

Abstract
Variants near the gene TRIB1 are significantly associated with several plasma lipid traits, circulating liver enzymes, and the development of coronary artery disease in humans; however, it is not clear how its protein product tribbles-1 regulates lipid metabolism. Here, we evaluated mice harboring a liver-specific deletion of Trib1 (Trib1_LSKO) to elucidate the role of tribbles-1 in mammalian hepatic lipid metabolism. These mice exhibited increased hepatic triglyceride (TG) content, lipogenic gene transcription, and de novo lipogenesis. Microarray analysis revealed altered transcription of genes that are downstream of the transcription factor C/EBPα, and Trib1_LSKO mice had increased hepatic C/EBPα protein. Hepatic overexpression of C/EBPα in WT mice phenocopied Trib1_LSKO livers, and hepatic knockout of Cebpa in Trib1_LSKO mice revealed that C/EBPα is required for the increased lipogenesis. Using ChIP-Seq, we found that Trib1_LSKO mice had increased DNA-bound C/EBPα near lipogenic genes and the Trib1 gene, which itself was transcriptionally upregulated by C/EBPα overexpression. Together, our results reveal that tribbles-1 regulates hepatic lipogenesis through posttranscriptional regulation of C/EBPα, which in turn transcriptionally upregulates Trib1. These data suggest an important role for C/EBPα in mediating the lipogenic effects of hepatic Trib1 deletion and provide insight into the association between TRIB1 and plasma lipids, and liver traits in humans.
Introduction
Genome-wide association studies have identified over 150 genomic loci that are associated with plasma lipid traits in humans, and more than half of these loci have no previously appreciated role in lipid metabolism (1–3). One of the most interesting of these lipid loci is the genomic region 8q24, at which a cluster of noncoding variants are significantly associated with all major plasma lipid traits, including triglycerides (TG), total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), and high-density cholesterol (HDL-C) (2, 3). The same variants are also significantly associated with plasma concentrations of alanine transaminase (ALT) (4) and with coronary artery disease (5). However, the molecular mechanisms underpinning these genetic associations are poorly understood.
The 8q24 genomic locus harbors the gene TRIB1, which encodes the protein tribbles-1 (TRIB1). We previously reported that hepatic overexpression of Trib1 in mice reduced lipogenesis, VLDL secretion, and plasma lipid levels (6). However, the mechanism through which gain-of-function of hepatic TRIB1 regulated lipid metabolism was not determined. Here, we reported the investigation of mice with a liver-specific deletion of the Trib1 gene and observed that these mice have increased hepatic lipogenesis and steatosis, and grossly dysregulated hepatic gene expression. These effects are largely due to the posttranscriptional increase in hepatic abundance of the transcription factor C/EBPα and the resultant increased DNA occupancy of this protein at or near lipogenic genes.
Results
The liver-specific Trib1 KO mouse has elevated plasma ALT and lipids.
We obtained mice on a C57BL/6 background with flanking loxP sites around the second exon of Trib1 (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/JCI77095DS1). Since the liver is the main site of lipid and lipoprotein metabolism, we pursued tissue-specific deletion of hepatic Trib1. Trib1 was deleted in the liver of Trib1fl/fl mice using 2 complementary approaches. Trib1fl/fl mice were either injected with adeno-associated viral vector serotype 8 (AAV8) expressing Cre recombinase under the control of the liver-specific thyroxine-binding globulin (TBG) promoter (AAV-TBG-Cre), or they were crossed with transgenic mice expressing Cre under the control of the albumin promoter. Trib1fl/fl mice receiving AAV8-TBG-Cre (herein referred to as Trib1_LSKO) displayed >95% deletion of the allele as early as 1 week after injection, as compared with Trib1fl/fl mice treated with AAV containing empty vector (herein referred to as Trib1_fl/fl) (Figure 1A). Trib1fl/fl crossed with albumin-Cre mice (Trib1fl/fl Alb-Cre +) had similar levels of Trib1 deletion (Figure 1B). In the setting of Trib1 deletion, we observed no compensatory upregulation of Trib2 (not detectable in liver) or Trib3 (Figure 1A). Interestingly, hepatic deletion of Trib1 caused significantly increased Trib1 message in brown adipose tissue and trends toward increased expression in white adipose tissue and skeletal muscle of Trib1_LSKO mice (Supplemental Figure 1B). Trib1_LSKO mice also had significant increases in plasma ALT levels, and these were more notable in males than females. This observation was made in both AAV-Cre mice(Figure 1C) and Alb-Cre mice (Figure 1D), indicating that this finding was not due to the administration of the viral vector.
Figure 1. Efficient deletion of Trib1, both somatic and germ-line, causes increased plasma ALTs.

(A) Hepatic transcript levels of Trib1-3 in mice (n = 5) receiving 1.5 × 1011 gc of either AAV_Null (Trib1_fl/fl) or AAV-TBG-Cre (Trib1_LSKO) 4 weeks after injection. Transcript levels were measured by TaqMan real-time RT-PCR of cDNA made from 1 μg liver total RNA. ND, not detectable. (B) Hepatic message levels of Trib1-3 in Trib1fl/fl mice crossed onto the albumin-Cre background causing germline deletion of hepatic Trib1. Mice were all aged 8–10 weeks, n = 5. (C) Plasma ALTs in male and female Trib1_fl/fl and Trib1_LSKO mice 4 weeks after injection, as measured by Cobas-Mira autoanalyzer. (D) Plasma ALTs of Trib1fl/fl Alb-Cre – and Trib1fl/fl Alb-Cre + males and females (n = 5), aged 8–10 weeks. Significance was determined in all panels by Student’s t test (**P ≤ 0.01, ***P ≤ 0.001).
Trib1_LSKO animals had significantly increased plasma TC (23%), non-HDL cholesterol (37%), and TG (52%) 4 weeks after AAV-Cre injection as compared with control Trib1fl/fl animals treated with AAV-Null (Figure 2, A and B). The cholesterol and non-HDL cholesterol levels in Trib1_LSKO mice increased over time out to at least 20 weeks after injection (Figure 2, C and D). Similar plasma lipid results were also observed in Trib1fl/fl Alb-Cre mice (Supplemental Figure 2).
