

Abstract
Cytokines and metabolic pathway–controlling enzymes regulate immune responses and have potential as powerful tools to mediate immune tolerance. Blockade of the interaction between CD40 and CD40L induces long-term cardiac allograft survival in rats through a CD8+CD45RClo Treg potentiation. Here, we have shown that the cytokine IL-34, the immunoregulatory properties of which have not been previously studied in transplantation or T cell biology, is expressed by rodent CD8+CD45RClo Tregs and human FOXP3+CD45RCloCD8+ and CD4+ Tregs. IL-34 was involved in the suppressive function of both CD8+ and CD4+ Tregs and markedly inhibited alloreactive immune responses. Additionally, in a rat cardiac allograft model, IL-34 potently induced transplant tolerance that was associated with a total inhibition of alloantibody production. Treatment of rats with IL-34 promoted allograft tolerance that was mediated by induction of CD8+ and CD4+ Tregs. Moreover, these Tregs were capable of serial tolerance induction through modulation of macrophages that migrate early to the graft. Finally, we demonstrated that human macrophages cultured in the presence of IL-34 greatly expanded CD8+ and CD4+ FOXP3+ Tregs, with a superior suppressive potential of antidonor immune responses compared with non–IL-34–expanded Tregs. In conclusion, we reveal that IL-34 serves as a suppressive Treg–specific cytokine and as a tolerogenic cytokine that efficiently inhibits alloreactive immune responses and mediates transplant tolerance.
Introduction
Organ transplantation has undergone substantial improvements in both the prevention and treatment of acute rejection, but subclinical episodes and chronic graft dysfunction still heavily impact medium- and long-term graft survival (1). Emerging therapeutic strategies, among them induction of tolerance to donor antigens, are moving to the clinical stage after years of experimental model work (2, 3). Among natural mechanisms and tolerance-inductive strategies, the use of different types of regulatory cells are among the most promising ones (4). The uses of CD8+ Tregs have been highlighted in recent years by our group and others in the transplantation field, but also in other pathological situations (5–8). Cytokines, enzymes controlling metabolic pathways, and cell surface molecules capable of inducing tolerance have also been described as new mediators of immune tolerance.
IL-34 was identified in 2008 (9). Studies showed that IL-34 shares homology with M-CSF and that they act through a common receptor, CD115, also called CSF-1R (9), expressed on the cell surface of monocytes, and in the brain through a newly described receptor, receptor-type protein-tyrosine phosphatase ζ (PTP-ζ) (10). However, studies have demonstrated that IL-34 and M-CSF display distinct biological activity and signal activation (11), in part due to their differing spatial and temporal expression (12). Up to now, IL-34 function has been mainly linked with the survival and function of monocytes and macrophages (osteoclasts, microglia), as well as with DCs (12). IL-34 protein expression in resting cells has been observed in keratinocytes, hair follicles, neurons, proximal renal tubule cells, and seminiferous tubule germ cells (12) and also in heart, brain, lung, liver, kidney, spleen, thymus, testicles, ovaries, prostate, colon, small intestine, spleen red pulp, and osteoclasts (9). Upon inflammation, other cells, such as fibroblasts and articular synovial cells, upregulate IL-34 expression (13, 14). So far, expression of IL-34 by other lymphoid cells, and particularly by T cells, has not been described or demonstrated. Similarly, IL-34 has not been linked to the effects of DCs or T cells on immune function (12). Finally, there is no description to date of a role for IL-34 in transplant tolerance.
CD45RC has been shown in rats, mice, and humans to be a marker of both CD4+ and CD8+ Tregs (15–20). In a rat cardiac allograft model, we have previously shown that blockade of CD40-CD40L interaction by CD40Ig treatment induces long-term allograft survival through the generation of CD8+CD45RClo Tregs (termed CD8+CD40Ig Tregs). This is in contrast to natural CD8+CD45RClo Tregs, which do not inhibit cardiac allograft rejection (18). We have shown that these CD8+CD40Ig Tregs impose allogeneic tolerance partially through production of IFN-γ and fibrinogen-like protein 2 (FGL2) (18, 21, 22) and recognition of a dominant MHC II–derived donor peptide presented by recipient MHC I (23). A potential role for FGL2 as an immune tolerogenic mechanism was first suspected when pan-genomic transcriptomic comparison of CD8+CD40Ig Tregs versus CD8+CD45RClo Tregs from naive animals showed increased Fgl2 mRNA expression (ArrayExpress database accession number E-MTAB-3535) (21). Results from the same transcriptomic analysis revealed that the cytokine IL-34 is overexpressed in CD8+CD40Ig Tregs from long-term recipients compared with that seen in CD8+CD45RClo Tregs from naive animals.
In this study, we investigate the expression and functional role of IL-34 produced by CD45RClo Tregs in rats and humans, evaluate the immunoregulatory potential of IL-34 ex vivo in humans and in vivo in an organ transplant model in rats, and elucidate the mechanisms involved. We provide here the first demonstration to our knowledge that IL-34 has immunosuppressive properties. We found that IL-34 was expressed in tolerated cardiac allografts and by CD8+CD40Ig Tregs. We also show the involvement of IL-34 in CD8+ Treg–suppressive function ex vivo and demonstrate that IL-34 has a significant suppressive function in allogeneic immune responses both ex vivo and in vivo and induces transplant tolerance. We believe we have unraveled the mechanism of this tolerance induction and demonstrate that IL-34–modified macrophages that migrate early to the graft induce highly suppressive and dominant Tregs. Significantly, we demonstrate the specific expression of IL-34 by natural human CD45RCloFOXP3+ CD4+ and CD8+ Tregs, not effector T cells, as well as the potential of IL-34 to suppress alloreactive responses and its involvement in CD4+CD25hiCD127lo and CD8+CD45RClo Treg–mediated suppression, providing the first evidence to our knowledge of a role for IL-34 in immune tolerance in humans. Finally, we provide proof of concept that human IL-34–differentiated macrophages expand and potentiate human FOXP3+ Tregs. Altogether, we have identified IL-34 as a tolerogenic cytokine that efficiently inhibits antidonor immune responses, thus providing a new mediator of transplant tolerance.
Results
IL-34 expressed by splenic CD8+CD40Ig Tregs and tolerated allograft.
RNA microarray analysis of CD8+CD40Ig Tregs versus naive CD8+CD45RClo Tregs from spleen showed that Il34 is among the most upregulated genes by CD8+CD40Ig Tregs, with a 4.05-fold change. This upregulation was confirmed by qPCR, with a greater than 11-fold increase in expression of Il34 mRNA in splenic CD8+CD40Ig Tregs from long-term surviving recipients compared with that in natural CD8+CD45RClo Tregs (P < 0.05, Figure 1A).
Figure 1. IL-34 and CD115, but not M-CSF, are involved in CD8+CD45RClo Treg–mediated suppression.

(A). FACSAria-sorted CD8+CD45RClo Tregs from spleen of naive or 120-day-old AdCD40Ig-treated recipients (n = 6) were analyzed for Il34 mRNA expression by qPCR. Mann-Whitney U test, **P < 0.01. (B) Il34 mRNA expression levels in cardiac grafts from AdCD40Ig-treated recipients on days 5 (n = 3) and 120 (n = 7) after transplantation were compared with those in grafts from nontreated (NT) recipients on days 5 (n = 8), 7 (n = 8) and 120 (n = 6) and with levels in native hearts from naive animals (n = 7). Mann-Whitney U test, *P < 0.05, **P < 0.01, ***P < 0.001. (C–E) The relative proportion of CFSE-labeled LEW.1A dividing CD4+CD25– T cells stimulated with donor LEW.1W pDCs was analyzed after 6 days of culture in the presence of LEW.1A CD8+CD40Ig Tregs at a 1:1 effector/suppressor ratio. Proliferation after the addition of anti–IL-34–blocking Ab (C), anti–M-CSF–blocking Ab (D), or anti-CD115–blocking Ab (E) was evaluated and compared with that of the appropriate isotypic control (n = 4 experiments performed in triplicate). The proportion of dividing CD4+CD25– T cells in the control proliferation condition with pDCs only representing approximately 80% of the cells on day 5 was given a value of 100 in each experiment. Results are expressed as the mean ± SEM of the relative proportion of dividing CD4+CD25– T cells. A representative raw CFSE profile is shown in C (right panel). Kruskal-Wallis and Dunn’s post tests, *P < 0.05 (C–E).
Analysis of mRNA from whole organs showed that Il34 mRNA was expressed constitutively in spleen and heart of naive animals, as observed by Lin et al. (ref. 24, Figure 1B, and Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI81227DS1) and was significantly decreased during acute allograft rejection on day 5 in the grafts of nontreated recipients (NT D5, Figure 1B). In correlation with our previous observations that CD8+CD40Ig Tregs accumulate in the graft during the first week (21), Il34 mRNA expression levels were significantly greater on day 5 in AdCD40Ig-treated grafts compared with levels in naive and nontreated recipients (Figure 1B) and had returned to normal by day 120 after transplantation.
