium (Heng et al., 2008; Miller et al., 2012). To analyze gene expression data, we log transformed normalized levels as provided in the original publication and databases. We used median expression to summarize microarray probe-level data and used average expression values between replicates of each DC subset. For visualization, we z-transformed the data.
Statistics and Data Representation
Results are presented as mean ± standard error unless noted and groups were compared by unpaired Student’s t test using GraphPad Prism software. See Supplemental Experimental Procedures for additional details.
Supplementary Material
Highlights.
γδ T cell production of IL-17A inhibits cutaneous Candida albicans infection
CD301b+ dDC production of IL-23 drives IL-17A production by dermal γδ T cells
Sensory neurons directly sense C. albicans and augment IL-23 production
CGRP from TRPV1-positive fibers drives IL-23 production from CD301b+ dDCs
Acknowledgments
We thank the laboratories of Marc Jenkins and Daniel Mueller (University of Minnesota) for Tcra−/− mice, Nico Ghilardi (Genentech) for Il23a−/− mice, Lloyd Miller (Johns Hopkins) for Tcrd−/− mice, Akiko Iwasaki and Yosuke Kumamoto (Yale University) for Mgl2-DTR mice, and Immo Prinz (Hannover Medical School) and Bryce Binstadt (University of Minnesota) for Il17af−/− mice. B. Chicoine provided technical assistance. We also thank the University of Minnesota Research Animal Resources staff for expert animal care and P. Champoux and T. Martin of the Flow Cytometry Core Facility at the Center for Immunology for assistance flow cytometry experiments. This work was supported by grants from the NIH (AR067187 and AR060744) to D.H.K., who is also supported by the Al Zelickson Family endowed professorship. S.W.K. was supported by the University of Minnesota NIH MSTP grant T32 GM008244, Immunology Training Grant T32 AI007313, and University of Minnesota CTSI Translational Research Development Program Grant UL1TR000114.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2015.08.016.
AUTHOR CONTRIBUTIONS
S.W.K. and D.H.K. designed experiments. S.W.K. and C.Y. performed experiments. S.W.K. and D.H.K. wrote the manuscript. M.S.R., C.N.H., and L.V. designed and performed DRG experiments and offered neuroscience expertise. All authors participated in discussions of experimental results and edited the manuscript.
References
- Assas BM, Miyan JA, Pennock JL. Cross-talk between neural and immune receptors provides a potential mechanism of homeostatic regulation in the gut mucosa. Mucosal Immunol. 2014;7:1283–1289. doi: 10.1038/mi.2014.80. [DOI] [PubMed] [Google Scholar]
- Augustyniak D, Nowak J, Lundy FT. Direct and indirect antimicrobial activities of neuropeptides and their therapeutic potential. Curr Protein Pept Sci. 2012;13:723–738. doi: 10.2174/138920312804871139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi A, Riol-Blanco L, Jäger A, Korn T, Pot C, Galileos G, Bettelli E, Kuchroo VK, Oukka M. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol. 2009;182:5904–5908. doi: 10.4049/jimmunol.0900732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon AW, Malmberg AB. Models of nociception: hot-plate, tail-flick, and formalin tests in rodents. Curr Protoc Neurosci. 2007;8:9. doi: 10.1002/0471142301.ns0809s41. [DOI] [PubMed] [Google Scholar]
- Bär E, Whitney PG, Moor K, Reis e Sousa C, LeibundGut-Landmann S. IL-17 regulates systemic fungal immunity by controlling the functional competence of NK cells. Immunity. 2014;40:117–127. doi: 10.1016/j.immuni.2013.12.002. [DOI] [PubMed] [Google Scholar]
- Bertin S, Aoki-Nonaka Y, de Jong PR, Nohara LL, Xu H, Stanwood SR, Srikanth S, Lee J, To K, Abramson L, et al. The ion channel TRPV1 regulates the activation and proinflammatory properties of CD4+ T cells. Nat Immunol. 2014;15:1055–1063. doi: 10.1038/ni.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Xue F, Fleming C, Yang J, Ding C, Ma Y, Liu M, Zhang HG, Zheng J, Xiong N, et al. Differential developmental requirement and peripheral regulation for dermal Vγ4 and Vγ6T17 cells in health and inflammation. Nat Commun. 2014 doi: 10.1038/ncomms4986. Published online June 9, 2014 http://dx.doi.org/10.1038/ncomms4986. [DOI] [PMC free article] [PubMed]
