

Abstract
Background
Tobacco (Nicotiana tabacum L.) is an important model system, which has been widely used in plant physiological studies and it is particularly useful as a bioreactor. Despite its importance, only limited molecular marker resources are available for genome analysis, genetic mapping and breeding. Restriction-site associated DNA sequencing (RAD-seq) is a powerful new method for targeted sequencing across the genomes of many individuals. This approach has broad potential for genetic analysis through linkage mapping.
Results
We constructed a RAD library using genomic DNA from a BC1 backcross population. Sequencing of 196 individuals was performed on an Illumina HiSeq 2500. Two linkage maps were constructed, one with a reference genome and another, termed as de novo identification of single nucleotide polymorphism (SNP) by RAD-seq, without a reference genome. Overall, 4138 and 2162 SNP markers with a total length of 1944.74 and 2000.9 cM were mapped to 24 linkage groups in the genetic maps based on reference genome and without reference, respectively.
Conclusions
Using two different SNP discovery methods based on next generation RAD sequencing technology, we have respectively mapped 2162 and 4318 SNPs in our backcross population. This study gives an excellent example for high density linkage map construction, irrespective of genome sequence availability, and provides saturated information for downstream genetic investigations such as quantitative trait locus analyses or genomic selection (e.g. bioreactor suitable cultivars).
Electronic supplementary material
The online version of this article (doi:10.1186/s40709-015-0034-3) contains supplementary material, which is available to authorized users.
Keywords: Tobacco, Linkage map, Nicotiana tabacum L., RAD sequencing, SNP
Background
Tobacco (Nicotiana tabacum L., 2n = 4x = 48) is an important model system in plant biotechnology [1], due to its unique advantages over other plant species. It not only has relatively short generation time and high protein content, but also can be easily genetically transformed [2, 3]. For this reason, tobacco has been widely used in studies on plant response to pathogens [4], pyridine alkaloid (like nicotine) biosynthesis [5], cell cycle [6, 7], oxidative stress [8] and pollen tube development [9]. More importantly, tobacco is an attractive green bioreactor proved to be able to produce a wide range of therapeutic proteins including antibodies [10–12], vaccines [13, 14] and immunomodulatory molecules such as cytokines [15, 16].
Despite the prospective applications of tobacco in pharmaceutical production, limited cultivars exist with low nicotine and alkaloid contents. Breeding new cultivars suitable for pharmaceutical production is further complicated by the paltry genomic information available to the public. Genetic linkage mapping based on molecular markers permits the elucidation of genome structure and organization [17]. It provides critical information for quantitative trait locus (QTL) marker assisted selection. For some economic plants, including potato (Solanum tuberosum), tomato (Solanum lycopersicum), eggplant (Solanum melongena), pepper (Capsicum species) and Petunia (Petunia hybrida), whole genome sequencing and genetic linkage maps have elucidated their genome structures and assisted breeding cultivars with molecular markers [18]. Therefore, a high density genome-based linkage map of the tetraploid tobacco will improve current genetic research tools in search of new cultivars. Thus far, linkage maps for tobacco have been constructed by using low-throughput molecular markers like simple sequence repeats (SSRs), which resulted in low density linkage maps [19, 20].
Single nucleotide polymorphisms (SNPs) as the most abundant type of DNA variations are currently used as genetic markers for their wide distribution in the genome [21]. Compared to genetic markers based on size discrimination or hybridization, SNPs directly interrogate sequence variation and possess the potential of reducing genotyping errors [22]. SNP discovery is amenable to high-throughput next-generation sequencing (NGS) technologies, which produce DNA sequences at a rate several orders of magnitude faster than conventional sequencing methods [17].
According to unpublished data, the genome size of tobacco is approximately 4.5 Gb. Because of the huge genome, great challenges must be faced up to. Reduced representation library sequencing is an energetic approach, which has been used for many genome studies [23]. Restriction site associated DNA sequencing (RAD-seq) technology [24–26] facilitates genetic variant discovery by allowing ortholog sequences to be targeted in multiple individuals [27]. This method relies on sequencing of DNA regions flanking the restriction sites of specific restriction enzymes. In brief, DNA fragments from the digestion of a chosen restriction enzyme are ligated with an adapter, which contains a molecular identifying sequence (MID) unique to each sample. The DNA sequences flanking each restriction site are sequenced via the massively parallel Illumina sequencing technology [28]. RAD sequencing is highly successful in re-identifying genomic regions controlling known phenotypes [29–31].
To generate a high density genome linkage map for tobacco, we have developed here 4138 SNP markers using the Illumina HiSeq 2500 high-throughput platform. The mapping population was generated by crossing two tobacco (N. tabacum L.) cultivars. The F1 progeny was back-crossed to the parents. A total of 193 progenies were generated and all individuals were used for linkage map construction. We conducted SNP detection both with and without a reference genome, the latter referred to as de novo identification of SNP by RAD-seq (DISR). We compared these two methods and constructed a genetic map of tobacco based on a backcross (BC1) population.
Results
RAD library preparation and sequencing
A total of 196 sampled individuals from three generations, HD (Hong hua Da jin yuan), RBST (Resistance to Black Shank Tobacco), F1 (HD × RBST) and 193 BC1 progenies were used in the construction of 10 libraries used for RAD-sequencing (Table 1). In summary, 2641 Gb of raw data containing 26.4 billion pair-end 2 × 100 bp raw reads for approximately 2640 billion base pairs were obtained. Library detail information is provided in Additional file 1. We removed the following types of reads: (a) reads with >10 % unidentified nucleotides (N), (b) reads with >40 bases having Phred quality ≤7, and (c) putative PCR duplicates generated by PCR amplification in the library construction process (i.e., read 1 and read 2 of two paired-end reads that were completely identical). These reads were stringently filtered from the index sequences to get clean data for each sample (Fig. 1). Totally, 2481 Gb clean data contain 24.8 billion clean reads after filtering with an average volume of 12.11 Gb for each sample, at an average sequencing depth of 2.7× (the unpublished tobacco genome size is approximately 4.5 Gb).
