

Abstract
Purpose:
The treatment of triple-negative breast cancer remains a daunting challenge with the standard-of-care treatments eventually failing due to acquired drug resistance, toxic side effects and the presence of a deregulated immune response. New treatments for overcoming these drawbacks include the use of plant extracts.
Study design:
In this study, the efficacy of betulinic acid, a naturally abundant phytochemical exhibiting anti-inflammatory and anti-proliferative activity, has been evaluated for the treatment of triple-negative breast cancer MDA-MB-231 and MDA-MB-468 cell lines. Furthermore, the ability of betulinic acid to inhibit angiogenesis was also determined.
Results:
Here, we report that betulinic acid was able to inhibit the inflammatory response, inhibit angiogenesis and cause cell cycle arrest ultimately causing apoptosis in triple-negative breast cancer cells. Our findings support that the identification of naturally occurring anti-tumour compounds may provide a chemotherapeutic approach for the treatment of triple-negative breast cancer.
Conclusion:
Overall, our results provide a molecular basis for the ability of betulinic acid to mediate apoptosis, suppress inflammation and inhibit angiogenesis in triple-negative breast cancer cell lines.
Keywords: Triple-negative breast cancer, drug resistance, toxicity, phytochemical, betulinic acid
Introduction
Breast cancer is the most common malignancy in women, accounting for approximately one-third of all female cancers. The aetiology of breast cancer is multifactorial and the period of development can span decades and clinical course is highly variable. Triple-negative breast cancer (TNBC) (oestrogen receptor negative, progesterone receptor negative and human epidermal growth factor receptor 2 (Her2) negative) accounts for 10%–17% of all breast cancers.1 They tend to be more aggressive than other types of breast cancer and have poor prognosis.2 The cause of death of patients with TNBC is often recurrence (30%–40% of cases), which presents as distant metastasis.3 Unfortunately, the anti-tumour efficacy of commonly used chemotherapeutic agents for TNBC, including platinum-based chemotherapy, is limited due to acquired drug resistance and toxicities.4 Therefore, the need for new strategies in the fight against cancer is clearly warranted.
Although cancer is always associated with genetic mutations, extensive evidence has lately emerged indicating that most chronic diseases, including cancer, are also caused by a deregulated inflammatory response.5 The deregulation of the inflammatory response is particularly involved in breast cancer.6 The identification of transcription factors such as NF-κB, AP-1 and STAT3 and their gene products such as tumour necrosis factor (TNF), interleukin-1 (IL-1), interleukin-6 (IL-6) and vascular endothelial growth factor (VEGF) has provided the molecular basis for the role of inflammation in cancer. The activation of these inflammatory pathways has been implicated in the transformation, survival, proliferation, invasion, angiogenesis, metastasis, chemo-resistance and radio-resistance of cancer cells.7
A number of botanical medicines and their isolates have been shown to impact cytokines and inflammatory markers offering a great potential in the fight against cancer by inhibiting the process of carcinogenesis, inducing cell cycle arrest and inhibiting signal transduction pathways. Betulinic acid (BetA) (Figure 1), a pentacyclic triterpene discovered in 1995 from the stem bark of the plant Zizyphus mauritiana, was initially reported to be a melanoma-specific cytotoxic agent.8 Since then, BetA has been found to exhibit a variety of biological and medicinal properties such as anti-bacterial, anti-malarial, anti-inflammatory and anti-cancer activities in addition to the ability to inhibit HIV.9 It has been found to be effective in many cancer cell lines including brain, breast, cervix, colorectal, lung and prostate cancers.10–12
Figure 1.
Structure of betulinic acid (BetA).
In this study, we investigated the cytotoxic effect of BetA on TNBC MDA-MB-231 and MDA-MB-468 cell lines. Furthermore, the anti-inflammatory and anti-angiogenic potential of BetA was also examined. We hypothesized that BetA could induce apoptosis through cell cycle arrest and inhibit the pro-inflammatory response present in the breast carcinoma cell lines and possibly inhibit angiogenesis in human mammary microvascular endothelial cells (HMMECs). The findings of this study suggest that the use of BetA may serve as a therapeutic approach targeting inflammatory factors and cell cycle genes to help prevent the progression and metastasis of breast carcinoma cells.
Materials and methods
Materials
BetA was obtained from Tianjin Zhongxin Pharmaceutical Group Corporation Limited (China; purity >98%). Human breast carcinoma MDA-MB-231 and MDA-MB-468 cell lines were obtained from Foleibao Biological Technology Development Co. Ltd (China).13 HMMECs, endothelial cell medium (ECM), foetal bovine serum (FBS), endothelial cell growth supplement (ECGS) and penicillin/streptomycin (P/S) for cell culture were purchased from ScienCell Research Laboratories (USA). L15, F15 and RPMI 1640 media for cell culture were purchased from Hyclone (USA).
Trypsin (0.25%) and ethylenediaminetetraacetic acid (EDTA; 0.02%) were purchased from Gibco (China), and dimethyl sulfoxide (DMSO) was purchased from Solarbio (China). Matrigel was obtained from BD Biosciences (USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) for MTT assay was purchased from Solarbio (China). RNA was extracted using TRIzol reagent and TRIzol plus RNA purification kit purchased from Tiangen (China). Agarose gel (1%) and real-time polymerase chain reaction (RT-PCR) kit were obtained from Takara (China). Fibronectin (FN) was obtained from Sigma (USA). Dulbecco’s Phosphate-Buffered Saline (DPBS) was obtained from Hyclone (USA).