- Chiu IM, Heesters BA, Ghasemlou N, Von Hehn CA, Zhao F, Tran J, Wainger B, Strominger A, Muralidharan S, Horswill AR, et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature. 2013;501:52–57. doi: 10.1038/nature12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JS, Pietras EM, Garcia NC, Ramos RI, Farzam DM, Monroe HR, Magorien JE, Blauvelt A, Kolls JK, Cheung AL, et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J Clin Invest. 2010;120:1762–1773. doi: 10.1172/JCI40891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Gaffen SL. Host responses to Candida albicans: Th17 cells and mucosal candidiasis. Microbes Infect. 2010;12:518–527. doi: 10.1016/j.micinf.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Baker O, Freeman AF, Jang WS, Holland SM, Li RA, Edgerton M, Gaffen SL. New mechanism of oral immunity to mucosal candidiasis in hyper-IgE syndrome. Mucosal Immunol. 2011;4:448–455. doi: 10.1038/mi.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Peterson AC, Brane L, Huppler AR, Hernández-Santos N, Whibley N, Garg AV, Simpson-Abelson MR, Gibson GA, Mamo AJ, et al. Oral-resident natural Th17 cells and γδ T cells control opportunistic Candida albicans infections. J Exp Med. 2014;211:2075–2084. doi: 10.1084/jem.20130877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, Moon C, Albring JC, Ise W, Michael DG, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med. 2010;207:823–836. doi: 10.1084/jem.20091627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Karim IA, Linden GJ, Orr DF, Lundy FT. Antimicrobial activity of neuropeptides against a range of micro-organisms from skin, oral, respiratory and gastrointestinal tract sites. J Neuroimmunol. 2008;200:11–16. doi: 10.1016/j.jneuroim.2008.05.014. [DOI] [PubMed] [Google Scholar]
- Engel MA, Izydorczyk I, Mueller-Tribbensee SM, Becker C, Neurath MF, Reeh PW. Inhibitory CB1 and activating/desensitizing TRPV1-mediated cannabinoid actions on CGRP release in rodent skin. Neuropeptides. 2011;45:229–237. doi: 10.1016/j.npep.2011.03.005. [DOI] [PubMed] [Google Scholar]
- Farmer MA, Taylor AM, Bailey AL, Tuttle AH, MacIntyre LC, Milagrosa ZE, Crissman HP, Bennett GJ, Ribeiroda-Silva A, Binik YM, Mogil JS. Repeated vulvovaginal fungal infections cause persistent pain in a mouse model of vulvodynia. Sci Transl Med. 2011;3:101ra91. doi: 10.1126/scitranslmed.3002613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14:585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghilardi N, Kljavin N, Chen Q, Lucas S, Gurney AL, De Sauvage FJ. Compromised humoral and delayed-type hypersensitivity responses in IL-23-deficient mice. J Immunol. 2004;172:2827–2833. doi: 10.4049/jimmunol.172.5.2827. [DOI] [PubMed] [Google Scholar]
- Gladiator A, LeibundGut-Landmann S. Innate lymphoid cells: new players in IL-17-mediated antifungal immunity. PLoS Pathog. 2013;9:e1003763. doi: 10.1371/journal.ppat.1003763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladiator A, Wangler N, Trautwein-Weidner K, LeibundGut-Landmann S. Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J Immunol. 2013;190:521–525. doi: 10.4049/jimmunol.1202924. [DOI] [PubMed] [Google Scholar]
- Graham DM, Huang L, Robinson KR, Messerli MA. Epidermal keratinocyte polarity and motility require Ca2+ influx through TRPV1. J Cell Sci. 2013;126:4602–4613. doi: 10.1242/jcs.122192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Suzuki K, Cyster JG. Cutting edge: Identification of a motile IL-17-producing gammadelta T cell population in the dermis. J Immunol. 2011;186:6091–6095. doi: 10.4049/jimmunol.1100427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Ramírez-Valle F, Xu Y, Wu S, Wu Z, Karjalainen KE, Cyster JG. Deficiency in IL-17-committed Vγ4(+) γδ T cells in a spontaneous Sox13-mutant CD45.1(+) congenic mouse substrain provides protection from dermatitis. Nat Immunol. 2013;14:584–592. doi: 10.1038/ni.2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas JD, Ravens S, Düber S, Sandrock I, Oberdörfer L, Kashani E, Chennupati V, Föhse L, Naumann R, Weiss S, et al. Development of interleukin-17-producing γδ T cells is restricted to a functional embryonic wave. Immunity. 2012;37:48–59. doi: 10.1016/j.immuni.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Haley K, Igyártó BZ, Ortner D, Bobr A, Kashem S, Schenten D, Kaplan DH. Langerhans cells require MyD88-dependent signals for Candida albicans response but not for contact hypersensitivity or migration. J Immunol. 