

Abstract
Myeloproliferative neoplasms (MPN) are chronic myeloid cancers thought to arise at the level of CD34+ hematopoietic stem/progenitor cells. They include essential thrombocythemia (ET), polycythemia vera (PV) and primary myelofibrosis (PMF). All can progress to acute leukemia, but PMF carries the worst prognosis. Increasing evidences indicate that deregulation of microRNAs (miRNAs) might plays an important role in hematologic malignancies, including MPN. To attain deeper knowledge of short RNAs (sRNAs) expression pattern in CD34+ cells and of their possible role in mediating post-transcriptional regulation in PMF, we sequenced with Illumina HiSeq2000 technology CD34+ cells from healthy subjects and PMF patients. We detected the expression of 784 known miRNAs, with a prevalence of miRNA up-regulation in PMF samples, and discovered 34 new miRNAs and 99 new miRNA-offset RNAs (moRNAs), in CD34+ cells. Thirty-seven small RNAs were differentially expressed in PMF patients compared with healthy subjects, according to microRNA sequencing data. Five miRNAs (miR-10b-5p, miR-19b-3p, miR-29a-3p, miR-379-5p, and miR-543) were deregulated also in PMF granulocytes. Moreover, 3’-moR-128-2 resulted consistently downregulated in PMF according to RNA-seq and qRT-PCR data both in CD34+ cells and granulocytes. Target predictions of these validated small RNAs de-regulated in PMF and functional enrichment analyses highlighted many interesting pathways involved in tumor development and progression, such as signaling by FGFR and DAP12 and Oncogene Induced Senescence. As a whole, data obtained in this study deepened the knowledge of miRNAs and moRNAs altered expression in PMF CD34+ cells and allowed to identify and validate a specific small RNA profile that distinguishes PMF granulocytes from those of normal subjects. We thus provided new information regarding the possible role of miRNAs and, specifically, of new moRNAs in this disease.
Background
Philadelphia-negative chronic myeloproliferative neoplasms (MPNs) are a heterogeneous group of clonal hematopoietic stem cell (HSC) disorders associated with overproduction of mature myeloid cells[1,2].
MPNs are a group of chronic myeloid cancers that include essential thrombocythemia (ET), polycythemia vera (PV) and primary myelofibrosis (PMF), associated with an increased risk of thrombosis and/or hemorrhage and a propensity to develop acute myeloid leukemia.
Myelofibrosis, the most symptomatic and associated with the worst survival, can arise primarily or as a progression of PV or ET (post-PV/ET MF). In primary myelofibrosis (PMF) the increased proliferation of megakaryocytes is accompanied by deposition of fibrosis in the bone marrow, abnormal stem cell trafficking, and extramedullary hematopoiesis (myeloid metaplasia).[1,2]
In 2005, a new somatic mutation in the Janus Kinase 2 (JAK2)[3–7] has been discovered, providing the first genetic insight into the MPN pathogenesis. The JAK2V617F mutation is present in approximately 95% of patients with PV, and in 50% to 60% of those with ET or PMF. Subsequently, other mutations have been discovered, including mutation in MPL gene or in epigenetic regulators.[2,8–10]
In 2013, somatic mutations of CALR, the gene encoding calreticulin, have been found in 20% to 25% of patients with essential thrombocythemia (ET) or PMF[11,12]. Like JAK2 and MPL mutations, somatic mutations of CALR behave as driver mutations responsible for the myeloproliferative phenotype.
Despite the fact that the mutational background of MPNs has been extensively investigated, the molecular etiology of the disease has not been fully elucidated. Indeed several lines of evidence indicate that the identified mutations are not sufficient for disease initiation and progression. Although murine models have provided unequivocal evidence that JAK2 V617F is able to cause MPNs[13], disease phenotype is significantly heterogeneous between different murine lines and even within the same line, suggesting that disease phenotype is affected by other unknown genetic or epigenetic factors[14].
MicroRNAs are endogenous small non-coding RNAs, approximately 22 nt in length, crucial for post-transcriptional gene regulation. They are loaded into the RNA-induced silencing complex (RISC), directing the complex (including Argonaute proteins) to downregulate target mRNA expression by either triggering mRNA degradation or translational repression.[15] Recent studies show that mRNA destabilization explains most (66%–>90%) miRNA-mediated repression[16].
It is known that deregulation of miRNAs plays an important role in both solid and hematologic malignancies[17,18]. Indeed, hematopoietic differentiation is tightly governed by gene expression that is strictly regulated at multiple cell-fate decision levels. miRNAs regulate hematopoiesis acting both in HSC and in committed progenitor cells[18–20]. At the stem cell level, some miRNAs evolutionally conserved are responsible for expanding HSCs by inhibiting apoptosis[21–23]. At the progenitor cell level, miRNAs regulate the developmental fate of the megakaryocyte-erythroid progenitor (MEP) cell, the common progenitor of the erythroid and megakaryocytic lineages[24,25]. At the more committed hematopoietic cell level, specific miRNAs are expressed in different blood cell lineages and in different stages of hematopoietic differentiation. For example Chen et al.[26] reported that miR-142s expression was lower in the erythroid and T-lymphoid lineages and higher in B-lymphoid and myeloid lineages, while miR-223 expression was confined to myeloid lineages, with a very low detectable expression in T- and B-lymphoid and erythroid lineages.
miRNAs have an important role in regulation of hematopoiesis[27–30]. miR-16, miR-451 upregulation and miR-150, miR-155, miR-221 and miR-222 downregulation are associated with different stages of erythropoiesis[31,32]. miR-223 expression level, determined by two regulatory regions on its gene, fine-tunes lineage commitment of myeloid precursor[33]. miR-181 family was detected during granulocytic and macrophage-like differentiation and its level decreases along the hematopoietic lineage. It modulates differentiation by targeting and negatively regulating PRKCD mRNA, an upstream regulator of a pathway of the myeloid differentiation, and CAMKK1 mRNA, involved in the granulocytic and PMA-induced macrophage-like differentiation[34,35].
Recent studies highlighted aberrant miRNA expression in MPNs, and specific miRNA signatures that distinguish MPN granulocytes from those of healthy donors[18,36]. We also previously demonstrated that abnormal expression of miR-16-2 in vitro and in vivo contributes to the expansion of erythroid lineage in polycythemia vera.[20]
High-throughput array-based analysis of miRNA expression levels in MPN CD34+ cells were previously reported only by Lin et al.[35,37] A recent study characterized both gene and microRNA (miRNA) expression profiles in CD34+ cells from PMF patients[38]. It identified several biomarkers and putative molecular targets such as FGR, LCN2, and OLFM4. By means of miRNA-gene expression integrative analysis, the study suggested that JARID2 downregulation, mediated by miR-155-5p overexpression, might contribute to MK hyperplasia in PMF.
In a preliminary study, we performed short RNA massive sequencing and extensive bioinformatic analysis in the JAK2V617F-mutated SET2 cell line[39], detected and quantified 652 known mature miRNAs, of which 21 were highly expressed, thus being responsible of most of miRNA-mediated gene repression. In the same study, we showed that the majority of miRNAs were mixtures of sequence variants (isomiRs) and we identified 78 novel miRNAs. Indeed, we discovered that SET2 cells express a number of miRNA-offset RNAs (moRNAs), short RNAs derived from genomic regions flanking mature miRNAs, whose biological role needs to be elucidated.
In the present study, we characterized miRNA and moRNA expression in CD34+ stem cells using massive small RNA-seq. The observed specificities in small RNAs expression of PMF CD34+ cells were subsequently confirmed considering granulocytes from PMF, PV and ET patients and from healthy controls. We thus provided new information regarding the possible role of miRNAs and, specifically, of new moRNAs in the disease.
Materials and Methods
Cell-sample preparation and RNA extraction
CD34+ cells were purified from 30 to 50 mL PB collected from PMF patients or from 5 mL BM aspirate obtained in preservative-free heparin of healthy donors. All subjects provided informed written consent. The study, conducted in accordance with the Declaration of Helsinki, was approved by the Ethics Committee of the Azienda Ospedaliero-Universitaria Careggi of Firenze (Largo Brambilla 3, 50134 Florence, Italy), the 04/22/2011 (protocol n. 2011/0014777–37/11) in relation to the protocol "AGIMM (AIRC-Gruppo Italiano Malattie Mieloproliferative) Research Project".
Density gradient–separated mononuclear cells were processed through two sequential steps of immunomagnetic CD34+ selection (Miltenyi Biotec, Bergisch Gladbach, Germany, http://miltenyibiotec.com); final purity was evaluated by flow cytometry after labeling with PE-HPCA2 anti-CD34 monoclonal antibody (BD Biosciences, San Jose, CA, USA, http://www.bdbiosciences.com), and found to be >97% in all instances.
Total RNA was extracted using Trizol. Disposable RNA chips (Agilent RNA 6000 Nano LabChip kit; Agilent Technologies, Waldbrunn, Germany, http://www.home.agilent.com) were used to determine concentration and purity/integrity of RNA with Agilent 2100 Bioanalyzer.
Granulocytes were separated by differential centrifugation over a Ficoll-Paque gradient, starting from 20 mL PB; contaminating red cells were removed by hypotonic lysis, and cell pellets were resuspended in Trizol (Invitrogen Ltd, Paisley, UK, http://www.invitrogen.com).
Small RNA-seq library construction and sequencing
For each sample, a small RNA library was prepared starting from 1 μg total RNA, using the TruSeq Small RNA Sample Preparation Kits and protocols (Illumina, San Diego, CA, USA, http://www.illumina.com). Library quality was checked using High Sensitivity DNA chip (Agilent Technologies, Waldbrunn, Germany, http://www.home.agilent.com). The purified cDNA libraries was used for cluster generation on Illumina's Cluster Station and sequenced on an Illumina HiSeq2000 instrument, producing single reads from 49 to 57 bp.
Small RNA data analysis: preprocessing
After the adapter removal preprocessing step, reads “adapter-only”, too short or unclipped have been discarded. Unclipped reads are discarded because they can't represent miRNA or miRNA like short RNAs.
