

Abstract
Optical imaging of whole, living animals has proven to be a powerful tool in multiple areas of preclinical research and has allowed noninvasive monitoring of immune responses, tumor and pathogen growth, and treatment responses in longitudinal studies. However, fluorescence-based studies in animals are challenging because tissue absorbs and autofluoresces strongly in the visible light spectrum. These optical properties drive development and use of fluorescent labels that absorb and emit at longer wavelengths. Here, we present a far-red absorbing fluoromodule–based reporter/probe system and show that this system can be used for imaging in living mice. The probe we developed is a fluorogenic dye called SC1 that is dark in solution but highly fluorescent when bound to its cognate reporter, Mars1. The reporter/probe complex, or fluoromodule, produced peak emission near 730 nm. Mars1 was able to bind a variety of structurally similar probes that differ in color and membrane permeability. We demonstrated that a tool kit of multiple probes can be used to label extracellular and intracellular reporter–tagged receptor pools with 2 colors. Imaging studies may benefit from this far-red excited reporter/probe system, which features tight coupling between probe fluorescence and reporter binding and offers the option of using an expandable family of fluorogenic probes with a single reporter gene.
Introduction
Imaging of live animal models has advanced the development and refinement of many oncologic therapies by allowing investigators to specifically and noninvasively monitor clinically relevant parameters such as tumor size and rate of growth, degree of vascularization, immune infiltrates, and responses to therapeutic agents over time (1–3). The ability of in vivo imaging to provide informative data relies upon the ability to tag specific targets of interest with labels that generate signals detectable through tissue. Targeted labeling can be achieved by using reporter/probe systems, a number of which are presently used in different imaging modalities. Human herpes simplex virus type 1 thymidine kinase/[18F]FHBG and sodium iodide symporter/124I reporter/probe pairs are examples of existing methods of selectively accumulating radionuclides in transgene-expressing cells for PET imaging (4–7). Reporter/probe systems are also utilized for optical imaging modalities to target bioluminescent and fluorescent signals. However, most probes generate signals regardless of whether they have localized to their target. Signals from circulating probe can undesirably degrade contrast between the target and its surroundings. A powerful strategy for minimizing this source of background is to use activatable probes that produce signals only in response to specific cues such as low pH (8, 9), endogenous enzyme activity (10, 11), or interaction with reporter gene products. Luciferins, for example, generate light only after luciferase-mediated oxidation, providing exceptional contrast between reporter activity and circulating probe (12). Fluorogenic SNAP-tag substrates provide similar functions for fluorescence imaging, but most of these absorb and fluoresce at wavelengths nonideal for tissue imaging (13–16).
While fluorescence imaging is a staple resource for addressing biological and pharmacological questions in vitro and in cultured cell systems, its potential in animal studies remains underexploited because many existing fluorescent tools are incompatible with the optical properties of tissue. Tissues present overwhelming absorbance, scattering, and autofluorescence, which confound and degrade the quality of data obtained from labels that absorb and emit light in the visible spectrum (17–19). Tissues are more transparent to longer wavelengths of light (700–1000 nm), and this spectral window allows excitation and detection of even deeply embedded optical probes. Ideal reporter/probe systems for in vivo optical imaging should absorb and emit maximally within this window, generate signals specifically from the target, and produce bright signals to minimize detection thresholds and image acquisition times.
Fluoromodules are a class of reporter/probe systems that feature robustly activatable probes that could be optimized, with respect to wavelength, for in vivo imaging. A fluorogenic synthetic dye (a fluorogen) serves as the probe, and a cognate scFv-based fluorogen activating protein (FAP) functions as a reporter. Each component alone is dark. When combined, probe and reporter form a highly fluorescent complex that is similar in molecular weight (24.4 kDa) to monomeric fluorescent proteins such as GFP (20). This fluorescence activation is thought to arise when dyes are prevented from twisting into nonradiative excited state conformations upon binding to FAPs, increasing fluorescence intensity by up to 18,000-fold (20, 21). Fluoromodules are versatile, since an FAP can bind and activate a host of structurally similar but functionally distinct probes. This property has been exploited to create membrane-permeant and -impermeant fluorogens, amplify fluoromodule brightness, and build activatable pH-reporting ratiometric probes that can all be activated by the same FAP (20, 22–24). All previously described fluoromodules, with one exception (K7:TO-PRO-5), absorb and fluoresce maximally in the visible light spectrum, which limits their effectiveness for animal imaging experiments (20, 25–28).
Here we describe a fluoromodule-based reporter/probe system that absorbs and emits at wavelengths well suited for in vivo imaging. The reporter FAP, Mars1, possesses nanomolar affinity for a far-red absorbing probe: a fluorogen called SC1. Like other fluorogens, SC1 is an activatable probe: negligibly fluorescent in solution, but intensely fluorescent, with excitation and emission maxima at 701 and 731 nm, respectively, upon binding to its cognate FAP. The corresponding fluoromodule, Mars1:SC1, assembles rapidly, and its fluorescence is stable across a broad pH range. We also discuss Mars1Cy, a mutant of Mars1 with improved expression in mammalian cells, and the design of SCi1, a membrane-impermeant version of SC1 for labeling cell-surface proteins. Both reporters can be used to tag a variety of intracellular and cell-surface proteins. They can also bind and activate malachite green–based (MG-based) fluorogens that absorb and fluoresce at different wavelengths, which enables 2-color labeling of intracellular and extracellular protein populations. Cells that express Mars1Cy:SC1 can be imaged through the skin of mice, and fluorogens can be delivered to FAP-bearing tumors via i.p. and i.v. injection. Imaging with this reporter/probe system is feasible in a small animal model, even when restricted to nonoptimal excitation and emission channels. The tissue-compatible excitation and emission wavelengths, activatable nature of fluorogens, and ability to selectively label extracellular proteins may be advantageous for many animal imaging investigations.
Results
Fluorogen design.
SC1 (Figure 1A) was designed to structurally mimic MG while incorporating lengthened charge delocalization, since studies of cyanine dyes show that extended charge delocalization corresponds to increased absorption wavelengths (29). From its similarity to triarylmethane dyes and MG-based fluorogens, we predicted that SC1 would be nonfluorescent in water, but would become fluorescent if bound and rigidized by a compatible FAP. SC1 was expected to be membrane permeant and therefore useful for labeling intracellular FAP-tagged targets due to its compositional and structural similarity to MG (30). Mars1 and Mars1Cy bind and activate fluorescence from MG-based fluorogens such as MG-ester and MG-2p, in addition to SC1, indicating that variation in probe structure, particularly in the form of flexible linkers on the end distal to the diarylmethane group, is tolerated. We therefore attached a short linker and sulfonate group to SC1 to create a membrane-impermeant fluorogen, SCi1 (Figure 1A), intended for selective labeling of extracellular FAP-tagged targets such as surface-displayed receptors. The sulfonate group has previously been shown to impart polarity and consequently inhibit dye movement across lipid bilayers when attached to fluorescent dyes and other fluorogens (22, 31, 32).
Figure 1. Properties of Mars1- and Mars1Cy-based fluoromodules.

(A) Chemical structures of fluorogens SC1 (membrane permeant) and SCi1 (membrane impermeant). (B) Absorbance spectra: SC1 in acidic (5% glacial acetic acid) methanol (mean, 4 samples) and Mars1:SC1 in PBS, pH 7.4 (mean, 2 samples); spectra are scaled to have peak absorbance of 1.0 units. Free and bound forms of SCi1 are similar but with a 3 nm bathochromic shift. (C) Excitation (dashes, mean of 5 samples) and emission (solid, mean of 4 samples) spectra of SCi1 in PBS and FBS with and without Mars1Cy; spectra of Mars1 and SC1 are virtually identical. Intensities are expressed as a fraction of the maximum signal. (D) Binding and fluorescence activation traces of Mars1:SC1, Mars1:SCi1, Mars1Cy:SC1, and Mars1Cy:SCi1 (1 acquisition per fluoromodule) recorded from solutions where [FAP] = KD and [fluorogen] = 10 KD. Respective on rates (3.3 × 106, 2.8 × 106, 3.7 × 105, and 1.5 × 106 M-1s-1) were calculated from activation observed at 4 different concentrations of fluorogen. (E) Fluorescence of Mars1 fluoromodules in buffers spanning pH 2.0 to 10.0 measured 1 and 30 hours after sample preparation. Points indicate mean sample fluorescence (n = 5: Mars1:SC1/1 hour, n = 4: others). Bars indicate SD. Values are scaled such that the highest mean signal is 1.0. Mars1Cy exhibits similar pH insensitivity. (F) Fluorescence from HEK293 cells that express cytoplasmic Mars1Cy-eGFP, and untransfected control cells maintained at 37°C prior to (0 minutes) and at intervals after addition of SC1 or SCi1. Points denote average of median population intensities (n = 3 separate samples prepared identically) analyzed by flow cytometry.
Selection of Mars1 and Mars1Cy.