Table 1.
Library information and data output
| Lib | Raw reads (M) | Raw bases (Gb) | Clean reads (M) | Clean bases (Gb) | GC rate (%) | Q20 rate (%) |
|---|---|---|---|---|---|---|
| 1 | 2251.27 | 225.13 | 2173.69 | 217.37 | 42.25 | 97.54 |
| 2 | 2612.37 | 261.24 | 2322.90 | 232.29 | 40.56 | 97.15 |
| 3 | 2622.27 | 262.23 | 2497.09 | 249.71 | 40.49 | 96.44 |
| 4 | 2456.33 | 245.63 | 1931.97 | 193.20 | 40.71 | 95.93 |
| 5 | 2765.69 | 276.57 | 2658.79 | 265.88 | 41.37 | 96.35 |
| 6 | 2750.33 | 275.03 | 2658.93 | 265.89 | 40.48 | 96.46 |
| 7 | 2410.54 | 241.05 | 2334.68 | 233.47 | 40.59 | 97.20 |
| 8 | 2867.82 | 286.78 | 2757.00 | 275.70 | 41.18 | 96.53 |
| 9 | 2912.57 | 291.26 | 2802.38 | 280.24 | 40.86 | 95.85 |
| 10 | 2761.11 | 276.11 | 2673.15 | 267.31 | 40.96 | 96.93 |
| Total | 26,410.31 | 2641.03 | 24,810.56 | 2481.06 | 40.95 | 96.64 |
Basic statistical information about the RAD sequencing is presented, which contain the raw reads, raw bases, clean reads and clean base after filtered, GC rate mean value and Q20 mean value
Fig. 1.
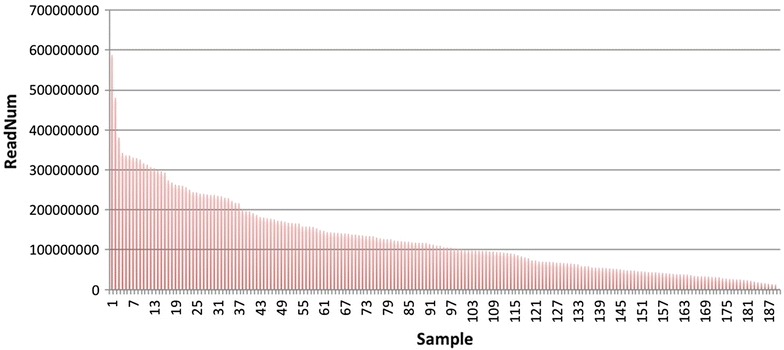
The statistic of read number for each sample
SNP calling and genotyping
Two distinct protocols were executed in SNP calling and genotyping: the first was with a reference genome; the second was without a reference genome, which we refer to as DISR. In the first protocol, 24.8 billion clean reads were aligned to the reference sequences (unpublished data) using SOAPaligner [32] (Release 2.21, http://soap.genomics.org.cn/). The mapping results were processed with Samtools [33]. Variations were called using the Unified Genotyper (Version 3.1, Genome Analysis Tool Kit) [34]. Any nucleotide difference between reads and the reference genome was initially called as variant. A large volume output of 7,343,419 raw SNPs suggested improvement in data assemblage. Three parameters (genotype coverage, genotype quality, and SNP quality) generated by the Unified Genotyper were used as criteria for filtering variant output.
Using a maximum missing data (MMD) threshold of 45 % in the BC1 population for each locus, a total of 8664 SNPs (p < 0.01) were recovered. Although the criteria are much looser than many other studies [31], the effective genotype size is larger than 100, which is sufficient for linkage map construction. In total, 5286 markers (χ2 < 15) were selected for genetic map construction by using JoinMap 4.0 [35] (Table 2).
Table 2.
Statistics for SNPs based on the two different methods
| Method of SNPcalling | Raw SNPs | Clean SNPs | χ 2 < 15 |
|---|---|---|---|
| DISRa | 181,770 | 7457 | 3282 |
| Based on reference genome | 7,343,419 | 8664 | 5286 |
Shown are the number of raw SNPs, the number of SNPs remained after filtering and the number of SNPs by a Chi square test
aDe novo identification of SNP by RAD-seq
In the second protocol (DISR), 181,770 raw SNPs were obtained after the clean reads were processed. Using the same MMD threshold as the first protocol, a total of 7457 SNPs (p < 0.01) were recovered. In total, 3282 markers were then selected (by the χ2 test) for the construction of genetic map in JoinMap 4.0 [35] (Table 2).
Linkage mapping
The first linkage map from sequence with reference genome was constructed with a total of 8664 SNPs (p < 0.01) which generated 4138 markers and mapped 24 linkage groups (LGs) successfully with a total length of 1944.74 cM. The LGs ranged from 33.58 to 129.176 cM in length. Six LGs contained over 220 marker loci. LG09, LG23 and LG24 were the shortest LGs, spanning 73.937–107.485 cM, respectively, and comprising 65 loci, whereas LG05 was the largest LG, spanning 60.73 cM, containing 494 loci with marker density of 0.123 cM/locus. The marker densities ranged from 0.117 cM/locus in LG12 to 1.679 cM/locus in LG23, resulting in an average distance of 0.712 cM between markers for the entire map (Table 3; Fig. 2).