We admitted a read length range between 15 and 30 nt, slightly wider than the human annotated miRNAs length in miRBase to conserve also possible new longer isomiRs. We therefore discarded raw reads out of the range 15–30 bps in length. We then filtered out low quality reads, keeping all those reads displaying a mean base quality higher than 30, and allowing no more than 2 nucleotides per read with quality under 20. To complete data preprocessing we eliminated ground noise, considered as reads belonging to unique sequences with less than 10 reads counts each.
Small RNA data analysis: reads mapping and comparative filtering
Reads have been mapped using Bowtie v. 1.1.0 both to the GRCh38 genome assembly and the known hairpins sequences extended in both directions by additional 30 bp to accommodate moRNAs mapping at the extremities of known hairpins. Reads mapping to more than 5 different loci on the genome, out of miRNA hairpins, are unlikely to be real miRNAs, and they have been thus discarded.
Barplot on Fig A in S1 File shows filtering effects on absolute reads counts for each sample.
We processed each sample data with our in-house pipeline miR&moRe. The output consists of lists of known miRNA read counts, lists of new miRNAs and moRNAs and lists of variants (isomiRs) for all the small RNAs found in each sample.
Expression data normalization and sample cluster analysis
Sample merging and carefully conducted steps of data normalization and transformation are needed to guarantee the comparability of samples, to allow descriptive unsupervised analyses and differential expression tests. We performed normalization using R/Bioconductor package DESeq. Inference of differential expression in DESeq relies on the estimation of the typical relationship between the data variance and their mean, or, equivalently, between the data dispersion and their mean. Variance dependence to the mean can be modeled following different ways using DESeq, with several algorithms and very different results. The selection of the method used is crucial, since variance estimation influences unsupervised classification, differential expression and all following analyses. We tried two different methods for fitting data variance: 1) a parametric model, 2) a local regression model.
The first is the recommended default but in some data sets could fail to give optimal results. Sum of square of residuals for local regression is 15311.52 whereas for GLM is 156849.3, so we can conclude that local regression fits better our data.
We performed cluster analysis using R to check whether samples were correctly classified in their own biological class. We clustered samples using both full sRNAs expression matrix (917 sRNAs) and filtered, computing Euclidean distance and complete linkage as clustering method.
To filter the expression matrix at different levels of sRNAs expression we calculated, for each sRNA in the matrix, the sum of expression vector values. Then we performed clustering analysis under the following conditions 1) considering all the sRNAs found 2) selecting only sRNAs over the median (474 miRNAs and 26 moRNAs), 3) filtering only sRNAs over the third quartile (237 miRNAs and 12 moRNAs).
Differentially expressed sRNAs
We performed a differential expression analysis using DESeq R/Bioconductor package. We considered those short RNAs that had a total expression throughout all samples higher than the median. We performed a multiple test correction according to the Benjamini Hochberg method (FDR). We considered a corrected p-value of 0.05 as threshold to identify differentially expressed elements.
Validation of differentially expressed sRNAs
We performed individual miRNAs assay by Taqman quantitative real-time PCR (QRT-PCR) for quantification of abnormally expressed miRNAs in PMF and control granulocytes and in CD34+ cells. cDNA was synthesized from total RNA using microRNA-specific RT primers contained in the TaqMan microRNA Human Assays (Applied Biosystems). Briefly, single-stranded cDNA was synthesized from 10 ng total RNA in 15-μL reaction volume with the High-Capacity cDNA Archive Kit (Applied Biosystems) using 1 mM deoxyribonucleoside triphosphates, 50 U Multiscribe reverse transcriptase, 3.8 U RNase Inhibitor, and 50 nM of miR-specific RT primers. The reaction was incubated at 16°C for 30 minutes followed by 30 minutes at 42°C, and inactivation at 85°C for 5 minutes. Each generated cDNA was amplified by QRT-PCR with sequence-specific primers from the TaqMan microRNA Assays on an ABI Prism 7300 real-time PCR system (Applied Biosystems). PCR reactions included 10 μL 2× Universal PCR Master Mix (No AmpErase UNG), 2 μL each 10× TaqMan MicroRNA Assay Mix and 1.5 μL reverse-transcribed product; they were incubated in a 96-well plate at 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Expression variations were calculated using the RQ method and a t-test p-value of 0.05 was used as threshold to identify differentially expressed elements.
Target prediction of validated sRNAs and functional enrichment
The complexity of miRNA-mRNA interactions causes ambiguity in target prediction results. Target genes identification is indeed challenging and many algorithms have been developed. Target prediction programs can be divided in two classes, distinguished on the basis of the use or not of the information about evolutionary conservation of interaction[40]. We chose to perform a target prediction using two different programs, miRanda[41] and PITA[42], which implement orthogonal target prediction strategies. Our choice was determined also by code availability, which allowed us to make custom predictions using as query sequences also isomiRs and moRNA sequences.
We performed a target prediction using both miRanda 3.3a and PITA executable version 6 (31-Aug-08). We applied default parameters of miRanda to predict target of selected miRNAs and moRNA sequences since these settings are reported to optimize the dynamic programming miRanda algorithm. We used default parameters for PITA target prediction too. According to PITA documentation, we considered a binding site with score ≤ -10 likely to be functional in endogenous microRNA expression levels. We performed a hypergeometric test using an in-house modified version of the R Category package of Bioconductor, which supports Reactome annotation maps via the reactome.db R package.
qRT-PCR analysis of target gene expression
For the analysis of expression level of MECOM, MEIS1, AGO1, CAV1, AKR1C1, TIMP3, AGO3, FSTL1, JAKMIP2, TNSF10, BRCA1, PTPN4, MME, TCF4, FKBP10, SYS1, TRPS1 and RAN in CD34+ cells, RNA was reverse-transcribed with random hexamers and Murine Leukemia Virus (MuLV) reverse transcriptase (Thermo Fisher Scientific). qRT-PCR was carried out with TaqMan Universal PCR master mix, using TaqMan gene expression assays (MECOM: Hs00602795_m1, MEIS1: Hs01017441_m1, AGO1: Hs01084653_m1, CAV1: Hs00971716_m1, AKR1C1: Hs04230636_sH, TIMP3: Hs00165949_m1, AGO3: Hs01087121_m1, FSTL1: Hs00907496_m1, JAKMIP2: Hs00207662_m1, TNSF10: Hs00921974_m1, BRCA1: Hs01556193_m1, PTPN4: Hs00267762_m1, MME: Hs00153510_m1, TCF4: Hs00162613_m1, FKBP10: Hs0022557_m1, SYS1: Hs01110991_m1, TRPS1: Hs00936363_m1 and RAN: Hs03044733_g1; Thermo Fisher Scientific) by means of StepOne real-time PCR system (Applied Biosystems). Assays were performed in quadruplicate. Gene expression profiling was achieved using the RQ method as above, using RNase-P as the housekeeping gene.
Results and Discussion
Small RNA expression in CD34+ cells of patients with PMF
We considered small RNA sequencing data of 6 CD34+ cells, including 3 samples collected from 3 pools of bone marrow CD34+ cells of healthy subjects (CTR), and 3 samples of circulating CD34+ cells of patients affected by primary myelofibrosis (PMF), two of which were from individual patients while the third constituted a pool obtained by mixing equal amounts of RNA from 4 PMF patients. Samples and raw reads information are summarized in Table A in S1 File. The Illumina 2000 sequencing produced a total of 787,913,722 raw reads (131,318,954 per sample on average) that were deposited in Gene Expression Omnibus (GEO Series GSE69089).
After a stringent filtering during the preprocessing and quality control steps, 349,372,534 were aligned to GRCh38 genome “extended” hairpins (Fig A in S1 File). Aligned reads corresponded to 44.3% of initial raw reads, but due to high sequencing depth, the number of considered reads was still high.
We detected a total of 917 sRNAs, including 784 know miRNAs, expressed in at least one of the 6 considered CD34+ samples. Notably, 8 known miRNAs (miR-10a-5p, miR-181a-5p, miR-191-5p, miR-92a-3p, let-7a-5p, miR-146b-5p, miR-26a-5p and let-7f-5p) are highly expressed and contribute to the total miRNA expression throughout all samples from 2.5% to 25%, representing all together the 80% of the total expression. Moreover, we detected 133 new sRNAs, including 34 new miRNAs produced from known hairpins, and 99 microRNA-offset RNAs (moRNAs). Table 1 reports a summary of different types of sRNAs detected in the considered samples, according to current miRNA annotations.
Table 1. Summary of small RNAs expressed in CD34+ cells.
| sRNAs | CTR | PMF | Total |
|---|---|---|---|
| known miRNAs | 568 | 760 | 784 |
| new miRNAs | 20 | 31 | 34 |
| moRNAs | 52 | 96 | 99 |
| Total new | 72 | 127 | 133 |
| Total | 640 | 887 | 917 |
Cluster analysis and heatmaps of pairwise sample correlations are represented in Fig B in S1 File. Two heatmap plots were generated, one by considering normalized expression profiles of all the sRNAs expressed and another considering only those expressed over the median level. Both unsupervised analyses indicate that samples are correctly clustered according to sRNAs expression, with CTR samples clustered together and separately from PMF samples. Cluster analyses and heatmaps show that patients and controls sRNAs profiles are significantly different from each other and highlight a characteristic miRNA and moRNA expression profile in PMF.
New miRNAs in CD34+ cells of PMF
In addition to 784 miRNAs annotated in miRBase, our in-house pipeline miR&moRe let us discover 34 new miRNAs expressed in considered samples (Table B in S1 File).
