

The 2015 Lasker~DeBakey Clinical Medical Research Award honors James P. Allison, PhD, for pioneering a new approach to cancer immunotherapy (Figure 1). Allison has made many seminal contributions to immunology, including the identification of the receptor on T cells that recognizes and binds antigens; the discovery that T cells require a second molecular signal from the costimulatory molecule CD28 to launch a response to a bound antigen; elucidation of the function of cytotoxic T lymphocyte antigen-4 (CTLA-4), which acts as a built-in off-switch on T cells; and the development of a CTLA-4–blocking antibody, which unleashes T cells, allowing them to eliminate cancer cells. A therapeutic CTLA-4 antibody (ipilimumab, also known as Yervoy) is now used to treat advanced melanoma and is currently under investigation for the treatment of many other malignancies. Allison’s work has caused a paradigm shift in the treatment of cancer in which treatment is focused on helping the patient’s immune system defeat the tumor instead of targeting the tumor itself.
Figure 1. James P. Allison is the winner of the 2015 Lasker~DeBakey Clinical Medical Research Award for pioneering a new field of cancer immunotherapy.

His achievements include elucidation of the structure of the T cell antigen receptor, elucidation of the function of CTLA-4, and the development of a CTLA-4–blocking antibody, which led to the development of therapeutic CTLA-4 antibodies (ipilimumab) that are now used to treat advanced melanoma.
The T cell mechanic
As early as the 1960s, it was clear that the functions of T lymphocytes were necessary for normal immunity; however, no one knew how these cells recognized and responded to antigen. Around 1970, researchers began to understand that T cells express some form of cell surface receptor that is involved in antigen recognition. Allison caught the first real glimpse of this T cell receptor (TCR) in 1982 (1). Working under the assumption that each T cell expressed a unique antigen receptor with both constant and antigen-specific variable regions, Allison and colleagues made monoclonal antibodies that recognized different T cell lymphoma clones. The molecules present on the surface of the monoclonal T cells were identical to the cell surface proteins on polyclonal T cells, except for the antigen-binding site of the TCR; thus, monoclonal antibodies specific for each clone would recognize the variable regions of the TCR. Indeed, biochemical analysis of the structures recognized by the clone-specific antibodies revealed the presence of a glycoprotein composed of two disulfide-bonded subunits (1). Shortly thereafter, several other groups identified similar structures in mice and humans (2–6). Peptide fingerprinting of TCRs isolated from T cell hybridomas showed that two subunits, the α and β chains, make up the antigen-recognition site of the TCR (7, 8).
In 1984, Mark Davis and Tak Mak cloned the mouse TCR α chain gene and the human TCR β chain gene, respectively (9, 10), allowing characterization of the TCR to be completed rapidly. In the majority of T cells, the extracellular antigen-recognition site of the TCR complex is composed of variable α and β chains and an invariant CD3 chain. A single TCR recognizes an antigenic peptide bound to an MHC molecule (11), while the intracellular portions of the CD3 chain mediate recruitment of signaling molecules that are necessary for T cell responses (12, 13).
Shortly after the structure of the TCR was elucidated, it became clear that the TCR/antigen interaction was not sufficient for full T cell activation (14); a second, costimulatory signal from antigen-presenting cells (APCs) was required. Additionally, T cells could be turned off, even in the presence of antigen, a state known as anergy (15). The first T cell costimulatory “on” molecule, CD28, was first identified in 1986 and was found to enhance T cell proliferation and IL-2 secretion (16) upon binding to ligands expressed on APCs, known as B7 molecules (17). Allison’s group, now at University of California, Berkeley, embarked on a series of experiments to show that CD28 was necessary and sufficient to provide the second costimulatory signal required for full T cell activation (18, 19).