Figure 2. Trib1_LSKO mice have increased plasma TC, HDL and non-HDL cholesterol, and plasma TGs.

(A) Plasma TC, non-HDL cholesterol, and TG in male Trib1_fl/fl or Trib1_LSKO mice (n = 10) 4 weeks after injection. Plasma samples were collected after mice were fasted for 4 hours. (B) Plasma TC, non-HDL cholesterol, and TG after 4-hour fast in female Trib1_fl/fl and Trib1_LSKO mice (n = 10) 4 weeks after injection. (C) Plasma TC in male Trib1_fl/fl and Trib1_LSKO mice (n = 5) out to 20 weeks after injection. (D) Change from baseline for non-HDL cholesterol in the same male Trib1_fl/fl and Trib1_LSKO mice from C. All plasma levels were measured by Cobas-Mira autoanalyzer with commercially available reagents. Significance was determined in all panels by Student’s t test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001).
Trib1_LSKO mice have increased liver TG and increased de novo lipogenesis.
Trib1_LSKO animals exhibited a 25% increase in liver weight (P < 0.01) relative to body weight (Figure 3A). Examination of hepatic lipids revealed a 78% increase in hepatic TG content (P < 0.001) of Trib1_LSKO mouse livers compared with controls (Figure 3B), while no significant change in hepatic cholesterol was observed (Figure 3C). Histological analysis of livers from Trib1_LSKO mice revealed increased microvesicular lipid deposition, hepatocellular hypertrophy, and presence of Mallory bodies, consistent with a finding of steatohepatitis (Figure 3, D and E). Increased circulating liver enzymes are also an indicator of steatosis, so in addition to ALTs, we investigated circulating ASTs and GGTs in Trib1_LSKO mice and found increased AST activity but not GGT activity (Supplemental Figure 3). Real-time PCR analysis revealed >2-fold increases in the hepatic transcription of lipogenic genes such as Acaca, Fasn, and Scd1 (Figure 3F). Transcript levels of known regulators of these genes, such as Chrebp and Srebf1, remained unaffected (Figure 3F), suggesting a differential regulation by another transcriptional mechanism. Additionally, the transcription of downstream targets of ChREBP and LXR also were not significantly affected, implying a lack of involvement of these pathways (Supplemental Figure 4). To determine whether the increased liver TG was due to increased production, we measured in vivo de novo lipogenesis using [3H]-acetate as a precursor. Trib1_LSKO animals exhibited significantly increased production of fatty acids (2.2-fold, P = 0.02), diacylglycerol (1.8-fold, P < 0.01), and TG (3.6-fold, P < 0.001) (Figure 3G).
Figure 3. Trib1_LSKO mice have increased hepatic TG content, transcription of fatty acid synthetic genes, and de novo lipogenesis.

(A) Liver mass of Trib1_fl/fl and Trib1_LSKO mice (n = 5) represented as percentage of body mass. (B) Liver TG content measured by colorimetric assay using 20 mg whole liver homogenized in PBS from Trib1_fl/fl and Trib1_LSKO mice 4 weeks after injection. (C) Liver cholesterol content measured by colorimetric assay from same liver homogenates used in B. (D) H/E staining of liver sections from Trib1_fl/fl and Trib1_LSKO mice 4 weeks after injection shows increased lipid deposition and breakdown of hepatocyte organization. (E) Enlarged image of steatotic liver from Trib1_LSKO mice. The white arrowhead indicates a hypertrophic hepatocyte with microvesicular steatosis, and the black arrowhead indicates a typical Mallory body. (F) Lipogenic gene transcription in livers of Trib1_fl/fl and Trib1_LSKO mice measured by TaqMan real-time RT-PCR (n = 5). (G) De novo lipogenesis in mice (n = 6) fasted overnight, then refed and injected with 100 μCi of [3H]-Acetate for labeling of newly synthesized fatty acids. Lipids were extracted from 100 mg liver, and values are CPM/mg liver. Scale bars: 20 µm. Significance was determined by Student’s t test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001).
Trib1_LSKO mice have altered hepatic gene expression and increased liver C/EBPα protein levels.
To investigate the extent to which hepatic transcription is differentially regulated in our model, we performed microarray analysis of Trib1_LSKO livers compared with controls 4 weeks after AAV injection. A total of 2,095 genes (1,478 upregulated, 617 downregulated) were significantly altered between the 2 groups (absolute fold change>1.5, FDR<10%). Gene set enrichment analysis of this array data suggested that the altered gene set was enriched for genes downstream of multiple transcriptional effectors, including the transcription factor C/EBPα (P = 6.22 × 10–12) (Table 1). Western blot analysis of liver extracts showed a dramatic increase in C/EBPα protein in Trib1_LSKO mice compared with controls, while mRNA abundance of Cebpa transcript was in fact decreased (–37%, P = 0.01), suggesting a posttranscriptional effect of Trib1 deletion on turnover of C/EBPα (Figure 4). We observed similar patterns with C/EBPβ protein and message as well, albeit to a lesser extent (Figure 4). These observations suggest that the increase in C/EBPα protein is, at least in part, contributing to the phenotypes observed in the Trib1_LSKO mice.
Table 1. Trib1_LSKO mice have increased transcription of C/EBP target genes as identified by pathway analysis.