To find new miRNAs, we considered all the hairpins precursors annotated in miRBase, to be used as reference for read mapping and small RNA detection and quantification. Some of the listed hairpin precursors were known to only generate one mature miRNA with only a handful of reads reported in miRBase across all NGS experiments surveyed. After identifying the hairpin region that would most likely pair with annotated mature we classified as new miRNAs all the clusters of reads that map there. A consistent number of reads were attributed to new miRNAs. Since these reads passed stringent quality filtering and mapping criteria, it is improbable that they originate from sequencing errors. Furthermore, two of these new miRNAs, miR-2110* and miR-548ag-2*, resulted highly expressed and showed a mean expression over the median of expression values calculated on all small RNAs.
miRNAs are mixtures of isoforms contributing to miRNAs expression
Microarray technology, relying on sequence hybridization to appropriately designed annealing probes, can only detect annotated miRNA sequences. Sequencing-based technology reveals instead all the repertoire of expressed small RNAs, both the unknown and the annotated miRNAs, and is able to detect isomiRs, that contribute to miRNA expression and qualitative characteristics. Our and other previous NGS studies showed that miRNAs are mixtures of sequences, slightly different from the official mature miRNA, called isomiRs [39,43].
In our dataset, miRNA expression counts indeed consider for each miRNA a group of reads that not only perfectly match the annotated miRNA (“exact”), but also match the precursor with a 1–2 nt shorter or longer sequence than the mature miRNA in the 3’ region (“shorter or longer at 3’”), in the 5’ end (“shorter or longer at 5’”) or at both the ends (“both”).
Expressed miRNAs with unique sequence are very few, only 165 out of the 784 annotated miRNAs detected (21%). Unique sequence miRNAs are, in general, weakly expressed: they were detected at level under the median of the mean expression values distribution. Notably, the remaining 619 expressed miRNAs have more than one isomiR. We also detected reads aligning to hairpin precursors with one or two mismatches but we excluded these reads from the total miRNAs expression estimation.
Fig 1A shows that the annotated sequence, called “exact” isomiR, contributes on average to 36.6% of the miRNA expression, whereas a considerable part of the total expression contribute is given by shorter or longer isomiR sequences (37.3% of expression, in average). Reads aligning with mismatches are also represented in display items for comparison (Fig 1A). We investigated if sequence differences observed in non “exact” isomiRs involve the 5’ half of the miRNA, including the seed region crucial for target recognition, the 3’ half, or both. Fig 1B shows that, considering all expressed miRNAs, most differences regard the 3’ half of the miRNAs, and isomiRs with 5’ region different from the “exact” sequence are very rare. Considering isomiR type and miRNA region involvement together, we observed that mismatch isomiRs impacting on the 5’ region were not detected at all (Fig C in S1 File). Considering the top 25% most expressed miRNAs, the picture is basically unchanged (Fig D-A-C in S1 File). Looking at specific miRNAs, the ratio between isomiR types changes. For example, miR-10b-5p is mainly detected in its “shorter or longer” variant (Fig 1C and 1D).
Fig 1. Distribution of reads per isomiR category (A) and per type (B) across samples, considering all 818 expressed miRNAs.

“Exact” reads are identical to the mature miRNA sequence annotated in miRBase, whereas “mismatch” reads present respectively one or two nucleotides different from the annotated sequence but identical length; the last category includes reads perfecly matching the miRNA precursor (and genomic) sequence but shorter or longer than the annotated mature miRNA. IsomiR types indicate if the sequence difference fall in the 5’ region of the miRNA, in the 3’ region, or in both regions. Abundance of reads falling in different isomiR categories (C) and types (D) for miR-10b-5p, showing that isomiRs different from the annotated mature miRNA sequence are very abundant.
IsomiRs, initially discarded as sequencing/alignment artifacts or poor quality RNAs,[44–47] were lately demonstrated to be highly expressed with an elevated degree of confidence. They were experimentally validated and characterized as tissues, conditions and cell types specific[48–50]. Despite isomiR biogenesis being still unclear, tissue specificity and differential expression during different development stages suggest to be ruled by a fine-tuned mechanism. Several lines of evidence suggest that isomiRs could be processed by an alternative Drosha/Dicer cleavage of the pre-miRNA[51–53] or by editing/nucleotide addition of the mature annotated miRNA[54].
Regarding isomiR biological role, they were demonstrated to be biologically functional and to work similarly to the canonical miRNA. In fact, Azuma-Mukai et al. [48] and Chin Tan et al.[50] co-immunoprecipitated isomiRs with Ago proteins, supporting isomiRs biological activity. Cloonan et al.[55] biotin-labeled miRNAs and isomiRs, to pull down endogenous mRNA targets: after ultra-deep miRNA-seq in different tissues, isomiRs expression levels resulted comparable to the canonical miRNAs. Reportedly, isomiRs are incorporated into RISC complexes binding endogenous mRNAs. The fact that 5’ isomiRs are also under selection during evolution witnesses their functional importance[50].
Anyway, sequence differences, and notably those in the seed region can potentially direct different isomiRs to diverse target mRNAs. Indeed 5’isomiRs target different mRNAs than their canonical counterparts[50]. Chan et al.[56] showed that different miR-31 isomiRs are expressed in different cell types and, notably, that even sharing the same seed region, slightly different miR-31 isomiRs exert different degree of repression of verified target genes.
Considering that different isomiRs can characterize different tissues or conditions, as tumor respect to normal tissues, and that diverse isomiR could impact differently on target genes and pathways, isomiR deregulated expression could be implicated in disease. We thus considered the possibility that peculiar genetic characteristics of the considered PMF cells would result in specific isomiR sequences, or could be related to variations of isomiRs expression level in disease. We searched for mismatch isomiRs expressed in PMF samples and not in CTR samples, possibly due to genetic mutations: 44 isomiRs belonging to 30 expressed miRNAs were identified only in PMF samples, but they resulted weakly expressed, with a read count lower than 400, and/or not accounting for at least the 25% of the corresponding miRNA expression (data not shown).
We searched also for differentially expressed isomiRs comparing PMF and CTR samples. 133 isomiRs from 59 miRNAs were associated to a Log of Fold change at least 1 and adjusted t-test p-value lower than 0.05. Considering all isomiRs accounting each for at least 10% of mature miRNA expression, we observed that isomiRs and miRNA expression profiles were highly correlated (75% of pairwise correlation values over 0.699, median correlation of 0.917; Fig E-A in S1 File). Similarly, also differentially expressed isomiRs resulted highly correlated with miRNAs (75% of pairwise correlation values over 0.743, median correlation of 0.895; Fig E-B in S1 File).
Anyway, since miRNAs are de facto mixtures of isomiRs, specific variations of isomiRs expression impact also on miRNAs expression. Thus, we considered isomiR counts for miRNA expression calculations and for the following analyses aiming at investigating the impact of miRNAs and on moRNAs in specific functions and pathways.
moRNAs discovery
As anticipated, we also detected in our samples sequences aligning to hairpins outside known and novel miRNAs, that correspond to expressed microRNA-offset RNAs, called moRNAs (Table 1). moRNAs sequences partially overlap miRNA regions but generally span the Drosha cutting sites, letting us hypothesize a non canonical processing of the hairpin precursor in moRNA biogenesis[39].
A complete list of all the detected moRNAs is in Table C in S1 File. Noteworthy, 28 moRNAs were highly expressed, 26 of them over the median of the short RNAs expression values distribution and 12 of them even over the third quartile (Table 2).
Table 2. List of most abundant moRNAs in considered CD34+ samples, which are expressed over the third quartile of all sRNAs expression.
For each moRNA, the table reports expression (per sample group normalized read count), position and sequence.
| moRNA | Average read count CTR | Average read count PMF | Strand | Position | Sequence |
|---|---|---|---|---|---|
| moR-128-2-3p | 2493 | 0 | + | chr3:35744548–35744568 | CCCTACTGTGTCACACTCCTA |
| moR-21-5p | 2946 | 2437 | + | chr17:59841243–59841271 | ACATCTCCATGGCTGTACCACCTTGTCGG |
| moR-24-2-5p | 5971 | 1719 | - | chr19:13836350–13836376 | TGCCTGGCCTCCCTGGGCTCTGCCTCC |
| moR-27a-5p | 1752 | 312 | - | chr19:13836510–13836534 | CGAAGCCTGTGCCTGGCCTGAGGAG |
| moR-3651-5p | 0 | 1266 | - | chr9:92292537–92292565 | ATGGACAGCTCTCCAGTGGATTCGATGGG |
| moR-421-5p | 848 | 2784 | - | chrX:74218449–74218472 | CCTAATCCGGTGCACATTGTAGGC |
| moR-6724-1-5p | 683 | 280 | + | chr21:8205298–8205332 | TGTGGGGGAGAGGCTGTCGCTGCGCTTCTGGGCCC |
| moR-6724-2-5p | 683 | 280 | + | chr21:8249488–8249522 | TGTGGGGGAGAGGCTGTCGCTGCGCTTCTGGGCCC |
| moR-6724-3-5p | 683 | 280 | + | chr21:8388345–8388379 | TGTGGGGGAGAGGCTGTCGCTGCGCTTCTGGGCCC |
| moR-6724-4-5p | 683 | 280 | + | chr21:8432513–8432547 | TGTGGGGGAGAGGCTGTCGCTGCGCTTCTGGGCCC |
| moR-941-4-5p | 6564 | 2531 | + | chr20:63919746–63919768 | CACCCGGCTGTGTGCACATGTGC |
| moR-941-5-5p | 9780 | 3800 | + | chr20:63919858–63919880 | CACCCGGCTGTGTGCACATGTGC |
We classified moRNAs on the basis of the hairpin precursor arm they where processed from: 5’-moRNAs mapping to the 5’ hairpin arm, and 3’-moRNAs spanning over the 3’ hairpin arm. 5’-moRs were significantly more abundant respect to 3’-moRs. Out of 99 moRNAs expressed in considered samples, only 16 (16.2%) were processed from the 3’ hairpin arm, while 83 (83.8%) were 5’-moRs. According to our data, seven hairpins were processed producing two moRNAs each (Table C in S1 File).
5’-moRs estimated expression values were 10 times higher than 3’-moRs, ranging from summed up normalized values over all samples of 5 to 40,739, compared to a 3’-moRs range of 6 to 7,478. Both 3’-moRs and 5’-moRs are more expressed in controls than in PMF patient samples.