Altogether, these results demonstrate that IL-34 is expressed by CD8+CD40Ig Tregs. Moreover, the early expression of IL-34 in graft and spleen suggests its early involvement in the inhibition of acute graft rejection and the possible establishment of allograft tolerance.
IL-34 expressed by CD8+CD40Ig Tregs, but not M-CSF, is involved in suppression of alloreactive CD4+ T cell proliferation.
We have previously demonstrated that CD8+CD40Ig Tregs suppress the antidonor proliferation of CD4+ effector T cells in response to allogeneic plasmacytoid DCs (pDCs) ex vivo (21). In addition, we have demonstrated the involvement of IFN-γ and FGL2 in this process; however, some suppression remains after blockade of the IFN-γ and FGL2 inhibitory effect (18, 21, 22). To address whether IL-34 is involved in CD8+CD40Ig Treg suppression, we tested a neutralizing anti–IL-34 Ab in the suppressive mixed lymphocyte reaction (MLR) assay (Figure 1C and Supplemental Figure 2A). The addition of increasing concentrations of neutralizing anti–IL-34 Ab resulted in a dose-dependent increase in CD4+CD25– T cell proliferation, reversing the CD8+CD40Ig Treg–mediated inhibition by up to 59% compared with isotype control Abs used at the highest concentration. Given that IL-34 has homologies with M-CSF, that we observed a significant expression of M-CSF by CD8+CD40Ig Tregs compared with natural CD8+CD45RClo Tregs (Supplemental Figure 3A), and that IL-34 and M-CSF use the same receptor in the macrophage lineage cells and DCs (24, 25), we investigated the possibility that M-CSF could play a role in the suppression of effector CD4+CD25– T cell proliferation. First, we tested the suppressive potential of M-CSF in the MLR assay described above. Interestingly, we observed that M-CSF efficiently suppressed, in a dose-dependent manner, up to 93.5% of CD4+CD25– T cell proliferation (Supplemental Figure 3B), suggesting that M-CSF–mediated suppression, like that for IL-34, acts through pDCs expressing the CD115 receptor (26, 27). However, the addition of a blocking anti–M-CSF Ab or of an isotype control Ab in coculture suppressive assays in the presence of CD8+ Tregs did not restore CD4+CD25– T cell proliferation, demonstrating that M-CSF is not involved in CD8+CD40Ig Treg–mediated suppression (Figure 1D).
We next tested the involvement of CD115, until now the only receptor for IL-34 described outside the CNS (24, 25), which is only expressed by monocytes and macrophages, conventional DCs (cDCs), and pDCs, and not by CD4+ T cells (26, 27). We used an anti-CD115–blocking Ab that has been previously shown to inhibit M-CSF action in both rats and mice (28). We demonstrated that blocking CD115 significantly, although not completely, abrogated, in a dose-dependent manner, CD8+ Treg–mediated suppression of CD4+ T cell proliferation in the presence of pDCs, in contrast to isotype control Ab (Figure 1E).
In conclusion, we have demonstrated the involvement of IL-34–CD115 interaction, but not M-CSF–CD115 interaction, in the suppressive effect of CD8+CD40Ig Tregs on effector CD4+ T cell proliferation.
IL-34 is expressed by human FOXP3+ T cells and possesses a strong suppressive potential.
Since IL-34 has never been demonstrated to be expressed by Tregs, to inhibit antidonor immune responses in humans, or to be involved in Treg-mediated suppression, we sought to assess the potential and applicability of our findings in humans. It has been suggested that human CD45RClo T cells are associated with Tregs, while CD45RChi T cells are associated with naive and effector T cells (20, 29). We first analyzed IL-34 protein expression on CD8+ and CD4+ CD45RClo cells versus CD45RChi T cells by multicolor flow cytometry (Figure 2A). Interestingly, we observed that both human CD4+CD45RClo and CD8+CD45RClo T cells expressed significant amounts of IL-34 protein in contrast to CD45RChi T cells. Moreover, we observed that CD8+CD45RClo T cells expressed significantly more IL-34 than did CD4+CD45RClo T cells. To further discriminate IL-34 expression by Tregs inside the CD45RClo cells, we analyzed IL-34 expression within FOXP3+ Tregs (Figure 2, B and C and Supplemental Figure 4). We observed that IL-34 expression was specific to CD4+CD45RCloFOXP3+ and CD8+CD45RCloFOXP3+ Tregs, but not in CD4+CD45RCloFOXP3–, CD8+CD45RCloFOXP3–, CD4+CD45RChi, or CD8+CD45RChi T cell subsets (Figure 2B), indicating that IL-34 is a Treg-specific cytokine. More precisely, we observed that approximately half of the FOXP3+ Tregs (CD4+ or CD8+) expressed IL-34 (Figure 2C).
Figure 2. IL-34 is a human FOXP3+ Treg–specific cytokine involved in the suppressive activity of antidonor immune responses.

The percentage of IL-34+ cells in healthy individuals was evaluated in CD4+ or CD8+ CD45RClo or CD45RChi T cells (A) or in FOXP3+ versus FOXP3– CD45RClo CD8+ or CD4+ T cells (B and C). (A) The mean ± SEM of 27 healthy individuals is represented. Mann-Whitney U test, *P < 0.05, ***P < 0.001. Representative histogram and plot of healthy individuals (B) showing the mean of expression ± SEM of 10 healthy individuals (C). (D) Soluble IL-34 was tested for suppression of CD4+CD25– T cell proliferation in response to allogeneic T cell–depleted PBMCs and analyzed by flow cytometry for CFSE dilution after 5 days of culture (n = 2–5 experiments performed in duplicate). Results are expressed as the mean ± SEM of the relative proportion of dividing CD4+CD25– T cells. Representative histogram of 1 experiment: Proliferation of CFSE-labeled CD4+CD25– T cells cocultured with T cell–depleted PBMCs with increased concentrations of IL-34. Two-way ANOVA with Bonferroni’s post test versus M-CSF, ***P < 0.001. (E) The relative proportion of CFSE-labeled dividing CD4+CD25– T cells stimulated with allogeneic T cell–depleted PBMCs was analyzed after 5 days of culture, in the presence of CD8+CD45RClo or CD4+CD25hiCD127– Tregs at 1:1 effector/suppressor ratios. The proliferation after addition of anti–IL-34–blocking Ab was evaluated and compared with isotypic control (n = 5–6 experiments performed in triplicate). The proportion of dividing CD4+CD25– T cells in the control proliferation condition with allogeneic T cell–depleted PBMCs only represented approximately 60% of the cells on day 5 and was given a value of 100 in each experiment. Results are expressed as the mean ± SEM of the relative proportion of dividing CD4+CD25– T cells. A representative raw CFSE profile is shown in the right panel. Wilcoxon test (versus proliferation without Tregs = 100), **P < 0.01.
Since we suspected a suppressive potential of IL-34 in humans, we added different doses of soluble human IL-34 to an MLR, whereby CD4+CD25– CFSE–labeled effector T cells were cultured in the presence of T cell–depleted allogeneic peripheral blood mononuclear cells (PBMCs) as antigen-presenting cells (APCs) (Figure 2D and Supplemental Figure 2B). We observed significant, dose-dependent inhibition of effector T cell proliferation in the presence of IL-34, thus confirming the suppressive potential of IL-34 on antidonor immune responses. Finally, to demonstrate the involvement of IL-34 in CD4+CD25hiCD127lo and CD8+CD45RClo Treg–mediated suppressive activity of antidonor immune responses, we added either anti-human IL-34–blocking Ab or a control isotype Ab to an MLR, in which CFSE-labeled CD4+CD25– effector T cell proliferation in the presence of allogeneic T cell–depleted PBMCs was inhibited by Tregs (Figure 2E). We observed that blocking IL-34 significantly reverted Treg-mediated suppression for both CD4+ and CD8+ Tregs compared with isotype control Ab, demonstrating the key role of IL-34 in the suppressive activity of Tregs.
Altogether, these data prove the relevance of our findings and provide proof of concept that IL-34 is a Treg-specific protein and a potential therapeutic target for manipulating the antidonor immune response.
Generation of an adeno-associated viral vector for sustained expression of IL-34.
We sought to demonstrate the in vivo suppressive activity of IL-34 and further understand IL-34 mechanisms. Since recombinant IL-34 rat cytokine is not commercially available and difficult to produce for in vivo experiments, we generated a recombinant adeno-associated viral (AAV) vector encoding rat IL-34, as we have done for other molecules in primates (30) and rats (22, 31). In this vector, the rat Il34 cDNA was fused with a C-terminal Myc tag, and both the plasmid (pIL-34) and AAV were used to respectively transfect or transduce cells of the HEK293 T cell line (Figure 3A and Supplemental Figure 5). IL-34 expression was analyzed by flow cytometry for the Myc tag. Myc staining was not detectable on pGFP-transfected or AAV-GFP–transduced HEK293 T cells, nor on pIL-34–transfected or AAV–IL-34–transduced HEK293 T cells stained with isotypic control Ab (Figure 3A and Supplemental Figure 5). However, HEK293 T cells transfected with pIL-34 or transduced with AAV–IL-34 displayed strong, dose-dependent expression levels of IL-34 (Figure 3A and Supplemental Figure 5), demonstrating the functionality of the vector.