In 1991, Peter Linsley and colleagues found an additional molecule on T cells, CTLA-4, that binds B7 molecules (20, 21), but researchers in the field were puzzled about its function, particularly because CTLA-4’s avidity for B7 was so much higher than that of CD28 (22). Studies of a CTLA-4/Ig fusion molecule (CTLA-4-Ig) in mice indicated that CTLA-4 inhibits T cell activity. Jeff Bluestone’s group found that CTLA-4-Ig enhanced the survival of pancreatic islet grafts in mice by blocking CD28/B7 interactions (23) and that CD28-dependent clonal expansion of antigen-specific cells is inhibited by CTLA-4-Ig.
As for the physiological role of CTLA-4, Linsley and colleagues proposed that CD28 and CTLA-4 acted synergistically to promote T cell proliferation (24). However, both Allison’s group and Bluestone’s group subsequently analyzed CD28 and CTLA-4 signaling in purified T cells and concluded that CD28 and CTLA-4 have opposing effects on the response of T cells to TCR stimulation (Figure 2 and refs. 25, 26). This conclusion was supported by the subsequent finding that CTLA-4 knockout mice developed fatal lymphoproliferative disorders (27–29).
Figure 2. Mechanism of action of ipilimumab.
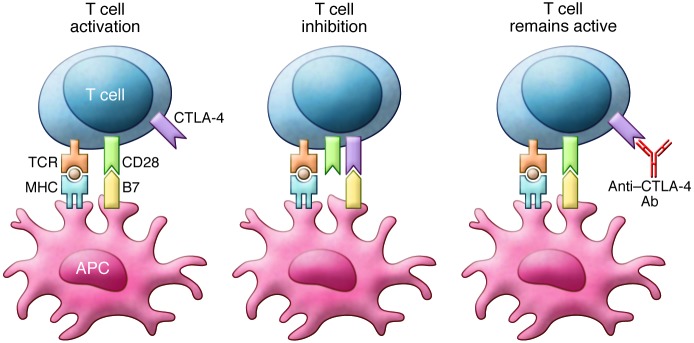
T cell activation requires stimulation through both the TCR and CD28. Binding of B7 family member proteins to CTLA-4 inhibits T cell function. Notably, CTLA-4 expression increases in parallel with TCR stimulation, thereby serving as a break on T cell responses. Anti–CTLA-4 antibodies such as ipilimumab block CTLA-4 binding to B7 proteins and prevent inhibition of T cell function. Reproduced from ref. 54.
These studies suggest that T cell function can be viewed as analogous to that of a car, with antigen binding to the TCR acting in a similar manner to the key in the ignition. Stimulation via CD28 hits the gas, inducing proliferation and differentiation, while CTLA-4 serves as the brakes, abrogating T cell antigen responses. The requirement of a second signal for T cell activation means that the duration and strength of T cell responses can be fine-tuned, ensuring that the immune system only eliminates pathogens or defective cells and avoids harming normal tissues. Thus, CTLA-4 serves as a “checkpoint” to ensure that the immune system does not mistakenly attack healthy “self” tissue. However, this protection can be co-opted by unhealthy tissue, including cancer cells.
The immune surveillance hypothesis
As early as the mid-1800s, Rudolf Virchow noted the presence of immune cells in many tumors. Roughly fifty years later, Paul Erhlich proposed the idea that large numbers of mutated or damaged cells are generated during both prenatal and postnatal development and that the body possesses the capacity to remove these cells to protect against cancer (30). Likewise, in the late 1950s, Lewis Thomas noted the evolutionary significance of cellular immunity as a defense against neoplasia (31), a function termed by Sir MacFarlane Burnet as “immunological surveillance” (32). All somatic cells display surface antigens that are continuously sampled and analyzed by the cells of the immune system. The immune system has been tolerized to these antigens and, thus, does not attack normal tissues. Cancer cells express mutant antigens, which allow them to be recognized and destroyed by the immune system; however, cancer cells can undergo an immunoediting process, in which a variety of mechanisms are employed that allow the cancer cells to escape immune surveillance and grow into clinically detectable tumors.