Considering the 9 most expressed moRNAs, Fig 2A shows expression estimations in PMF and CTR samples of all the expressed small RNAs that are produced from the same hairpins precursor. Specific hairpins, as hsa-mir-421 and hsa-mir-941-1, reported in Fig 2A, express two moRNAs each.
Fig 2. A) Expression, in CD34+ cells of PMF patients and controls, of miRNAs and moRNAs produced from the same hairpin, considering the hairpins expressing most abundant moRNAs; the boxplot in panel (B) shows the distribution of Pearson correlation values calculated pairwise between expression profiles of moRNAs and miRNAs produced from the same hairpin arm, considering all moRNAs detected in CD34+ cells.

Considering the miRNA and the moRNA produced from the same hairpin arm, we noted that in 4 out the 9 reported in Fig 2A the moRNA is more expressed than the miRNA and in one the miRNA close to the moRNA is not detected at all. Moreover, some hairpins show a similar trend toward up- or down-regulation for both the miRNA and the moRNA, whereas in others the two sRNAs show opposite behavior. We then compared the expression behavior of all expressed moRNAs with that of the miRNA coming from the same hairpin arm. The boxplot in Fig 2B shows the distribution of 93 Pearson correlation values calculated pairwise between expression profiles of detected moRNAs and miRNAs from the same hairpin arm: the values range from -0.93 to +1, with median and mean values of 0.17 and 0.21, indicating a very slight tendency toward positive correlation, the presence of abundant anticorrelated pairs (36, 38.7%). Considering the statistical significance of correlation, only 7 pairs (7.5%) resulted significantly correlated (with a q-value at most 0.1); since they include an anticorrelated pair (5’moR-93 and miR-93-5p) and do not overlap with the group of highly expressed moRNAs, we can conclude that moRNA expression is largely independent from that of the close miRNA.
Even if moRNAs mechanism of action is still unknown, their considerable high expression level in this dataset together with previously reported observations [39,43,57] offers an indirect but intriguing indication of a biological role.
Identification of sRNA differentially expressed in PMF vs CTR
Since myeloproliferative disorders are clonal hematopoietic stem cell neoplasias, miRNA and moRNA deregulation can be implied in tumor physiopathology.
We recognized 37 sRNAs significantly differentially expressed (DE) in PMF patient samples respect to control CD34+ (Table D in S1 File). Fig 3A shows the logarithm of the mean expression ratio in PMF and control cells for DE miRNAs and moRNAs. While only five small RNAs are downregulated, the majority of DE sRNAs resulted upregulated in PMF patients.
Fig 3. Differential expression of small RNAs in PMF vs CTR CD34+.

A) Log2 FC of 37 small RNA differentially expressed considering PMF vs CTR CD34+, according to RNA-seq data (FDR<0.05). When a small RNA was not expressed in one sample category, the ratio was infinite and we represent it as the arbitrary maximum value of 15. B) RT-PCR expression calculation of five selected miRNAs in granulocytes collected from an independent cohort of normal controls (n = 10) and of PMF (50) samples; ***, ** and * indicate respectively a p-value <0.001, <0.01 or <0.05.
Noteworthy, among the differentially expressed sRNAs, 2 moRNAs (5’-moR-542 and 3’-moR-128-2) are included. Incidentally, the two miRNAs (miR-542-5p and miR-128-3p) that are produced by the same hairpin arm of differentially expressed moRNAs are not differentially expressed in the same sample comparison. 5’-moR-542 is up-regulated in PMF with a log2FC of 3.5. 3’-moR-128-2 is highly expressed in normal CD34+ cells, at levels over the third quartile of the overall small RNA expression distribution, and dramatically downregulated in PMF patients: the moRNA was not detected in considered PMF samples. We mapped 3’-moR-128-2 sequence to the whole human genome to exclude multiple matching loci and to rule out mapping or annotations artifacts. We can thus exclude that moRNA-associated reads could come from different or contaminating RNAs. An additional UCSC Blat[58] analysis confirmed that the moRNA sequence only aligned to a single genomic locus (chr3:35786042–35786062). We are therefore confident that the detected small RNA is a moRNA derived from the non-canonical processing of the human mir-128-2 hairpin.
Validations of selected differentially expressed miRNAs in PMF granulocytes
We selected the most significantly deregulated and highly expressed miRNAs for further analysis. Specifically, we considered 6 miRNAs among the differentially expressed for a quantification with Real-time PCR (RT-PCR) in granulocytes collected from an independent and sizeable cohort of normal controls (N = 10) and PMF (N = 50), PV (N = 30) or ET (N = 30) patients. miR-10b-5p and moR-128-2 resulted significantly downregulated in PMF granulocytes samples, as according to RNA-seq data in CD34+ cells (Figs 3B and 4B). Two additional miRNAs (miR-379-5p and miR-543) showed in PMF granulocytes the same trend toward upregulation in PMF granulocytes (Fig 3B) as in CD34+ cells by RNA-seq data. Both miR-29a-3p and miR-19b-3p resulted significantly downregulated in PMF granulocytes (Fig 3B).
Fig 4. Differential expression of 3’-moR-128-2 in PMF (n = 3) vs CTR (3) cells.
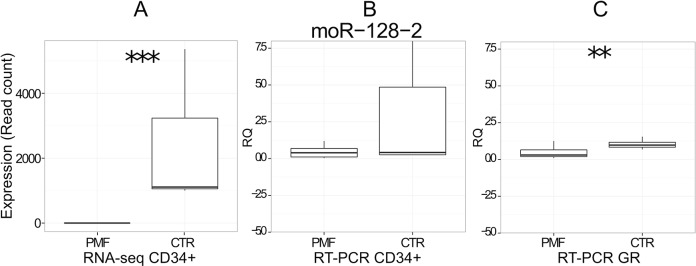
A) moR expression in PMF and CTR CD34+ according to RNA-seq data. B) RT-PCR expression (RQ) in CD34+ cells from independent cohort of normal controls (n = 8) and of PMF (20) samples. C) RT-PCR expression (RQ) in granulocytes from independent cohort of normal controls (n = 10) and of PMF (50) samples; ***, ** and * indicate respectively a p-value <0.001, <0.01 or <0.05.
Furthermore, among the six small RNAs considered for validation in PMF granulocytes, 5 showed the same trend of changes also in ET or PV granulocytes, whereas miR-543 resulted upregulated in PMF and ET, and downregulated in PV, with small variations (Fig F in S1 File). miR-379-5p was upregulated in ET and in PV, as in PMF; miR-543 was slightly increased in ET, as in PMF, but not in PV; miR-19b-3p and moR-128-2 showed a similar trend, being decreased both in ET and in PV, but at a lower extent than in PMF; miR-10b-5p was decreased in ET and in PV, also more than in PMF. Notably, considering the statistical significance of the observed variation, miR-10b-5p is significantly downregulated both in PV and ET samples, indicating that it is associated with MPN in general more than being specific for PMF.
Of interest, miR-10b-5p resulted downregulated both in PMF CD34+ and granulocytes. Accordingly, miR-10b-5p has been previously reported to be deregulated in breast cancer[59–61]and involved in chemoresistance related pathway[62]. It has been validated as downregulated in endometrial carcinoma[63], bladder cancer [64], in advanced stage of small cell carcinoma of the cervix (SCCC)[65] and in clear cell renal cell carcinoma (ccRCC) and its expression level has been also included in a linear model that capture the metastatic tumor signature and patient prognosis[66].
We found miR-29a-3p upregulated in patients CD34+ respect to controls, but downregulated in the same comparison when considering committed granulocytes. This observation is in accordance with results previously obtained by Han et al.[67] who showed that miR-29a-3p is expressed at lower levels in hematopoietic progenitors compared to lineage-committed progenitors, including granulocytes, overall indicating that its expression level increases along with commitment. Therefore, we point to miR-29a as a deregulated small RNA in PMF, whose expression is uniquely modulated along myeloid differentiation of PMF CD34+ cells. Consistently with our finding, Han et al. also showed also that sustained expression of miR-29a-3p in mouse HSC/progenitors pushed myeloid progenitors to self-renewal capacity, to biased myelopoiesis and to the onset of a myeloproliferative disorder that progressed to acute myeloid leukemia. Additional data supporting miR-29a-3p deregulation derives from a previous study by Norfo et al.[38], in which miRNA expression profiling was obtained by Affymetrix miRNA 2.0 array analysis in a cohort of 42 PMF patients CD34+ cells and 31 healthy donors. In that study, miR-29a-3p was found upregulated in CD34+ from PMF patients as well as, miR-29a-3p upregulation in PMF CD34+ cells was validated by RT-PCR (with TaqMan probes) in an independent set of CD34+ cells from 10 PMF patients and 8 healthy subjects.
In the study of Norfo et al., also the upregulation of miR-379-5p, miR-543 and miR-19b-3p were validated in PMF CD34+. In present study, we were unable to confirm a statistically significant upregulation of these miRNAs in PMF, although there was a trend toward for miR-379-5p and miR-543. Interestingly, Norfo et al. validated microarray-based observations using two sets of qRT-PCR experiments, conducted on CD34+ cells and on granulocytes, showing that PMF-specific variations of a few miRNAs may be observed in CD34+ cells unlike in granulocytes. Indeed, miR-486-3p was significantly downregulated in PMF granulocytes and upregulated in PMF in CD34+ cells.
In summary, available evidence indicate that miR-10b-5p and moR-128-2 are downregulated in PMF CD34+ cells, whereas miR-19b-3p, miR-379-5p and miR-543 are upregulated. For these miRNAs the evidence of differential expression in PMF was robust, since it was detected by NGS and also validated technically (by qRT-PCR) and biologically (in independent samples).