Figure 3. AAV-mediated expression of rat IL-34 inhibits antidonor cellular responses.

(A) Cells were transduced with AAV–IL-34 or control AAV-GFP at the indicated MOI and analyzed for IL-34 expression 24 hours later (solid thick black line: AAV–IL-34 MOI 102 labeled with anti-Myc; solid thin gray line: AAV–IL-34 MOI 10; dotted black line: AAV-GFP MOI 102 labeled with anti-Myc Ab; and filled gray area: AAV–IL-34 MOI 102 with isotypic control Ab; 1 representative experiment of 4). (B) IL-34 protein was quantified by ELISA in AAV–IL-34 versus AAV-GFP–treated rats 15 days after infection. Serum of nontreated naive animals was used as a control for IL-34 endogenous expression (n = 3). (C) Supernatants of AAV–IL-34 or AAV-GFP–transduced cells were tested for suppression of CD4+CD25– T cell proliferation in response to allogeneic pDCs and analyzed for CFSE dilution after 5 days. CD8+ Tregs were used as a positive control for suppression (n = 3 experiments performed in duplicate). Results are expressed as the mean ± SEM of the normalized percentage of proliferation versus proliferation in the absence of CD8+ Tregs (100%). Representative histogram is shown. Two-way repeated-measures ANOVA with Bonferroni’s post test versus AAV-GFP–transduced cell supernatant dilution, *P < 0.05. (D) Serial dilution of sera from AAV–IL-34 or AAV-GFP–treated (not shown) rats or naive animals (dotted line) were tested for suppression of CD4+CD25– T cell proliferation in response to allogeneic pDCs and analyzed for CFSE dilution after 5 days. The proportion of dividing CD4+CD25– T cells with pDCs only represented 80% of the cells on day 5 and was given a value of 100 in each experiment. Results are expressed as the mean ± SEM of the relative proportion of dividing CD4+CD25– T cells. CD8+ Tregs were used as positive control of suppression (n = 3 experiments performed in duplicate). Two-way repeated-measures ANOVA with Bonferroni’s post test versus serum from naive rats, *P < 0.05, ***P < 0.001.
We next assessed IL-34 production in vivo by ELISA. First, rats were injected i.v. with 1 × 1012 vg/rat of AAV, and sera were collected on day 15 after injection. We observed an increase in IL-34 protein in the sera of AAV–IL-34–treated rats compared with that in AAV-GFP–treated rats on day 15 (Figure 3B), demonstrating the efficient expression of IL-34 in vivo, in accordance with the expression of genes vectorized by AAVs that we have previously analyzed (30).
We then tested the suppressive potential of AAV–IL-34–transduced HEK293 T cell culture supernatants (Figure 3C) and sera from AAV–IL-34–treated rats (Figure 3D), both of which contain high amounts of IL-34 protein (Figure 3B and data not shown). We observed that both supernatants from AAV–IL-34–transduced cells and sera from AAV–IL-34–treated rats significantly inhibited, in a dose-dependent manner, the proliferative response of CD4+ effector T cells stimulated by allogeneic pDCs, whereas supernatants from AAV-GFP–transduced cells and sera from AAV-GFP–treated rats did not (Figure 3, C and D and data not shown).
Altogether, these results demonstrate the functionality of the vector and the suppressive ex vivo efficacy of IL-34 in inhibiting rat effector T cell proliferation, as we have shown in humans (Figure 2D).
Therapeutic effect of IL-34 on allograft tolerance induction.
To further determine the suppressive potential of IL-34 in vivo as a therapeutic strategy, we treated recipients with either AAV–IL-34 (1 × 1012 vg/rat) or a control noncoding AAV, delivered i.v. 1 month before transplantation to obtain optimal transgene expression through the kinetics of AAV vector expression (22, 31). Such treatment with IL-34 alone resulted in a significant prolongation of allograft survival (mean survival time 32.6 ± 7.8 days) compared with controls injected with noncoding AAV (10.4 ± 1.4 days) or untreated recipients (7.8 ± 0.6 days) (Figure 4A). To improve allograft survival, recipients were then treated with a suboptimal dose of rapamycin (0.4 mg/kg/day for 10 days) in addition to the AAV vector. Ten days of rapamycin treatment alone did not significantly extend allograft survival (Figure 4A). In contrast, we observed an indefinite allograft survival in 64% of the recipients given the combined therapy of AAV–IL-34 and rapamycin (n = 13, Figure 4A). Analysis of grafts from long-surviving recipients for signs of chronic rejection revealed a complete absence of vascular lesions, i.e., normal vessel structure and absence of leukocyte infiltration into the myocardium in all recipients analyzed (Figure 4B). In addition, analysis of the antidonor Ab presence in the sera from long-surviving recipients revealed a complete inhibition of total IgG, IgG1, IgG2a, and IgG2b antidonor Ab production in contrast to the sera from recipients treated with the noncoding AAV (Figure 4C).
Figure 4. Il34 gene transfer prolongs allograft survival alone and in combination with a suboptimal dose of rapamycin.

(A) Recipients given i.v. 1012 vg/rat AAV–IL-34 or noncoding AAV or no treatment were grafted 30 days later with no additional treatment or in combination with a suboptimal dose of rapamycin (10 days, starting on day 0). Graft survival was evaluated by palpation through the abdominal wall. Log-rank test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (B) Representative image of the anatomopathological analysis of graft from AAV–IL-34 plus rapamycin–treated, day-120 long-term surviving recipients. Original magnification, ×100. (C) IgG alloantibody production was evaluated in naive and treated long-term recipients more than 120 days after transplantation. Graph represents MFI ± SEM. Two-way ANOVA with Bonferroni’s post test, *P < 0.05, ***P < 0.001, ****P < 0.0001 for noncoding AAV–IL-34 versus noncoding AAV.
Altogether, we were able to demonstrate that treatment with IL-34, in combination with a suboptimal dose of rapamycin, is a valuable therapeutic strategy for tolerance induction and results in abrogation of all allogeneic immune responses.
Induction of Tregs capable of infectious tolerance following IL-34 treatment.
As demonstrated above, IL-34 is produced specifically by CD8+CD40Ig Tregs, but we also wanted to assess whether regulatory cells were induced in the context of IL-34 treatment and involved in the long-term allograft survival generated by the AAV–IL-34 and rapamycin combination. To do so, we performed adoptive cell–transfer experiments, putting splenocytes of long-surviving recipients into naive grafted, sublethally irradiated recipients, as we have done previously (18). First, adoptive transfer of 1.5 × 108 splenocytes into secondary naive grafted, irradiated recipients resulted in significant prolongation of allograft survival in 60% of the recipients (Figure 5A), demonstrating that IL-34 efficiently induces regulatory cells. We investigated the anatomopathological status of the graft of first adoptively transferred long-term splenocyte recipients and observed a complete absence of vascular lesions and obstructions (i.e., no signs of chronic rejection) (data not shown). We then determined that this prolongation of allograft survival can be serially transferred at least 3 times into naive grafted, irradiated recipients (Figure 5A, second and third transfers).
Figure 5. Serial tolerance mediated by Tregs following IL-34 induction.

(A) LEW.1A recipients were sublethally irradiated (4.5 Gy) on day –1 and received heart allografts and i.v. injections of 1.5 × 108 splenocytes from long-surviving recipient or naive animals on day 0. Graft survival was monitored by abdominal palpation. (B) LEW.1A recipients were sublethally irradiated (4.5 Gy) on day –1 and received heart allografts and i.v. injections of total splenocytes or purified cell subpopulations (T cells: 4 × 107; B cells: 6 × 107; CD11b/c+ cells: 1.5 × 107; CD8+CD45RClo Tregs: 4 × 106; CD4+CD25hi Tregs: 4 × 106) from long-surviving recipients on day 0. Graft survival was monitored by abdominal palpation. Log-rank test, *P < 0.05, **P < 0.01.
We investigated the regulatory cell population allowing serial adoptive tolerance transfer, including that of macrophages (32). To do so, we purified subpopulations of the different main cell subsets (B cells, T cells, and macrophages) from tolerant recipients treated with IL-34 (Supplemental Figure 2C) and performed adoptive cell transfer into naive grafted, irradiated recipients (Figure 5B). Surprisingly, we observed that tolerance transfer was achieved only with T cell transfer, demonstrating for the first time to our knowledge that Tregs are induced following IL-34 treatment. Although macrophages did not transfer tolerance, IL-34 may induce Tregs through macrophages (see below). To further determine which Treg population (i.e. CD4+CD25hi or CD8+CD45RClo T cells) could confer tolerance to newly grafted recipients, we sorted CD4+CD25hi and CD8+CD45RClo Tregs and performed adoptive cell transfer (Figure 5B). We observed that adoptive transfers of both CD4+CD25hi and CD8+CD45RClo Tregs resulted in long-term allograft survival in 50% of recipients, suggesting that both populations of Tregs had been equally potentiated by the IL-34–modified macrophages.