Table 3.
Statistics of 24 linkage groups with the reference genome
| Linkage group | Marker no. | Size (cM) | Marker density (cM/loci) |
|---|---|---|---|
| LG01 | 190 | 67.024 | 0.355 |
| LG02 | 116 | 76.268 | 0.663 |
| LG03 | 280 | 33.580 | 0.120 |
| LG04 | 161 | 105.310 | 0.658 |
| LG05 | 494 | 60.780 | 0.123 |
| LG06 | 402 | 50.368 | 0.126 |
| LG07 | 206 | 52.656 | 0.257 |
| LG08 | 125 | 129.176 | 1.042 |
| LG09 | 65 | 73.937 | 1.155 |
| LG10 | 294 | 37.134 | 0.127 |
| LG11 | 279 | 37.254 | 0.134 |
| LG12 | 227 | 26.454 | 0.117 |
| LG13 | 199 | 115.937 | 0.586 |
| LG14 | 209 | 150.580 | 0.724 |
| LG15 | 115 | 106.013 | 0.930 |
| LG16 | 105 | 126.780 | 1.219 |
| LG17 | 103 | 47.001 | 0.461 |
| LG18 | 100 | 93.132 | 0.941 |
| LG19 | 97 | 90.641 | 0.944 |
| LG20 | 81 | 130.057 | 1.626 |
| LG21 | 81 | 74.543 | 0.932 |
| LG22 | 79 | 76.465 | 0.980 |
| LG23 | 65 | 107.485 | 1.679 |
| LG24 | 65 | 76.166 | 1.190 |
| Total | 4138 | 1944.741 | 0.712 |
Shown are the marker number, linkage size (cM), and marker density of each linkage group. LG5 contains a maximum marker number of 494. The LGs distance ranged from 33.58 to 129.176 cM. Six LGs contained over 220 marker loci. For these LGs Haldane’s map unit is used while for other LGs we used Kosambi’s map unit
Fig. 2.

Linkage maps based on the reference genome. This was constructed with a total of 8664 SNPs (p < 0.01) which generated 4138 markers mapping 24 linkage groups (LGs) successfully with a total length of 1944.74 cM. The LGs distance ranged from 33.58 to 129.176 cM. Six LGs contained over 220 marker loci and for these LGs Haldane’s map unit is used while for other LGs we used Kosambi’s map unit. The LG09, LG23 and LG24 were the shortest LGs, spanning 73.937–107.485 cM, respectively, and comprising 65 loci, whereas LG05 was the longest LG, spanning 60.73 cM and containing 494 loci with a marker density of 0.123 cM/locus. The marker densities ranged from 0.117 cM/locus in LG12 to 1.679 cM/locus in LG23, resulting in an average distance of 0.712 cM between markers for the entire map
The second linkage map from DISR was constructed with 7457 SNPs that gave 3282 markers. Out of those, 2162 markers successfully mapped 24 LGs with a total length of 2700.9 cM. The LGs ranged from 58.1 to 238.4 cM in length, and only one LG contained over 220 marker loci. LG24 was the shortest LG, comprising only 13 loci, whereas LG01 was the largest LG, spanning 159.9 cM and containing 224 loci with marker density of 0.7 cM/locus. The marker densities ranged from 0.5 cM/locus in LG02 to 5.6 cM/locus in LG24, resulting in an average distance of 1.8 cM between markers for the entire map (Table 4; Fig. 3).
Table 4.
Statistics of 24 linkage groups without the reference genome (DISR)
| Linkage group | Marker no. | Size (cM) | Marker density (cM/loci) |
|---|---|---|---|
| LG01 | 224 | 159.9 | 0.7 |
| LG02 | 186 | 92.5 | 0.5 |
| LG03 | 160 | 181.8 | 1.1 |
| LG04 | 159 | 129.6 | 0.8 |
| LG05 | 132 | 97.2 | 0.7 |
| LG06 | 150 | 147.4 | 1.0 |
| LG07 | 123 | 138.8 | 1.1 |
| LG08 | 136 | 129.5 | 1.0 |
| LG09 | 89 | 75.9 | 0.9 |
| LG10 | 87 | 142.7 | 1.7 |
| LG11 | 84 | 116.1 | 1.4 |
| LG12 | 71 | 75.9 | 1.1 |
| LG13 | 57 | 70.9 | 1.3 |
| LG14 | 54 | 58.1 | 1.1 |
| LG15 | 120 | 238.4 | 2.0 |
| LG16 | 45 | 117.7 | 2.7 |
| LG17 | 64 | 127.2 | 2.0 |
| LG18 | 43 | 98.7 | 2.4 |
| LG19 | 41 | 74.8 | 1.9 |
| LG20 | 32 | 78.6 | 2.5 |
| LG21 | 30 | 93.3 | 3.2 |
| LG22 | 23 | 72.0 | 3.3 |
| LG23 | 39 | 116.9 | 3.1 |
| LG24 | 13 | 67.0 | 5.6 |
| Total | 2162 | 2700.9 | 1.8 |
Shown are the marker number, linkage size (cM), and marker density of each linkage group. LG24 was the shortest LG, comprising only 13 loci, whereas LG01 was the longest, spanning 159.9 cM and containing 224 loci with a marker density of 0.7 cM/locus (map unit determined by Haldane’s distance while for other LGs Kosambi’s was used)
Fig. 3.