3'-moR-128-2
3’-moR-128-2, a newly annotated small RNA, resulted expressed in CD34+ while it was not detected in PMF. We evaluated its expression level with qRT-PCR both in CD34+ cells and in granulocytes. RT-PCR validation in CD34+ cells were conducted considering 8 normal controls and 20 PMF patients, whereas the validation in granulocytes was done with the same design and samples used for miRNA validation. Fig 4 shows the consistent downregulation of 3’-moR-128-2 in PMF patients according to RNA-seq (Fig 4A) and RT-PCR data in CD34+ (Fig 4B) and to RT-PCR data in granulocytes (Fig 4C). Moreover, 3’-moR-128-2 was decreased, but at a lower extent, without reaching statistical significance, also in PV and ET granulocytes (data not shown).
Intrigued by the striking expression pattern of the newly discovered 3'-moR-128-2 we looked in details into sequence, structure, expression and functional differences of 3'-moR-128-2 and miR-128-3p.
In Fig 5 we show additional sequence information regarding miR-128-3p and 3’-moR-128-2. Since the moRNA sequence is not contained in the canonical hairpin (Fig 5A), the moRNA probably derives from the processing of an alternative hairpin precursor. In Fig 5B we show the RNAfold predicted minimum free energy (MFE) folding structure of the canonical hairpin and of a longer sequence from which the moRNA can be derived. Fig 5C shows that mir-128 locus is inside an intron of ARPP21 gene, and displays the genomic region Mammals UCSC base-wise PhyloP conservation score.
Fig 5. The 3’-moR-128-2 is produced by the precursor sequence of miR-128-3p.

A) The moRNA is derived from a region of the primary miRNA sequence exceeding the canonical hairpin precursor sequence, and it is not exaclty adjactent to the annotated miRNA. B) Minimum free energy (MFE) folding structure, predicted by RNAfold, for the canonical hairpin sequence and for the longer one, from which the moRNA is probably derived. C) Both the considered small RNAs are conserved in evolution through vertebrates.
moRNA biological roles and mechanisms of function still deserve investigation. Very likely, moRNAs can function as miRNAs in post-transcriptional gene silencing, guiding RISC to complementary target mRNAs. This was first demonstrated by Umbach and colleagues, that used a luciferase-based indicator assay to demonstrate that a viral moRNA (moR-rR1-3-5p) has inhibitory activity against an artificial mRNA bearing a perfect target site [68,69]. Beyond this proof of principle experiment, a recent study reported moRNA specific expression in human embryonic stem cells[70] (hESCs). In the same study, moRNA and miRNA transfection experiments and microarray quantification of gene expression were conducted and identified gene silenced by moR-103a-2-3p, one of the most abundantly expressed moRNAs in hESCs, and by miR-103a. In line with these previous studies, we assumed that 3’-moR-128-2 can act as a miRNA, and investigated its possible impact on target gene silencing and on specific pathways or biological processes. To this end, we considered how sequence variants (isomiRs) of these two small RNAs relate to each other (Fig 6). For miR-128-3p, we identified 7 variants expressed in considered CD34+ samples: one exact isomiR, corresponding to the miRBase annotated mature form, and 6 “shorter or longer” variants (miR-128-3p-SL-1 to miR-128-3p-SL-6), whereas only 2 3'-moR-128-2 isomoR were found out (3'-moR-128-2-1 and 3'-moR-128-2-2)(Fig 6A).
Fig 6. Origin, sequence variability, and relations beween 3’-moR-128-2 and the adjacent miR-128-3p.

A) 3’-moR-128-2 and miR-128-3p map to the same locus and both shows sequence variability (isomiRs and isomoRs). Both the major and the minor isomoRs are found in normal CD34+ cells and not in PMF samples. Red and blue colors indicate isomiR and isomoR groups that can be produced with an unique sequence cutting sites. The most expressed isomoR is not associated to the corresponding most expressed isomiR. Moreover, expression levels, in CTR and PMF samples, of isomiRs and isomoRs are poorly correlated intragroup. These observations, point against the moRNA being simply a by-product of the miRNA biogenesis. A similar indication is given by the fact that some abundant isomiRs are not associated to detected isomoR sequences. B) 3’-moR-128-2 and miR-128-3p have different, poorly overlapping, sets of predicted targets. C) 3’-moR-128-2 sequence can stably bind a target site in the 3’UTR of the RAN mRNA, causing post transcriptional silencing.
According to the conservative hypothesis that interprets moRNA as byproducts of Drosha cleavage[71] one should expect comparable mean levels of miRNA and moRNA cognate variants (i.e. obtained from a single endonucleolytic cleavage cut) in PMF and CTR samples. The observation that 3'-moR-128-2-1, the most abundant isomoR, is expressed only in CTR samples and its only viable cognate partner is the miR-128-3p-exact variant that is highly and nearly equally expressed in CTR and PMF samples (with 3,739 and 2,256 normalized reads, respectively)(Fig 6A) does not point in this direction. Neither do the poor correlation of expression levels, in CTR and PMF samples, of cognate isomiRs and isomoRs, the fact that some abundant isomiRs are not associated to detected isomoR sequences, the good conservation of moRNA sequence (Fig 5C), and the previously reported observation that both isomoRs are not contained in the canonical hairpin (Figs 5A and 5B and 6A).
Genes and pathways targeted by the sRNAs deregulated in PMF
We conducted a preliminary functional characterization of the possible biological role of sRNAs DE in PMF, by a double strategy. First we investigated possible target genes and pathways of the group of validated DE sRNAs, considered as a whole. Then, we focused on one of the most novel elements emerged by our results, 3’-moR-128-2, to get specific insights on its possible functions in CD34+ and, in turn, in PMF disease.
Target predictions of miR-10b-5p, miR-19b-3p, miR-29a-3p, miR-379-5p, miR-543 and of moR-128-2 were performed by using two different programs, miRanda[41] and PITA[42], which implement orthogonal target prediction strategies, and for which the code availability allowed us to make custom predictions of possible miRNA and moRNA target genes, by using as query sequences the identified isomiR and isomoR sequences.
Among different isomiRs detected for each considered miRNA, we considered the most expressed, even if it was different from the annotated sequence (Table E in S1 File). We also considered those isomiRs that were significantly contributing to miRNA total expression, and which were differently expressed in patients respect to controls (t-test p-value <0.05 and |log2FC|>1), according to RNA-seq data. Accordingly, both isomoRs were considered for moR-128-2.
A functional enrichment analysis, based on Reactome annotation maps, of targets predicted by both methods was obtained using a hypergeometric test.
Table F in S1 File reports, the Reactome pathways enriched (q-value < = 0.05) among the union of predicted target genes of miRNAs and moRNAs differentially expressed in PMF vs CTR samples, giving some hints on the pathway more specifically targeted, collectively, by short RNAs deregulated in PMF. These enrichments are based on the subset (1,521; 36%) of 4,222 predicted target genes that are currently annotated in Reactome.
Table G in S1 File reports Reactome pathways resulting enriched among predicted targets of each of the considered isomiRs belonging to differentially expressed sRNAs, giving details of pathways most enriched among the targets of specific miRNAs and also indicates that different miRNAs target genes converge to the same enriched pathways, as “Signaling in FGFR in disease”, “DAP12 signaling”, “Oncogene induced senescence” and “Post-transcriptional silencing by small RNAs”.
Human fibroblast grow factor receptors (FGFRs) are a family of four tyrosine kinase receptors (FGFR1–4), which are involved, in a variety of cellular processes. They are indeed key regulators of fibrogenesis, embryogenesis, angiogenesis, metabolism, and many other processes of proliferation and differentiation[72,73]. Deregulation of FGFR signaling has been observed in numerous tumors.[74,75]
DAP12 is an immunoreceptor tyrosine-based activation motif (ITAM)-bearing transmembrane adapter molecule and it is reported to be signaling partner of activating natural killer receptors. DAP12 complex to TREM-1 and MDL-1 receptors to form receptor complexes involved in macrophage differentiation[76] and apoptosis in M1 leukemia cells[77], significant monocytic activation of myeloid cell, calcium mobilization and inflammatory response[78,79]. Its elevated expression levels are associates with enhanced cytotoxic characteristics in large granular lymphocyte leukemia[80].
Senescence is the stable cell growth arrest. Oncogene senescence (OIS) occurs when the activation of an oncogene is triggered; in this case it is termed oncogene-induced senescence. OIS acts as a barrier against tumour progression by driving stable growth arrest of cancer progenitor cells [81–83].
As anticipated, we then considered 3'-moR-128-2 and miR-128-3p targets for comparison. We predicted the targets of the four most expressed miR-128-3p isomiRs (miR-128-3p-exact, miR-128-3p-SL-2, miR-128-3p-SL-3, and miR-128-3p-SL-4) and targets of the two 3'-moR-128-2 isomoRs (3'-moR-128-2-1 and 3'-moR-128-2-2)(Fig 6A), assuming that they would act as miRNA, as indicated by the available experimental data[69,70]. We compared the union of predicted targets of miR-128-3p variants and the union of predicted targets of 3-moR-128-2 variants, to understand if and how much the moRNA function can be related to that of the cognate miRNA, as previously supposed[70]. As shown in the Venn diagram in Fig 6B, only a small fraction of 3-moR-128-2 target genes, less than 17%, is putatively targeted also by at least one of the miR-128-3p isomiRs.
According to Reactome-based functional enrichments, performed as explained in the previous paragraph, different pathways are enriched in predicted targets of 3’-moR-128-2 and of miR-128-3p. miR-128-3p targets are enriched in genes that are part of cellular pathways for the most part related to NGF, FGFR, ERBB4, ERBB2 signaling and transduction and to calcium ion homeostasis and signal transduction.
Targets of 3’-moR-128-2 are enriched too in genes part of several, distinct, pathways related to cellular signaling in growth and proliferation as “Signaling by Notch“, “Signaling by ERBB4”, “Signaling by FGFR in disease” but also, quite interestingly, in genes part of the “Post-transcriptional silencing by small RNAs” and of the more general “Regulatory RNA pathways” (Table H in S1 File).