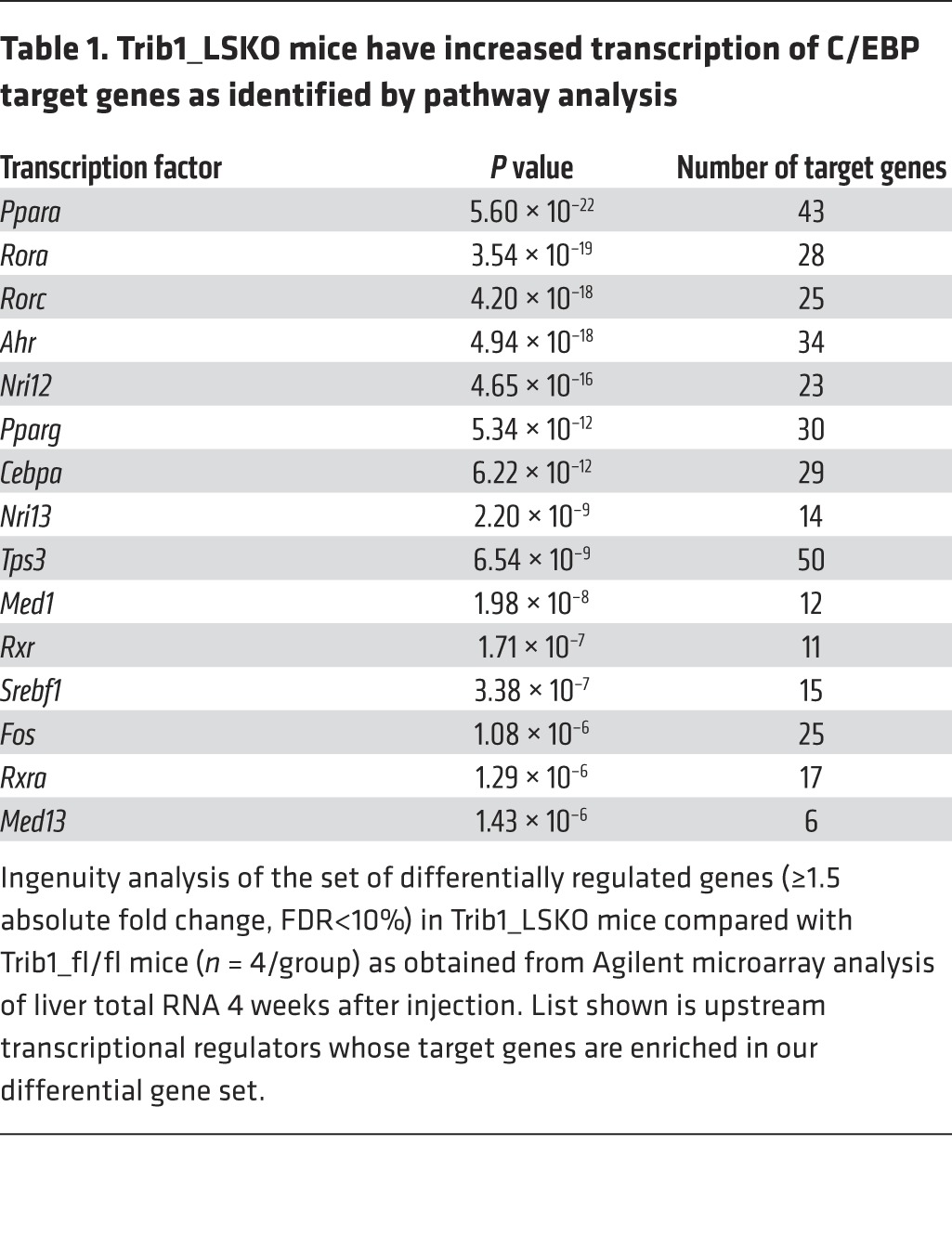
Figure 4. Loss of Hepatic Trib1 leads to increased C/EBPa and C/EBPb protein levels, as well as decreased transcript levels.

(A) Western blot analysis of C/EBPα, C/EBPβ, and β-actin levels in livers of Trib1_fl/fl and Trib1_LSKO mice. Whole-liver protein extracts were made in PBS + protease inhibitors, and proteins were size-separated by SDS-PAGE. (B) Real-time RT-PCR analysis of Cebpa and Cebpb hepatic transcript levels in Trib_fl/fl and Trib1_LSKO mice (n = 5) 4 weeks after injection. TaqMan Realtime RT-PCR was performed using commercially available gene-expression probes on cDNA made from 1 μg total liver RNA. Results in B were analyzed by Student’s t test (**P ≤ 0.01).
Overexpression of mCebpa is sufficient to phenocopy the lipogenesis phenotype of the Trib1_LSKO mice but not the plasma lipid findings.
To determine if increased C/EBPα protein was sufficient to drive increased de novo lipogenesis, we overexpressed murine C/EBPα (mCebpa) via AAV8 in the livers of C57BL/6 mice (Supplemental Figure 5A). AAV_mCebpa mice had increased hepatic TG content as compared with control mice receiving AAV_Null 2 weeks after injection (Figure 5A). Mice overexpressing mCebpa also had a similar gene expression profile to Trib1_LSKO mice with respect to lipogenic genes; Fasn, Scd1, and Acaca were all significantly increased, while Chrebp and Srebf1 were not (Figure 5B). De novo lipogenesis in mCebpa-overexpressing mice was significantly increased in production of fatty acids (1.7-fold, P < 0.001), diacylglycerol (1.5-fold, P < 0.01), and TG (3.5-fold, P < 0.001) as compared with control mice receiving AAV_Null (Figure 5C). This closely mimics the phenotype seen in the Trib1_LSKO mice and strongly suggests that increased levels of C/EBPα are responsible for the changes in lipogenic gene expression, hepatic TG content, and de novo lipogenesis observed in the setting of hepatic loss of Trib1. AAV_mCebpa mice displayed increased plasma ALT levels, as well (Supplemental Figure 5B); however, in contrast to the Trib1_LSKO mice, they surprisingly did not exhibit increased plasma lipids, even 10 weeks after injection (Figure 5D). We observed no significant differences in plasma TC or TG between the groups, along with a small but significant decrease in plasma non-HDL levels in the AAV_mCebpa animals (Figure 5D). The lack of increased plasma lipids in the AAV_mCebpa mice suggests that the plasma lipid phenotype in Trib1_LSKO mice is due to an alternative mechanism independent of C/EBPα.
Figure 5. Overexpression of mCebpa in livers of mice phenocopies the increased hepatic TG synthesis observed in Trib1_LSKO mice.

(A) Liver TG content in mice receiving 1 × 1012 AAV_Null or AAV_mCebpa 2 weeks after injection after overnight fast (n = 5). (B) Hepatic lipogenic gene expression in AAV_Null and AAV_mCebpa mice 2 weeks after injection, as measured by TaqMan real-time RT-PCR from 1 μg total RNA. Values are relative expression compared with GAPDH housekeeping gene. (C) De novo lipogenesis in mice from A after overnight fast, refeeding, and fatty acid labeling with [3H]-Acetate. (D) Plasma TC, non-HDL cholesterol, and TG were measured after a 4-hour fast in mice (n = 6) 10 weeks after AAV_Null or AAV_mCebpa injection. Significance was determined in all panels by Student’s t test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001).