Mars1 was developed from a previously reported scFv, designated HL9-MG, which was one of the first FAPs isolated from a naive scFv library for its ability to activate fluorescence in MG-based fluorogens (20). The light chain of HL9-MG was previously found sufficient for fluorogen activation (33). Studies of similar light chain–only FAPs have shown that the assembled fluoromodule contains 1 fluorogen molecule bound between 2 light chains and that tethering of 2 light chains by a flexible linker increases binding affinity (28, 34). A gene encoding a peptide-linked dimer of light chains from HL9-MG was generated, randomly mutagenized, and screened using a yeast expression system and cell sorting in hopes of finding clones with improved binding and fluorescence activation of a membrane-permeant MG derivative, MG-ester. One mutant recovered from screening, designated Mars1, was found to exhibit increased expression and activity and, upon sequencing, was found to host a V70A mutation in the N-terminal light chain. This clone was also identified as activating fluorescence from SC1.
Seeking to further improve the expression characteristics of Mars1 and simultaneously explore the possibility of adjusting its affinity toward SC1, we propagated the V70A mutation to both light chains of Mars1 to generate a symmetric template. This template was subjected again to PCR-mediated random mutagenesis in order to produce a library suitable for SC1-activation screening. This process led to isolation of Mars1Cy, a mutant that hosts Y96H mutations on both light chains in addition to both V70A mutations present in its parent. Expression of Mars1 and Mars1Cy from otherwise identical vectors in the cytoplasm of mammalian cells indicated that the latter clone presents more robust activity in this environment (Supplemental Figure 1).
Characterization of fluoromodule properties.
The spectral and binding properties of Mars1 and Mars1Cy with SC1 and SCi1 were studied in vitro using purified protein recovered from either yeast (Saccharomyces cerevisiae) or bacterial (E. coli) expression systems. The Mars1:SC1 fluoromodule absorbs light in the far-red spectrum (Figure 1B), possesses a dissociation constant of 7.9 nM, and produces fluorescence with peak excitation and emission wavelengths of 702 and 731 nm, respectively. The excitation spectrum is broad and includes a secondary peak at 615 nm (Figure 1C). The other 3 fluoromodules are highly similar both in spectral properties (Figure 1C) and binding affinities (Table 1). Mars1Cy binds slightly less tightly to both fluorogens when examined in vitro. Critically, the fluorogens SC1 and SCi1 exhibit negligible fluorescence in the absence of cognate FAP, both in aqueous buffer and in whole serum (Figure 1C). Fluorescence enhancements of 3,300- (n = 16, SD = 650) and 2,000-fold (n = 16, SD = 210) for SC1 and SCi1, respectively, were obtained by comparing free versus Mars1Cy-bound fluorogen in PBS. Mars1 produced similar enhancements: 3,200- (n = 16, SD = 620), 2,100-fold (n = 16, SD = 220). Binding occurred rapidly upon addition of fluorogen to cognate FAP (Figure 1D). These FAPs also retained the ability to bind and induced shorter wavelength fluorescence from MG-based fluorogens (Supplemental Figure 2) that have been well characterized with use in different fluoromodules (20, 35).
Table 1. Physical and spectral properties of Mars1 and Mars1Cy complexed with SC1 and SCi1.
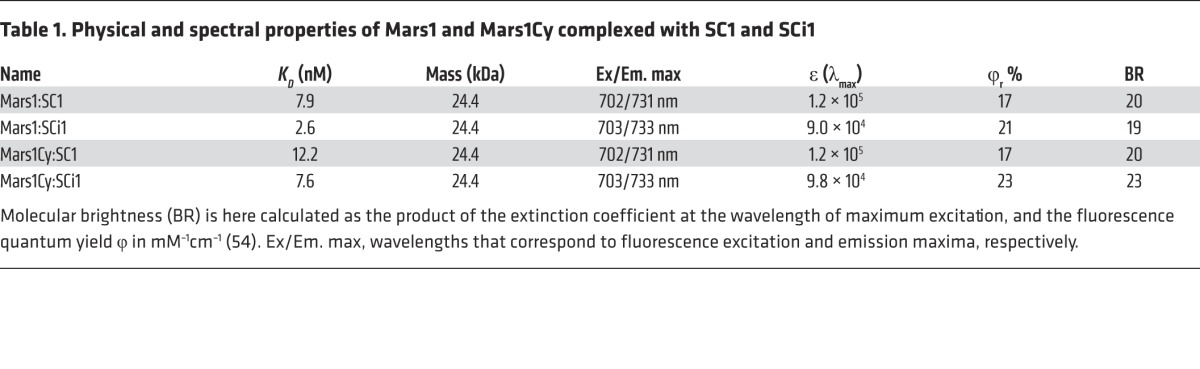
The fluorogens SC1 and SCi1 had extinction coefficients of 120,000 and 97,000 M-1cm-1, respectively, when dissolved in methanol with 5% (v/v) glacial acetic acid, with absorbance maxima at 691 and 696 nm. The spectral properties of these fluorogens bound to Mars1 and Mars1Cy in PBS at pH 7.4 are summarized in Table 1.
A limitation exhibited by many fluorescent protein reporters is the dependence of fluorescence intensity upon environmental factors such as pH, a characteristic that can confound quantitative measurements. The stability of Mars1 and Mars1Cy across a broad range of pH values was investigated by incubating the respective SC1 and SCi1 fluoromodules in a Carmody buffer series spanning 2.0 to 10.0 (36). Fluorescence intensities at 730 nm remained broadly stable, only showing substantial signal loss above pH 9.0 (Figure 1E). No pH dependence in signal intensity was observed even after incubation at room temperature for 30 hours.
Membrane permeability of SC1 and SCi1.
We expressed Mars1 and Mars1Cy in the cytoplasm of mammalian cells to confirm that they remain active in the intracellular environment. The resulting cell strains provided an opportunity to verify the predicted abilities of SC1 and SCi1 to cross cell membranes. We reasoned that a membrane-permeant fluorogen would gain entry to these cells and become fluorescent upon binding intracellular FAP, while membrane-impermeant fluorogens would be excluded from cells, which would consequently remain dark. HEK293 cells expressing cytosolic Mars1Cy-eGFP (where eGFP indicates enhanced GFP) were therefore incubated with either SC1 or SCi1. Fluoromodule and eGFP emissions were recorded by flow cytometry prior to and at intervals after the application of fluorogen (200 nM) to cell suspensions maintained at 37°C. Addition of SC1 produced an approximately 10,000-fold increase in fluorescence, relative to pretreatment and nontransfected cells, after 10 minutes. SCi1 produced a 2-fold signal increase within the same interval (Figure 1F). Similar trends in signal intensity change over time were observed from cells incubated at 20°C and on ice (Supplemental Figure 3, A and B).
Comparison of brightness between Mars1Cy and IFP1.4 in living cells.
HEK293 cells expressing cytosolic Mars1Cy and IFP1.4 with eGFP in the form of a fusion protein or as separate polypeptides expressed at stoichiometric levels via a P2A sequence (37) were used to compare the in vivo brightness of Mars1Cy:SC1 with that of IFP1.4 via microscopy. IFP1.4-expressing cells grown in media supplemented with 50 μM biliverdin-IX for 48 hours prior to imaging were 6- to 16-fold dimmer than cells expressing an equal amount of Mars1Cy labeled with 400 nM SC1 (normalization was carried out using coexpressed eGFP), as observed through a 731/137 nm emission filter under 640 nm excitation (Figure 2, A and B). The observed fluoromodule brightness exhibited a dependence on expression context: the C-terminal eGFP fusion construct yielded approximately 2-fold greater mean brightness over nonfused Mars1Cy.
Figure 2. Fluoromodules appear brighter than IFP1.4 and are functional when expressed within mammalian cells.

Channels are labeled DIC or formatted excitation:emission/bandwidth. (A) HEK293 cells expressing Mars1Cy-eGFP or IFP1.4-eGFP fusion proteins, from otherwise identical vectors, were imaged in the presence of 400 nM SC1 and 50 μM biliverdin, respectively, to compare observed brightness under identical conditions and display settings. Images are representative samples of data analyzed in B. (B) Box plot of fluoromodule/eGFP signal intensity ratios measured from protein coexpressed with eGFP in HEK293 cells as fusion proteins or separate polypeptides (P2A). Whiskers indicate maximum and minimum values; for boxes, top, line, and bottom indicate first, second, and third quartiles, respectively. Differences between mean ratios were analyzed by 1-way ANOVA: F(3,614) = 5332.14, P < 0.001. A Games-Howell post-hoc test shows differences between paired group means as denoted by lowercase letters: a (IFP1.4-eGFP fusion versus separate IFP1.4/eGFP peptides, P = 0.650); other pairs (ab, ac, bc) describe different means with high confidence, P < 0.001. Number of measurements (n) per group (IFP1.4/eGFP: 159, IFP1.4-eGFP: 180, Mars1Cy/eGFP: 102, Mars1Cy-eGFP: 177) indicate the number of unique cells measured. (C) Mars1Cy anchored to the plasma membrane by a CD80 transmembrane domain (TMD) can be labeled with membrane-impermeant SCi1. (D) Signal peptide–prefixed (COXIV) Mars1Cy-eGFP is directed to the mitochondria and becomes labeled upon addition of the membrane-permeant fluorogen SC1. (E–H) Mars1- and Mars1Cy-based fluoromodules can be used to tag proteins such as β-actin, vimentin, histone H1.1, and IGF1R in living cells. Images in C–H represent at least 10 fields of view per transgene-hosting cell population. Scale bars: 16 μm.
Use of Mars1 and Mars1Cy as fusion tags in various cellular contexts.