Remembering that 3’-moR-128-2 is highly expressed in normal and not detected in PMF CD34+, it is worth notice that, 4 out of 7 genes of the “Post-transcriptional silencing by small RNAs” path, namely AGO1, AGO3, TNRC6A, and TNRC6B can be targeted by at least one isomoR of 3-moR-128-2 (Table 3). Moreover, both considered 3-moR-128-2 isomoRs could also target RAN (Table 3), the RAS-related nuclear protein, member of the RAS Oncogene Family, which is required for RNA export from the nucleus (Fig 6C).
Table 3. Putative targets of 3’-moR-128-2 variants in the “Regulatory RNA” pathway.
| Short RNA variant | Sequence | Target Genes |
|---|---|---|
| 3'-moR-128-2-1 | CCCTACTGTGTCACACTCCTAAT | AGO3—Argonaute 3 |
| 3'-moR-128-2-1 | CCCTACTGTGTCACACTCCTAAT | RAN—RAN, member RAS oncogene family |
| 3'-moR-128-2-1 | CCCTACTGTGTCACACTCCTAAT | POLR2H - Polymerase (RNA) II (DNA directed) polypeptide H |
| 3'-moR-128-2-2 | CCTACTGTGTCACACTCCTAAT | AGO1—Argonaute 1 |
| 3'-moR-128-2-2 | CCTACTGTGTCACACTCCTAAT | RAN—RAN, member RAS oncogene family |
| 3'-moR-128-2-2 | CCTACTGTGTCACACTCCTAAT | TNRC6A - Trinucleotide repeat containing 6A |
| 3'-moR-128-2-2 | CCTACTGTGTCACACTCCTAAT | TNRC6B - Trinucleotide repeat containing 6B |
In principle 3’-moR-128-2, where it is expressed, as in CD34+ hematopoietic stem cells, could affect the expression of genes important for the entire process of miRNA-based silencing. It can indeed target genes essential for post-transcriptional silencing both by translation repression, as AGO1/3, and by mRNA degradation, as TNRC6A/B. AGO1 and AGO3 are required for post-transcriptional translation repression activity; AGO1 is also involved in transcriptional silencing of promoters[84], and AGO3 is also putatively involved into the modulation of mature miRNA incorporation to the RISC complex, thus controlling the ratio between microRNA guide and passenger strand [85]. TNRC6A, and TNRC6B play a role in miRNA-dependent translation repression and endonucleolytic cleavage, by recruiting specific deadenylase complexes.
The multifunctional protein RAN is involved in many processes and diseases: it controls cell cycle progression and it is a potential therapeutic target for treatment of cancers with activation of the PI3K/Akt/mTORC1 and Ras/MEK/ERK pathways[86]. Specifically in relation to the above mentioned findings, as known, RAN play a key role in RNA export from the nucleus and for the biogenesis of all miRNAs. Thus, RAN silencing by 3’-moR-128-2 can impair pre-miRNA transportation to the cytoplasm and output a reduction of miRNA biogenesis. A similar situation was documented by a recent study that identified, in B. mori, a virus-encoded miRNA that suppresses the host miRNA biogenesis, exactly by targeting the host exportin-5 RAN cofactor [87].
Validation of potential mRNA targets
Bioinformatics predictions yielded several hundreds putative target genes for each selected small RNA (moR-128-2, miR-379-5p, miR10b-5p, miR-19b-3p, miR-29a-3p and miR-543). Because only a few predicted targets have been experimentally validated in vitro or in vivo, and in order to narrow the analysis to a manageable number of variables, we choose to focus on possible targets eventually selected based on their potential pathogenetic role in PMF.
TaqMan assays were carried out for 18 putative target genes in an independent cohort of CD34+ cells from 20 PMF patients and 10 healthy subjects (Table 4). Using real-time RT-PCR, we found that TNSF10, MME, TCF4, TRPS1 and SYS1 were all significantly reduced in PMF CD34+ compared with healthy controls, whereas BRCA1 and FKBP10 were increased. Other genes, probably due to the limited number of samples, did not reach a statistically significant difference yet they showed the expected trend of expression changes. In particular, we found that MECOM, MEIS1, AGO1, AGO3, RAN, CAV1, AKR1C1, TIMP3, FSTL1 were increased in CD34+ of PMF patients compared with normal subjects, while JAKMIP2 and PTPN4 were reduced.
Table 4. Expression pattern of 18 mRNAs potentially targeted by miRNAs, according to qRT-PCR data.
For each mRNA, miRNA and moRNA, we report if it is increased (I) or decreased (D) in the PMF VS CTR sample comparison; the mRNAs p-values associated to a significant difference are listed; the last column indicates if the expression of observed target mRNA variation is inversely related to the corresponding miRNA or moRNA.
| miRNA name | miRNA I/D | mRNA target gene name | mRNA I/D | mRNA DE p-value | Opposite variation |
|---|---|---|---|---|---|
| moR-128-2 | D | MECOM | I | NS | YES |
| moR-128-2 | D | MEIS1 | I | NS | YES |
| moR-128-2 | D | AGO1 | I | NS | YES |
| moR-128-2 | D | AGO3 | I | NS | YES |
| moR-128-2 | D | RAN | I | NS | YES |
| moR-128-2 | D | CAV1 | I | NS | YES |
| moR-128-2 | D | TNSF10 | D | 0.0001 | NO |
| miR-379-5p | I | MME | D | 0.0001 | YES |
| miR-379-5p | I | SYS1 | D | 0.0005 | YES |
| miR-379-5p | I | TCF4 | D | 0.05 | YES |
| miR-379-5p | I | BRCA1 | I | 0.05 | NO |
| miR-379-5p | I | AKR1C1 | I | NS | NO |
| miR-379-5p | I | TIMP3 | I | NS | NO |
| miR-19b-3p | I | TRPS1 | D | 0.04 | YES |
| miR-19b-3p | I | PTPN4 | D | NS | YES |
| miR-19b-3p | I | FKBP10 | I | 0.02 | NO |
| miR-29a-3p | I | AGO3 | I | NS | NO |
| miR-543 | I | TRPS1 | D | 0.04 | YES |
| miR-10b-5p | D | FSTL1 | I | NS | YES |
| miR-10b-5p | D | AGO3 | I | NS | YES |
| miR-10b-5p | D | JAKMIP2 | D | NS | NO |
| miR-10b-5p | D | PTPN4 | D | NS | NO |
According to miRNAs expression levels, and considering only significantly differentially expressed genes, an opposite behavior of expression variation was found for miR-379-5p and MME, TCF4 and SYS1; miR-19b-3p and miR-543 with TRPS1. Moreover, miR-10b-5p and FSTL1 showed opposite behavior and 5 tested genes (AGO1, RAN, MECOM, MEIS1, and CAV1) putative targets of moR-128-2 showed a trend toward increase in PMF, opposite to the moRNA.
Table 4 reports results of mRNA expression tests, indicating, for each miRNA-mRNA pair, if the observed variations are in the same or in the opposite sense. However, before one can reliably conclude that these predicted interactions do play a role in PMF cell abnormalities and disease pathogenesis, functional studies of miRNA modulation will be required.
Conclusions
In this study, we reported the results of the first RNA-seq project aiming at studying small RNAs in PMF CD34+ cells compared with control CD34+ cells. We detected 784 know miRNAs expressed in CD34+ cells and discovered 34 new miRNAs produced from known hairpins. We showed that expressed miRNAs with unique sequence are rare and that most miRNAs are isomiR mixtures, whose expression needs to be considered for miRNA expression estimation and for differential expression detection. Moreover, we discovered 99 microRNA-offset RNAs (moRNAs) probably produced from alternative microRNA precursors.
Cluster analyses and heatmaps of samples demonstrated that patients and controls small RNAs profiles are significantly different and highlighted a characteristic miRNA and moRNA expression profile in PMF. In fact, according to RNA-seq data, we identified several sRNAs expressed at significantly different level in patient compared to control CD34+. Subsequent analysis in granulocytes from independent patients and controls samples strengthened the evidence of 6 differentially expressed sRNAs, including 3’-moR-128-2. Predicted targets of these differentially expressed miRNAs are enriched in many remarkable pathways involved in tumor development and progression, as “signaling by FGFR”, “DAP12 signaling” and “Oncogene Induced Senescence”. Moreover, we validated the differential expression of 7 predicted target mRNAs, and identified 5 miRNA-target pairs that show variations in the opposite sense in the PMF vs control CD34+ cell comparison.
We investigated in detail sequence, structure, expression and possible functions of the newly discovered 3'-moR-128-2, also by comparison with the cognate miR-128-3p, showing that the miRNA and moRNA expression profiles are poorly correlated. At the functional level, assuming that moRNAs may act as miRNAs, we noticed that 3'-moR-128-2 and miR-128-3p potential target genes and pathways are markedly different. Interestingly, 3'-moR-128-2, that was expressed in normal CD34+ cells and resulted absent in PMF cells, may target pathways related to the control of cell growth and proliferation and, strikingly, target several genes involved in microRNA biogenesis or in miRNA-mediated silencing.
Overall, in this study, we provided novel data regarding the expression profile of small RNA expressed in PMF CD34+ as well as in healthy control cells, by NGS analysis and their possible impact on specific genes and pathways; in addition, we described for the first time new moRNAs as possible contributors to disease pathogenesis. This information may represent the basis for further studies aimed at a deeper knowledge of the role on miRNAs and moRNAs in normal and pathological hematopoiesis.
Supporting Information
(DOCX)
Data Availability
Sequencing and expression data have been deposited into NCBI GEO (Series GSE69089).