Hepatic deletion of C/EBPα in Trib1_LSKO mice completely attenuates the lipogenic phenotype.
While increased C/EBPα protein levels are sufficient to drive increased lipogenesis in Trib_LSKO mice, we next sought to test if C/EBPα was required for this phenotype. We obtained a previously reported Cebpafl/fl conditional KO mouse (7) and crossed this with our Trib1fl/fl mouse to generate 3 experimental groups: WT, Trib1fl/fl, and Trib1fl/fl Cebpafl/fl mice. We administered AAV-TBG-Cre to all 3 groups to generate WT, Trib1_LSKO, and Trib1/Cebpa double-LSKO (herein referred to as Trib1/Cebpa_dLSKO) mice, the last of which had >95% deletion of Cebpa (Figure 6A). Trib1_LSKO mice exhibited the expected increases in lipogenic gene transcription, while Trib1/Cebpa_dLSKO exhibited transcription at WT levels or lower (Figure 6B). Production of fatty acids, diacylglycerol, and TG in Trib1/Cebpa_dLSKO mice were returned to WT levels as compared with Trib1_LSKO mice (Figure 6C). These data confirm that C/EBPα is required for the increased lipogenesis phenotype observed in Trib1_LSKO mice.
Figure 6. C/EBPα is required for increased lipogenesis in Trib1_LSKO mice.

(A) Hepatic transcript levels of Cebpa in WT mice, Trib1_LSKO mice, or Trib1/Cebpa_dLSKO mice (n = 6) 2 weeks after injection with 1.5 × 1011 gc of AAV-TBG-Cre. (B) Transcription of lipogenic genes in all 3 experimental groups. Gene expression values are relative to the Gapd housekeeping gene, with the WT group set to 1. (C) Production of fatty acids, diacylglycerol, and TG is shown in WT, Trib1_LSKO, and Trib1/Cebpa_dLSKO mice after injection with [3H]-acetate. Significance was determined in all panels by ANOVA followed by Tukey’s post-hoc test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001).
Deletion of hepatic Trib1 leads to increased C/EBPα binding near upregulated lipogenic genes, as well as Trib1 itself.
While the above data reveal a causal role for C/EBPα in the lipogenic phenotype observed in the Trib1_LSKO mice, they do not address whether it functions directly or indirectly through the regulation of another transcription factor to induce lipogenic gene transcription. To investigate this, we performed ChIP-Seq for C/EBPα in biological replicates of Trib1_LSKO mice, as well as for the AAV_mCebpa-treated mice, and compared the results to the respective controls from each experiment. We identified approximately 48,000 C/EBPα-binding sites in liver tissue from the Trib1_fl/fl and Trib1_LSKO mice. Of these, 4,480 and 1,361 sites had increased and decreased C/EBPα occupancy, respectively, in the Trib1_LSKO versus Trib1_fl/fl mice; C/EBPα occupancy was affected similarly at these sites by forced expression of C/EBPα (Figure 7A), suggesting that differential occupancy is a direct consequence of increased levels of C/EBPα. Motif analysis at sites with stronger C/EBPα binding in the Trib1_LSKO mice revealed robust enrichment for the canonical C/EBP motif (Figure 7B), and gene-set enrichment analysis of these peaks showed major enrichment of genes involved in lipid metabolism as one of the top findings (Supplemental Figure 6A). Moreover, differentially expressed genes in the Trib1_LSKO mice were highly correlated with the direction of altered C/EBPα occupancy (Figure 7C), and differentially expressed genes had 1.8-fold higher frequency of association with at least one differential C/EBPα peak than nondifferential genes (Supplemental Figure 6B). Multiple genes involved in fatty acid and triglyceride synthesis showed both upregulated expression in the livers of Trib1_LSKO mice and the presence of at least one substantially enriched C/EBPα-binding site within 100 kb of their transcription start sites (TSSs) (Figure 7D). As a whole, these data demonstrate a strong association between the lipogenic transcriptome and C/EBPα cistrome in liver, and suggest a direct role for C/EBPα in inducing lipogenic genes in the absence of Trib1.
Figure 7. Trib1_LSKO mice have increased C/EBPα occupancy near lipogenic genes.

(A) Pie chart of all C/EBPα binding sites identified in liver by ChIP-seq showing the number of sites that were unchanged (constitutive), increased or decreased in Trib1_LSKO mice compared with Trib1_fl/fl mice. Density heat maps (lower panel) of raw sequence tags at regions differentially occupied by C/EBPα in Trib1_LSKO mice are shown for Trib1_fl/fl, Trib1_LSKO, AAV_Null, and AAV_mCebpa mice. (B) HOMER de novo motif analysis of constitutive and differential C/EBPα-binding sites in Trib1_LSKO mice. (C) Relationship of gene expression to C/EBPα occupancy. Left panel shows the relative expression of all genes in the microarray ordered from highest to lowest fold-change in Trib1_LSKO vs. fl/fl control. Right heat map depicts C/EBPα occupancy in Trib1_fl/fl and Trib1_LSKO mice at sites within 100 kb of the TSS. (D) Heat map of differential C/EBPα binding sites within 100 kb of the TSS for genes involved in hepatic lipogenesis. The number of C/EBPα sites and the microarray fold change are indicated on the right for each gene.
To identify genes strongly associated with differential C/EBPα peaks, we mapped clusters of Trib1_LSKO-induced C/EBPα peaks that were within 50 kb of each other (Supplemental Table 1). Interestingly, the second-ranked cluster was approximately 40 kb downstream of the Trib1 locus, where 10 peaks in an approximately 20-kb region had substantially more C/EBPα binding in both the Trib1_LSKO and AAV_mCebpa mice as compared with controls (Figure 8A). Indeed, Trib1 gene expression was significantly increased in livers of AAV_mCebpa mice (Figure 8B). These findings suggest that C/EBPα transcriptionally upregulates TRIB1, while the TRIB1 protein posttranscriptionally downregulates C/EBPα protein, and that these 2 proteins participate in a strong negative feedback loop (Figure 8C).