2012;188:4334–4339. doi: 10.4049/jimmunol.1102759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, Wasan PS, Wang XN, Malinarich F, Malleret B, et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity. 2012;37:60–73. doi: 10.1016/j.immuni.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng TSP, Painter MW Immunological Genome Project Consortium. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 2008;9:1091–1094. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- Hernández-Santos N, Huppler AR, Peterson AC, Khader SA, McKenna KC, Gaffen SL. Th17 cells confer long-term adaptive immunity to oral mucosal Candida albicans infections. Mucosal Immunol. 2013;6:900–910. doi: 10.1038/mi.2012.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjo M, Elbe A, Steiner G, Assmann I, Wolff K, Stingl G. Thymus-independent generation of Thy-1+ epidermal cells from a pool of Thy-1- bone marrow precursors. J Invest Dermatol. 1990;95:562–567. doi: 10.1111/1523-1747.ep12505543. [DOI] [PubMed] [Google Scholar]
- Hsieh YL, Lin CL, Chiang H, Fu YS, Lue JH, Hsieh ST. Role of peptidergic nerve terminals in the skin: reversal of thermal sensation by calcitonin gene-related peptide in TRPV1-depleted neuropathy. PLoS ONE. 2012;7:e50805. doi: 10.1371/journal.pone.0050805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppler AR, Bishu S, Gaffen SL. Mucocutaneous candidiasis: the IL-17 pathway and implications for targeted immunotherapy. Arthritis Res Ther. 2012;14:217. doi: 10.1186/ar3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igyártó BZ, Haley K, Ortner D, Bobr A, Gerami-Nejad M, Edelson BT, Zurawski SM, Malissen B, Zurawski G, Berman J, Kaplan DH. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity. 2011;35:260–272. doi: 10.1016/j.immuni.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itohara S, Mombaerts P, Lafaille J, Iacomini J, Nelson A, Clarke AR, Hooper ML, Farr A, Tonegawa S. T cell receptor δ gene mutant mice: independent generation of α β T cells and programmed rearrangements of γ δ TCR genes. Cell. 1993;72:337–348. doi: 10.1016/0092-8674(93)90112-4. [DOI] [PubMed] [Google Scholar]
- Joly S, Ma N, Sadler JJ, Soll DR, Cassel SL, Sutterwala FS. Cutting edge: Candida albicans hyphae formation triggers activation of the Nlrp3 inflammasome. J Immunol. 2009;183:3578–3581. doi: 10.4049/jimmunol.0901323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TB, Jones C. Th17 pathway genes are primarily downregulated by spinal cord injury. FASEB J. 2014;28:729–4. [Google Scholar]
- Joseph T, Kurian J, Warwick DJ, Friedmann PS. Unilateral remission of psoriasis following traumatic nerve palsy. Br J Dermatol. 2005;152:185–186. doi: 10.1111/j.1365-2133.2005.06330.x. [DOI] [PubMed] [Google Scholar]
- Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagami S, Rizzo HL, Kurtz SE, Miller LS, Blauvelt A. IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans. J Immunol. 2010;185:5453–5462. doi: 10.4049/jimmunol.1001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DH. In vivo function of Langerhans cells and dermal dendritic cells. Trends Immunol. 2010;31:446–451. doi: 10.1016/j.it.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DH, Jenison MC, Saeland S, Shlomchik WD, Shlomchik MJ. Epidermal langerhans cell-deficient mice develop enhanced contact hypersensitivity. Immunity. 2005;23:611–620. doi: 10.1016/j.immuni.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Kashem SW, Igyártó BZ, Gerami-Nejad M, Kumamoto Y, Mohammed J, Jarrett E, Drummond RA, Zurawski SM, Zurawski G, Berman J, et al. Candida albicans morphology and dendritic cell subsets determine T helper cell differentiation. Immunity. 2015;42:356–366. doi: 10.1016/j.immuni.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasabova IA, Simone DA, Seybold VS. Cannabinoids attenuate depolarization-dependent Ca2+ influx in intermediate-size primary afferent neurons of adult rats. Neuroscience. 2002;115:613–625. doi: 10.1016/s0306-4522(02)00449-9. [DOI] [PubMed] [Google Scholar]