Altogether, these in vivo results demonstrate that efficient Tregs are generated following IL-34 treatment in the context of reduced inflammation and transplantation and that those Tregs can induce serial tolerance in a dominant fashion.
Treg induction is mediated by IL-34–modified macrophages infiltrating the graft.
We further characterized the effect of IL-34 on macrophages in the context of induction of tolerance to an allograft. We first sorted macrophages from spleen, blood, and graft of AAV–IL-34–treated recipients on day 15 following transplantation (i.e., day 45 after AAV injection) and macrophages from naive rats and analyzed by qPCR a number of genes described in the literature (refs. 33, 34, and Figure 6). Interestingly, we observed that, compared with naive macrophages, macrophages in the graft from AAV–IL-34–treated recipients strongly upregulated arginase 1 and inducible NO synthase (Inos), both of which are implicated in the metabolism of essential amino acids and described as common mechanisms of immunoregulation of suppressive macrophages by limiting the proliferation of T lymphocytes (35, 36). We also observed increased expression of Cd14 and decreased expression of Cd23, Cd86, and Il10, mostly in the graft but also in the spleen and the blood of AAV–IL-34–treated macrophages, compared with naive macrophages. Finally, we observed no significant differences in Cd16, Cd32, Il1, Tgfb, or Tnfa expression. It is also interesting to note that there were significant differences between macrophages located in the blood versus those in the graft (Arg1, P = 0.0243; Cd14, P = 0.0357; Cd23, P = 0.0303; Il10, P = 0.0470), suggesting that IL-34–modified regulatory macrophages migrate and locate in the graft quickly after transplantation.
Figure 6. Transcript accumulation in macrophages following IL-34 treatment.

mRNA expression was assessed by qPCR on sorted macrophages from untreated spleen or AAV–IL-34–treated spleen, blood, and graft of recipients on day 15 after transplantation. Results are normalized to Hprt and expressed as 2–ΔΔCt ± SEM. Kruskal-Wallis and Dunn’s post tests, *P < 0.05.
To prove that IL-34–modified macrophages play a role in tolerance induction, macrophages were depleted using clodronate-loaded liposomes, from day –25 before transplantation to day 3 after transplantation, and treated simultaneously with IL-34 or noncoding AAV plus a suboptimal dose of rapamycin over a period of 10 days. As a result, AAV–IL-34 injected 30 days before transplantation could not act through macrophages that were depleted at that same time. Significantly, by the time the liposome depletion started, most of the AAV serotype 8 had been integrated into the hepatocytes. We observed that recipients treated with clodronate-loaded liposomes rejected their graft more quickly after transplantation than did the control group animals, demonstrating that macrophages are essential in tolerance induction by IL-34 (Figure 7).
Figure 7. Macrophage depletion results in allograft rejection following IL-34 treatment.

On day –30, allograft recipient animals received i.v. 1012 vg/rat AAV–IL-34 or noncoding AAV or were untreated, with or without weekly i.p. administration of clodronate liposomes from day –25 to day 3, and were grafted on day 0 in combination with a suboptimal dose of rapamycin (10 days, starting on day 0). Graft survival was evaluated by palpation through the abdominal wall. Log-rank test, **P < 0.01.
IL-34–differentiated macrophages efficiently induce and potentiate human CD4+ and CD8+ FOXP3+ Tregs.
Given that we have demonstrated a modified macrophage phenotype following IL-34 administration to rats and induction of potentiated Tregs able to adoptively transfer tolerance to grafted recipients, we wondered whether these results could be reproduced ex vivo in humans. To investigate this, CD14+ monocytes from healthy volunteers were cell sorted and differentiated in the presence of IL-34 for 6 days, and the phenotype was analyzed (Supplemental Figure 6). We observed that IL-34–differentiated macrophages expressed higher levels of CD163, CD14, HLA-DR, CD86, and CD80 than did fresh monocytes. The differentiated macrophages were then added to allogeneic PBMCs for 15 days, then the proportion, number, and suppressive capacity of Tregs were analyzed (Figure 8). Interestingly, we observed that, following culture with IL-34–differentiated macrophages, FOXP3+CD45RCloCD4+ and FOXP3+CD45RCloCD8+ T cells increased as a percentage of CD4+ or CD8+ T cells by 5- and 8.2-fold, respectively (Figure 8, A and B). This increase in percentage was also accompanied by an increase in the number of FOXP3+CD45RCloCD4+ and FOXP3+CD45RCloCD8+ T cells of 83.4- and 100.6-fold, respectively (Figure 8C). Most important, we observed that the IL-34–expanded CD4+CD25hiCD127lo Tregs and CD8+CD45RClo Tregs displayed a highly potent suppressive capacity up to a 16:1 effector/suppressor ratio, where we observed approximately 50% of the suppression compared with unstimulated and polyclonally stimulated CD4+CD25hiCD127lo Tregs and CD8+CD45RClo Tregs (Figure 8D and Supplemental Figure 2D).
Figure 8. Efficient human FOXP3+ Treg expansion and potentiation following IL-34–induced macrophage differentiation.

CD14+ monocytes were differentiated for 6 days with IL-34 or not and added to total allogeneic PBMCs for 21 days. The percentage of FOXP3+ cells was evaluated in PBMCs among CD4+ or CD8+ CD45RClo or CD45RChi T cells. A representative plot (A) and graph (B) are shown before and after culture for 3 healthy individuals. Mϕ, macrophages. (C) Fold expansion was evaluated for FOXP3+ CD4+ or CD8+ Tregs. (D) Unstimulated, stimulated, or IL-34–expanded CD4+CD25hiCD127lo and CD8+CD45RClo Tregs were tested for suppression of CFSE-labeled CD4+CD25– T cell proliferation in response to allogeneic T cell–depleted PBMCs and analyzed by flow cytometry for CFSE dilution after 5 days of culture (n = 3). The proportion of dividing CD4+CD25– T cells in the control proliferation condition with allogeneic T cell–depleted PBMCs only represented approximately 60% of the cells on day 5 and was given a value of 100 in each experiment. Results are expressed as the mean ± SEM of the relative proportion of dividing CD4+CD25– T cells. A representative raw CFSE profile is shown. Two-way repeated-measures ANOVA, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Altogether, these results demonstrate that IL-34–differentiated monocytes have the capacity to selectively expand and to not only maintain, but potentiate, the suppressive capacity of FOXP3+ Tregs.
Discussion
The biological importance of IL-34 remains, to date, largely unknown and debatable. The current understanding of the role of IL-34 was mostly driven by the study of pathological situations in which IL-34 was found to exert inflammatory effects, as M-CSF does. Studies have shown that M-CSF administration increases inflammation in a model of collagen-induced rheumatoid arthritis (37) and that IL-34 correlates with the severity of synovitis (13) and can be induced by TNF-α, as M-CSF can (38). Furthermore, both IL-34 and M-CSF induce proinflammatory cytokines such as IL-6, IP-10/CXCL10, IL-8/CXCL8, and MCP1/CCL2 (39). In contrast, it has also been shown that M-CSF and, more recently, IL-34, alone or in combination with other cytokines, can induce regulatory macrophages (32, 40–43). In transplantation, it has been demonstrated that pretreatment of mice with M-CSF expands macrophages and inhibits graft-versus-host disease (GVHD) (44). In addition, a combination of M-CSF and IFN-γ differentiates monocytes into regulatory macrophages capable of prolonging heart allograft survival in an iNOS-dependent manner (36). These studies highlight the paradoxical role of both M-CSF and IL-34. In our study, in an attempt to unravel the complex mechanisms of tolerance induction in transplantation, we provide evidence, for the first time to our knowledge, of the unexpected properties of IL-34 as a master regulator of immune responses and tolerance. We also provide what we believe to be the first proof that IL-34 can be expressed by rat CD8+CD45RClo Tregs and human FOXP3+ Tregs and, most important, that potent Tregs are induced by IL-34 treatment. This induction of Tregs is due to the generation of regulatory macrophages and is associated in vivo with a reduction of both inflammation and antigen presentation.
We have previously demonstrated that treatment of cardiac graft recipients with an adenovirus encoding CD40Ig leads to indefinite allograft survival in 93% of recipients and that this acceptance is mediated by CD8+CD45RClo Tregs in an IFN-γ–, IDO-, and FGL2-dependent manner. We more recently demonstrated that CD8+CD45RClo Tregs recombined a biased restricted Vβ11 repertoire to recognize a dominant MCH class II–derived peptide and that this peptide induces Tregs and induces tolerance (23). In the present study, we show that IL-34 is expressed at high levels by tolerated grafts from AdCD40Ig-treated recipients, and importantly, also by splenic CD8+CD45RClo Tregs from the same recipients. Furthermore, CD8+CD45RClo Treg–mediated suppression can be partially abrogated by blockade of IL-34. Thus, IL-34 possesses immunosuppressive properties that have never, to our knowledge, been studied and acts in complement with FGL2, IDO, and IFN-γ in the CD40Ig model of suppression mediated by CD8+CD45RClo Tregs (21, 22).