Figure 8. C/EBPα and TRIB1 are in a regulatory feedback loop that is both transcriptional and posttranslational.

(A) Differential C/EBPα binding peaks in the downstream region of Trib1 in Trib1_fl/fl, Trib1_LSKO, AAV_Null, and AAV_mCebpa mice, as measured by C/EBPα ChIP-Seq. n = 2 for this experiment, while only one mouse/group is shown. (B) Hepatic TRIB1 expression in AAV_Null and AAV_mCebpa mice (n = 5) 2 weeks after injection. (C) Schematic of the TRIB1- C/EBPα regulatory feedback loop. Results in B were analyzed by Student’s t test (**P ≤ 0.01).
Discussion
Recent genome-wide association studies performed in extremely large patient cohorts have allowed for the identification of over 150 genomic loci as being implicated in human plasma lipid metabolism (3). One of the most strongly associated of these loci is nearest to the gene TRIB1, and this locus was subsequently associated with liver transaminases (4), as well as with coronary artery disease (5). Here, we used liver-specific deletion of Trib1 in mice to gain greater insight into the mechanisms by which TRIB1 influences hepatic lipid metabolism.
We previously reported that AAV8-mediated hepatic overexpression of Trib1 decreased hepatic lipogenesis and plasma lipids in mice (6). In this study, we report that hepatic-specific deletion of Trib1 resulted in upregulation of the transcription of genes involved in fatty acid synthesis, de novo lipogenesis, increased hepatic triglyceride and plasma transaminases, and elevated plasma lipids. We found a dramatic increase in the hepatic protein levels of C/EBPα, despite no increase in Cebpa mRNA abundance. Overexpression of C/EBPα in mice phenocopied the hepatic lipogenesis, steatosis, and increased transaminase phenotypes observed in the Trib1_LSKO mice. Deletion of Cebpa in the livers of Trib1_LSKO mice returned the levels of transcription of lipogenic genes and de novo lipogenesis back to WT levels or lower. ChIP-Seq verified that increased occupancy of C/EBPα occurred near lipogenic genes. These data strongly support a role for C/EBPα in de novo lipogenesis via upregulation of fatty acid–synthesis genes, and this regulation appears to be independent of canonical regulators of fatty acid synthesis, such as ChREBP, LXR, and SREBP1, the latter of which C/EBPα is reported to regulate the transcription of in adipose (8). C/EBPα protein abundance in liver is regulated by TRIB1, and thus the TRIB1-C/EBPα axis may be an underappreciated moderator of human hepatic lipogenesis.
The tribbles protein was originally identified in Drosophila as a regulator of cell migration and mitosis during oogenesis (9–11), with its role in oogenesis determined to be regulating the proteasomal degradation of Slbo, the Drosophila homolog of the human gene C/EBPα (12). There are 3 mammalian tribbles genes (tribbles1-3), all of which are known as pseudokinases; they have a protein domain homologous to a kinase domain yet are lacking critical amino acids in the catalytic region that would allow for phosphorylation of proteins (13). Work in the myeloblast 32D cell line has shown that overexpression of TRIB1 or TRIB2 induces the proteasomal degradation of C/EBPα and C/EBPβ by promoting their ubiquitination by the E3 ligase COP1 through direct binding to both targets and the ligase (14, 15). This function is critical for tribbles-mediated leukemogenesis (16, 17) and also coordinates TRIB1 regulation of macrophage polarization and differentiation (18). Earlier work has identified partially defined roles for other tribbles proteins in human metabolism, particularly TRIB3, which is involved in regulation of insulin signaling through interactions with AKT/Protein Kinase B (19, 20) and as a binding partner for ATF4 in pancreatic β cells (21). Adipose TRIB3 can also facilitate the turnover of the protein acetyl–coenzyme A carboxylase (ACACA), the rate-limiting enzyme in fatty acid synthesis (22).
C/EBPα belongs to the C/EBP family of transcription factors known to play wide-ranging roles in transcriptional regulation in adipose and the liver (23). C/EBPα has been long known to regulate lipogenesis in adipocytes, and multiple studies have implicated C/EBPα in regulation of lipogenic genes in the setting of mice deficient for leptin signalling (24, 25). An age-associated isoform of C/EBPα has been shown to promote steatosis in older mice, as well through regulation of TG-synthetic enzymes (26). However, a role for C/EBPα in hepatic lipogenesis in WT mice, especially in the context of TRIB1-mediated regulation, has not been investigated. We showed that Trib1_LSKO mice exhibiting increased lipogenesis also have increased hepatic C/EBPα and C/EBPβ protein levels. C/EBPα and β have been shown experimentally to have nearly completely overlapping ChIP-Seq binding profiles in both mouse liver (27) and adipose (28), and thus we suspect any role for C/EBPβ in the Trib1_LSKO mice is redundant to that of C/EBPα. Through AAV-mediated overexpression, we showed that increased C/EBPα levels are sufficient to cause increased lipogenic gene expression and fatty acid synthesis. We also deleted Cebpa in the livers of Trib1_LSKO mice, which completely ablated the lipogenic effects of hepatic deficiency of Trib1, showing that C/EBPα is also necessary for the lipogenic effects of hepatic Trib1 deletion. It remains possible that TRIB1 affects the function of other drivers of lipogenic gene expression (i.e., SREBP, ChREBP) via increased C/EBPα DNA binding, which may in turn facilitate increased binding of other transcription factors. Interestingly, our AAV_mCebpa mice did not display increased plasma lipids, suggesting that the mechanism governing the increased plasma lipids in the Trib1_LSKO mice is independent of the increased C/EBPα protein levels, and the elucidation of this mechanism remains a focus of our ongoing studies. The AAV_mCebpa mice did, however, exhibit increased plasma ALT levels, in keeping with the steatotic phenotype of these mice.