Linkage maps based on DISR. This map was constructed with 7457 SNPs that produced 3282 markers. Out of those, 2162 markers successfully mapped 24 LGs with a total length of 2700.9 cM. The LGs ranged from 58.1 to 238.4 cM in length. LG24 was the shortest LG, comprising only 13 loci, whereas LG01 was the longest, spanning 159.9 cM and containing 224 loci with a marker density of 0.7 cM/locus (map unit determined by Haldane’s distance while for other LGs Kosambi’s distance was used). The marker densities ranged from 0.5 cM/locus in LG02 to 5.6 cM/locus in LG24, resulting in an average distance of 1.8 cM between markers for the entire map
Comparison of the DISR and the reference genome methods
Comparison was performed by presenting the ratio of the marker overlaps between the genetic maps based on reference genome and DISR. The consensus sequence was mapped back to the reference genome to mark the loci of the SNPs. After this process, the markers from the DISR method were compared with the markers generated from the reference genome method. Consistent markers were recorded and presented as a Venn diagram. In total, 677 overlapping markers, constituting 30 % of the DISR map and 16 % of the map based on reference genome were observed. All in all, 1535 makers were specified for the DISR map and 3461 markers for the map based on reference genome (Fig. 4).
Fig. 4.
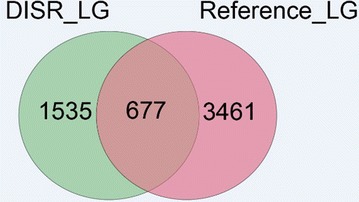
Comparison of the two map versions. In total, 677 overlapping markers, constituting 30 % of the DISR map and 16 % of the map based on the reference genome were observed. All in all, 1535 makers were specified for the DISR map and 3461 markers for the map based on the reference genome
Discussion
Although tobacco has been proved to be an attractive green bioreactor for the production of therapeutic proteins, the paucity of cultivars with low nicotine and alkaloid contents has blocked its movement from bench to field scale. A high density genetic map can provide sufficient information to accelerate the genome breeding. Previous attempts for genetic linkage map construction for tobacco were achieved by using molecular marker based techniques, including restriction fragment length polymorphism (RFLP) [36], conserved ortholog sequences (COS) [37] and simple sequence repeat (SSR) markers [19, 20]. As the best of the three linkage maps, the SSR linkage map comprises 2318 SSR markers mapping to 2363 loci in 24 clearly defined LGs with a total length of 3270 cM [19] (Table 5). In comparison, our technique generated 4138 SNP markers for tobacco that defined 24 LGs with a total coverage of 1944.7 cM. This result is not only an improvement over those of previous reports, but also a confirmation of SNPs in providing excellent marker density for linkage mapping and genomic selection [38]. To our knowledge, the tobacco linkage maps from this study, particularly the map generated with a reference genome, provide the highest number of markers among all available population-specific linkage maps.
Table 5.
Comparison of linkage maps for tobacco
| Cross combination | Population type | Type of markers | No. of markers | Map length (cM) | Groups | References |
|---|---|---|---|---|---|---|
| Hicks Broadleaf × Red Russian | F2 | SSRa | 286 | 1920 | 26 | [20] |
| Flue-cured tobacco Taiyan 7 × burley tobacco cultivar Bailei 21 | F2 | SRAPb/ISSRc | 112 | 1560.2 | 26 | [47] |
| Burley37 × Burley21 | DHd | SRAP/AFLPe | 118 | 1953.6 | 22 | [48] |
| Hicks Broadleaf × Red Russian | F2 | SSR | 2317 | 3270 | 24 | [19] |
| Hicks Broadleaf × HHDJY(HD) | DH | SSR/DArTf | 851 | 2291 | 24 | [49] |
| Honghua Dajinyuan (HD) × HBL | DH | SSR | 611 | 1882.1 | 24 | [50] |
| (HD × RBST) × HD | BC1 | SNP | 4138 | 1944.7 | 24 | This study |
| (HD × RBST) × HD | BC1 | SNP | 2162 | 2700.9 | 24 | This study |
aSimple sequence repeats
bSequence related amplified polymorphism
cInter-simple sequence repeat
dDoubled haploid population
eAmplified fragment length polymorphism
fDiversity arrays technology
The Mendelian basis of quantitative traits provides a genetic framework for the dissection of polygenic traits [39] and can pave the way for the identification of candidate loci controlling the inheritance of complex traits. NGS technology makes it possible to achieve dense SNP marker coverage of genomes without the need for a reference sequence [24, 26]. An example of this is restriction-associated DNA sequencing (RAD-seq), which was originally developed as a tool for genetic mapping in fish and fungi [29] and later expanded to many other species, including plants (Lolium perenne L., Momordica charantia, Corchorus olitorius L.) [25, 30, 40, 41]. In this study, a separate linkage map via the DISR method was also obtained, which did not need a reference genome. The DISR linkage map contains 2162 markers with a total coverage of 2700.9 cM and an average distance of 1.8 cM between markers. It demonstrates that these two high density linkage maps are compelling tools for gene (Table 5) and QTL mapping and marker-assisted breeding [42].
A comparison of the two maps showed an overlap of 677 markers (Fig. 4). We compared the ratios of overlaps between the two protocols and found that the use of a reference genome was more efficient than without a reference genome. In the method of DISR, the information of only one end of the pair reads is used for the SNP calling. However, if we conduct the SNP calling with a reference genome, whole genome information is used. This kind of experiment is often required in nature, particularly in building linkage maps for species that do not have a complete genome sequence database. However, an integration of the two protocols could result in a higher density map and thus, assist in the breeding of other low nicotine and alkaloid content cultivars.