While we observed that M-CSF was also significantly upregulated by CD8+CD40Ig Tregs and that M-CSF significantly inhibited antidonor immune responses ex vivo (P = 0.0159 and P < 0.05, respectively), there was no involvement of M-CSF in this model, demonstrating that this property is specific to IL-34. This might be due to a lesser amount of M-CSF being secreted compared with IL-34 and to a lower suppressive capacity of M-CSF (250 ng/ml of rat M-CSF required for 75% suppression versus 24 pg/ml of rat IL-34). Accumulating evidence suggest that IL-34 and M-CSF exhibit specific and nonredundant properties. This is supported by structural analysis comparisons showing that IL-34 and M-CSF bind differently to CD115 (11, 25). The recent identification of a second distinct receptor for IL-34 reinforces this interpretation (10). Very recently, IL-34–deficient mice have been generated that exhibit an absence of certain cell subsets such as Langerhans cells and microglia (12), effects that have not been observed in M-CSF–KO mice. This demonstrates not only a different temporal and spatial expression role, but also different functional effects with IL-34 compared with M-CSF.
In order to emphasize the significance of our findings, we analyzed IL-34 expression by human FOXP3+CD45RClo CD4+ and CD8+ Tregs. Strikingly, we observed a specific and uniquely high level of expression of IL-34 by FOXP3+CD45RClo CD4+ and CD8+ Tregs that did not occur for CD45RChi and FOXP3–CD45RClo CD4+ and CD8+ T cells, demonstrating that among T cells, Tregs specifically express IL-34. In addition, we also demonstrated the ex vivo–suppressive potential of IL-34 alone on the antidonor immune response, suggesting its potential as a target therapeutic in transplantation and, by extension, in autoimmune diseases. However, we observed that 5 μg/ml human IL-34 was needed to significantly suppress in vitro human antidonor immune responses, while it is known that IL-10 and TGF-β act in the ng/ml range to suppress effector T cell responses, suggesting that either the conditions we used to test the suppressive capacity of IL-34 were not optimal or that IL-34 is less potent at inhibiting antidonor immune responses than IL-10 or TGF-β. Interestingly, we managed to demonstrate that blocking IL-34 in a suppressive assay with CD4+CD25hiCD127lo or CD8+CD45RClo Tregs restored effector T cell proliferation, confirming the important contribution of IL-34 to human Treg–mediated activity. The concentration of the anti–IL-34 Ab was in a relatively high range, but we cannot exclude that the affinity of this Ab is not optimal, and the adequate isotype- and species-negative control validates this observation. The generation of IL-34–deficient rats using engineered nucleases as previously done in our laboratory (45, 46) is under way and will help to further address the key role of IL-34 in Treg function. Similarly, the generation of FOXP3-GFP rats will help to define whether FOXP3+ Tregs are the only Tregs that express IL-34 and to further explore the role of IL-34 in Treg biology. The therapeutic value of this molecule is evidenced by the generation of an AAV encoding IL-34. With this vector, we were able to show the potent immunosuppressive properties of IL-34 in vitro and, more important, in vivo, where we obtained tolerance in 64% of the recipients when IL-34 was combined with a suboptimal dose of rapamycin. We also demonstrated that such therapy results in abrogation of all allogeneic immune responses and of tolerance induction. Previous studies have demonstrated that both M-CSF and IL-34 can differentiate monocytes into regulatory macrophages (32, 36) and that the regulatory macrophages induced in vitro by M-CSF and IFN-γ can be used in vivo to prolong heart allograft survival in mice (36). Another study has demonstrated that administration of M-CSF before transplantation into mice can expand macrophages and limit donor T cell expansion and GVHD (44). Chen et al. showed an increase in the CD11b+ cell population in mice treated with soluble IL-34 protein (47). Other studies revealed a decrease in pDCs and cDCs in CD115-deficient osteopetrotic mice (27) and an M-CSF–induced increase in DC numbers (26). Surprisingly, in our study, in contrast with other studies, an in vivo IL-34 tolerogenic effect following administration was mainly transferred by Tregs (either CD4+CD25hi or CD8+CD45RClo Tregs). We demonstrated that the tolerance achieved in AAV–IL-34–treated recipients can be transferred to newly grafted, irradiated recipients for at least 3 generations and that this effect is mediated by Tregs but, despite an increase in the CD11b+ cell population (data not shown), not by splenic macrophages. However, since T cells do not express the CD115 receptor, we hypothesized and demonstrated that IL-34 mediated its effect on Tregs through macrophages. Regulatory macrophages can anergize CD4 effector T cells (48), convert T cells into Tregs (49), and inhibit other APCs (50). We demonstrate that IL-34–induced regulatory macrophages migrate early to the graft and express high levels of arginase 1 and iNOS, suggesting that immunoregulatory mechanisms limit the proliferation and survival of effector T lymphocytes (35, 36). These results highlight the functional differences between IL-34 and M-CSF and add to the debate on this topic, as several studies have shown a proinflammatory role of M-CSF, which increases macrophage proliferation and accumulation in rejected renal allografts (51).
To provide the proof of concept that a similar mechanism exists in humans, we differentiated monocytes in the presence of IL-34. We were able to demonstrate that IL-34–differentiated macrophages that are added to total allogeneic PBMCs efficiently expand and potentiate the ability of FOXP3+ Tregs to inhibit antidonor immune responses. It should be noted that both IL-34 and FOXP3 coexpression in Tregs remained stable. Altogether, these data suggest a retroactive loop of IL-34–secreting Tregs that are able to induce regulatory macrophages that in turn induce new IL-34–secreting Tregs.
In conclusion, we describe here the role of a new cytokine, IL-34, in transplant tolerance, and we reveal its potential as a therapy in transplantation and, by extension, in other diseases. We also demonstrate for the first time to our knowledge that this cytokine can be produced by Tregs and that, in turn, administration of IL-34 results in the potent induction of Tregs capable of tolerance induction in a dominant manner, opening new possibilities for the generation of Tregs that could be transferable to the human setting.
Methods
Animals and cardiac transplantation models.
Heart allotransplantation was performed between whole MHC-incompatible male LEW.1W (donors) and LEW.1A (recipients) rats as previously described (18). Heart survival was evaluated by palpation through the abdominal wall, and heart beat was graded from +++ to –.
Healthy volunteer blood collection and PBMC separation.
Blood was diluted 2-fold with PBS before PBMCs were isolated by Ficoll-Paque density gradient centrifugation (Eurobio) at 770 g for 20 minutes at room temperature without braking. Collected PBMCs were washed in 50 ml PBS at 620 g for 10 minutes.
Rat splenocyte purification.
Spleens were digested by collagenase D for 15 minutes at 37°C, stopped with 0.01 mM EDTA, and red cells were lysed.
Cell sorting.
Macrophages were sorted on TCRαβ– (R7/3) and TCRγδ– (V65) cells and CD45RA-FITC– (OX33) and CD11b/c-APC+ (OX42) cells. Naive LEW.1A CD4+CD25– T cells and LEW.1W pDC and LEW.1A CD8+CD45RClo Treg subsets were sorted as previously described (21). The Abs used for the sorting of T cells (TCRαβ, clone R7/3), CD4+CD25– T cells (clones OX35 and OX39), CD8+CD45RClo T cells (clones OX8 and OX22), and TCR–CD45RA–CD4+CD45R+ pDCs (clones His24 and OX35) were obtained from the European Collection of Cell Culture. Biotin-labeled mAbs were visualized using streptavidin-phycoerythrin-Cy7 or streptavidin-Alexa Fluor 405 (BD Biosciences). Human CD4+CD25–T cells were sorted by gating on CD3+CD4+CD25– cells (clones SKY7, L-200, and MA251; BD Biosciences), CD4+ Tregs by gating on CD25hi and CD127lo cells (clone HIL7-R M21; BD Biosciences), and CD8+ Tregs by gating on CD3+CD4–CD45RClo cells (clone MT2; IQ Products). A FACSAria I cell sorter (BD Biosciences) was used to sort cells.
mAbs and flow cytometry.
Rat IL-34–Myc was detected using an anti-Myc Ab (9E10; Sigma-Aldrich). Blocking Abs against IL-34 (PAB16574; Abnova and clone 578416; R&D Systems); CD115 (MCA1898; Serotec); and M-CSF (AB-416-NA; R&D Systems) were used in MLR assays.
Abs against MHC II (OX6); CD11b/c (OX42); and CD45RA+ B cells (OX33) were analyzed to characterize rat cell phenotypes.
Abs against CD3-PeCy7 (SKY7; BD Biosciences); CD4-PercPCy5.5 (L200; BD Biosciences); CD25-APCCy7 (M-A251; BD Biosciences); CD127-PE (HIL7-R M21; BD Biosciences); CD45RC-FITC (MT2; IQ Product); FOXP3-APC (236A/E7; eBioscience); and IL-34–PE (578416; R&D Systems) were used to characterize human cell phenotypes.