The utility of Mars1 and Mars1Cy fluoromodules to act as fluorescent tags in living cells was further explored by expressing the FAPs in a variety of cellular environments, either by appending a signal sequence that directs a specific subcellular localization or by fusing the FAP to a native protein. Targeting of Mars1 to the plasma membrane and Mars1Cy to the mitochondrial matrix by utilizing Igκ- and COXIV-1–derived signal peptides, respectively, was demonstrated in HEK293 cells (Figure 2, C and D). Mars1Cy–β-actin, Mars1Cy-vimentin, histone H1.1–Mars1Cy, and Mars1Cy-IGF1R fusion proteins were expressed in HeLa cells (Figure 2, E–H). In these examples, cells expressing CMV-driven FAP constructs were used to establish stably expressing clonal lines that were maintained for weeks in the absence of chemical selection.
The feasibility of long-term labeling of living cells was also explored. An HEK293 strain expressing cytosolic Mars1Cy-eGFP was cultured continuously in media supplemented with 200 nM SC1 over a 52-day period, during which the proportion of FAP-expressing cells was periodically monitored by flow cytometry and observed to remain stable. The population positive for fluorescence in both channels exhibited little fluctuation over 10 measurements, with a mean value of 98% positive for both eGFP and FAP fluorescence (SD, 0.39%).
Two-color labeling of extracellular and intracellular protein populations.
We tested the use of fluorogens that excite and emit at different colors as a means of differentiating between external and internal populations of Mars1Cy-labeled arginine vasopressin receptor 2 (AVPR2, Homo sapiens) in cultured cells. A CMV promoter–driven gene encoding Mars1Cy-AVPR2 was introduced into HEK293 cells via lentiviral transduction in order to obtain a stably expressing clonal cell line that expresses FAP-tagged AVPR2. A membrane-impermeant fluorogen such as MG-2p or SCi1 was used to saturate FAP-tagged receptors on the exterior of the cell. We found that receptors labeled in this manner remained responsive to stimulation by desmopressin (Figure 3A). Initial labeling of cells with membrane-impermeant fluorogen for a brief period (10 minutes), followed by addition of a differently colored membrane-permeant fluorogen such as MG-ester or SC1 to SCi1- or MG-2p–labeled cells, respectively, allowed specific labeling of the intracellular protein pool by the membrane-permeant fluorogen. Mars1Cy:MG-2p and Mars1Cy:SCi1 fluoromodules produce easily separable colors, and we observed that addition of SCi1 followed by MG-ester or addition of MG-2p followed by SC1 allowed 2-color labeling of extracellular versus intracellular protein prior to and during desmopressin-stimulated endocytosis (Figure 3B).
Figure 3. Mars1Cy-tagged AVPR2 is responsive to desmopressin-stimulated (DDAVP) endocytosis and demonstrates the feasibility of 2-channel separation of external and internal protein pools using impermeant and membrane-permeant fluorogens that emit in the far-red.

(A) HEK293 cells expressing Mars1Cy-AVPR2 were incubated at 37°C with the indicated membrane-impermeant fluorogens for 5 minutes prior to stimulation with DDAVP. Images were acquired immediately upon agonist application and at 20 minutes after addition. (B) Sequential addition of impermeant fluorogen (either SCi1 or MG-2p, 10 minutes) followed by permeant fluorogen MG-ester (MGE) or SC1, respectively, for 30 minutes. Each panel is representative of 3 replicates. Scale bars: 16 μm. Magnification is constant across all time points.
Imaging of FAPs in the presence of nonbinding dyes.
Various red to near-infrared fluorophores, TMR isothiocyanate (TRITC), Lissamine Green B, Oxazine 1, methylene blue, Cy5.18, and Cy7, were incubated with Mars1-expressing cells, which were imaged to examine whether Mars1 can bind different dyes. Binding was expected to produce signal enrichment at the plasma membrane (for nonmembrane permeant dyes) or in the secretory pathway as well as the plasma membrane in the case of membrane-permeant dyes. SCi1 was then added to the samples, which were imaged again to assess whether the presence of other dyes prevents fluorogen binding and activation. None of the dyes tested appeared to bind to Mars1, in that they failed to enrich signals at sites of FAP expression, nor did they prevent binding and activation of SCi1 (Supplemental Figure 4).
Imaging of fluoromodules in a small animal model.
The optical properties of these fluoromodules make them excellent candidates for animal imaging–based studies, so we examined their performance in various in vivo imaging scenarios. HCT116 cells were transduced with lentivirus to afford expression of either iRFP713-eGFP or Mars1Cy-eGFP in the cytoplasm. A similar vector bearing an Igκ signal sequence and transmembrane anchor was used to express Mars1Cy on the plasma membrane surface. Single cell–derived populations expressing each protein were isolated, and protein expression was verified by flow cytometry (Supplemental Figure 5). To gauge the performance of our fluoromodules relative to iRFP713 within the context of living animals, we performed s.c. injection of defined numbers of cells into the flanks of athymic nude mice. Transduced cells hosting either protein were visible through the skin and easily distinguished from tissue autofluorescence in all cases, down to the minimum injected dose of 300 cells (Figure 4A). We found that the fluorescent protein and fluoromodule yielded comparable signals for all amounts of injected cells (Figure 4B and Supplemental Figure 6), though the 680 channel of the FMT2500LX instrument more effectively excited and collected light from iRFP713 than Mars1Cy.
Figure 4. Application and quantitation of fluorescence from fluoromodules in living mice.

(A) Example of a nude mouse implanted with 300 cells of iRFP713-expressing HCT116 cells in the left flank and an equal number of Mars1Cy-expressing cells, prelabeled with SC1, in the right flank. Image is representative of data quantified in B. The fluoromodule-hosting cells used here express less protein than the cells hosting iRFP713 (Supplemental Figure 5). (B) Fluorescence measured from mice injected and imaged as described in A, using different numbers of cells. Points denote means; bars indicate SEM (n = 3: 1 × 105 Mars1Cy-expressing cells, n = 4: others, total animals treated and imaged: 16). Analysis of the same data in terms of relative fluoromodule signal yields similar results (Supplemental Figure 6). (C) Fluorescence image of a tumor section (3.2 by 1.7 mm) composed of HCT116 cells that express Mars1Cy on the cell surface, labeled by SC1 given i.p. Scale bar: 16 μm. Field is near the center of the section. (D and E) s.c. FAP-bearing tumors that express Mars1Cy on the cell surface were labeled by SC1 (400 nM final) administered i.p. and i.v. (tail vein), respectively. (F and G) s.c. tumors as in D and E were labeled with SCi1 (400 nM final), injected i.p. or i.v., respectively. (H) i.p. injection of SC1 produces no significant background signal when FAP-bearing cells are not present. (I and J) Mars1Cy-bearing tumors can be imaged in the i.p. cavity of mice after delivery of SC1 or SCi1, respectively, via i.p. injection. Data presented in C–J represent results obtained from 3 samples/animals. Scales in mouse images are given in millimeters.
Feasibility of systemic fluorogen delivery to tumors.
Recognizing that ex vivo labeling of samples is impractical for many animal imaging experiments, we next inquired whether these fluorogens can be delivered to FAP-hosting cells in living mice via injection into the i.p. cavity or administration via tail vein. In one experiment, HCT116 cells that express plasma membrane–directed Mars1Cy were used to form tumors in mice. Membrane-permeant fluorogen was then delivered via i.p. injection, after which the host animal was sacrificed and the tumor extracted and sectioned. Fluorescence from SC1 bound to Mars1Cy was detected within the tumor sections via confocal microscopy (Figure 4C), even near the center of the tumor. We further imaged s.c. tumors generated using the same cell line and found that labeling can be obtained from administration of membrane-permeant fluorogen via i.p. and i.v. routes (Figure 4, D and E, respectively) and impermeant fluorogen via i.p. and i.v. routes (Figure 4, F and G, respectively). Finally, to determine whether these fluoromodules allow visualization of targets deeper within a host animal, we generated FAP-bearing tumors in the i.p. cavity of mice. Once tumors reached 10 to 25 mm3, SC1 and SCi1 were delivered via i.p. injection, and fluorescence was quantified (Figure 4, H–J). Labeled tumors were also extracted, sectioned, and imaged to assess the degree of labeling throughout the cell mass with different fluorogens (Supplemental Figure 7).
Discussion
In this work, we present an activatable fluorescent reporter/probe system with properties suited for whole animal imaging. These fluoromodules, composed of Mars1 and Mars1Cy FAPs paired with SC1 and SCi1 fluorogens, maximally absorb and emit near 700 and 730 nm respectively, where tissue absorbance and autofluorescence subside, and provide stable fluorescence intensity across a broad range of pH conditions. The fluorescence quantum yields measured for Mars1 and Mars1Cy bound to SC1 and SCi1 are several-fold greater than reported values for spectrally similar fluorescent reporter proteins (38–41), and we observed that Mars1Cy appears substantially brighter when imaged alongside IFP1.4 in cultured cells. We have shown here that the far-red absorbing dyes SC1 and SCi1 are fluorogenic: dark in solutions such as PBS and FBS as well as suspensions of cultured mammalian cells and highly fluorescent upon binding to Mars1 or Mars1Cy. The thousands-fold difference in fluorescence between bound and unbound fluorogen allows FAP-tagged proteins, expressed in cultured cells, to be imaged without washing away excess fluorogen. This property could also be advantageous for in vivo imaging in mice because it reduces signals contributed by probes in circulation.