We also observed that overexpression of C/EBPα induces the transcription of Trib1 in WT mice. A previous report investigating C/EBPα genomic binding sites that are highly conserved between 5 different species identified 4 sites near TRIB1 that ranked in the top 25 most evolutionarily conserved (29). This data is bolstered by our own ChIP-Seq findings that, in Trib1_LSKO mice and AAV_mCebpa mice, some of the most highly enriched binding peaks for C/EBPα are in fact downstream of the Trib1 gene. Given these data, it appears that TRIB1 and C/EBPα participate in a tight feedback loop, wherein more C/EBPα protein leads to increased Trib1 transcription, whereupon TRIB1 targets C/EBPα for degradation (Figure 8C). This feedback loop is completely posttranscriptional, with respect to C/EBPα, and would not be identified by looking solely at transcript levels. Interestingly, Trib1 has a circadian pattern of expression, with message levels peaking at the start of dark hours (30). It follows then that C/EBPα protein levels, and therefore C/EBPα-mediated lipogenesis, are likely circadian, as well, and hepatic lipogenesis is in fact known to be circadian (31). The highly induced C/EBPα binding peaks near the Trib1 gene in our Trib1_LSKO mice are in a region with high synteny to the downstream region of TRIB1 in humans, where the plasma lipid GWAS signal is located (3). It is an attractive hypothesis that common variation in that region is causing defects in this circadian rhythm, and our group hopes to elucidate the functionally relevant SNPs in this region with further studies that are already underway.
The same variants at the TRIB1 locus associated with plasma lipid traits are also associated with circulating liver transaminases (4). Elevated ALT and AST levels are some of the strongest indicators of hepatic steatosis and nonalcoholic fatty liver disease (NAFLD) in humans (32), as the increased lipid deposition causes hepatocellular damage, which in turn increased serum transaminase levels (33). Interestingly, our Trib1_LSKO mice have increased ALTs, a finding replicated in the Trib1fl/fl Alb-Cre mice, suggesting that increased ALTs are a consequence of decreased Trib1 levels and not an artifact of the virus administration. The observation that Trib1_LSKO mice have increased histopathological findings consistent with steatohepatitis as well as elevated plasma ALT and AST levels, combined with the genetic association between TRIB1 and ALTs in humans, suggests that there is a role for altered TRIB1 levels in NAFLD etiology. It has been previously shown that increased hepatic de novo lipogenesis can contribute to NAFLD (34), and thus TRIB1 regulation of hepatic TG synthesis could be a part of disease pathology. Genome-wide scans for genes associated with NAFLD in humans have not identified TRIB1 as a disease locus. Some of these studies, however, have relied on coding variation that would not capture the noncoding SNPs downstream of TRIB1 (35, 36), and all of these studies were performed in patient cohorts significantly smaller than those used to identify the association of TRIB1 with transaminases (37). More recently, a targeted study investigating the association of 3 SNPs in the region downstream of TRIB1 with NAFLD in a Japanese cohort of patients (n = ~2,200) found significant associations between that region and hepatic TG content in those patients (38). Future genome-wide studies of increasing power performed on larger subject cohorts will likely identify TRIB1 as an NAFLD locus. Regardless of the exact role for TRIB1 in NAFLD pathology, it is an attractive therapeutic target for the treatment of such disorders, as one might anticipate that, in the setting of fatty liver, increased levels of TRIB1 might mitigate the excess of hepatic lipids. Indeed, a study aimed at identifying compounds that upregulate transcript levels of TRIB1 in hepatocytes was recently published, suggesting that this may be a viable therapeutic mechanism (39).
In summary, our loss-of-function studies indicate that hepatic TRIB1 is a key posttranscriptional regulator of C/EBPα protein, which in turn is a major regulator of hepatic lipogenesis. In addition, C/EBPα strongly promotes the transcription of its regulator, TRIB1. TRIB1 likely plays many roles in human metabolic diseases, and further research in other TRIB1 mechanisms of action in the liver and in other tissues may present even more innovative avenues for intervention in cardiovascular disease and metabolic disorders.
Methods
Animals.
Trib1 genetically engineered mice C57BL/6-Trib1tm1.1mrl were provided by Merck and were produced for Merck by contract with Taconic. Details of the design of the Trib1 mice may be found at http://www.taconic.com/10265 Previously reported mice harboring a conditional allele of Cebpa (Cebpatm1Dgt/J) were obtained from The Jackson Laboratory (stock number 006447) (7). Albumin-Cre mice were also obtained from The Jackson Laboratory. All mice were fed ad libitum with chow diet. All mice were fasted for 4 hours prior to collection of plasma for lipid measurements, unless stated otherwise. Blood was collected retroorbitally and centrifuged, and plasma was obtained. Fasting plasma lipids and ALTs were analyzed on a Roche Cobas Mira autoanalyzer using commercially available reagents (Sigma-Aldrich). Plasma ASTs (MAK055, Sigma-Aldrich) and GGTs (MAK089, Sigma-Aldrich) were measured using commercially available kits. Liver-lipid measurements (TG and TC) were obtained by homogenizing approximately 250 mg of whole liver in PBS and then assaying 1 mg via colorimetric assay. AAV vectors containing an empty expression cassette (AAV_Null) or encoding Cre recombinase (AAV-TBG-Cre) or mouse C/EBPα protein (AAV_mCebpa) were generated by the University of Pennsylvania Vector Core (Philadelphia, Pennsylvania, USA). For experiments utilizing AAV-TBG-Cre, animals were injected with AAV vectors at a dose of 1.5 × 1011 genome copies (GC) via i.p. injection and examined at the stated timepoints. For mCebpa overexpression experiments, mice received 1 × 1012 GC via i.p. injection of either AAV_mCebpa or AAV_Null.
De novo lipogenesis.
Animals were fasted overnight (~16 hours) and then fed a chow diet for 3 hours. Mice were then i.p. injected 100 μCi [3H]-Acetate (PerkinElmer) diluted in PBS, and allowed to continue feeding for 1.5 hours. Mice were sacrificed, livers were harvested, and lipids were extracted from 250 mg homogenized liver via Bligh-Dyer (40). Lipid extracts were dried and resuspended in 100 μl chloroform, half of which was run on a thin-layer chromatography plate in Hexanes/Diethyl Ether/Acetic Acid (70:30:1) solution. Lipid fractions were visualized by placing plates into an iodine chamber, and bands were cut out and counted. Values presented are counts normalized to mg liver.