Fluorescence was measured with a FACSCanto II flow cytometer (BD Biosciences), and FlowJo software was used to analyze data. Cells were first gated by their morphology, excluding dead cells by selecting DAPI-viable cells (Supplemental Figure 2).
Adoptive cell transfer.
Rat cells were sorted as previously described (ref. 5, 8, and Supplemental Figure 2C) using a FACSAria cell sorter (BD Biosciences) by gating on TCRαβ-APC (R7/3), CD45RA-FITC (OX33), and CD11b/c-biotin-streptavidin PECy7–positive (OX42) cells. Recipients of splenocytes from IL-34–treated rats were defined as first transfers, and then iterative transfers were defined as second to third transfers. Total splenocytes (1.5 × 108 cells) and FACSAria-sorted CD45RA+ B cells (6 × 107); T cells (4 × 107); CD11b/c+ cells (1.5 × 107); CD4+CD25hi Tregs (4 × 106); or CD8+CD45RClo Tregs (4 × 106) were adoptively transferred i.v. the day before heart transplantation into naive LEW.1A recipients that had received 4.5 Gy whole-body irradiation the same day.
MLR.
Sorted rat or human CD4+CD25– T cells were plated in triplicate with allogeneic rat pDCs or allogeneic human T cell–depleted PBMCs, respectively, in 200 μl complete RPMI-1640 medium in round- or conic-bottomed 96-well plates, respectively, at 37°C and 5% CO2. Proliferation of CFSE-labeled CD4+CD25– T cells was analyzed by flow cytometry 6 days later by gating on TCR+CD4+ cells among living cells (DAPI–) (Supplemental Figure 2A). Sera from AAV–IL-34–treated recipients, AAV-GFP–treated recipients, and naive rats were added in coculture to reach final concentrations of 3.12%, 6.25%, and 12.5%. Supernatent of transduced cells was added to CD4+T cells and pDCs at a final concentration of 20% for the suppressive activity test. Rat IL-34–, CD115-, or M-CSF–blocking Abs were tested for blocking action at 1.25 to 30 μg/ml in the presence or absence of CD8+CD40Ig Tregs. Human IL-34 Ab was used at 50 μg/ml, and variable numbers of Tregs were added. Isotype control Abs were used at the highest concentration displayed in the respective graphs. M-CSF protein (ab56288; Abcam) was tested from 0.1 to 2 μg/ml. Soluble human IL-34 (eBioscience) was added at a concentration of 1, 2, or 5 μg/ml for the suppressive activity test.
qPCR.
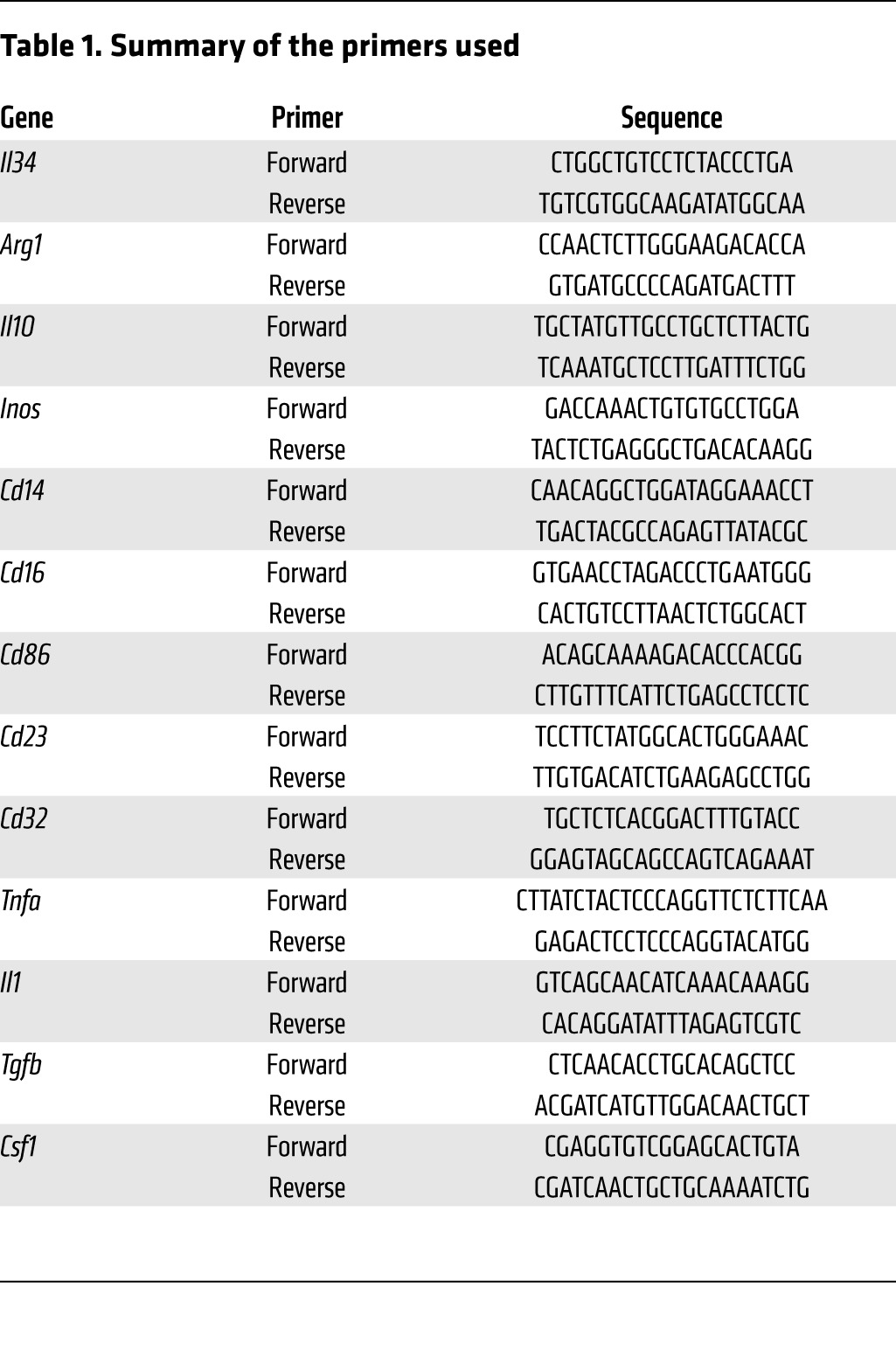
Total RNA was isolated from cells using TRIzol reagent (Invitrogen) or an RNeasy Mini Kit (QIAGEN). RNA from macrophages was amplified with a MessageAmp II aRNA Amplification Kit according to the manufacturer’s instructions (Life Technologies) and reverse transcribed with random primers and M-MLV reverse transcriptase (Life Technologies). qPCR was performed using the Fast SYBR Green technology in a 20-μl final reaction volume containing 10 μl Master Mix 2X (Life Technologies), 0.6 μl primers (10 μM), 1 μl cDNA, and 8.4 μl water. The reaction was performed on the Applied Biosystems StepOne system (Life Technologies). Thermal conditions were as follows: 3 seconds at 95°C, 30 seconds at 60°C, and 15 seconds at –5°C of the melting temperature with a final melting curve stage. The oligonucleotides used in this study are listed in Table 1.
Table 1. Summary of the primers used.
AAV generation and use in vitro and in vivo.
Complete cDNA sequences of rat IL-34 (9), or GFP as a control, were positioned downstream of an RSV promoter. Plasmids were first tested in HEK293T cells transfected with lipofectamine reagent (Life Technologies). Cells were analyzed for IL-34–Myc expression 48 hours later by FACS with anti-Myc Ab. Then, plasmids were used to produce AAV vectors of serotype 8 (Viral Vector Core, INSERM UMR 1089, Nantes, France). HEK293T cells were transduced with 10 or 100 MOI vector genome copies/cell of AAV–IL-34 or AAV-GFP and 5 MOI AdLacZ. Twenty-four hours later, the cells were harvested and analyzed for IL-34–Myc expression by FACS, and supernatant was tested for suppression activity on CD4+ T cells responding to allogeneic pDCs, at 1:10 and 1:5 dilutions. Recombinant AAV–IL-34, AAV-GFP, and noncoding AAV (1 × 1012 vector genomes/rat) vectors were injected i.v. into 4-week-old rats 1 month before transplantation to allow optimal expression from AAV vectors (30). Recipients were orally administered 0.4 mg/kg/day rapamycin from the day of transplantation (day 0) until 10 days after transplantation. Blood samples were taken on day 120 for donor-allospecific Ab quantification and IL-34 protein quantification in serum by ELISA (EL011657RA; Cusabio).
Clodronate liposome in vivo treatment.
Clodronate liposomes for macrophage depletion were purchased from Vrije University (Netherlands) (www.clodronateliposomes.org) and prepared as recommended (52). Briefly, 2.5 ml suspended solution was administered i.p. weekly from day –25 to day 3.