The fluorescence excitation spectra of Mars1 and Mars1Cy-based fluoromodules are broad and often, to our benefit during this study, allow excitation from a variety of readily available light sources, including 633 and 640 nm diode lasers in addition to the 670 nm excitation line of the FMT2500LX small animal imaging instrument used here for mouse studies. We were unable to excite these fluoromodules near their optimal absorbance wavelength for our imaging experiments. However, use of commercially available 690 and 705 nm lasers, which further bypass tissue absorption and autofluorescence while offering more efficient excitation of these fluoromodules, may yield different results, particularly for animal imaging applications.
We demonstrated the feasibility of imaging fluoromodules in vivo using nude mice and a commercially available small animal imager. Fluorescence could be detected from 300 FAP-expressing cells implanted underneath the skin. The observed signal is at least comparable to an equivalent number of cells that express iRFP713, a fluorescent reporter (40, 41), though we did not correct for differences in protein expression between the cell strains used. We observed during work with cultured cell strains that Mars1Cy expression under a CMV promoter could remain stable for weeks in the presence of SC1 and in the absence of chemical selection agents. Mars1Cy was therefore employed to generate FAP-bearing tumors in mice, which were used to assess systemic fluorogen delivery. Our data show that both membrane-permeant and membrane-impermeant fluorogens can label FAP-bearing tumors in the i.p. cavity upon administration by i.p. and i.v. injection routes and that these labeled tumors can be noninvasively visualized. SCi1 more robustly labels tumors compared with SC1 at lower doses. A number of factors might contribute to this behavior; the membrane-permeant fluorogen could be sequestered inside cells proximal to the injection site, become bound by serum proteins, or undergo rapid clearance.
Mars1:SC1 and the other fluoromodules presented here differ in several ways from existing optical reporter/probe systems that feature similar reporter-dependent signal activation — a property shared by luciferin/luciferase and that is available to the SNAP-tag approach. The former system is based upon luminescence at shorter wavelengths, rather than fluorescence, and light output is coupled to oxidation of a substrate rather than excitation and emission cycles of a fluorophore. Most fluorogenic SNAP-tag substrates absorb and fluoresce at shorter wavelengths than the fluoromodules discussed here upon activation and consequently may experience greater interference from autofluorescence (14–16). Relative to existing fluorogenic SNAP-tag substrates, SC1 and SCi1 exhibit faster signal activation and much greater fluorescence enhancements upon binding, though the FAP-fluorogen activation rates observed here are slower than the exceptional binding rates of nonfluorogenic tetramethylrhodamine (TMR) substrates with HaloTag variants (42).
In contrast to fluorescent gene reporters, imaging of fluoromodules requires addition of an extrinsic reagent — the fluorogen; however, systemic delivery via the 2 routes demonstrated here makes application of this tool no more laborious than use of other reporter/probe systems. One advantage of using a synthetic probe is the ability to use different probes with a single reporter. We demonstrate with SCi1 that membrane permeability is a parameter that can be rationally engineered to allow selective probing of extracellular or whole-cell protein populations. We further used the ability of Mars1Cy to bind and activate fluorescence from MG-2p, MG-ester, SC1, and SCi1 to show that extracellular and intracellular pools of a FAP-tagged receptor can be labeled with different-colored probes — a capability that may be useful for protein trafficking or pulse-chase studies. This particular labeling scheme could be difficult to apply in vivo, since MG-2p and MG-ester are less spectrally compatible with tissue. However, it illustrates one manner in which multiple probes can be employed. We are excited to further explore whether other chemical groups or compounds could be coupled in place of the short linker and sulfonate on SCi1 without ablating binding or fluorescence activation. Neither far-red fluorogen was designed with consideration of biodistribution in animals, and we have not yet explored parameters such as the rate of clearance in mice. We expect that the in vivo labeling effectiveness of these fluorogens could be improved by attaching chemical substituents known to alter small molecule retention within living animals (43). Coupling to polyethylene glycol, for example, may increase the solubility and circulation time of the molecule and consequently decrease the quantity of fluorogen required for effective labeling. Additional fluorogen modification strategies to increase probe brightness or allow the probe to report pH have been demonstrated in vitro and on cultured cells (23, 24) and may be adapted to SC1-based fluorogens to further extend their usefulness.
Methods
Cell lines and strains.
HEK293, HeLa, and HCT116 cell lines used in this study originated from ATCC. The Phoenix-GP retroviral packaging strain was provided by Margaret Fuhrman (Carnegie Mellon University). The 293FT strain used for lentiviral packaging was purchased from Invitrogen. E. coli strain NEB10β was obtained from New England Biolabs; Rosetta-gami 2 was from Novagen. Yeast plasmids (pPNL6, pPNL9) and S. cerevisiae strain YVH10 are components of an scFv library obtained from Pacific Northwest National Laboratory (http://www.sysbio.org/dataresources/index.stm). JAR200 cells, provided by Andrew Rakestraw (Massachusetts Institute of Technology, Cambridge, Massachusetts, USA), were obtained via Christopher Szent-Gyorgyi (Carnegie Mellon University).
Mars1.
The light chain of HL9-MG was converted into a tandem dimer by attachment of a glycine-serine linker followed by a codon-scrambled L9 gene via PCR. The L9 tandem dimer gene was subjected to PCR-mediated random mutagenesis and cloned into pPNL9 to generate an expression library suitable for high-speed cell sorting (44). Library elements were induced in shaking culture flasks for 5 days at 20°C in secretion media (20) prior to being screened for fluorescence activation of MG-ester on a FACSVantage SE flow cytometer with FACSDiva Option (BD) by 633 nm laser excitation, viewed through a 685/70 nm band-pass filter. The brightest 0.3% of activators in the presence of 50 nM MG-ester were enriched over 4 rounds of sorting. Single cells from round 5 were picked, grown, and induced in the same manner as used to screen the library described above, and their induction media was assessed for FAP activity by assessing MG-ester fluorescence activation using a Tecan Safire 2 plate reader. A clone with particularly high yields hosting a V70A mutation in the N-terminal light chain was designated Mars1.
Mars1Cy.
The V70A mutation was propagated to the C-terminal light chain by PCR-based site-directed mutagenesis, and the product was subjected to an additional round of random mutagenesis. Amplicons were recombined into pPNL6, and the resulting library was screened by flow cytometry for activation of SC1 via 633 nm laser excitation, with fluorescence detected through a 755/75 nm band-pass filter. The brightest 0.5% of activators in the presence of 50 nM SC1 was enriched over 4 rounds of cell sorting prior to cloning. Cloned cells were induced and screened for fluorescence activation of SC1 in 96-well plates using a Tecan Safire 2. Subsequent sequencing of bright clones identified a mutant bearing Y96H mutations in both N- and C-terminal chains. This clone was later found to exhibit higher activity when expressed in mammalian cell cytosol.
S. cerevisiae expression.
Mars1 and Mars1Cy genes were cloned into pPNL9 to incorporate a C-terminal hexahistidine tag for immobilized metal ion affinity chromatography. The resulting plasmids were hosted and expressed in yeast strain YVH10 (45). Protein expression and purification protocols followed those previously described (20); however, buffer (Wash Buffer G) was composed of 50 mM Tris, pH 7.4, 500 mM NaCl, 30 mM imidazole, pH 8.0, and 0.015% Triton X-100. Elution buffer was identical, with the exception of 250 mM imidazole.
E. coli expression.
Expression and purification were conducted in the Rosetta-gami 2 strain of E. coli. FAP genes were cloned into a modified pET21a vector (a gift from Josef Franke, Carnegie Mellon University) that encodes an N-terminal decahistidine tag, followed by GST and an HRV3C protease cleavage site. Following IPTG induction at 20°C, cells were washed in 25 mM Tris, pH 8.0, with 150 mM NaCl. Washed cells were pelleted and frozen (–80°C). Cells were thawed in Wash Buffer G and lysed by sonication in the presence of DNase, lysozyme, and PMSF. Lysate was clarified by centrifugation (4°C, 22,789 g, 45 minutes) and transferred to NiNTA agarose (QIAGEN) for 1 to 4 hours of incubation at 4°C. Beads were transferred to a column and washed prior to addition of His6-tagged HRV3C protease. Following overnight incubation, fresh NiNTA agarose was added, and liberated protein was washed from the column following 1 hour of incubation.
Absorbance spectroscopy.
Data were acquired using a PerkinElmer Lambda 45 UV/Vis spectrophotomer at room temperature, set to a spectral width of 2.0 nm. All sample path lengths were 1.0 cm.
Molar extinction coefficient measurements.
HPLC-purified SC1 was dissolved in HPLC-grade methanol and transferred into preweighed vessels. The fluorogen samples were dried in a SpeedVac (AES-1000, Savant) at 43°C immediately prior to weighing. Known amounts were dissolved in methanol supplemented with 5.0% acetic acid, and a dilution series was generated from these master stocks. Absorbance spectra were obtained for each dilution series element (5 per dilution), and the molar extinction coefficient at λmax was determined by linear regression on the absorbance versus concentration as calculated from the measured mass. Measurement of the SCi1 extinction coefficient was impeded by its indeterminate assortment of counter ions. A proxy fluorogen, SKC-672, featuring a carboxylic acid in place of the sulfonate moiety, was instead used to determine the extinction coefficient of the SCi1 chromophore. Dried SKC-672 (55.5 mg) was transferred to a 500 ml volumetric flask and dissolved in methanol to generate a master stock of fluorogen. The master stock was used to generate a dilution series in methanol supplemented with 5.0% acetic acid. Measurement and analysis proceeded as described for SC1.