Conclusions
Using next generation RAD sequencing technology for two distinct SNP discovery methods, we have respectively mapped 2162 and 4318 SNPs in tobacco. This study gives an excellent example for high density linkage map construction, irrespective of reference genome sequence availability, and provides saturated information for downstream genetic investigations such as QTL analyses or genomic selection (e.g. bioreactor suitable cultivars).
Methods
Mapping population
Two tobacco varieties, Hong hua Da jin yuan (HD) and Resistance to Black Shank Tobacco (RBST) were used to develop the BC1 inbred population. HD is a high leaf mass cultivar from southwest of China. RBST has high resistance to tobacco black shank disease. The BC1 inbred population was generated through a (HD × RBST) × HD crossing in a breeding unit in Yuxi of Yunnan Province.
RAD library preparation and sequencing
Fresh young leaves were collected from HD, RBST, F1 (HD × RBST) and 193 individuals of BC1 (F1 × HD) population. Leaf samples were snap frozen in liquid nitrogen and stored at −80 °C. Genomic DNA isolation and purification were conducted using a DNA extraction kit (Qiagen). DNA quality was analyzed in 1 % agarose gel. The concentration of extracted DNA was determined by a spectrophotometer. Approximately 15 μg of purified DNA was processed to obtain 10 RAD libraries, each including about 20 individuals following the protocol of Baird et al. [29] and the instructions of the reagent manufacturers. Genomic DNA from individual samples was digested with EcoRI (New England Biolabs). Individual specific barcodes were ligated with an adaptor by T4 DNA ligase for sample multiplexing. Ligated DNA samples were pooled and sheared, and consequently electrophoresed to isolate DNA fragments with sizes of 300–700 bp in 1.5 % agarose gel. Quick Blunting Kit (New England Biolabs) was used to generate phosphorylated blunt ends. Klenow Fragment (3′ → 5′ exo-; New England Biolabs) was used to add adenosine to the 3′ end. An adapter with divergent ends (P2 adapter) was ligated to enable selective PCR. The samples were PCR-amplified and the libraries purified with MinElute column (Qiagen) to obtain approximately 100 μl (>50 ng μl−1) of sequencing libraries. The obtained RAD libraries were sequenced on an Illumina HiSeq 2500 in 100 bp pair-end reads.
SNP calling with reference genome
The raw reads were removed using the following criteria: (a) reads with >10 % unidentified nucleotides (N), (b) reads with >40 bases having Phred quality ≤7, and (c) putative PCR duplicates generated by PCR amplification in the library construction process (i.e., read 1 and read 2 of two paired-end reads that were completely identical). All the obtained short clean reads were aligned to reference sequences (unpublished data) using SOAPaligner (Release 2.21, http://soap.genomics.org.cn/) [32]. During alignment, long reads with high error rates at 3′-ends were substituted with 5′ 32 bp subsequence as seeds. The entire lengths of the reads were used. Five mismatches in one read were allowed (important arguments: -l 32 -v 5). The mapping results SAM files were converted with Samtools [33]. Variations were called using the Unified Genotyper (Version 3.1, Genome Analysis Tool Kit) [34]. Any nucleotide difference between reads and the reference genome was identified as a variant. This criterion generated a large variant output, which was filtered by three parameters generated with the Unified Genotyper, including genotype coverage, genotype quality, and SNP quality.
SNP calling without reference genome (DISR)
Besides, the method based on reference, we have attempted to call SNPs by DISR. Instead, we used a multistep process to identify RAD tag loci within populations, assign a consensus sequence to each individual at each RAD tag locus, and align consensus sequences across populations (Fig. 5). A flowchart is also provided for clarity in Additional file 2.
Fig. 5.
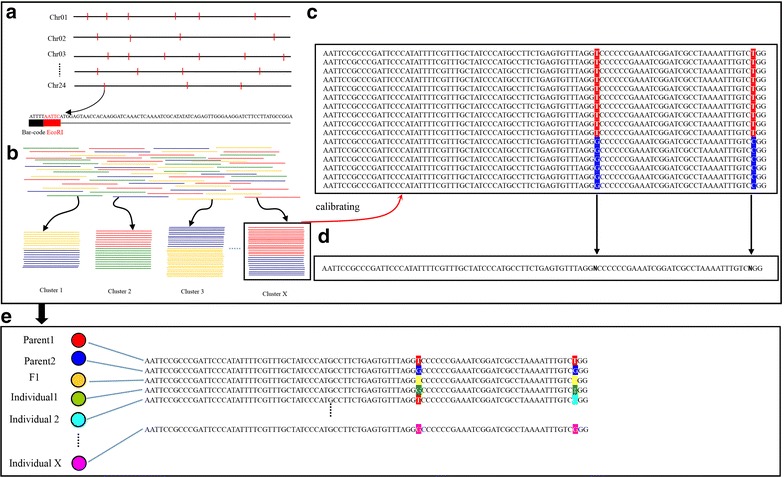
SNP calling based on DISR. a Nicotiana tabacum L. has 24 nuclear chromosomes, each of which contains multiple EcoRI cut sites (red marks). The genomic DNA is digested, bar coded with a population-specific sequence, and amplified resulting in multiple sequence reads from each of the RAD tag sites in the genome. Each sequence consists of a population-specific 5-bp barcode (black), the enzyme-recognition sequence (red), and the downstream sequence. b The de novo RAD tag pipeline compares all the sequenced reads and builds clusters of exactly matching tags. c Pair wise comparisons are made between all clusters. d There is a cluster in the locus that is SNP. e The consensus sequence for that RAD tag site within the population
Within each individual, identical reads were aligned together into clusters (other study termed it as stacks) (Fig. 5b–d). The pairwise sequence divergence among clusters was used to group them into putative loci (Fig. 5e). Loci were defined as a set of clusters such that for each cluster there is another cluster in the locus that is at most one nucleotide divergent. Clusters containing excessive numbers of sequence reads can occur when multiple, repetitive sites in the genome are all within a single nucleotide of one another. For this analysis, all clusters with a depth of coverage greater than two standard deviations above the mean cluster depth were removed and the remaining clusters were merged into a locus. For each nucleotide site in a locus, a likelihood ratio test of the read counts of alternative nucleotides was used to test whether the allele frequency of the most observed nucleotide was significantly larger than a threshold p following the method of Emerson et al. [43]. After these processes, an in-house perl script was used to integrate the clusters of parents and F1 progeny into a catalog and create a set of all possible loci in a mapping cross. Then, clusters of BC1 progenies are matched against the catalog to determine the genotype at each locus in every individual in the cross population.