Western blot analysis.
Protein extracts were prepared from liver tissue homogenized in PBS with protease inhibitors added (Halt Protease Inhibitors, Thermo Electron Corp.), and 50 μg protein was separated via SDS Page using the NuPage system (Invitrogen). CEBPα, CEBPβ, and β-actin were all visualized using primary antibodies from Santa Cruz Biotechnology Inc. (sc-61, sc-150, and sc-81178, respectively), HRP-conjugated secondary antibodies from Roche Applied Science, and the Crescendo ECL reagent (Millipore).
Gene-expression profiling.
RNA was collected from approximately 50 mg flash-frozen liver tissue using Trizol solution (Invitrogen) following the manufacturer’s protocol. cDNA was produced from 1 μg of total RNA with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) following the manufacturer’s instructions. Real-time PCR analysis was performed using standard TaqMan Gene Expression probes (Invitrogen) on an ABI7900 RT-PCR machine. The relative quantity of each mRNA was calculated using the delta CT method with Gapdh as the housekeeping gene. Total liver RNA was used for microarray analysis on the Agilent 4 × 44 Whole Mouse Genome Array. Microarray and data analysis were performed by the Penn Functional Genomics Core (Philadelphia, Pennsylvania, USA). Differential expression was determined using a False Discovery Rate of 10% and a minimum fold change of 1.5. All array data is publicly available on the NCBI Gene Expression Omnibus (GEO) database under accession number GSE70848.
ChIP-Seq analysis of C/EBPα binding in mouse liver.
C/EBPα ChIP was performed with sonicated, nuclear extract prepared from formaldehyde-crosslinked liver and 14AA (sc-61, Santa Cruz Biotechnology Inc.) antibody. Each experimental condition was assayed in duplicate with separate mice. ChIP-Seq libraries were produced and sequenced according to Illumina protocols as previously described (41), with the addition that libraries were multiplexed for sequencing. Peak calling and de novo motif analyses were performed using HOMER (42). Peaks were required to meet a cutoff of 1 RPM (average tag density of the replicate conditions) in either the Trib1_fl/fl or Trib1_LSKO condition. Peak mapping to annotated genes and gene ontology analyses were conducted with CEAS (43) and GREAT (44). Data generated from these ChIP-Seq experiments are publicly available in the NCBI GEO online resource under accession number GSE70848.
Statistics.
All data are reported as the mean ± SD except for gene expression results, which are mean ± SEM. Results were analyzed by 2-tailed Student’s t test, or ANOVA with Tukey’s post-hoc test when multiple comparisons were made. Statistical significance was defined as P < 0.05.
Study approval.
Animal experiments were reviewed and approved by the Institutional Animal Care and Use Committees of the University of Pennsylvania, Philadelphia, Pennsylvania, USA.
Supplementary Material
Acknowledgments
We acknowledge Thomas F. Vogt, John S. Mudgett, Sajesh Parathath, and Merck for providing the Trib1fl/fl mouse. We acknowledge Dr. Endre Kiss-Toth from the University of Sheffield (Sheffield, UK) for helpful discussions. We also acknowledge Emma E. Furth for providing her expertise on histopathological analysis of Trib1_LSKO livers. We acknowledge the Penn CVI Histology Core for H/E staining of liver tissue, the Penn Vector Core for production of all relevant vectors, and the Penn Functional Genomics Core for performing microarray and sequencing for the ChIP-Seq experiments. These studies were funded by NIH R01-HL109489 (to D.J. Rader), a Transatlantic Network of Excellence grant 10CVD03 from the Fondation Leducq (to D.J. Rader), NIH R01-DK098542 (to D.J. Steger), and American Heart Association postdoctoral fellowship 12POST12040456 (to R.C. Bauer).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(10):3809–3818. doi:10.1172/JCI77095.
References
- 1.Willer CJ, et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40(2):161–169. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teslovich TM, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466(7307):707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Global Lipids Genetics Consortium. et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45(11):1274–1283. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chambers JC, et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat Genet. 2011;43(11):1131–1138. doi: 10.1038/ng.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.CARDIoGRAMplusC4D Consortium. et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45(1):25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burkhardt R, et al. Trib1 is a lipid- and myocardial infarction-associated gene that regulates hepatic lipogenesis and VLDL production in mice. J Clin Invest. 2010;120(12):4410–4414. doi: 10.1172/JCI44213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang P, et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBPα. Immunity. 2004;21(6):853–863. doi: 10.1016/j.immuni.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 8.Payne VA, et al. C/EBP transcription factors regulate SREBP1cgene expression during adipogenesis. Biochem J. 2009;425(1):215–223. doi: 10.1042/BJ20091112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seher TC, Leptin M. Tribbles, a cell-cycle brake that coordinates proliferation and morphogenesis during Drosophila gastrulation. Curr Biol. 2000;10(11):623–629. doi: 10.1016/S0960-9822(00)00502-9. [DOI] [PubMed] [Google Scholar]
- 10.Grosshans J, Wieschaus E. A genetic link between morphogenesis and cell division during formation of the ventral furrow in Drosophila. Cell. 2000;101(5):523–531. doi: 10.1016/S0092-8674(00)80862-4. [DOI] [PubMed] [Google Scholar]
- 11.Mata J, Curado S, Ephrussi A, Rørth P. Tribbles coordinates mitosis and morphogenesis in Drosophila by regulating string/CDC25 proteolysis. Cell. 2000;101(5):511–522. doi: 10.1016/S0092-8674(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 12.Rørth P, Szabo K, Texido G. The level of C/EBP protein is critical for cell migration during Drosophila oogenesis and is tightly controlled by regulated degradation. Mole Cell. 2000;6(1):23–30. doi: 10.1016/s1097-2765(05)00008-0. [DOI] [PubMed] [Google Scholar]