Extinction coefficients of Mars1 and Mars1Cy fluoromodules.
Master stocks used for free fluorogen extinction coefficient measurements were diluted to known concentrations (between 500 nM and 1.0 μM in order to remain well above the estimated fluoromodule dissociation constant) in PBS, pH 7.4, supplemented with 1.0 mM EDTA. An excess of purified FAP was added and mixed. Absorbance scans were collected until absorbance at λmax stabilized.
Quantum yield of Mars1 and Mars1Cy fluoromodules.
Relative quantum yield measurements were conducted using Cy5.18 (PBS, pH 7.4) and oxazine-1 (ethanol) by the gradient method (46–48). For each replicate, at least 5 fluoromodule samples were constituted using various fluorogen concentrations at least 20-fold greater than the dissociation constant in the presence of excess purified protein. The solutions were allowed to complex at 4°C for at least 1 hour prior to reequilibration and measurement at room temperature. Fluorescence spectra were obtained using a Quantamaster fluorometer (Photon Technology International) equipped with dual excitation and emission monochromators set to 2.00 mm (approximately 5.0 nm spectral windows). Emission spectra were integrated using Spekwin32, version 1.71.6.1 (http://www.effemm2.de/spekwin/), and relative quantum yield was determined via linear fit to area versus absorbance plots.
Fluoromodule binding properties.
Fluoromodule dissociation constants KD were measured using a ligand depletion assay. 5.0 nM FAP was titrated against each fluorogen at concentrations from 10 pM to 1.0 μM. Each complex was allowed to equilibrate at 4°C, and fluorescence of these complexes was measured using a fluorescence plate reader (Infinite M1000, Tecan) at the emission maximum. The curve obtained using this method was fit to a ligand depletion model to yield the KD. For on-rate measurements, FAP solutions were prepared in PBS, pH 7.4, supplemented with 0.01% Pluronic F127 at a concentration equal to the KD of the fluoromodule. These solutions were taken in a 4 ml quartz cuvette, and fluorescence was measured using a fluorometer (Photon Technology International). Time dependence of fluorescence was measured at the respective emission peaks after the addition of the fluorogen. For each FAP, fluorogen was used at concentrations of 5, 10, 50, and 100 equivalents of the experimentally determined KD. On-rate curves were fit using an exponential growth model. The association rates (kon*) determined from these curves were plotted against the fluorogen concentration and the slope of the curve yielded the on-rate for the complex.
Fluorescence enhancement measurement.
Fluorogens SC1 and SCi1 were diluted to 500 nM in PBS, pH 7.4, supplemented with 1.0 mM EDTA. Purified Mars1Cy or Mars1 was added to achieve a 10-fold excess of protein to fluorogen. Control solutions were prepared in the same manner, but with no FAP. Solutions were mixed and distributed in a black 96-well flat-bottom plate (Greiner) with 160 μl of each solution per well, 12 replicates per sample type. The samples were allowed to equilibrate at room temperature for 1 hour. Measurements were conducted with a Tecan Safire 2 plate reader using instrument-optimized gain in extended dynamic range mode, 40 μs integration time, exciting at 700 nm and collecting at 730 nm, and with bandwidths set to 8 nm.
Characterization of fluoromodule pH sensitivity.
Fluoromodules were prepared as 50-fold stocks in 10 mM Tris, pH 7.4, with 300 mM NaCl prior to dilution into each buffered pH condition, yielding 500 nM fluoromodule (fluorogen-limited). Five replicate wells were prepared for each pH condition. Measurements were obtained after 60 minutes of room temperature incubation and again after 30 hours using a Tecan Safire 2 plate reader, exciting each sample at 695 nm and recording fluorescence at 730 nm. A parallel set of samples with FAPs omitted exhibited negligible fluorescence under identical measurement conditions.
Cloning and use of FAP-expressing vectors.
Restriction enzymes were purchased from New England Biolabs Inc. as were polymerase (Q5 DNA polymerase) and ligase (Quick Ligation Kit). Primers were purchased from Life Technologies, Bioneer Inc., and Integrated DNA Technologies. Transformations and DNA propagation steps were conducted in the commercial E. coli strain NEB10β.
Plasma membrane display of Mars1 and Mars1Cy.
An ORF bearing an Igκ signal sequence and HA epitope, followed by a murine CD80 transmembrane anchor (49) flanked by 5′ XbaI and XmnI, and 3′ SpeI and XmaI cloning sites, was inserted into pEGFP-C2 (Clontech) between NheI and XmaI sites. External display plasmids pOIMars1N and pOIMars1CyN were generated by cloning the respective FAP genes between the XbaI and XmnI sites. Transfection of HEK293 cells with these constructs was accomplished via use of FuGENE 6 (Roche Applied Science) in accordance with manufacturer-supplied protocols. Stable cells were selected by application of G418 for 1 week. Clones were isolated by flow cytometry and expanded. Postexpansion expression stability was reverified by flow cytometry. Unless noted, other mammalian expression plasmids were used to generate stable strains in a similar fashion. The authors were unable to confirm expression by fluorescence signal from cells transfected with analogous iRFP713 constructs.
Cytoplasmic expression of Mars1Cy-eGFP fusions.
Overlapping PCR primers were used to assemble and amplify an ORF encoding Mars1Cy tethered to eGFP by a peptide linker (GGRASGGGASGGGSGGSG) flanked by NheI and XmaI sites. Amplicons were digested and ligated into like-digested plasmid backbone prepared from pOIMars1N to generate pMars1Cy-eGFP.
Cytoplasmic expression of eGFP and Mars1Cy from a bicistronic transcript.
Plasmid pP2AeGFPMars1Cy was constructed by fusing a nucleic acid sequence encoding a P2A site between eGFP and Mars1Cy fragments via overlapping primer PCR. The resulting amplicon was inserted in place of Mars1Cy-eGFP in the plasmid pMars1Cy-eGFP.
Mitochondrial expression of Mars1Cy-eGFP.
Overlapping primers were used to fuse a mitochondrial localization signal that consists of the N-terminal 23 amino acids of cytochrome c oxidase subunit IV (H. sapiens) to the N terminus of Mars1Cy-eGFP via PCR. The amplicon was used to replace Mars1Cy-eGFP in pMars1Cy-eGFP, yielding pMTMars1Cy-eGFP.
Retrovirus for stable expression of Mars1 and Mars1Cy fusion proteins.
A pBABE retroviral vector was modified to host a 1.5-kb fragment of A2UCOE (50), followed immediately by a CMV promoter and an ORF, between existing BamHI and SalI restriction sites. The ORF of this derivative vector, designated pUB, was flanked by AgeI and SalI sites, between which various FAP fusion proteins were cloned. A modified plasmid containing a 3′ SbfI cloning site was also generated. Retroviral packaging was accomplished using the Phoenix-GP cell line (51) by cotransfection of pMD2.g with FAP-hosting pUB vectors. Viral supernatant, supplemented with polybrene (0.6 μg/ml), was used to transduce HeLa cells in order to produce strains used for imaging of FAP-tagged proteins.
Lentiviral vectors for stable transduction of tumorigenic cell lines.
A parent vector, pLenti-CaMKIIa-hChR2(E123T-H134R)-EYFP (52) (a gift from the Deisseroth lab, Stanford University, Stanford, California, USA) was modified by substitution of all content between PacI and EcoRI sites with an insert encoding the promoter and ORF from pOIMars1N fused upstream of an IRES and puromycin resistance gene. The Mars1Cy-eGFP fusion protein ORF was amplified from pMars1Cy-eGFP using primers that maintained the 5′ AgeI restriction site and appended a 3′ BsrGI site. The digested amplicon was cloned into like-digested lentiviral vector to generate pLenCMV-Mars1Cy-eGFPiPac. The analogous vector, pLenCMV-iRFP713eGFPiPac, was produced by amplifying iRFP713 with primers that introduced flanking AgeI and AscI restriction sites, followed by standard digestion/ligation cloning. The plasma membrane–targeted Mars1Cy ORF from pOIMars1CyN was amplified with primers that preserved the 5′ AgeI site and appended a sequence for fusion with eGFP at 7 amino acids after the predicted end of the membrane-spanning segment. This PCR product was further amplified in the presence of eGFP template DNA and primers that yielded 5′ AgeI and 3′ BsrGI restriction sites that flanked the assembled product. Cloning into the pLenCMV-Mars1Cy-eGFPiPac vector backbone at these sites afforded pLenCMV-PMMars1Cy-eGFPiPac for plasma membrane–directed expression of a Mars1Cy-eGFP fusion protein. Viral particles were packaged using the ViraPower Lentiviral Packaging Mix (Invitrogen K4975) and 293FT cells in accordance with product-supplied protocols. Transduced cells were sorted for FAP activity via fluorogen activation or iRFP713 activity by flow cytometry. Clones from sorting were maintained in the presence of puromycin (5 μg/ml).