Genotyping and linkage mapping
Distorted markers (p < 0.01) were filtered off to construct a genetic map by a Chi square test (χ2 < 15 was selected for JoinMap 4.0) [35]. LGs were identified with an independent logarithm of odds (LOD) threshold of 7. Due to the large number of markers segregating in the population, if the number of the linkage group is more than 220, we used (in JoinMap 4.0) a maximum likelihood algorithm mapping the marker order for calculation efficiency [44]. We also calculated genetic distances (cM) using Haldane’s mapping function. However, the scope of corresponding linkage groups (3000–6000 cM) exceeded JoinMap 4.0 and therefore, the linkage length was divided by 100 for map presentation. In other linkage groups whose maker number was equal or less than 220, a linear regression algorithm and Kosambi’s mapping function was used for map construction and genetic distance estimation [45]. Following the initial mapping, potential errors that appeared as doubtful double-recombinants were identified using genotype probabilities function of JoinMap 4.0 [35] (p < 0.001). The suspicious genotype was replaced by a missing value as suggested by Isidore et al. [46] and Van Ooijen [35]. A linkage map was then constructed afresh using the corrected dataset. Potential error elimination and linkage map construction was iterated until no dubious genotype was identified. Markers with >45 % missing values or distorted (χ2 test, p < 0.001, d.f. = 2) were removed in each step of the iteration.
Authors’ contributions
BX: constructed map population, extracted tobacco DNA, performed sequencing and wrote part of the text. YT: performed most of the bioinformatic analysis and wrote part of the text. NL: performed data preprocess. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by Grants from CNTC [110201201003 (JY-03), 110201301006 (JY-06)], and YNTC (2012YN01, 2013YN01).
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interests.
Additional files
Additional file 1. Library detail information.
Additional file 2. A flowchart for bioinformatic analysis procedure in this study.
Footnotes
Bingguang Xiao and Yuntao Tan contributed equally to this work
Contributor Information
Bingguang Xiao, Email: xiaobg@263.net.
Yuntao Tan, Email: tanyt@dongyang-lab.org.
Ni Long, Email: longni@dongyang-lab.org.
Xuejun Chen, Email: cxjkm@163.com.
Zhijun Tong, Email: tzj861@163.com.
Yang Dong, Email: dongyang@dongyang-lab.org.
Yongping Li, Email: liyp_yatas@163.com.
References
- 1.Olmstead RG, Bohs L, Migid HA, Santiago-Valentin E, Garcia VF, Collier SM. A molecular phylogeny of the Solanaceae. Taxon. 2008;57:1159–1181. [Google Scholar]
- 2.Komori T, Imayama T, Kato N, Ishida Y, Ueki J, Komari T. Current status of binary vectors and superbinary vectors. Plant Physiol. 2007;145:1155–1160. doi: 10.1104/pp.107.105734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sparkes IA, Runions J, Kearns A, Hawes C. Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat Protoc. 2006;1:2019–2025. doi: 10.1038/nprot.2006.286. [DOI] [PubMed] [Google Scholar]
- 4.Romeis T. Protein kinases in the plant defence response. Curr Opin Plant Biol. 2001;4:407–414. doi: 10.1016/S1369-5266(00)00193-X. [DOI] [PubMed] [Google Scholar]
- 5.Häkkinen ST, Tilleman S, Swiatek A, De Sutter V, Rischer H, Vanhoutte I, et al. Functional characterisation of genes involved in pyridine alkaloid biosynthesis in tobacco. Phytochemistry. 2007;68:2773–2785. doi: 10.1016/j.phytochem.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 6.Mironov V, Veylder LD, Montagu MV, Inzéa D. Cyclin-dependent kinases and cell division in plants—the nexus. Plant Cell. 1999;11:509–522. doi: 10.1105/tpc.11.4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakagami H, Sekine M, Murakami H, Shinmyo A. Tobacco retinoblastoma-related protein phosphorylated by a distinct cyclin-dependent kinase complex with Cdc2/cyclin D in vitro. Plant J. 1999;18:243–252. doi: 10.1046/j.1365-313X.1999.00449.x. [DOI] [PubMed] [Google Scholar]
- 8.Langebartels C, Kerner K, Leonardi S, Schraudner M, Trost M, Heller W, et al. Biochemical plant responses to ozone: I. Differential induction of polyamine and ethylene biosynthesis in tobacco. Plant Physiol. 1991;95:882–889. doi: 10.1104/pp.95.3.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor LP, Hepler PK. Pollen germination and tube growth. Annu Rev Plant Physiol Plant Mol Biol. 1997;48:461–491. doi: 10.1146/annurev.arplant.48.1.461. [DOI] [PubMed] [Google Scholar]
- 10.Tremblay R, Wang D, Jevnikar AM, Ma S. Tobacco, a highly efficient green bioreactor for production of therapeutic proteins. Biotechnol Adv. 2010;28:214–221. doi: 10.1016/j.biotechadv.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villani ME, Morgun B, Brunetti P, Marusic C, Lombardi R, Pisoni I, et al. Plant pharming of a full-sized, tumour-targeting antibody using different expression strategies. Plant Biotechnol J. 2009;7:59–72. doi: 10.1111/j.1467-7652.2008.00371.x. [DOI] [PubMed] [Google Scholar]