- Kinnebrew MA, Buffie CG, Diehl GE, Zenewicz LA, Leiner I, Hohl TM, Flavell RA, Littman DR, Pamer EG. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. 2012;36:276–287. doi: 10.1016/j.immuni.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisand K, Bøe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, Ersvaer E, Perheentupa J, Erichsen MM, Bratanic N, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med. 2010;207:299–308. doi: 10.1084/jem.20091669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto Y, Linehan M, Weinstein JS, Laidlaw BJ, Craft JE, Iwasaki A. CD301b+ dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity. 2013;39:733–743. doi: 10.1016/j.immuni.2013.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Wetzel-Strong SE, Hua X, Tilley SL, Oswald E, Krummel MF, Caron KM. Deficiency of RAMP1 attenuates antigen-induced airway hyperresponsiveness in mice. PLoS ONE. 2014;9:e102356. doi: 10.1371/journal.pone.0102356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod AS, Hemmers S, Garijo O, Chabod M, Mowen K, Witherden DA, Havran WL. Dendritic epidermal T cells regulate skin antimicrobial barrier function. J Clin Invest. 2013;123:4364–4374. doi: 10.1172/JCI70064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald DR. TH17 deficiency in human disease. J Allergy Clin Immunol. 2012;129:1429–1435. doi: 10.1016/j.jaci.2012.03.034. quiz 1436–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern N, Schlitzer A, Gunawan M, Jardine L, Shin A, Poyner E, Green K, Dickinson R, Wang XN, Low D, et al. Human dermal CD14+ cells are a transient population of monocyte-derived macrophages. Immunity. 2014;41:465–477. doi: 10.1016/j.immuni.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J, Ovsepian SV, Wang J, Pickering M, Sasse A, Aoki KR, Lawrence GW, Dolly JO. Activation of TRPV1 mediates calcitonin gene-related peptide release, which excites trigeminal sensory neurons and is attenuated by a retargeted botulinum toxin with anti-nociceptive potential. J Neurosci. 2009;29:4981–4992. doi: 10.1523/JNEUROSCI.5490-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, Pandey G, Leboeuf M, Elpek KG, Helft J, et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol. 2012;13:888–899. doi: 10.1038/ni.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombaerts P, Clarke AR, Rudnicki MA, Iacomini J, Itohara S, Lafaille JJ, Wang L, Ichikawa Y, Jaenisch R, Hooper ML, et al. Mutations in T-cell antigen receptor genes α and β block thymocyte development at different stages. Nature. 1992a;360:225–231. doi: 10.1038/360225a0. [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992b;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmüller W, Deming C, Quinones M, Koo L, Conlan S, et al. Compartmentalized control of skin immunity by resident commensals. Science. 2012;337:1115–1119. doi: 10.1126/science.1225152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, Conlan S, Himmelfarb S, Byrd AL, Deming C, et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature. 2015;520:104–108. doi: 10.1038/nature14052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash MS. Known and plausible modulators of depressed immune functions following spinal cord injuries. J Spinal Cord Med. 2000;23:111–120. doi: 10.1080/10790268.2000.11753518. [DOI] [PubMed] [Google Scholar]
- Ostrowski SM, Belkadi A, Loyd CM, Diaconu D, Ward NL. Cutaneous denervation of psoriasiform mouse skin improves acanthosis and inflammation in a sensory neuropeptide-dependent manner. J Invest Dermatol. 2011;131:1530–1538. doi: 10.1038/jid.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Döffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, Cobat A, Ouachée-Chardin M, Toulon A, Bustamante J, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med. 2010;207:291–297. doi: 10.1084/jem.20091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332:65–68. doi: 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren K, Dubner R. Inflammatory models of pain and hyperalgesia. ILAR J. 1999;40:111–118. doi: 10.1093/ilar.40.3.111. [DOI] [PubMed] [Google Scholar]
- Riol-Blanco L, Ordovas-Montanes J, Perro M, Naval E, Thiriot A, Alvarez D, Paust S, Wood JN, von Andrian UH. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature. 2014;510:157–161. doi: 10.1038/nature13199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggenkamp D, Köpnick S, Stäb F, Wenck H, Schmelz M, Neufang G. Epidermal nerve fibers modulate keratinocyte growth via neuropeptide signaling in an innervated skin model. J Invest Dermatol. 2013;133:1620–1628. doi: 10.1038/jid.2012.464. [DOI] [PubMed] [Google Scholar]