Igκ-Mars1Cy was amplified with primers that preserved the 5′ AgeI site and appended a sequence that encodes a 16-residue C-terminal linker (GTAAAGAGAAGGAAAG). This PCR amplicon was further amplified using the original forward primer and a reverse primer that appended a sequence encoding the full first exon of H. sapiens AVPR2 and the first five 5′ nucleotides of exon 2. Separately, human genomic DNA was amplified using primers that annealed to the 5′ terminus of AVPR2 exon 2 and the 3′ terminus of exon 3, including the native stop codon. These primers flanked the intervening DNA with a 5′ extension that complements the 3′ terminus of the extended Mars1Cy amplicon and a 3′ extension encoding a BsrGI restriction site. The 2 resulting fragments were amplified together using their respective 5′ and 3′ terminal primers to produce Igκ-Mars1Cy-AVPR2 (including introns 2-3), which was cloned into a vector backbone of AgeI- and BsrGI-digested pLenCMV-PMMars1Cy-eGFPiPac to produce pLenCMV-PMMars1CyAVPR2.
Analysis of fluorogen membrane permeance.
HEK293 cells expressing Mars1Cy or untransfected controls were grown in DMEM supplemented with 10% FBS and 500 μg/ml G-418 (736 μg/mg potency). Cells were expanded onto three 100-mm dishes and grown to 50% confluence prior to collection by application of 1.0 ml 0.25% trypsin-EDTA (Gibco; Life Technologies). Detached cells were washed and resuspended with fresh DMEM to yield 1.0 ml per condition. Analysis was performed on the previously described BD instrument using 488:530/30, 488:610/20, 633:685/70, and 633:755/75 channels. Basal fluorescence was recorded for transfected and untransfected cells at room temperature, while 37°C and on-ice samples were allowed to equilibrate. Stock fluorogen in methanol or DMSO was diluted 10-fold in DMEM prior to addition to cell suspensions. Sixty-second acquisitions (recording approximately 10,000–40,000 cells) were performed immediately upon addition of fluorogen to 200 nM and at intervals afterwards. Fluorescence calibration beads (Bangs Laboratories Inc., product 822A, lot 10376) were measured during each replicate. Scatter was used to designate cell-sized and bead-sized populations in FlowJo X (LLC) for which median fluorescence values were calculated in eGFP (488:530/30) and fluoromodule (633:755/75) channels. Median intensity values were converted to molecules of equivalent soluble fluorophore (MESF) using QuickCal 2.3 (Bangs Laboratories Inc.). Data are presented as the normalized mean MESF for 3 replicates per condition with associated SD.
Comparison of protein brightness in living cells.
Cells were plated in imaging dishes (MatTek Corp.) 24 hours prior to each experiment. Cells expressing IFP1.4 were cultured in media supplemented with 50 μM biliverdin-IX for at least 48 hours prior to imaging, while cells expressing Mars1Cy were labeled 4 to 24 hours prior to imaging by adding fluorogen directly to growth media. Imaging was conducted on an Andor Revolution XD spinning disk confocal microscope using a ×60 1.49 NA oil immersion objective. Signal from eGFP was acquired during excitation at 488 nm using a 525/50 nm band-pass filter (Semrock FF03-525/50-25), while IFP1.4 and Mars1Cy were excited by laser at 640 nm and emission collected through a 731/137 nm band-pass filter (Semrock FF01-731/137-25). Images were processed in ImageJ (53): mean pixel intensities from eGFP and fluoromodule emission channels were measured from ROIs drawn around transfected cells and a blank area in each frame. Blank ROI values were subtracted from cell ROI values measured from the same frame to generate corrected means for eGFP and fluoromodule channels from which fluoromodule/eGFP signal ratios were calculated for every cell. Box plots were generated using RStudio. Ratios were analyzed by 1-way ANOVA followed by a Games-Howell post-hoc test in SPSS v23 (IBM).
Labeling of Mars1Cy-AVPR2.
HEK293 cells expressing Mars1Cy-AVPR2 were labeled with impermeant fluorogen SCi1 or MG-2p (200 nM, 5 minutes at 37°C) and stimulated with desmopressin (Sigma-Aldrich, 10 μM). Samples were sequentially excited with 488 nm and 640 nm lasers, and emission was collected through 685/70 and 794/160 nm band-pass filters for each respective channel. For dual-color labeling with spectrally separated external and internal far-red labels, SCi1 was applied at 200 nM for 10 minutes, followed by addition of MG-ester at 100 nM for 30 minutes prior to imaging. To achieve the inverse labeling scheme, MG-2p was applied at 200 nM for 10 minutes, followed by addition of SC1 at 200 nM for 30 minutes to label the extracellular receptor pool far red and the intracellular pool farther red.
Cell culture in the presence of SC1.
A stable HEK293 clone expressing cytosolic Mars1Cy-eGFP was cultured in DMEM and 10% FBS supplemented with 200 nM SC1 from a 1,000× stock in DMSO. Cells were split 1:10 every 48 to 72 hours, and percentages of positive cells were monitored by flow cytometry every 4 to 7 days from day 6 to day 52.
Imaging Mars1-expressing cells with nonbinding dyes.
HEK293 cells stably expressing Mars1 on the plasma membrane were plated onto glass bottom dishes 24 hours prior to imaging. A dye (TRITC, Lissamine Green B, oxazine 1, methylene blue, Cy5.18, or Cy7) was added to a final concentration of 500 nM in each dish. Cells were incubated under normal growth conditions (37°C, 5% CO2) for 60 minutes prior to imaging using an Andor Revolution XD spinning disk confocal microscope equipped with a ×60 1.49 NA oil immersion objective. Samples were imaged under 4 channels (bright field, 560:607/36, 640:685/70, 640:794/160). Each dish of cells was supplemented with SCi1 to a final concentration of 500 nM and returned to a tissue culture incubator for 15 minutes prior to a second round of imaging using identical settings.
SC1: [(E)-N-(4-(1,3-bis(4-(dimethylamino)phenyl)allylidene)cyclohexa-2,5-dien-1-ylidene)-N-methylmethanaminium].
4-(Dimethylamino)benzaldehyde (149 mg, 1 mmol) and 4,4′–vinylidene bis (N,N- dimethylaniline) (300 mg, 1.12 mmol) were dissolved in 5 ml acetic anhydride, and 200 μl of 60% perchloric acid was added slowly. The solution was refluxed for 3 hours with stirring, then cooled to room temperature. Precipitated by diethyl ether, the blue residue was purified by silica gel column chromatography using 10% methanol in chloroform, yielding 242 mg deep blue solid as product (Supplemental Figure 8). The product had a chemical composition of C27H32N3+ with a MW of 398.56 g/mol (61% yield). The 1H-NMR (CD3OD) spectra were as follows: δ 7.72–7.85 (m, 2H); 7.53 (d, 2H); 7.45 (d, 1H); 7.12 (d, 1H); 6.52–6.98 (m, 8H); 3.24 (d, 12H); 2.9 (m., 6H), and electrospray ionization mass spectrometry (ESI-MS) (+) of 398.3.
SKC-657: [Ethyl 5-(methyl(phenyl)amino)pentanoate].
5.52 ml (48 mmol) of N-methyl aniline, 8 ml bromovalerate (50.2 mmol), and 2,6- lutidine (5.82 ml, 50.2 mmol) were taken in 100 ml acetonitrile and refluxed for 24 hours while stirring. Acetonitrile was removed under vacuum, and residues were dissolved in ether and water. The organic phase was collected and washed with water. After drying over magnesium sulfate and filtering, the solvent was removed, leaving a brown liquid. The residue was purified by flash chromatography on SiO2 using a 10% ethyl acetate in hexane as eluent, giving 10.1 g light yellow oil as product (Supplemental Figure 9). The product had a chemical composition of C14H21NO2 with a MW of 235.32 g/mol (90% yield). The 1H-NMR (CDCl3) spectra were as follows: δ 7.25 (m, 2H); 6.72 (m, 3H); 4.15 (m, 2H); 3.35 (m, 2H); 2.94 (s, 3H); 2.35 (m, 2H); 1.67 (m, 4H); 1.27 (m, 3H), and ESI-MS (+) of 235.62.
SKC-659: [Ethyl 5-((4-formylphenyl)(methyl)amino)pentanoate].
2.94 g (1.25 mmol) of SKC-657 was dissolved in 10 ml dry DMF and cooled to –5°C by salt ice bath. 5 ml of POCl3 was added drop wise over a period of 1 hour at –5°C, and the reaction mixture was stirred at room temperature for 20 hours. The temperature was raised to 90°C for 2.5 hours. This was cooled to room temperature after which we poured in 200 g of ice water, neutralized by solid K2CO3. The mass was extracted 3 times with 75 ml CHCl3. The combined organic layer was washed with 2 × 100 ml water and dried over anhydrous MgSO4. Concentration under vacuum resulted in 2.2 g light brown oil (Supplemental Figure 9). The product had a chemical composition of C15H21NO3 with a MW of 263.33 g/mol (66% yield). The 1H-NMR (CDCl3) spectra were as follows: δ 9.76 (s, 1H); 7.75 (d, 2H); 6.80 (d, 2H); 4.14 (m, 2H); 3.44 (m, 2H); 3.07 (s, 3H); 2.35 (m, 2H); 1.68 (m, 4H); 1.27 (m, 3H), and ESI-MS (+) of 263.23.
SKC-660.