- 12.Sack M, Paetz A, Kunert R, Bomble M, Hesse F, Stiegler G, et al. Functional analysis of the broadly neutralizing human anti-HIV-1 antibody 2F5 produced in transgenic BY-2 suspension cultures. FASEB J. 2007;21:1655–1664. doi: 10.1096/fj.06-5863com. [DOI] [PubMed] [Google Scholar]
- 13.Brandsma M, Wang X, Diao H, Kohalmi SE, Jevnikar AM, Ma S. A proficient approach to the production of therapeutic glucagon-like peptide-1 (GLP-1) in transgenic plants. Open Biotechnol J. 2009;3:9. doi: 10.2174/1874070700903010057. [DOI] [Google Scholar]
- 14.Burtin D, Chabre H, Olagnier D, Didierlaurent A, Couret MN, Comeau D, et al. Production of native and modified recombinant Der p 1 molecules in tobacco plants. Clin Exp Allergy. 2009;39:10. doi: 10.1111/j.1365-2222.2009.03201.x. [DOI] [PubMed] [Google Scholar]
- 15.Menassa R, Du C, Yin Z, Ma S, Poussier P, Brandle J, et al. Therapeutic effectiveness of orally administered transgenic low-alkaloid tobacco expressing human interleukin-10 in a mouse model of colitis. Plant Biotechnol J. 2007;5:50–59. doi: 10.1111/j.1467-7652.2006.00214.x. [DOI] [PubMed] [Google Scholar]
- 16.Wang D, Brandsma M, Yin Z, Wang A, Jevnikar A, Ma S. A novel platform for biologically active recombinant human interleukin-13 production. Plant Biotechnol J. 2008;6:11. doi: 10.1111/j.1467-7652.2008.00337.x. [DOI] [PubMed] [Google Scholar]
- 17.Zhou X, Xia Y, Ren X, Chen Y, Huang L, Huang S, et al. Construction of a SNP-based genetic linkage map in cultivated peanut based on large scale marker development using next-generation double-digest restriction-site-associated DNA sequencing (ddRADseq) BMC Genom. 2014;15:351. doi: 10.1186/1471-2164-15-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foolad MR, Panthee DR. Marker-assisted selection in tomato breeding. Crit Rev Plant Sci. 2012;31:93–123. doi: 10.1080/07352689.2011.616057. [DOI] [Google Scholar]
- 19.Bindler G, Plieske J, Bakaher N, Gunduz I, Ivanov N, Van der Hoeven R, et al. A high density genetic map of tobacco (Nicotiana tabacum L.) obtained from large scale microsatellite marker development. Theor Appl Genet. 2011;123:219–230. doi: 10.1007/s00122-011-1578-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bindler G, van der Hoeven R, Gunduz I, Plieske J, Ganal M, Rossi L, et al. A microsatellite marker based linkage map of tobacco. Theor Appl Genet. 2006;114:341–349. doi: 10.1007/s00122-006-0437-5. [DOI] [PubMed] [Google Scholar]
- 21.Brookes AJ. The essence of SNPs. Gene. 1999;234:177–186. doi: 10.1016/S0378-1119(99)00219-X. [DOI] [PubMed] [Google Scholar]
- 22.Oliver RE, Lazo GR, Lutz JD, Rubenfield MJ, Tinker NA, Anderson JM, et al. Model SNP development for complex genomes based on hexaploid oat using high-throughput 454 sequencing technology. BMC Genom. 2011;12:77. doi: 10.1186/1471-2164-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young AL, Abaan HO, Zerbino D, Mullikin JC, Birney E, Margulies EH. A new strategy for genome assembly using short sequence reads and reduced representation libraries. Genome Res. 2010;20:249–256. doi: 10.1101/gr.097956.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barchi L, Lanteri S, Portis E, Acquadro A, Valè G, Toppino L, et al. Identification of SNP and SSR markers in eggplant using RAD tag sequencing. BMC Genom. 2011;12:304. doi: 10.1186/1471-2164-12-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hegarty M, Yadav R, Lee M, Armstead I, Sanderson R, Scollan N, et al. Genotyping by RAD sequencing enables mapping of fatty acid composition traits in perennial ryegrass (Lolium perenne(L.)) Plant Biotechnol J. 2013;11:572–581. doi: 10.1111/pbi.12045. [DOI] [PubMed] [Google Scholar]
- 26.Bonaventure G, Barchi L, Lanteri S, Portis E, Valè G, Volante A, et al. A RAD tag derived marker based eggplant linkage map and the location of QTLs determining anthocyanin pigmentation. PLoS One. 2012;7:e43740. doi: 10.1371/journal.pone.0043740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baxter SW, Davey JW, Johnston JS, Shelton AM, Heckel DG, Jiggins CD, et al. Linkage mapping and comparative genomics using next-generation RAD sequencing of a non-model organism. PLoS One. 2011;6:e19315. doi: 10.1371/journal.pone.0019315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3:e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfender WF, Saha MC, Johnson EA, Slabaugh MB. Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne. Theor Appl Genet. 2011;122:1467–1480. doi: 10.1007/s00122-011-1546-3. [DOI] [PubMed] [Google Scholar]
- 31.Chutimanitsakun Y, Nipper RW, Cuesta-Marcos A, Cistué L, Filichkina T, Johnson EA, et al. Construction and application for QTL analysis of a restriction site associated DNA (RAD) linkage map in barley. BMC Genom. 2011;12:4. doi: 10.1186/1471-2164-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 33.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Ooijen JW. JoinMap ® 4, Software for the calculation of genetic linkage maps in experimental populations. Wageningen: Kyazma BV; 2006.