Donor-specific alloantibody quantification.
Donor spleens were digested by collagenase D, stopped with 0.01 mM EDTA, and red cells were lysed. Serum from recipients was added to donor splenocytes at a 1:8 dilution and incubated with either anti-rat IgG-FITC (Jackson ImmunoResearch Laboratories); anti-rat IgG1 (MCA 194; Serotec); anti-rat IgG2a (MCA 278; Serotec); or anti-rat IgG2b (STAR114F; Serotec) and anti-Ms Ig-FITC (115-095-164; Jackson ImmunoResearch Laboratories). A FACSCanto (BD Biosciences) was used to measure fluorescence, and data were analyzed using FlowJo software. The geometric mean of fluorescence (mean fluorescence intensity [MFI]) of tested sera was divided by the mean MFI of 5 naive LEW.1A sera as a control.
Anatomopathologic analysis of cardiac grafts.
Graft samples were fixed in paraformaldehyde and embedded in paraffin. Five-micrometer coronal sections were stained with H&E-saffron. Tissues were analyzed by a pathologist (K. Renaudin) blinded to the groups, and chronic rejection was evaluated as previously described (53).
Treg and monocyte differentiation protocol.
PBMCs from healthy volunteer (HV) blood were isolated by Ficoll gradient, and monocytes were elutriated according to forward scatter (FSC) and side scatter (SSC) morphologic parameters. CD14+ monocytes were then sorted by FACSAria, washed, and seeded at 1 × 106/ml in medium (RPMI 1640, 2 mM glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 10% FCS supplemented with 50 ng/ml hIL-34 (eBioscience). On day 6, cells were harvested, stimulated with 100 ng/ml LPS for 24 hours for phenotype analysis, or seeded at 4 × 105/ml with 1 to 5 allogeneic PBMCs in Iscove’s modified Dulbecco’s medium (IMDM) (2 mM glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 5% AB serum). IL-2 (25 U/ml) and IL-15 (10 ng/ml) were freshly added on days 10, 13, and 16. Macrophages were removed by successive transfers of floating cells to a new plate on days 19 and 21 for 48 hours and 2 hours, respectively. On day 21, cells were stimulated with PMA and ionomycin in the presence of brefeldin A for phenotype analysis, or T cells, CD8+CD45RClo T cells, and CD4+CD25hiCD127lo T cells were sorted by FACSAria for suppression assays. Fresh syngeneic CD4+CD25– T cells were used as responder T cells stimulated with allogeneic APCs isolated from the same donor as that of the CD14+ cells. Proliferation was assessed 5 days later by CFSE dilution, by gating on CD3+CD4+ cells after exclusion of DAPI-labeled dead cells and CPD405-labeled CD4+ Tregs.
Statistics.
Mann-Whitney U and Kruskal-Wallis tests were used for PCR analyses. ANOVA tests were used for coculture experiments, a 2-way ANOVA test and Bonferroni’s post tests were applied for donor-directed Ab detection, and a log-rank (Mantel-Cox) test was used to analyze survival curves. A P value of less than 0.05 was considered significant.
Study approval.
Animal experiments were approved by the ethics committee of the Ministère de l’Enseignement Supérieur et de la Recherche (Paris, France; CEEA.2013.52). Blood was collected from healthy donors, after informed consent was given, at the Etablissement Français du Sang (Nantes, France).
Supplementary Material
Acknowledgments
We thank Emmanuel Merieau and Bernard Martinet for technical assistance with animal models. We thank the Viral Vector Core of the CHU de Nantes, INSERM UMR 1089, which is supported by the Association Française Contre les Myopathies, for producing the viral vectors. We thank the Fondation Progreffe and the Société Francophone de Transplantation for financial support and the Crédit Agricole for the donation of the FACSAria. This work was performed as part of the LabEX ImmunoGraft Oncology (IGO) program supported by the National Research Agency via the investment of the future program ANR-11-LABX-0016-01. This work was also supported by funding from the Institut Hospitalo-Universitaire-Centre Européen des Sciences de la Transplantation et Immunothérapie project, which received French Government financial support and was managed by the National Research Agency via the investment of the future program ANR-10-IBHU-005. The IHU-Cesti project is also supported by Nantes Metropole and the Pays de la Loire region. C. Guillonneau was supported for this work by a Junior Basic Science grant from European Society for Organ Transplantation (ESOT). I. Anegon was supported by a Roche Organ Transplantation Research Foundation (ROTRF) grant. S. Bézie was supported by Progreffe and the Société Francophone de Transplantation. E. Picarda was supported by an INSERM–Region Pays de la Loire Fellowship. J. Ossart was supported by Progreffe and the Fondation pour la Recherche Médicale.
Footnotes
Conflict of interest: Carole Guillonneau, Ignacio Anegon, and Séverine Bézie have filed 3 patents that are pending and are entitled to a share in the net income generated from licensing of these patent rights for commercial development.
Reference information:J Clin Invest. 2015;125(10):3952–3964. doi:10.1172/JCI81227.
See the related Commentary beginning on page 3751.
References
- 1.Nankivell BJ, Borrows RJ, Fung CL, O’Connell PJ, Allen RD, Chapman JR. The natural history of chronic allograft nephropathy. N Engl J Med. 2003;349(24):2326–2333. doi: 10.1056/NEJMoa020009. [DOI] [PubMed] [Google Scholar]
- 2.Srinivas TR, Kaplan B. Transplantation in 2011: New agents, new ideas and new hope. Nat Rev Nephrol. 2012;8(2):74–75. doi: 10.1038/nrneph.2011.215. [DOI] [PubMed] [Google Scholar]
- 3.Londono MC, Danger R, Giral M, Soulillou JP, Sanchez-Fueyo A, Brouard S. A need for biomarkers of operational tolerance in liver and kidney transplantation. Am J Transplant. 2012;12(6):1370–1377. doi: 10.1111/j.1600-6143.2012.04035.x. [DOI] [PubMed] [Google Scholar]
- 4.Wood KJ, Bushell A, Hester J. Regulatory immune cells in transplantation. Nat Rev Immunol. 2012;12(6):417–430. doi: 10.1038/nri3227. [DOI] [PubMed] [Google Scholar]
- 5.Niederkorn JY. Emerging concepts in CD8(+) T regulatory cells. Curr Opin Immunol. 2008;20(3):327–331. doi: 10.1016/j.coi.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Picarda E, Anegon I, Guillonneau C. T-cell receptor specificity of CD8(+) Tregs in allotransplantation. Immunotherapy. 2011;3(4 suppl):35–37. doi: 10.2217/imt.11.37. [DOI] [PubMed] [Google Scholar]
- 7.Guillonneau C, Picarda E, Anegon I. CD8+ regulatory T cells in solid organ transplantation. Curr Opin Organ Transplant. 2010;15(6):751–756. doi: 10.1097/MOT.0b013e32834016d1. [DOI] [PubMed] [Google Scholar]
- 8.Menoret S, Guillonneau C, Bezie S, Caron L, Anegon I, Li XL. Phenotypic and functional characterization of CD8(+) T regulatory cells. Methods Mol Biol. 2011;677:63–83. doi: 10.1007/978-1-60761-869-0_5. [DOI] [PubMed] [Google Scholar]
- 9.Wei S, et al. Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. J Leukoc Biol. 2010;88(3):495–505. doi: 10.1189/jlb.1209822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nandi S, et al. Receptor-type protein-tyrosine phosphatase zeta is a functional receptor for interleukin-34. J Biol Chem. 2013;288(30):21972–21986. doi: 10.1074/jbc.M112.442731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chihara T, et al. IL-34 and M-CSF share the receptor Fms but are not identical in biological activity and signal activation. Cell Death Differ. 2010;17(12):1917–1927. doi: 10.1038/cdd.2010.60. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. 2012;13(8):753–760. doi: 10.1038/ni.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chemel M, et al. Interleukin 34 expression is associated with synovitis severity in rheumatoid arthritis patients. Ann Rheum Dis. 2012;71(1):150–154. doi: 10.1136/annrheumdis-2011-200096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baud’huin M, et al. Interleukin-34 is expressed by giant cell tumours of bone and plays a key role in RANKL-induced osteoclastogenesis. J Pathol. 2010;221(1):77–86. doi: 10.1002/path.2684. [DOI] [PubMed] [Google Scholar]
- 15.Spickett GP, Brandon MR, Mason DW, Williams AF, Woollett GR. MRC OX-22, a monoclonal antibody that labels a new subset of T lymphocytes and reacts with the high molecular weight form of the leukocyte-common antigen. J Exp Med. 1983;158(3):795–810. doi: 10.1084/jem.158.3.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xystrakis E, Bernard I, Dejean AS, Alsaati T, Druet P, Saoudi A. Alloreactive CD4 T lymphocytes responsible for acute and chronic graft-versus-host disease are contained within the CD45RChigh but not the CD45RClow subset. Eur J Immunol. 2004;34(2):408–417. doi: 10.1002/eji.200324528. [DOI] [PubMed] [Google Scholar]