- 13.Hegedus Z, Czibula A, Kiss-Toth E. Tribbles: a family of kinase-like proteins with potent signalling regulatory function. Cell Signal. 2007;19(2):238–250. doi: 10.1016/j.cellsig.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 14.Keeshan K, et al. Tribbles homolog 2 inactivates C/EBPα and causes acute myelogenous leukemia. Cancer Cell. 2006;10(5):401–411. doi: 10.1016/j.ccr.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dedhia PH, et al. Differential ability of Tribbles family members to promote degradation of C/EBPα and induce acute myelogenous leukemia. Blood. 2010;116(8):1321–1328. doi: 10.1182/blood-2009-07-229450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keeshan K, et al. Transformation by Tribbles homolog 2 (Trib2) requires both the Trib2 kinase domain and COP1 binding. Blood. 2010;116(23):4948–4957. doi: 10.1182/blood-2009-10-247361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoshida A, Kato JY, Nakamae I, Yoneda-Kato N. COP1 targets C/EBP for degradation and induces acute myeloid leukemia via Trib1. Blood. 2013;122(10):1750–1760. doi: 10.1182/blood-2012-12-476101. [DOI] [PubMed] [Google Scholar]
- 18.Satoh T, et al. Critical role of Trib1 in differentiation of tissue-resident M2-like macrophages. Nature. 2013;495(7442):524–528. doi: 10.1038/nature11930. [DOI] [PubMed] [Google Scholar]
- 19.Andreozzi F, et al. TRIB3 R84 variant is associated with impaired insulin-mediated nitric oxide production in human endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28(7):1355–1360. doi: 10.1161/ATVBAHA.108.162883. [DOI] [PubMed] [Google Scholar]
- 20.Du K, Herzig S, Kulkarni RN, Montminy M. TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science. 2003;300(5625):1574–1577. doi: 10.1126/science.1079817. [DOI] [PubMed] [Google Scholar]
- 21.Liew CW, et al. The pseudokinase tribbles homolog 3 interacts with ATF4 to negatively regulate insulin exocytosis in human and mouse β cells. J Clin Invest. 2010;120(8):2876–2888. doi: 10.1172/JCI36849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qi L, et al. TRB3 links the E3 ubiquitin ligase COP1 to lipid metabolism. Science. 2006;312(5781):1763–1766. doi: 10.1126/science.1123374. [DOI] [PubMed] [Google Scholar]
- 23.Schrem H, Klempnauer J, Borlak J. Liver-enriched transcription factors in liver function and development. Part II: the C/EBPs and D site-binding protein in cell cycle control, carcinogenesis, circadian gene regulation, liver regeneration, apoptosis, and liver-specific gene regulation. Pharmacol Rev. 2004;56(2):291–330. doi: 10.1124/pr.56.2.5. [DOI] [PubMed] [Google Scholar]
- 24.Matsusue K, et al. Hepatic CCAAT/enhancer binding protein alpha mediates induction of lipogenesis and regulation of glucose homeostasis in leptin-deficient mice. Mol Endocrinol. 2004;18(11):2751–2764. doi: 10.1210/me.2004-0213. [DOI] [PubMed] [Google Scholar]
- 25.Qiao L, MacLean PS, You H, Schaack J, Shao J. knocking down liver ccaat/enhancer-binding protein alpha by adenovirus-transduced silent interfering ribonucleic acid improves hepatic gluconeogenesis and lipid homeostasis in db/db mice. Endocrinology. 2006;147(6):3060–3069. doi: 10.1210/en.2005-1507. [DOI] [PubMed] [Google Scholar]
- 26.Jin J, et al. Increased expression of enzymes of triglyceride synthesis is essential for the development of hepatic steatosis. Cell Rep. 2013;3(3):831–843. doi: 10.1016/j.celrep.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jakobsen JS1, Waage J, Rapin N, Bisgaard HC, Larsen FS, Porse BT. Temporal mapping of CEBPA and CEBPB binding during liver regeneration reveals dynamic occupancy and specific regulatory codes for homeostatic and cell cycle gene batteries. Genome Res. 2013;23(4):592–603. doi: 10.1101/gr.146399.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lefterova MI, et al. PPARγ and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008;22(21):2941–2952. doi: 10.1101/gad.1709008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt D, et al. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science. 2010;328(5981):1036–1040. doi: 10.1126/science.1186176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hughes ME, et al. Harmonics of circadian gene transcription in mammals. PLoS Genet. 2009;5(4): doi: 10.1371/journal.pgen.1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Green CB, Takahashi JS, Bass J. The meter of metabolism. Cell. 2008;134(5):728–742. doi: 10.1016/j.cell.2008.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papadia FS, et al. Liver damage in severely obese patients: a clinical-biochemical-morphologic study on 1,000 liver biopsies. Obes Surg. 2004;14(7):952–958. doi: 10.1381/0960892041719644. [DOI] [PubMed] [Google Scholar]
- 33.Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. 2010;5(1):145–171. doi: 10.1146/annurev-pathol-121808-102132. [DOI] [PubMed] [Google Scholar]
- 34.Hooper AJ, Adams LA, Burnett JR. Genetic determinants of hepatic steatosis in man. J Lipid Res. 2011;52(4):593–617. doi: 10.1194/jlr.R008896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Romeo S, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40(12):1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kozlitina J, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46(4):352–356. doi: 10.1038/ng.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011;7(3): doi: 10.1371/journal.pgen.1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishizuka Y, et al. TRIB1 downregulates hepatic lipogenesis and glycogenesis via multiple molecular interactions. J Mol Endocrinol. 2014;52(2):145–158. doi: 10.1530/JME-13-0243. [DOI] [PubMed] [Google Scholar]
- 39.Nagiec MM, et al. Modulators of hepatic lipoprotein metabolism identified in a search for small-molecule inducers of tribbles pseudokinase 1 expression. PLoS One. 2015;10(3): doi: 10.1371/journal.pone.0120295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37(8):911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 41.Steger DJ, et al. Propagation of adipogenic signals through an epigenomic transition state. Genes Dev. 2010;24(10):1035–1044. doi: 10.1101/gad.1907110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shin H, Liu T, Manrai AK, Liu XS. CEAS: cis-regulatory element annotation system. Bioinformatics. 2009;25(19):2605–2606. doi: 10.1093/bioinformatics/btp479. [DOI] [PubMed] [Google Scholar]
- 44.McLean CY, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28(5):495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.