- 36.Fulton TM, Van der Hoeven R, Eannetta NT, Tanksley SD. Identification, analysis, and utilization of conserved ortholog set markers for comparative genomics in higher plants. Plant Cell. 2002;14:1457–1467. doi: 10.1105/tpc.010479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu F, Mueller LA, Crouzillat D, Pétiard V, Tanksley SD. Combining bioinformatics and phylogenetics to identify large sets of single-copy orthologous genes (COSII) for comparative, evolutionary and systematic studies: a test case in the euasterid plant clade. Genetics. 2006;174:1407–1420. doi: 10.1534/genetics.106.062455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poland J, Endelman J, Dawson J, Rutkoski J, Wu S, Manes Y, et al. Genomic selection in wheat breeding using genotyping-by-sequencing. Plant Gen. 2012;5:103–113. doi: 10.3835/plantgenome2012.06.0006. [DOI] [Google Scholar]
- 39.Jansen RC. Complex plant traits: time for polygenic analysis. Trends Plant Sci. 1996;1:89–94. doi: 10.1016/S1360-1385(96)80040-9. [DOI] [Google Scholar]
- 40.Matsumura H, Miyagi N, Taniai N, Fukushima M, Tarora K, Shudo A, et al. Mapping of the gynoecy in bitter gourd (Momordica charantia) using RAD-seq analysis. PLoS One. 2014;9:e87138. doi: 10.1371/journal.pone.0087138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kundu A, Chakraborty A, Mandal NA, Das D, Karmakar PG, Singh NK, et al. A restriction-site-associated DNA (RAD) linkage map, comparative genomics and identification of QTL for histological fibre content coincident with those for retted bast fibre yield and its major components in jute (Corchorus olitorius L., Malvaceae s. l.) Mol Breed. 2015;35:19. doi: 10.1007/s11032-015-0249-x. [DOI] [Google Scholar]
- 42.Xu Y, Crouch JH. Marker-assisted selection in plant breeding: from publications to practice. Crop Sci. 2008;48:391. doi: 10.2135/cropsci2007.04.0191. [DOI] [Google Scholar]
- 43.Emerson KJ, Merz CR, Catchen JM, Hohenlohe PA, Cresko WA, Bradshaw WE, et al. Resolving postglacial phylogeography using high-throughput sequencing. Proc Natl Acad Sci USA. 2010;107:16196–16200. doi: 10.1073/pnas.1006538107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haldane J. The combination of linkage values and the calculation of distances between the loci of linked factors. J Genet. 1919;8:299–309. doi: 10.1007/BF02983270. [DOI] [Google Scholar]
- 45.Kosambi DD. The estimation of map distances from recombination values. Ann Eugen. 1943;12:172–175. doi: 10.1111/j.1469-1809.1943.tb02321.x. [DOI] [Google Scholar]
- 46.Isidore E, van Os H, Andrzejewski S, Bakker J, Barrena I, Bryan GJ, et al. Toward a marker-dense meiotic map of the potato genome: lessons from linkage group I. Genetics. 2003;165:2107–2116. doi: 10.1093/genetics/165.4.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hong-Bo M, Jian-Min Q, Yan-Kun L, Jing-Xia L, Tao W, Tao L, et al. Construction of a molecular genetic linkage map of tobacco based on SRAP and ISSR markers. Acta Agron Sin. 2008;34:1958–1963. [Google Scholar]
- 48.Cai C-C, Chai L-G, Wang Y, Xu F-S, Zhang J-J, Lin G-P. Construction of genetic linkage map of burley tobacco (Nicotiana tabacum L.) and genetic dissection of partial traits. Acta Agron Sin. 2009;35:1646–1654. doi: 10.3724/SP.J.1006.2009.01646. [DOI] [Google Scholar]
- 49.Lu X, Gui Y, Xiao B, Li Y, Tong Z, Liu Y, et al. Development of DArT markers for a linkage map of flue-cured tobacco. Chin Sci Bull. 2012;58:641–648. doi: 10.1007/s11434-012-5453-z. [DOI] [Google Scholar]
- 50.Tong Z-J, Jiao F-C, Wu X-F, Wang F-Q, Chen X-J, Li X-Y, et al. Mapping of quantitative trait loci underlying six agronomic traits in flue-cured tobacco. Acta Agron Sin. 2013;38:1407–1415. doi: 10.3724/SP.J.1006.2012.01407. [DOI] [Google Scholar]