- 17.Xystrakis E, et al. Identification of a novel natural regulatory CD8 T-cell subset and analysis of its mechanism of regulation. Blood. 2004;104(10):3294–3301. doi: 10.1182/blood-2004-03-1214. [DOI] [PubMed] [Google Scholar]
- 18.Guillonneau C, et al. CD40Ig treatment results in allograft acceptance mediated by CD8CD45RC T cells, IFN-γ, and indoleamine 2,3-dioxygenase. J Clin Invest. 2007;117(4):1096–1106. doi: 10.1172/JCI28801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Powrie F, Mason D. OX-22high CD4+ T cells induce wasting disease with multiple organ pathology: prevention by the OX-22low subset. J Exp Med. 1990;172(6):1701–1708. doi: 10.1084/jem.172.6.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ordonez L, Bernard I, L’Faqihi-Olive FE, Tervaert JW, Damoiseaux J, Saoudi A. CD45RC isoform expression identifies functionally distinct T cell subsets differentially distributed between healthy individuals and AAV patients. PLoS One. 2009;4(4): doi: 10.1371/journal.pone.0005287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li XL, et al. Mechanism and localization of CD8 regulatory T cells in a heart transplant model of tolerance. J Immunol. 2010;185(2):823–833. doi: 10.4049/jimmunol.1000120. [DOI] [PubMed] [Google Scholar]
- 22.Bezie S, et al. Fibrinogen-like protein 2/fibroleukin induces long-term allograft survival in a rat model through regulatory B cells. PLoS One. 2015;10(3): doi: 10.1371/journal.pone.0119686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Picarda E, et al. MHC-derived allopeptide activates TCR-biased CD8+ Tregs and suppresses organ rejection. J Clin Invest. 2014;124(6):2497–2512. doi: 10.1172/JCI71533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin H, et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science. 2008;320(5877):807–811. doi: 10.1126/science.1154370. [DOI] [PubMed] [Google Scholar]
- 25.Garceau V, et al. Pivotal Advance: Avian colony-stimulating factor 1 (CS9F-1), interleukin-34 (IL-34), and CSF-1 receptor genes and gene products. J Leukoc Biol. 2010;87(5):753–764. doi: 10.1189/jlb.0909624. [DOI] [PubMed] [Google Scholar]
- 26.Fancke B, Suter M, Hochrein H, O’Keeffe M. M-CSF: a novel plasmacytoid and conventional dendritic cell poietin. Blood. 2008;111(1):150–159. doi: 10.1182/blood-2007-05-089292. [DOI] [PubMed] [Google Scholar]
- 27.MacDonald KP, et al. The colony-stimulating factor 1 receptor is expressed on dendritic cells during differentiation and regulates their expansion. J Immunol. 2005;175(3):1399–1405. doi: 10.4049/jimmunol.175.3.1399. [DOI] [PubMed] [Google Scholar]
- 28.Gilmore GL, Shadduck RK. Inhibition of day-12 spleen colony-forming units by a monoclonal antibody to the murine macrophage/monocyte colony-stimulating factor receptor. Blood. 1995;85(10):2731–2734. [PubMed] [Google Scholar]
- 29.Ordonez L, et al. A higher risk of acute rejection of human kidney allografts can be predicted from the level of CD45RC expressed by the recipients’ CD8 T cells. PLoS One. 2013;8(7): doi: 10.1371/journal.pone.0069791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toromanoff A, et al. Safety and efficacy of regional intravenous (r.i.) versus intramuscular (i.m.) delivery of rAAV1 and rAAV8 to nonhuman primate skeletal muscle. Mol Ther. 2008;16(7):1291–1299. doi: 10.1038/mt.2008.87. [DOI] [PubMed] [Google Scholar]
- 31.Le Texier L, et al. Immunoregulatory function of IL-27 and TGF-beta1 in cardiac allograft transplantation. Transplantation. 2012;94(3):226–233. doi: 10.1097/TP.0b013e31825b0c38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Foucher ED, et al. IL-34 induces the differentiation of human monocytes into immunosuppressive macrophages. antagonistic effects of GM-CSF and IFNγ. PLoS One. 2013;8(2): doi: 10.1371/journal.pone.0056045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6: doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murray PJ, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cobbold SP, et al. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc Natl Acad Sci U S A. 2009;106(29):12055–12060. doi: 10.1073/pnas.0903919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riquelme P, et al. IFN-gamma-induced iNOS expression in mouse regulatory macrophages prolongs allograft survival in fully immunocompetent recipients. Mol Ther. 2013;21(2):409–422. doi: 10.1038/mt.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Campbell IK, Rich MJ, Bischof RJ, Hamilton JA. The colony-stimulating factors and collagen-induced arthritis: exacerbation of disease by M-CSF and G-CSF and requirement for endogenous M-CSF. J Leukoc Biol. 2000;68(1):144–150. [PubMed] [Google Scholar]
- 38.Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8(7):533–544. doi: 10.1038/nri2356. [DOI] [PubMed] [Google Scholar]
- 39.Eda H, Zhang J, Keith RH, Michener M, Beidler DR, Monahan JB. Macrophage-colony stimulating factor and interleukin-34 induce chemokines in human whole blood. Cytokine. 2010;52(3):215–220. doi: 10.1016/j.cyto.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 40.Doyle AG, Halliday WJ, Barnett CJ, Dunn TL, Hume DA. Effect of recombinant human macrophage colony-stimulating factor 1 on immunopathology of experimental brucellosis in mice. Infect Immun. 1992;60(4):1465–1472. doi: 10.1128/iai.60.4.1465-1472.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakurai T, Wakimoto N, Yamada M, Shimamura S, Motoyoshi K. Effect of macrophage colony-stimulating factor (M-CSF) on mouse immune responses in vivo. Immunopharmacol Immunotoxicol. 1998;20(1):79–102. doi: 10.3109/08923979809034810. [DOI] [PubMed] [Google Scholar]
- 42.Sakurai T, Yamada M, Simamura S, Motoyoshi K. Recombinant human macrophage-colony stimulating factor suppresses the mouse mixed lymphocyte reaction. Cell Immunol. 1996;171(1):87–94. doi: 10.1006/cimm.1996.0177. [DOI] [PubMed] [Google Scholar]
- 43.Duluc D, et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood. 2007;110(13):4319–4330. doi: 10.1182/blood-2007-02-072587. [DOI] [PubMed] [Google Scholar]
- 44.Hashimoto D, et al. Pretransplant CSF-1 therapy expands recipient macrophages and ameliorates GVHD after allogeneic hematopoietic cell transplantation. J Exp Med. 2011;208(5):1069–1082. doi: 10.1084/jem.20101709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Menoret S, et al. Characterization of immunoglobulin heavy chain knockout rats. Eur J Immunol. 2010;40(10):2932–2941. doi: 10.1002/eji.201040939. [DOI] [PubMed] [Google Scholar]
- 46.Larcher T, et al. Characterization of dystrophin deficient rats: a new model for Duchenne muscular dystrophy. PLoS One. 2014;9(10): doi: 10.1371/journal.pone.0110371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Z, Buki K, Vaaraniemi J, Gu G, Vaananen HK. The critical role of IL-34 in osteoclastogenesis. PLoS One. 2011;6(4): doi: 10.1371/journal.pone.0018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tzachanis D, Berezovskaya A, Nadler LM, Boussiotis VA. Blockade of B7/CD28 in mixed lymphocyte reaction cultures results in the generation of alternatively activated macrophages, which suppress T-cell responses. Blood. 2002;99(4):1465–1473. doi: 10.1182/blood.V99.4.1465. [DOI] [PubMed] [Google Scholar]
- 49.Liu G, Duan K, Ma H, Niu Z, Peng J, Zhao Y. An instructive role of donor macrophages in mixed chimeras in the induction of recipient CD4(+)Foxp3(+) Treg cells. Immunol Cell Biol. 2011;89(8):827–835. doi: 10.1038/icb.2011.65. [DOI] [PubMed] [Google Scholar]
- 50.Holt PG, Schon-Hegrad MA, Oliver J. MHC class II antigen-bearing dendritic cells in pulmonary tissues of the rat. Regulation of antigen presentation activity by endogenous macrophage populations. J Exp Med. 1988;167(2):262–274. doi: 10.1084/jem.167.2.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jose MD, Le Meur Y, Atkins RC, Chadban SJ. Blockade of macrophage colony-stimulating factor reduces macrophage proliferation and accumulation in renal allograft rejection. Am J Transplant. 2003;3(3):294–300. doi: 10.1034/j.1600-6143.2003.00068.x. [DOI] [PubMed] [Google Scholar]
- 52.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174(1–2):83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 53.Guillonneau C, et al. The role of TNF-related activation-induced cytokine-receptor activating NF-κB interaction in acute allograft rejection and CD40L-independent chronic allograft rejection. J Immunol. 2004;172(3):1619–1629. doi: 10.4049/jimmunol.172.3.1619. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.