Allison had directly observed the ability of the immune system to eliminate cancer cells. As a graduate student, he used an enzyme called asparaginase to cure leukemia in mice. When he injected the cured mice with cancer cells a second time, the mice rejected the tumors. Allison’s was not the first attempt to harness the immune system’s ability to fight off cancer. In 1891, William Coley began injecting patients with sarcoma with bacterial products known as “Coley’s toxins,” which caused a raging infection and occasionally had the side effect of shrinking tumors (33). Due to a lack of understanding of the underlying mechanisms and poor reproducibility, Coley’s toxins never made it into widespread clinical use. Other strategies to enhance anticancer immune responses have included administration of the tuberculosis vaccine Bacillus Calmette-Guérin (34), which is a first-line therapy for superficial bladder cancer, and cytokines such as IL-2 and IFN-α, which are currently used as adjuvants, though they can have severe side effects. The identification of CTLA-4 as an attenuator of the immune response suggested to Allison that there might be other ways to augment anticancer immunity.
Cutting the brakes on the anticancer immune response
Many tumors are poorly immunogenic, making it difficult for the immune system to eliminate them. One reason for the poor immunogenicity may be that cancer antigen presentation is insufficient to activate T cells. Studies in which tumor cells transfected with the CD28 ligands B7-1 and B7-2 induced potent antitumor responses (35, 36) validated this hypothesis, suggesting that costimulatory signals are required to elicit tumor-specific T cells. Additionally, inhibitory signaling events, such as those mediated by CTLA-4, can potentially impair T cell–mediated antitumor responses. Because CTLA-4 expression increases with T cell activation, tumor-targeted T cells are eventually turned off by the increasing inhibitory signal. Reasoning that removal of T cell inhibitory signals might enhance and/or prolong T cell antitumor activity, Allison and colleagues tested the effects of CTLA-4 blockade on preestablished tumors in mice.
Allison saw the effect of CTLA-4 blockade on tumor growth for the first time in December 1995 and was shocked by the results. “I was expecting it [anti–CTLA-4] to slow the tumors a little bit, but the tumors completely melted,” said Allison in a recent interview with the JCI. “I had to immediately set up the experiment again, to do all the injections, label the cages A, B, C, and D, and then go away and wait.... For a couple of weeks I was really depressed. I didn’t know which mice were which, but I could tell that all of the tumors were growing. Then, all of a sudden, it looked like some of the cages were progressing, but others just stopped. Then, in the ones that had stopped, some of the tumors started necrosing and they just went away.”
In March of 1996, Allison and colleagues reported that in vivo administration of CTLA-4–targeted antibodies eliminated both B7-positive and B7-negative tumors and protected the mice against subsequent challenge with cancer cells (37). These results indicated that removing inhibitory signals in the T cell costimulatory pathways enhances antitumor immunity by unleashing the immune system’s anticancer response (Figure 2). Importantly, this study established the idea that the immune system, rather than the tumor, can be treated in order to eliminate cancer. Spurred on by this success, Allison’s group tested the effects of CTLA-4 antibodies in a variety of cancers and were nearly always able to eliminate the tumors by using CTLA-4 as a monotherapy or in combination with another therapeutic modality such as radiation (38–42).
Into the clinic
The successful preclinical trials made Allison anxious to translate the concept of CTLA-4 blockade into an anticancer therapy. He spent two years pitching the idea to drug companies until Medarex, a small company out of Princeton, New Jersey, decided to take a chance on the idea. Alan Korman and his colleagues at Medarex developed a human version of the CTLA-4–blocking antibody, and testing in nonhuman primates demonstrated that the antibody had a reasonable safety profile. Moreover, treatment with anti–CTLA-4 augmented immune responses to a melanoma vaccine (43). Based on these encouraging results, the Medarex antibody (MDX-010) was advanced to clinical evaluation and assigned the generic name of ipilimumab.