1.3 g (5 mmol) of SKC-659 and 1.6 g (6 mmol) of 4, 4′–vinylidene bis (N,N-dimethylaniline) were suspended in 15 ml acetic anhydride, and 400 μl of 60% perchloric acid was added slowly. The blue reaction mixture was stirred at room temperature for 20 hours and concentrated; the residue was purified by flash chromatography on SiO2 using a 5% methanol in chloroform as eluent, resulting in 600 mg of deep blue solid (Supplemental Figure 9). The product had a chemical composition of C33H42N3O2+ with a MW of 512.70 g/mol (24% yield). The 1H-NMR (CD3OD) spectra were as follows: δ 7.42–7.99 (m, 9H); 6.76–6.97 (m, 5H); 4.15 (m, 2H), 3.61 (m, 2H); 3.23 (s, 6H); 2.39 (m, 2H); 2.01 (s, 6H); 1.71 (m, 4H); 1.25 (m, 3H), and ESI-MS (+) of 512.3.
SKC-672.
450 mg of SKC-660 was suspended in 50 ml THF. 15% LiOH aqueous solution was added and stirred at room temperature for 24 hours. The organic layer was separated. The aqueous layer was extracted with 3 × 50 ml CHCl3 and combined with the organic layer. The organic fraction was dried over anhydrous MgSO4 and concentrated to 20 ml. 200 μl of 60% HClO4 was added slowly and stirred at room temperature for 1 hour. Upon concentration, the blue residue was purified by flash chromatography on SiO2 using 10% methanol in chloroform as eluent, which resulted in 200 mg (47% yield) of pure product (Supplemental Figure 10). The product had a chemical composition of C31H38N3O2+ with a MW of 484.65 g/mol. The 1H-NMR (CD3OD) spectra were as follows: δ 6.65–7.88 (m, 14H); 3.33 (m, 9H); 2.99 (m, 2H); 2.91 (m, 3H); 2.31 (m, 2H); 1.66 (m, 4H), and ESI-MS (+) of 484.3.
SKC-722.
SKC-672 (162 mg) was dissolved in 3 ml anhydrous DMF. 160 mg of TSTU and 25 μl of diisopropylethylamine were added. The mixture was stirred (room temperature, 3 hours). After concentration under vacuum, the residue was precipitated by diethyl ether and dried, yielding 173 mg of active ester SKC-722 (Supplemental Figure 10). The product had a chemical composition of C35H41N4O4+ with a MW of 581.72 g/mol (89% yield). The 1H-NMR (CD3OD) spectra were as follows: δ 6.53–7.82 (m, 14H); 3.21–3.38 (m, 12H); 3.12 (m, 4H); 2.89 (m, 2H); 2.31 (m, 2H); 1.42 (m, 4H), and ESI-MS (+) of 581.3.
SCi1.
SKC-722 (58 mg, 1 mmol) and DL-homocysteic acid (23 mg, 1.25 mmol) were dissolved in 2 ml anhydrous DMF. DIPEA (20 μl) was added, and the reaction was stirred at room temperature for 24 hours. The product was concentrated, and the residue was purified by flash chromatography on SiO2 using 20% methanol in chloroform as eluent, which provided 51 mg of pure product (Supplemental Figure 10). The product had a chemical composition of C35H45N4O6S+ with a MW of 649.81 g/mol (78% yield). The 1H-NMR (CD3OD) spectra were as follows: δ 6.78–7.99 (m, 14H); 2.72-3.33 (m, 14H); 3.39 (m, 4H); 2.35 (m, 2H); 1.31 (m, 4H), and ESI-MS (+) of 649.
Animal studies.
Athymic nude mice (Taconic Biosciences Inc., catalog NCRNU-F sp/sp), aged 6 to 8 weeks, were used for all in vivo imaging experiments and were maintained on alfalfa and soybean-free feed (Harlan Teklad, catalog 2916). Labeling of Mars1Cy-expressing HCT116 cells for comparison with iRFP713 was conducted ex vivo by incubation of cells with fluorogen (SC1) at 400 nM prior to collection and injection. The s.c. implantation of different numbers of cells was achieved using a total volume of 200 μl as a 1:1 suspension of cells and PBS in Matrigel (Corning Life Sciences). Four mice contributed data per each number of injected cells, with the exception of 1 × 105/Mars1Cy, for which 3 mice were analyzed. For other studies, tumorigenic cells suspended in PBS were implanted either s.c. or into the i.p. cavity as described in the figure legends, as are the cell lines used. For systemic FAP labeling, fluorogen was administered to approximately 400 nM via i.v. or i.p. injection. Imaging of live animals was conducted using an FMT2500LX (PerkinElmer), applying the built-in 680 fluorescence channel (670 nm excitation, 690–740 nm emission collection) in quantitative tomography mode, with mice held under 2% isoflurane. Data were acquired from 20–24 illumination points over the course of 3 to 5 minutes per animal. Quantitation was conducted with the instrument software suite (TrueQuant version 3.1, PerkinElmer) after calibration with purified protein solutions of known concentration. Fluorescence imaging of tumor sections was conducted with the previously described Andor microscope system, using 640 nm excitation coupled with a 794/160 emission filter through a ×60 1.49 NA oil immersion objective or a ×20 0.75 NA air objective (Figure 4C and Supplemental Figure 7, A–C, respectively). Andor IQ software was used to collect fluorescence images as a tiled set of fields with approximately 15% overlap between adjacent fields.
Statistics.
Statistical tests are described alongside the data to which they were applied. P < 0.05 was considered significant.
Study approval.
All studies were approved by the IACUC at the University of Pittsburgh.
Supplementary Material
Acknowledgments
Funding for this work was contributed by the NIH Technology Center for Networks and Pathways (5U54GM103529). This work also used University of Pittsburgh Cancer Institute core facilities, including the In Vivo Imaging Facility, supported in part by Cancer Center Support Grant award P30CA047904. The authors thank Matharishwan Naganbabu for assistance with purification of fluorogens and Yehuda Creeger for accommodations made to cell sorting instrumentation.
Footnotes
Conflict of interest: Marcel P. Bruchez and Alan S. Waggoner are founders of Sharp Edge Labs, a company commercializing assays based on fluorogen-activating peptides. Subhasish K. Chakraborty, Ming Zhang, and Alan S. Waggoner are authors on US patent application 13/920,775, which describes the use of these dyes and proteins as a fluorescent labeling method.
Reference information:J Clin Invest. 2015;125(10):3915–3927. doi:10.1172/JCI81086.
Saumya Saurabh’s present address is: Stanford University, Department of Chemistry, Stanford, California, USA.
References
- 1.Yang M, et al. Direct external imaging of nascent cancer, tumor progression, angiogenesis, and metastasis on internal organs in the fluorescent orthotopic model. Proc Natl Acad Sci U S A. 2002;99(6):3824–3829. doi: 10.1073/pnas.052029099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ntziachristos V, et al. Visualization of antitumor treatment by means of fluorescence molecular tomography with an annexin V-Cy5.5 conjugate. Proc Natl Acad Sci U S A. 2004;101(33):12294–12299. doi: 10.1073/pnas.0401137101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montet X, Ntziachristos V, Grimm J, Weissleder R. Tomographic fluorescence mapping of tumor targets. Cancer Res. 2005;65(14):6330–6336. doi: 10.1158/0008-5472.CAN-05-0382. [DOI] [PubMed] [Google Scholar]
- 4.Alauddin MM, Conti PS. Synthesis and preliminary evaluation of 9-(4-[18F]-fluoro-3-hydroxymethylbutyl)guanine ([18F]FHBG): a new potential imaging agent for viral infection and gene therapy using PET. Nucl Med Biol. 1998;25(3):175–180. doi: 10.1016/S0969-8051(97)00160-1. [DOI] [PubMed] [Google Scholar]
- 5.Yaghoubi SS, Gambhir SS. PET imaging of herpes simplex virus type 1 thymidine kinase (HSV1-tk) or mutant HSV1-sr39tk reporter gene expression in mice and humans using [18F]FHBG. Nat Protoc. 2007;1(6):3069–3074. doi: 10.1038/nprot.2006.459. [DOI] [PubMed] [Google Scholar]
- 6.Soghomonyan S, et al. Molecular PET imaging of HSV1-tk reporter gene expression using [18F]FEAU. Nat Protoc. 2007;2(2):416–423. doi: 10.1038/nprot.2007.49. [DOI] [PubMed] [Google Scholar]
- 7.Penheiter AR, Russell SJ, Carlson SK. The sodium iodide symporter (NIS) as an imaging reporter for gene, viral, and cell-based therapies. Curr Gene Ther. 2012;12(1):33–47. doi: 10.2174/156652312799789235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Urano Y, et al. Selective molecular imaging of viable cancer cells with pH-activatable fluorescence probes. Nat Med. 2009;15(1):104–109. doi: 10.1038/nm.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee H, et al. Near-infrared pH-activatable fluorescent probes for imaging primary and metastatic breast tumors. Bioconjug Chem. 2011;22(4):777–784. doi: 10.1021/bc100584d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weissleder R, Tung CH, Mahmood U, Bogdanov A. In vivo imaging of tumors with protease-activated near-infrared fluorescent probes. Nat Biotechnol. 1999;17(4):375–378. doi: 10.1038/7933. [DOI] [PubMed] [Google Scholar]
- 11.Kossodo S, et al. Noninvasive in vivo quantification of neutrophil elastase activity in acute experimental mouse lung injury. Int J Mol Imaging. 2011;2011: doi: 10.1155/2011/581406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mcelroy WD, Seliger HH, White EH. Mechanism of bioluminescence, chemiluminescence and enzyme function in the oxidation of firefly luciferin. Photochem Photobiol. 1969;10(3):153–170. doi: 10.1111/j.1751-1097.1969.tb05676.x. [DOI] [PubMed] [Google Scholar]