- Rubin-Asher D, Zeilig G, Klieger M, Adunsky A, Weingarden H. Dermatological findings following acute traumatic spinal cord injury. Spinal Cord. 2005;43:175–178. doi: 10.1038/sj.sc.3101697. [DOI] [PubMed] [Google Scholar]
- Saraceno R, Kleyn CE, Terenghi G, Griffiths CEM. The role of neuropeptides in psoriasis. Br J Dermatol. 2006;155:876–882. doi: 10.1111/j.1365-2133.2006.07518.x. [DOI] [PubMed] [Google Scholar]
- Satpathy AT, Briseño CG, Lee JS, Ng D, Manieri NA, Kc W, Wu X, Thomas SR, Lee WL, Turkoz M, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol. 2013;14:937–948. doi: 10.1038/ni.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, Ho AWS, See P, Shin A, Wasan PS, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. 2013;38:970–983. doi: 10.1016/j.immuni.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, Gorman DM, Bowman EP, McClanahan TK, Yearley JH, et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4−CD8− entheseal resident T cells. Nat Med. 2012;18:1069–1076. doi: 10.1038/nm.2817. [DOI] [PubMed] [Google Scholar]
- Siebenhaar F, Magerl M, Peters EMJ, Hendrix S, Metz M, Maurer M. Mast cell-driven skin inflammation is impaired in the absence of sensory nerves. J Allergy Clin Immunol. 2008;121:955–961. doi: 10.1016/j.jaci.2007.11.013. [DOI] [PubMed] [Google Scholar]
- Smeekens SP, Plantinga TS, van de Veerdonk FL, Heinhuis B, Hoischen A, Joosten LAB, Arkwright PD, Gennery A, Kullberg BJ, Veltman JA, et al. STAT1 hyperphosphorylation and defective IL12R/IL23R signaling underlie defective immunity in autosomal dominant chronic mucocutaneous candidiasis. PLoS ONE. 2011;6:e29248. doi: 10.1371/journal.pone.0029248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeekens SP, van de Veerdonk FL, Kullberg BJ, Netea MG. Genetic susceptibility to Candida infections. EMBO Mol Med. 2013;5:805–813. doi: 10.1002/emmm.201201678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stangenberg L, Burzyn D, Binstadt BA, Weissleder R, Mahmood U, Benoist C, Mathis D. Denervation protects limbs from inflammatory arthritis via an impact on the microvasculature. Proc Natl Acad Sci USA. 2014;111:11419–11424. doi: 10.1073/pnas.1410854111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumaria N, Roediger B, Ng LG, Qin J, Pinto R, Cavanagh LL, Shklovskaya E, Fazekas de St Groth B, Triccas JA, Weninger W. Cutaneous immunosurveillance by self-renewing dermal gammadelta T cells. J Exp Med. 2011;208:505–518. doi: 10.1084/jem.20101824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KHG. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Thompson M, Bywaters EG. Unilateral rheumatoid arthritis following hemiplegia. Ann Rheum Dis. 1962;21:370–377. doi: 10.1136/ard.21.4.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautwein-Weidner K, Gladiator A, Nur S, Diethelm P, LeibundGut-Landmann S. IL-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal Immunol. 2015;8:221–231. doi: 10.1038/mi.2014.57. [DOI] [PubMed] [Google Scholar]
- Welty NE, Staley C, Ghilardi N, Sadowsky MJ, Igyártó BZ, Kaplan DH. Intestinal lamina propria dendritic cells maintain T cell homeostasis but do not affect commensalism. J Exp Med. 2013;210:2011–2024. doi: 10.1084/jem.20130728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohn C, Ober-Blöbaum JL, Haak S, Pantelyushin S, Cheong C, Zahner SP, Onderwater S, Kant M, Weighardt H, Holzmann B, et al. Langerin(neg) conventional dendritic cells produce IL-23 to drive psoriatic plaque formation in mice. Proc Natl Acad Sci USA. 2013;110:10723–10728. doi: 10.1073/pnas.1307569110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward Davis AS, Bergsbaken T, Delaney MA, Bevan MJ. Dermal-resident versus recruited γδ T cell response to cutaneous vaccinia virus infection. J Immunol. 2015;194:2260–2267. doi: 10.4049/jimmunol.1402438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiki R, Kabashima K, Honda T, Nakamizo S, Sawada Y, Sugita K, Yoshioka H, Ohmori S, Malissen B, Tokura Y, Nakamura M. IL-23 from Langerhans cells is required for the development of imiquimod-induced psoriasis-like dermatitis by induction of IL-17A-producing γδ T cells. J Invest Dermatol. 2014;134:1912–1921. doi: 10.1038/jid.2014.98. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.