Funding Statement
The study received funding by Fondazione Cassa di Risparmio di Padova e Rovigo Progetti di Eccellenza 2011/2012 (http://www.fondazionecariparo.net/english-version/) to SB and PG; by Ministero dell’Istruzione, dell’Università e della Ricerca (http://www.istruzione.it/) PRIN 2010/11 (2010NYKNS7_002) and FIRB 2011 (RBAP11CZLK) to AMV and SB; and by “Special Program Molecular Clinical Oncology 5x1000” to Associazione Italiana per la Ricerca sul Cancro (http://www.airc.it/english/) Gruppo Italiano Malattie Mieloproliferative (http://www.progettoagimm.it/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: The 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2007;22: 14–22. [DOI] [PubMed] [Google Scholar]
- 2. Vannucchi AM, Guglielmelli P, Tefferi A. Advances in Understanding and Management of Myeloproliferative Neoplasms. 2009;59: 171–191. [DOI] [PubMed] [Google Scholar]
- 3. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. 2005;365: 1054–1061. [DOI] [PubMed] [Google Scholar]
- 4. Jones AV, Kreil S, Zoi K, Waghorn K, Curtis C, Zhang L, et al. Widespread occurrence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood. 2005;106: 2162–2168. [DOI] [PubMed] [Google Scholar]
- 5. James C, Ugo V, Le Couédic J, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434: 1144–1148. [DOI] [PubMed] [Google Scholar]
- 6. Kralovics R, Passamonti F, Buser AS, Teo S, Tiedt R, Passweg JR, et al. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N Engl J Med. 2005;352: 1779–1790. [DOI] [PubMed] [Google Scholar]
- 7. Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB, et al. Identification of an Acquired JAK2 Mutation in Polycythemia Vera. J Biol Chem. 2005;280: 22788–22792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klampfl T, Harutyunyan A, Berg T, Gisslinger B, Schalling M, Bagienski K, et al. Genome integrity of myeloproliferative neoplasms in chronic phase and during disease progression. Blood. 2011;118: 167–176. 10.1182/blood-2011-01-331678 [DOI] [PubMed] [Google Scholar]
- 9. Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27: 1861–1869. 10.1038/leu.2013.119 [DOI] [PubMed] [Google Scholar]
- 10. Vannucchi AM, Biamonte F. Epigenetics and mutations in chronic myeloproliferative neoplasms. Haematologica. 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369: 2391–2405. 10.1056/NEJMoa1312542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369: 2379–2390. 10.1056/NEJMoa1311347 [DOI] [PubMed] [Google Scholar]
- 13. Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell. 2010;17: 584–596. 10.1016/j.ccr.2010.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen E, Beer PA, Godfrey AL, Ortmann CA, Li J, Costa-Pereira A, et al. Distinct Clinical Phenotypes Associated with JAK2V617F Reflect Differential STAT1 Signaling. 2010;18: 524–535. 10.1016/j.ccr.2010.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116: 281–297. [DOI] [PubMed] [Google Scholar]
- 16. Eichhorn S, Guo H, McGeary S, Rodriguez-Mias R, Shin C, Baek D, et al. mRNA Destabilization Is the Dominant Effect of Mammalian MicroRNAs by the Time Substantial Repression Ensues. Mol Cell. 2014;56: 104–115. 10.1016/j.molcel.2014.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. 2012;4: 143–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guglielmelli P, Tozzi L, Pancrazzi A, Bogani C, Antonioli E, Ponziani V, et al. MicroRNA expression profile in granulocytes from primary myelofibrosis patients. Exp Hematol. 2007;35: 1708.e1-1708.e12. [DOI] [PubMed] [Google Scholar]
- 19. Hussein K, Theophile K, Dralle W, Wiese B, Kreipe H, Bock O. MicroRNA expression profiling of megakaryocytes in primary myelofibrosis and essential thrombocythemia. Platelets. 2009;20: 391–400. [DOI] [PubMed] [Google Scholar]
- 20. Guglielmelli P, Tozzi L, Bogani C, Iacobucci I, Ponziani V, Martinelli G, et al. Overexpression of microRNA-16-2 contributes to the abnormal erythropoiesis in polycythemia vera. Blood. 2011;117: 6923–6927. 10.1182/blood-2010-09-306506 [DOI] [PubMed] [Google Scholar]
- 21. Guo S, Lu J, Schlanger R, Zhang H, Wang JY, Fox MC, et al. MicroRNA miR-125a controls hematopoietic stem cell number. 2010;107: 14229–14234. 10.1073/pnas.0913574107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. O'Connell RM, Chaudhuri AA, Rao DS, Gibson WSJ, Balazs AB, Baltimore D. MicroRNAs enriched in hematopoietic stem cells differentially regulate long-term hematopoietic output.. 2010;107: 14235–14240. 10.1073/pnas.1009798107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ooi AGL, Sahoo D, Adorno M, Wang Y, Weissman IL, Park CY. MicroRNA-125b expands hematopoietic stem cells and enriches for the lymphoid-balanced and lymphoid-biased subsets. 2010;107: 21505–21510. 10.1073/pnas.1016218107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu J, Guo S, Ebert BL, Zhang H, Peng X, Bosco J, et al. MicroRNA-Mediated Control of Cell Fate in Megakaryocyte-Erythrocyte Progenitors. 2008;14: 843–853. 10.1016/j.devcel.2008.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kumar MS, Narla A, Nonami A, Mullally A, Dimitrova N, Ball B, et al. Coordinate loss of a microRNA and protein-coding gene cooperate in the pathogenesis of 5q- syndrome. Blood. 2011;118: 4666–4673. 10.1182/blood-2010-12-324715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen C, Li L, Lodish HF, Bartel DP. MicroRNAs Modulate Hematopoietic Lineage Differentiation. Science. 2004;303: 83–86. [DOI] [PubMed] [Google Scholar]
- 27. Zhan H, Cardozo C, Raza A. MicroRNAs in myeloproliferative neoplasms. Br J Haematol. 2013;161: 471–483. 10.1111/bjh.12276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang L, Sankaran VG, Lodish HF. MicroRNAs in erythroid and megakaryocytic differentiation and megakaryocyte-erythroid progenitor lineage commitment. Leukemia. 2012;26: 2310–2316. 10.1038/leu.2012.137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Báez A, Martín-Antonio B, Piruat J, Barbado M, Prats C, Álvarez-Laderas I, et al. Gene and miRNA Expression Profiles of Hematopoietic Progenitor Cells Vary Depending on Their Origin. 2014;20: 630–639. 10.1016/j.bbmt.2014.01.022 [DOI] [PubMed] [Google Scholar]
- 30. Raghavachari N, Liu P, Barb JJ, Yang Y, Wang R, Nguyen QT, et al. Integrated analysis of miRNA and mRNA during differentiation of human CD34+ cells delineates the regulatory roles of microRNA in hematopoiesis. Exp Hematol. 2014;42: 14–27.e2. 10.1016/j.exphem.2013.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bruchova H, Yoon D, Agarwal AM, Mendell J, Prchal JT. Regulated expression of microRNAs in normal and polycythemia vera erythropoiesis. Exp Hematol. 2007;35: 1657–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bruchova H, Merkerova M, Prchal JT. Aberrant expression of microRNA in polycythemia vera. Haematologica. 2008;93: 1009–1016. 10.3324/haematol.12706 [DOI] [PubMed] [Google Scholar]
- 33. Vian L, Di Carlo M, Pelosi E, Fazi F, Santoro S, Cerio AM, et al. Transcriptional fine-tuning of microRNA-223 levels directs lineage choice of human hematopoietic progenitors. 2013;21: 290–301. 10.1038/cdd.2013.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Su R, Lin H, Zhang X, Yin X, Ning H, Liu B, et al. MiR-181 family: regulators of myeloid differentiation and acute myeloid leukemia as well as potential therapeutic targets. Oncogene. 2014. [DOI] [PubMed] [Google Scholar]
- 35. Lin X, Rice KL, Buzzai M, Hexner E, Costa FF, Kilpivaara O, et al. miR-433 is aberrantly expressed in myeloproliferative neoplasms and suppresses hematopoietic cell growth and differentiation. Leukemia. 2013;27: 344–352. 10.1038/leu.2012.224 [DOI] [PubMed] [Google Scholar]
- 36.Slezak S, Jin P, Caruccio L, Ren J, Bennett M, Zia N, et al. Gene and microRNA analysis of neutrophils from patients with polycythemia vera and essential thrombocytosis: down-regulation of micro RNA-1 and -133a. 2009;7. [DOI] [PMC free article] [PubMed]
- 37. Zhan H, Cardozo C, Yu W, Wang A, Moliterno AR, Dang CV, et al. MicroRNA deregulation in polycythemia vera and essential thrombocythemia patients. 2013;50: 190–195. 10.1016/j.bcmd.2012.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Norfo R, Zini R, Pennucci V, Bianchi E, Salati S, Guglielmelli P, et al. miRNA-mRNA integrative analysis in primary myelofibrosis CD34+ cells unveils the role of miR-155/JARID2 axis in abnormal megakaryopoiesis. Blood. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bortoluzzi S, Bisognin A, Biasiolo M, Guglielmelli P, Biamonte F, Norfo R, et al. Characterization and discovery of novel miRNAs and moRNAs in JAK2V617F-mutated SET2 cells. Blood. 2012;119: e120–e130. 10.1182/blood-2011-07-368001 [DOI] [PubMed] [Google Scholar]
- 40. Witkos TM, Koscianska E, Krzyzosiak WJ. Practical aspects of microRNA target prediction. Curr Mol Med. 2011;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS, et al. MicroRNA targets in Drosophila. Genome Biol. 2004;5: R1–R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39: 1278–1284. [DOI] [PubMed] [Google Scholar]
- 43. Gaffo E, Zambonelli P, Bisognin A, Bortoluzzi S, Davoli R. miRNome of Italian Large White pig subcutaneous fat tissue: new miRNAs, isomiRs and moRNAs. Anim Genet. 2014;45: 685–698. 10.1111/age.12192 [DOI] [PubMed] [Google Scholar]