During this time, Allison received an offer from Harold Varmus to become the head of the immunology program at Memorial Sloan-Kettering Cancer Center (MSKCC). Eager to be closer to the clinicians who were bringing ipilimumab into the clinic, Allison made the decision to move to New York in 2004. His goal at MSKCC was not only to look at patient endpoint data from clinical trials, but also to examine the changes taking place in patient tumors at the cellular level, which allowed his research group to assess the combinations that were in trials in murine models of disease in order to elucidate the exact mechanism by which ipilimumab enhances antitumor immunity (44, 45).
The first early-stage clinical trials of ipilimumab were conducted in patients with several different tumor types, including prostate cancer, ovarian cancer, and advanced melanoma (summarized in refs. 46 and 47). Because these trials did not uncover any serious toxicities, in 2006, Bristol-Myers Squibb began sponsoring a phase II clinical trial program for ipilimumab monotherapy in patients with late-stage melanoma, which has long been considered a promising target for immunotherapy (48, 49).
The success of a new cancer therapeutic is measured by its ability to induce tumor regression within 12 weeks of administration. Based on this standard, the early results with ipilimumab were not promising — only 10% of patients exhibited tumor regression — which was not better than any other form of immunotherapy. In many patients, tumors actually grew during those first months after ipilimumab treatment; however, some patients reported that they were feeling better and many were living longer than expected. Follow-up scans revealed that the response to ipilimumab is frequently delayed and that it can take 6 to 12 months for the immune system to mount an effective anticancer response. These effects prompted Medarex and Bristol-Myers Squibb to change the primary measurement of the trial to overall survival.
Clinical success and FDA approval
The first placebo-controlled, randomized phase III trial of ipilimumab in patients with metastatic melanoma began in fall of 2004. There were three arms in the study: one group of patients received the gp100 melanoma peptide vaccine and placebo; the second group received the gp100 vaccine and ipilimumab; and the third group received ipilimumab as a monotherapy. Patients receiving gp100 in combination with ipilimumab or ipilimumab monotherapy lived 4 months longer than patients receiving gp100 alone (50). These results made ipilimumab the first treatment to significantly improve survival in patients with advanced melanoma. More importantly, long-term follow-up of 5,000 patients with melanoma who received ipilimumab found that 22% survived for at least 10 years (51). These results indicate that ipilimumab induces a persistent antitumor response that prevents disease recurrence.
Melanoma accounts for approximately 5% of all skin cancers in the US but is responsible for 75% of skin cancer deaths. In 2011, ipilimumab received US Food and Drug Administration approval for the treatment of metastatic melanoma. Because ipilimumab treats the patient’s immune system rather than the tumor, it is likely to be applicable to many forms of cancer. There are currently more than 200 clinical trials of ipilimumab alone and in combination with other therapies for the treatment of a variety of cancers, including prostate, lung, liver, pancreatic, and renal cancers.
Arming the immune system: a paradigm shift in cancer treatment
Allison cites three main reasons to target the immune system to treat cancer. (a) Specificity. Because cancer is caused by mutation or aberrant expression of proteins, cancer cells produce antigens that can trigger an immune response specific to the cancer, sparing healthy tissues. (b) Immune memory. If a cancer recurs, the immune system will be able to recognize and target it for destruction before it has a chance to take hold. (c) Adaptability. Even if the tumor changes, it is possible to tweak the immune system so that it attacks the latest iteration of the cancer. The clinical results with ipilimumab proved that the immune system could be induced to fight cancer and keep it from recurring.
In the years since the discovery of CD28 and CTLA-4, researchers have identified multiple interactions between T cells and APCs, known as checkpoints, that regulate the T cell response to antigen (52). Each of these interactions represents a new opportunity in immune checkpoint blockade therapy. Continued basic research in this area will elucidate the mechanisms by which the human immune system responds to tumors, helping to guide the development of novel therapies targeting the immune response.