- 13.Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2002;21(1):86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 14.Sun X, et al. Development of SNAP-tag fluorogenic probes for wash-free fluorescence imaging. Chembiochem. 2011;12(14):2217–2226. doi: 10.1002/cbic.201100173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu TK, et al. A rapid SNAP-tag fluorogenic probe based on an environment-sensitive fluorophore for no-wash live cell imaging. ACS Chem Biol. 2014;9(10):2359–2365. doi: 10.1021/cb500502n. [DOI] [PubMed] [Google Scholar]
- 16.Gong H, et al. Near-infrared fluorescence imaging of mammalian cells and xenograft tumors with SNAP-tag. PLoS One. 2012;7(3): doi: 10.1371/journal.pone.0034003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradley RS, Thorniley MS. A review of attenuation correction techniques for tissue fluorescence. J R Soc Interface. 2006;3(6):1–13. doi: 10.1098/rsif.2005.0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schnell SA, Staines WA, Wessendorf MW. Reduction of lipofuscin-like autofluorescence in fluorescently labeled tissue. J Histochem Cytochem Off J Histochem Soc. 1999;47(6):719–730. doi: 10.1177/002215549904700601. [DOI] [PubMed] [Google Scholar]
- 19.Shcherbo D, et al. Bright far-red fluorescent protein for whole-body imaging. Nat Methods. 2007;4(9):741–746. doi: 10.1038/nmeth1083. [DOI] [PubMed] [Google Scholar]
- 20.Szent-Gyorgyi C, et al. Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat Biotechnol. 2008;26(2):235–240. doi: 10.1038/nbt1368. [DOI] [PubMed] [Google Scholar]
- 21.Silva GL, Ediz V, Yaron D, Armitage BA. Experimental and computational investigation of unsymmetrical cyanine dyes: understanding torsionally responsive fluorogenic dyes. J Am Chem Soc. 2007;129(17):5710–5718. doi: 10.1021/ja070025z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan Q, et al. Near-instant surface-selective fluorogenic protein quantification using sulfonated triarylmethane dyes and fluorogen activating proteins. Org Biomol Chem. 2015;13(7):2078–2086. doi: 10.1039/C4OB02309A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szent-Gyorgyi C, Schmidt BF, Fitzpatrick JAJ, Bruchez MP. Fluorogenic dendrons with multiple donor chromophores as bright genetically targeted and activated probes. J Am Chem Soc. 2010;132(32):11103–11109. doi: 10.1021/ja9099328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grover A, Schmidt BF, Salter RD, Watkins SC, Waggoner AS, Bruchez MP. Genetically encoded pH sensor for tracking surface proteins through endocytosis. Angew Chem Int Ed. 2012;51(20):4838–4842. doi: 10.1002/anie.201108107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Özhalici-Ünal H, et al. A rainbow of fluoromodules: a promiscuous scFv protein binds to and activates a diverse set of fluorogenic cyanine dyes. J Am Chem Soc. 2008;130(38):12620–12621. doi: 10.1021/ja805042p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shank NI, Zanotti KJ, Lanni F, Berget PB, Armitage BA. Enhanced photostability of genetically encodable fluoromodules based on fluorogenic cyanine dyes and a promiscuous protein partner. J Am Chem Soc. 2009;131(36):12960–12969. doi: 10.1021/ja9016864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zanotti KJ, et al. Blue fluorescent dye-protein complexes based on fluorogenic cyanine dyes and single chain antibody fragments. Org Biomol Chem. 2011;9(4):1012–1020. doi: 10.1039/C0OB00444H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Senutovitch N, et al. A variable light domain fluorogen activating protein homodimerizes to activate dimethylindole red. Biochemistry (Mosc) 2012;51(12):2471–2485. doi: 10.1021/bi201422g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishchenko AA. The length of the polymethine chain and the spectral-luminescent properties of symmetrical cyanine dyes. Russ Chem Bull. 1994;43(7):1161–1174. doi: 10.1007/BF00698237. [DOI] [Google Scholar]
- 30.Liu J, Subir M, Nguyen K, Eisenthal KB. Second harmonic studies of ions crossing liposome membranes in real time. J Phys Chem B. 2008;112(48):15263–15266. doi: 10.1021/jp806690z. [DOI] [PubMed] [Google Scholar]
- 31.Loew LM, Cohen LB, Salzberg BM, Obaid AL, Bezanilla F. Charge-shift probes of membrane potential. Characterization of aminostyrylpyridinium dyes on the squid giant axon. Biophys J. 1985;47(1):71–77. doi: 10.1016/S0006-3495(85)83878-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kreder R, et al. Blue fluorogenic probes for cell plasma membranes fill the gap in multicolour imaging. RSC Adv. 2015;5(29):22899–22905. doi: 10.1039/C4RA16225K. [DOI] [Google Scholar]
- 33.Falco CN, Dykstra KM, Yates BP, Berget PB. scFv-based fluorogen activating proteins and variable domain inhibitors as fluorescent biosensor platforms. Biotechnol J. 2009;4(9):1328–1336. doi: 10.1002/biot.200900075. [DOI] [PubMed] [Google Scholar]
- 34.Szent-Gyorgyi C, et al. Malachite green mediates homodimerization of antibody VL domains to form a fluorescent ternary complex with singular symmetric interfaces. J Mol Biol. 2013;425(22):4595–4613. doi: 10.1016/j.jmb.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holleran JP, et al. Pharmacological rescue of the mutant cystic fibrosis transmembrane conductance regulator (CFTR) detected by use of a novel fluorescence platform. Mol Med Camb Mass. 2012;18:685–696. doi: 10.2119/molmed.2012.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carmody WR. Easily prepared wide range buffer series. J Chem Educ. 1961;38(11): doi: 10.1021/ed038p559. [DOI] [Google Scholar]
- 37.Kim JH, et al. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One. 2011;6(4): doi: 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Auldridge ME, Satyshur KA, Anstrom DM, Forest KT. Structure-guided engineering enhances a phytochrome-based infrared fluorescent protein. J Biol Chem. 2012;287(10):7000–7009. doi: 10.1074/jbc.M111.295121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shu X, et al. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science. 2009;324(5928):804–807. doi: 10.1126/science.1168683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Filonov GS, Piatkevich KD, Ting L-M, Zhang J, Kim K, Verkhusha VV. Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat Biotechnol. 2011;29(8):757–761. doi: 10.1038/nbt.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shcherbakova DM, Verkhusha VV. Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat Methods. 2013;10(8):751–754. doi: 10.1038/nmeth.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Encell LP. Development of a dehalogenase-based protein fusion tag capable of rapid, selective and covalent attachment to customizable ligands. Curr Chem Genomics. 2013;6(1):55–71. doi: 10.2174/1875397301206010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Monfardini C, Veronese FM. Stabilization of Substances in Circulation. Bioconjug Chem. 1998;9(4):418–450. doi: 10.1021/bc970184f. [DOI] [PubMed] [Google Scholar]
- 44.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006;1(2):755–768. doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 45.Feldhaus MJ, et al. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat Biotechnol. 2003;21(2):163–170. doi: 10.1038/nbt785. [DOI] [PubMed] [Google Scholar]
- 46.Mujumdar RB, Ernst LA, Mujumdar SR, Lewis CJ, Waggoner AS. Cyanine dye labeling reagents: sulfoindocyanine succinimidyl esters. Bioconjug Chem. 1993;4(2):105–111. doi: 10.1021/bc00020a001. [DOI] [PubMed] [Google Scholar]
- 47.Würth C, Pauli J, Lochmann C, Spieles M, Resch-Genger U. Integrating sphere setup for the traceable measurement of absolute photoluminescence quantum yields in the near infrared. Anal Chem. 2012;84(3):1345–1352. doi: 10.1021/ac2021954. [DOI] [PubMed] [Google Scholar]
- 48.Williams ATR, Winfield SA, Miller JN. Relative fluorescence quantum yields using a computer-controlled luminescence spectrometer. The Analyst. 1983;108(1290):1067–1071. doi: 10.1039/an9830801067. [DOI] [Google Scholar]
- 49.Chou WC, Liao KW, Lo YC, Jiang SY, Yeh MY, Roffler SR. Expression of chimeric monomer and dimer proteins on the plasma membrane of mammalian cells. Biotechnol Bioeng. 1999;65(2):160–169. doi: 10.1002/(sici)1097-0290(19991020)65:2<160::aid-bit5>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 50.Pfaff N, et al. A ubiquitous chromatin opening element prevents transgene silencing in pluripotent stem cells and their differentiated progeny. Stem Cells. 2013;31(3):488–499. doi: 10.1002/stem.1316. [DOI] [PubMed] [Google Scholar]
- 51.Swift S, Lorens J, Achacoso P, Nolan GP. Rapid production of retroviruses for efficient gene delivery to mammalian cells using 293T cell-based systems. Curr Protoc Immunol. 2001;Chapter 10: doi: 10.1002/0471142735.im1017cs31. [DOI] [PubMed] [Google Scholar]
- 52.Zhang F, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446(7136):633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
- 53.Preibisch S, Saalfeld S, Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 2009;25(11):1463–1465. doi: 10.1093/bioinformatics/btp184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2(12):905–909. doi: 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.