- 44. Reese TA, Xia J, Johnson LS, Zhou X, Zhang W, Virgin HW. Identification of novel microRNA-like molecules generated from herpesvirus and host tRNA transcripts. J Virol. 2010;84: 10344–10353. 10.1128/JVI.00707-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. de Hoon MJ, Taft RJ, Hashimoto T, Kanamori-Katayama M, Kawaji H, Kawano M, et al. Cross-mapping and the identification of editing sites in mature microRNAs in high-throughput sequencing libraries. Genome Res. 2010;20: 257–264. 10.1101/gr.095273.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sdassi N, Silveri L, Laubier J, Tilly G, Costa J, Layani S, et al. Identification and characterization of new miRNAs cloned from normal mouse mammary gland. BMC Genomics. 2009;10: 149-2164-10-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kim J, Cho IS, Hong JS, Choi YK, Kim H, Lee YS. Identification and characterization of new microRNAs from pig. Mamm Genome. 2008;19: 570–580. 10.1007/s00335-008-9111-3 [DOI] [PubMed] [Google Scholar]
- 48. Azuma-Mukai A, Oguri H, Mituyama T, Qian ZR, Asai K, Siomi H, et al. Characterization of endogenous human Argonautes and their miRNA partners in RNA silencing.. 2008;105: 7964–7969. 10.1073/pnas.0800334105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fernandez-Valverde S, Taft RJ, Mattick JS. Dynamic isomiR regulation in Drosophila development. RNA. 2010;16: 1881–1888. 10.1261/rna.2379610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tan GC, Chan E, Molnar A, Sarkar R, Alexieva D, Isa IM, et al. 5' isomiR variation is of functional and evolutionary importance. Nucleic Acids Res. 2014;42: 9424–9435. 10.1093/nar/gku656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jaskiewicz L, Zavolan M. Dicer partners expand the repertoire of miRNA targets. Genome Biol. 2012;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Neilsen CT, Goodall GJ, Bracken CP. IsomiRs- the overlooked repertoire in the dynamic microRNAome. 2012;28: 544–549. 10.1016/j.tig.2012.07.005 [DOI] [PubMed] [Google Scholar]
- 53. Fukunaga R, Han B, Hung J, Xu J, Weng Z, Zamore P. Dicer Partner Proteins Tune the Length of Mature miRNAs in Flies and Mammals. Cell. 2012;151: 533–546. 10.1016/j.cell.2012.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Morin RD, O'Connor MD, Griffith M, Kuchenbauer F, Delaney A, Prabhu A-, et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008;18: 610–621. 10.1101/gr.7179508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cloonan N, Wani S, Xu Q, Gu J, Lea K, Heater S, et al. MicroRNAs and their isomiRs function cooperatively to target common biological pathways. Genome Biol. 2011;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chan Y, Lin Y, Lin R, Kuo H, Thang W, Chiu K, et al. Concordant and Discordant Regulation of Target Genes by miR-31 and Its Isoforms. 2013;8 10.1371/journal.pone.0058169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Langenberger D, Bermudez-Santana C, Hertel J, Hoffmann S, Khaitovich P, Stadler PF. Evidence for human microRNA-offset RNAs in small RNA sequencing data. Bioinformatics. 2009;25: 2298–2301. 10.1093/bioinformatics/btp419 [DOI] [PubMed] [Google Scholar]
- 58. Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gee HE, Camps C, Buffa FM, Colella S, Sheldon H, Gleadle JM, et al. MicroRNA-10b and breast cancer metastasis. Nature. 2008;455: E8–E9. 10.1038/nature07362 [DOI] [PubMed] [Google Scholar]
- 60. Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449: 682–688. [DOI] [PubMed] [Google Scholar]
- 61. Chan M, Liaw CS, Ji SM, Tan HH, Wong CY, Thike AA, et al. Identification of Circulating MicroRNA Signatures for Breast Cancer Detection. 2013;19: 4477–4487. 10.1158/1078-0432.CCR-12-3401 [DOI] [PubMed] [Google Scholar]
- 62. Ouyang M, Li Y, Ye S, Ma J, Lu L, Lv W, et al. MicroRNA Profiling Implies New Markers of Chemoresistance of Triple-Negative Breast Cancer. 2014;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tsukamoto O, Miura K, Mishima H, Abe S, Kaneuchi M, Higashijima A, et al. Identification of endometrioid endometrial carcinoma-associated microRNAs in tissue and plasma. Gynecol Oncol. 2014;132: 715–721. 10.1016/j.ygyno.2014.01.029 [DOI] [PubMed] [Google Scholar]
- 64. Zaravinos A, Radojicic J, Lambrou GI, Volanis D, Delakas D, Stathopoulos EN, et al. Expression of miRNAs Involved in Angiogenesis, Tumor Cell Proliferation, Tumor Suppressor Inhibition, Epithelial-Mesenchymal Transition and Activation of Metastasis in Bladder Cancer. J Urol. 2012;188: 615–623. 10.1016/j.juro.2012.03.122 [DOI] [PubMed] [Google Scholar]
- 65. Huang L, Lin J, Yu Y, Zhang M, Wang H, Zheng M. Downregulation of Six MicroRNAs Is Associated with Advanced Stage, Lymph Node Metastasis and Poor Prognosis in Small Cell Carcinoma of the Cervix. 2012;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wu X, Weng L, Li X, Guo C, Pal SK, Jin JM, et al. Identification of a 4-microRNA Signature for Clear Cell Renal Cell Carcinoma Metastasis and Prognosis. 2012;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Han Y-, Park CY, Bhagat G, Zhang J, Wang Y, Fan J-, et al. microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. J Exp Med. 2010;207: 475–489. 10.1084/jem.20090831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Umbach JL, Strelow LI, Wong SW, Cullen BR. Analysis of rhesus rhadinovirus microRNAs expressed in virus-induced tumors from infected rhesus macaques. Virology. 2010;405: 592–599. 10.1016/j.virol.2010.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bortoluzzi S, Biasiolo M, Bisognin A. MicroRNA-offset RNAs (moRNAs): by-product spectators or functional players? Trends Mol Med. 2011;17: 473–474. 10.1016/j.molmed.2011.05.005 [DOI] [PubMed] [Google Scholar]
- 70. Asikainen S, Heikkinen L, Juhila J, Holm F, Weltner J, Trokovic R, et al. Selective MicroRNA-Offset RNA Expression in Human Embryonic Stem Cells. PLoS One. 2015;10: e0116668 10.1371/journal.pone.0116668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ma H, Wu Y, Choi JG, Wu H. Lower and upper stem-single-stranded RNA junctions together determine the Drosha cleavage site. Proc Natl Acad Sci U S A. 2013;110: 20687–20692. 10.1073/pnas.1311639110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Brooks AN, Kilgour E, Smith PD. Molecular Pathways: Fibroblast Growth Factor Signaling: A New Therapeutic Opportunity in Cancer. 2012;18: 1855–1862. [DOI] [PubMed] [Google Scholar]
- 73. Tiong KH, Mah LY, Leong C. Functional roles of fibroblast growth factor receptors (FGFRs) signaling in human cancers. Apoptosis. 2013;18: 1447–1468. 10.1007/s10495-013-0886-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Katoh M, Nakagama H. FGF Receptors: Cancer Biology and Therapeutics. Med Res Rev. 2014;34: 280–300. 10.1002/med.21288 [DOI] [PubMed] [Google Scholar]
- 75. Tan L, Wang J, Tanizaki J, Huang Z, Aref AR, Rusan M, et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Aoki N, Kimura S, Takiyama Y, Atsuta Y, Abe A, Sato K, et al. The Role of the DAP12 Signal in Mouse Myeloid Differentiation. 2000;165: 3790–3796. [DOI] [PubMed] [Google Scholar]
- 77. Aoki N, Kimura S, Oikawa K, Nochi H, Atsuta Y, Kobayashi H, et al. DAP12 ITAM Motif Regulates Differentiation and Apoptosis in M1 Leukemia Cells. Biochem Biophys Res Commun. 2002;291: 296–304. [DOI] [PubMed] [Google Scholar]
- 78. Gingras M, Lapillonne H, Margolin JF. TREM-1, MDL-1, and DAP12 expression is associated with a mature stage of myeloid development. Mol Immunol. 2002;38: 817–824. [DOI] [PubMed] [Google Scholar]
- 79. Bakker ABH, Baker E, Sutherland GR, Phillips JH, Lanier LL. Myeloid DAP12-associating lectin (MDL)-1 is a cell surface receptor involved in the activation of myeloid cells. 1999;96: 9792–9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chen X, Bai F, Sokol L, Zhou J, Ren A, Painter JS, et al. A critical role for DAP10 and DAP12 in CD8+ T cell-mediated tissue damage in large granular lymphocyte leukemia. Blood. 2009;113: 3226–3234. 10.1182/blood-2008-07-168245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Aird KM, Zhang R. Nucleotide metabolism, oncogene-induced senescence and cancer. Cancer Lett. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Grasso D, Vaccaro MI. Macroautophagy and the oncogene-induced senescence. 2014;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hills S, Diffley J. DNA Replication and Oncogene-Induced Replicative Stress. 2014;24: R435–R444. 10.1016/j.cub.2014.04.012 [DOI] [PubMed] [Google Scholar]
- 84. Romero-Cordoba SL, Salido-Guadarrama I, Rodriguez-Dorantes M, Hidalgo-Miranda A. miRNA biogenesis: Biological impact in the development of cancer. Cancer Biol Ther. 2014;15: 1444–1455. 10.4161/15384047.2014.955442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Winter J, Diederichs S. Argonaute-3 activates the let-7a passenger strand microRNA. RNA Biol. 2013;10: 1631–1643. 10.4161/rna.26424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yuen HF, Chan KK, Grills C, Murray JT, Platt-Higgins A, Eldin OS, et al. Ran is a potential therapeutic target for cancer cells with molecular changes associated with activation of the PI3K/Akt/mTORC1 and Ras/MEK/ERK pathways. Clin Cancer Res. 2012;18: 380–391. 10.1158/1078-0432.CCR-11-2035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Singh CP, Singh J, Nagaraju J. A baculovirus-encoded MicroRNA (miRNA) suppresses its host miRNA biogenesis by regulating the exportin-5 cofactor Ran. J Virol. 2012;86: 7867–7879. 10.1128/JVI.00064-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
Data Availability Statement
Sequencing and expression data have been deposited into NCBI GEO (Series GSE69089).