Coda
Allison moved to MD Anderson Cancer Center in 2012, where he is the Vivian L. Smith Distinguished Chair in Immunology and Executive Director of the Immunotherapy Platform for MD Anderson’s Moon Shots Program. The immunotherapy platform has three components: (a) preclinical studies to establish the feasibility and efficacy of new treatments; (b) immunologic monitoring to provide instrumentation and technical support for the cellular and molecular analysis of the effect of therapies on the immune system; and (c) immunopathology to develop an understanding of changes in the tumor microenvironment associated with immunotherapy (53). Findings from these studies will be of particular help in identifying combinatorial therapies that can improve the effects of immune checkpoint blockade therapy and applying it to different forms of cancer. Much in the same way that Allison worked to bring anti–CTLA-4 therapy into the clinic, the ultimate goal of this program is to accelerate the conversion of scientific discoveries into clinical advances that will reduce cancer deaths.
James Allison’s work on CTLA-4 and T cell responses has changed the way that we think about cancer therapy and spurred the development of new treatments in which the immune system is targeted rather than the cancer. Additionally, Allison’s work has served as a model for basic researchers. “I think the way to do research is to ask fundamentally important questions and, every now and then, kick your feet up on the desk, have a glass of wine, and say ‘How could I use this? What could we do with this information I’ve generated?’” said Allison. His investigations into the basic mechanisms that govern the function of T cells identified a new therapeutic target that resulted in a highly successful therapy and serve as the ultimate example of how a basic science finding can change the practice of medicine.
Footnotes
Reference information:J Clin Invest. 2015;125(10):3732–3736. doi:10.1172/JCI84236.
References
- 1.Allison JP, McIntyre BW, Bloch D. Tumor-specific antigen of murine T-lymphoma defined with monoclonal antibody. J Immunol. 1982;129(5):2293–2300. [PubMed] [Google Scholar]
- 2.Acuto O, et al. The human T cell receptor: appearance in ontogeny and biochemical relationship of α and β subunits on IL-2 dependent clones and T cell tumors. Cell. 1983;34(3):717–726. doi: 10.1016/0092-8674(83)90528-7. [DOI] [PubMed] [Google Scholar]
- 3.Kappler J, Kubo R, Haskins K, White J, Marrack P. The mouse T cell receptor: comparison of MHC-restricted receptors on two T cell hybridomas. Cell. 1983;34(3):727–737. doi: 10.1016/0092-8674(83)90529-9. [DOI] [PubMed] [Google Scholar]
- 4.Meuer SC, et al. Clonotypic structures involved in antigen-specific human T cell function. Relationship to the T3 molecular complex. J Exp Med. 1983;157(2):705–719. doi: 10.1084/jem.157.2.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samelson LE, Germain RN, Schwartz RH. Monoclonal antibodies against the antigen receptor on a cloned T-cell hybrid. Proc Natl Acad Sci U S A. 1983;80(22):6972–6976. doi: 10.1073/pnas.80.22.6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bigler RD, Fisher DE, Wang CY, Rinnooy Kan EA, Kunkel HG. Idiotype-like molecules on cells of a human T cell leukemia. J Exp Med. 1983;158(3):1000–1005. doi: 10.1084/jem.158.3.1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kappler J, et al. The major histocompatibility complex-restricted antigen receptor on T cells in mouse and man: identification of constant and variable peptides. 1983. J Immunol. 2006;176(5):2683–2690. [PubMed] [Google Scholar]
- 8.McIntyre BW, Allison JP. The mouse T cell receptor: structural heterogeneity of molecules of normal T cells defined by xenoantiserum. Cell. 1983;34(3):739–746. doi: 10.1016/0092-8674(83)90530-5. [DOI] [PubMed] [Google Scholar]
- 9.Chien Y, Becker DM, Lindsten T, Okamura M, Cohen DI, Davis MM. A third type of murine T-cell receptor gene. Nature. 1984;312(5989):31–35. doi: 10.1038/312031a0. [DOI] [PubMed] [Google Scholar]
- 10.Yanagi Y, Chan A, Chin B, Minden M, Mak TW. Analysis of cDNA clones specific for human T cells and the α and β chains of the T-cell receptor heterodimer from a human T-cell line. Proc Natl Acad Sci U S A. 1985;82(10):3430–3434. doi: 10.1073/pnas.82.10.3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dembić Z, et al. Transfer of specificity by murine α and β T-cell receptor genes. Nature. 1986;320(6059):232–238. doi: 10.1038/320232a0. [DOI] [PubMed] [Google Scholar]
- 12.Weiss A, Imboden J, Hardy K, Manger B, Terhorst C, Stobo J. The role of the T3/antigen receptor complex in T-cell activation. Annu Rev Immunol. 1986;4:593–619. doi: 10.1146/annurev.iy.04.040186.003113. [DOI] [PubMed] [Google Scholar]
- 13.Weiss A, Imboden JB. Cell surface molecules and early events involved in human T lymphocyte activation. Adv Immunol. 1987;41:1–38. doi: 10.1016/S0065-2776(08)60029-2. [DOI] [PubMed] [Google Scholar]
- 14.Jenkins MK, Pardoll DM, Mizuguchi J, Quill H, Schwartz RH. T-cell unresponsiveness in vivo and in vitro: fine specificity of induction and molecular characterization of the unresponsive state. Immunol Rev. 1987;95:113–135. doi: 10.1111/j.1600-065X.1987.tb00502.x. [DOI] [PubMed] [Google Scholar]
- 15.Schwartz RH, Mueller DL, Jenkins MK, Quill H. T-cell clonal anergy. Cold Spring Harb Symp Quant Biol. 1989;54(pt 2):605–610. doi: 10.1101/sqb.1989.054.01.072. [DOI] [PubMed] [Google Scholar]
- 16.Martin PJ, Ledbetter JA, Morishita Y, June CH, Beatty PG, Hansen JA. A 44 kilodalton cell surface homodimer regulates interleukin 2 production by activated human T lymphocytes. J Immunol. 1986;136(9):3282–3287. [PubMed] [Google Scholar]
- 17.Linsley PS, Clark EA, Ledbetter JA. T-cell antigen CD28 mediates adhesion with B cells by interacting with activation antigen B7/BB-1. Proc Natl Acad Sci U S A. 1990;87(13):5031–5035. doi: 10.1073/pnas.87.13.5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature. 1992;356(6370):607–609. doi: 10.1038/356607a0. [DOI] [PubMed] [Google Scholar]
- 19.Harding FA, Allison JP. CD28-B7 interactions allow the induction of CD8+ cytotoxic T lymphocytes in the absence of exogenous help. J Exp Med. 1993;177(6):1791–1796. doi: 10.1084/jem.177.6.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brunet JF, et al. A new member of the immunoglobulin superfamily — CTLA-4. Nature. 1987;328(6127):267–270. doi: 10.1038/328267a0. [DOI] [PubMed] [Google Scholar]
- 21.Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174(3):561–569. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell. 1992;71(7):1065–1068. doi: 10.1016/S0092-8674(05)80055-8. [DOI] [PubMed] [Google Scholar]
- 23.Lenschow DJ, et al. Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4lg. Science. 1992;257(5071):789–792. doi: 10.1126/science.1323143. [DOI] [PubMed] [Google Scholar]
- 24.Linsley PS, et al. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J Exp Med. 1992;176(6):1595–1604. doi: 10.1084/jem.176.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walunas TL, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1(5):405–413. doi: 10.1016/1074-7613(94)90071-X. [DOI] [PubMed] [Google Scholar]
- 26.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182(2):459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waterhouse P, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270(5238):985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 28.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3(5):541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 29.Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity. 1997;7(6):885–895. doi: 10.1016/S1074-7613(00)80406-9. [DOI] [PubMed] [Google Scholar]
- 30.Ehrlich P. Über den jetzigen Stand der Karzinomforschung. Beitr Zur Exp Pathol Chemother. 1909;5:117–164. [Google Scholar]
- 31. Thomas L. Discussion of cellular and humoral aspects of the hypersensitivity states. In: Cellular and Humoral Aspects of Hypersensitivity. New York, New York, USA: Hoeber-Harper; 1959:529–532. [Google Scholar]
- 32.Burnet FM. Immunological surveillance in neoplasia. Transplant Rev. 1971;7:3–25. doi: 10.1111/j.1600-065x.1971.tb00461.x. [DOI] [PubMed] [Google Scholar]
- 33.Thotathil Z, Jameson MB. Early experience with novel immunomodulators for cancer treatment. Expert Opin Investig. 2007;16(9):1391–1403. doi: 10.1517/13543784.16.9.1391. [DOI] [PubMed] [Google Scholar]
- 34.Herr HW, Morales A. History of bacillus Calmette-Guerin and bladder cancer: an immunotherapy success story. J Urol. 2008;179(1):53–56. doi: 10.1016/j.juro.2007.08.122. [DOI] [PubMed] [Google Scholar]
- 35.Townsend SE, Allison JP. Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science. 1993;259(5093):368–370. doi: 10.1126/science.7678351. [DOI] [PubMed] [Google Scholar]
- 36.Allison JP, Hurwitz AA, Leach DR. Manipulation of costimulatory signals to enhance antitumor T-cell responses. Curr Opin Immunol. 1995;7(5):682–686. doi: 10.1016/0952-7915(95)80077-8. [DOI] [PubMed] [Google Scholar]
- 37.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 38.Kwon ED, et al. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc Natl Acad Sci U S A. 1997;94(15):8099–8103. doi: 10.1073/pnas.94.15.8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hurwitz AA, Townsend SE, Yu TF, Wallin JA, Allison JP. Enhancement of the anti-tumor immune response using a combination of interferon-γ and B7 expression in an experimental mammary carcinoma. Int J Cancer. 1998;77(1):107–113. doi: 10.1002/(SICI)1097-0215(19980703)77:1<107::AID-IJC17>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 40.Hurwitz AA, Yu TF, Leach DR, Allison JP. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc Natl Acad Sci U S A. 1998;95(17):10067–10071. doi: 10.1073/pnas.95.17.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190(3):355–366. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hurwitz AA, et al. Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res. 2000;60(9):2444–2448. [PubMed] [Google Scholar]
- 43.Keler T, et al. Activity and safety of CTLA-4 blockade combined with vaccines in cynomolgus macaques. J Immunol. 2003;171(11):6251–6259. doi: 10.4049/jimmunol.171.11.6251. [DOI] [PubMed] [Google Scholar]
- 44.Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. 2006;116(7):1935–1945. doi: 10.1172/JCI27745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Engelhardt JJ, Sullivan TJ, Allison JP. CTLA-4 overexpression inhibits T cell responses through a CD28-B7-dependent mechanism. J Immunol. 2006;177(2):1052–1061. doi: 10.4049/jimmunol.177.2.1052. [DOI] [PubMed] [Google Scholar]
- 46.Hoos A, et al. Development of ipilimumab: contribution to a new paradigm for cancer immunotherapy. Semin Oncol. 2010;37(5):533–546. doi: 10.1053/j.seminoncol.2010.09.015. [DOI] [PubMed] [Google Scholar]
- 47.Wolchok JD, et al. Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci. 2013;129:1–13. doi: 10.1111/nyas.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Atkins MB, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- 49.Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH. Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol. 1996;14(1):7–17. doi: 10.1200/JCO.1996.14.1.7. [DOI] [PubMed] [Google Scholar]
- 50.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delyon J, Maio M, Lebbé C. The ipilimumab lesson in melanoma: achieving long-term survival. Semin Oncol. 2015;42(3):387–401. doi: 10.1053/j.seminoncol.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 52.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 54.Buchbinder E, Hodi FS. Cytotoxic T-lymphocyte antigen-4 and immune checkpoint blockade. J Clin Invest. 2015;125(9):3377–3383. doi: 10.1172/JCI80012. [DOI] [PMC free article] [PubMed] [Google Scholar]