Cell culture and drug preparation
Human breast carcinoma cell lines MDA-MB-231 and MDA-MB-468 were recovered in 25-cm2 tissue culture flasks in an incubator at 37°C in a humidified atmosphere consisting of 5% CO2 and 95% air (Forma 3110; Thermo, USA). The cells were maintained in logarithmic growth phase in complete medium consisting of F15, 10% FBS and 1% P/S. Once the cells were 80% confluent, they were seeded into appropriate multi-well plates depending on the assay. HMMECs were recovered in 1-mL ECM containing 5% FBS, 1% P/S and 1% ECGS and inoculated in 75-cm2 flasks pre-coated with 150 µL FN and 5 mL DPBS. The cells were maintained in culture medium and incubated at 37°C in a 5% CO2 incubator (Forma 3110; Thermo, USA) and the medium was changed every second day. After 90% confluency was reached, subculture was initiated. The medium was removed and the cells were washed gently once with D-Hank’s liquid without calcium and magnesium. The cells were resuspended in dissociation solution made from equal volume of 0.25% trypsin and 0.02% EDTA. The suspension was centrifuged at 1000 r/min for 5 min. The supernatant was removed followed by resuspending the cells in ECM made from 5% FBS, 1% P/S and 1% ECGS. The cells were then seeded in culture plates depending on the assays used. For the culture of breast stromal cells, breast cancer tissue of size 1 cm3 was trimmed to 1 mm3 after removing fatty tissue. The tissue was digested in culture medium containing 100 IU/mL collagenase and 150 IU/mL hyaluronidase for 12 h in 37°C incubator. Cells were passed through an 80-mesh screen, centrifuged at 80 g and filtered for 6 min. Finally, the cells were resuspended in high-glucose ECM containing 5% FBS, 1% P/S and inoculated in a flask at 37°C. BetA was first dissolved in DMSO and then diluted to the required concentrations with milli-Q (mQ) water. DMSO was used to enhance the solubility of the extracts with a final concentration of 3%. The concentration used in our experiments showed good solubility in cell culture medium and 3% DMSO. This was further verified microscopically with the absence of BetA precipitates.
Drug cytotoxicity
MDA-MB-231, MDA-MB-468 and HMMECs were seeded at a density of 5 × 104 cells/mL in flat-bottomed 96-well culture plate in FBS-free F15 culture medium and incubated for 24 h at 37°C in a humidified atmosphere. Treatments with BetA ranged between concentrations of 0.625 and 160 µg/mL depending on the cell type followed by the addition to equal volumes of cell culture into six wells per concentration. The plate was left to incubate for 24 h. Images of the cells and observations on the morphological changes after drug treatments were obtained using an inverted phase-contrast microscope (DC300F; Leica, Germany). The inhibition of cell growth was determined using the MTT assay. After 4 h of the addition of 200 µL of MTT (0.5 mg/mL), the supernatant was discarded and the yellow formazen crystals produced were dissolved in 200 µL/well of DMSO. Optical density (OD) values were determined using a microplate reader set at an absorbance wavelength of 570 nm on FlexStation 3 multi-functional microplate station (Molecular Devices, USA). The activity of BetA against the HMMECs and breast carcinoma cell lines MDA-MB-231 and MDA-MB-468 was evaluated according to OD values. The inhibitory rates were calculated using the following formula
The messenger RNA expression of inflammatory factors and cell cycle–related genes
RT-PCR was used to determine the expression of inflammatory and cell cycle–related genes in human breast carcinoma cell lines MDA-MB-231 and MDA-MB-468 following treatment with BetA. The messenger RNA (mRNA) expression levels of VEGF and basic fibroblast growth factor (bFGF) in HMMECs were also determined using RT-PCR to identify the effect of BetA on angiogenesis. MDA-MB-231 and MDA-MB-468 cells were seeded at a density of 5 × 104 cells/mL, and HMMECs were seeded at 1 × 105 cells/mL in a flat-bottomed 12-well culture plate in L15 culture medium containing 10% FBS and 1% P/S. The cells were incubated under normal growth conditions until the cells were 80% confluent. When in co-culture with breast stromal cells, the plate was put into transwell sleeve containing stromal cells. BetA at 1, 10 and 20 µg/mL was added to triplicate wells. After 24 h of incubation, the cellular RNA was isolated using TRIzol reagent and TRIzol plus RNA purification kit (Invitrogen, USA). RNA concentration was determined by the measurement of OD with the nucleic acid protein analyser at 260 nm on Nucleic acid spectrometer (Du530; Beckman, USA) and OD260/OD280 > 1.8 ensured high-quality RNA. The degree of RNA degradation was analysed by electrophoresis; 4.5 µL RNA was mixed with 0.5 µL sample buffer and the sample was electrophoresed on 1% agarose gel. The degree of RNA degradation was determined using Bio Imaging System (Syngene, UK). Then, the mRNA expression levels of inflammatory factors TNF-α, TLR4, NF-κB1, IL-6, STAT3, HIF1A and i-NOS and cell cycle–related genes Cipl/P21, Kipl/p27, CDK2, CDK6 and cyclin Dl with β-actin used as an internal control were determined using RT-PCR (Tables 1 and 2). The reverse transcription reaction mixture (final volume of 20 µL) was prepared according to the RT-PCR kit protocol. Amplification was carried out in ABI 7500 Real Time PCR System using a single incubation at 95°C for 15 min, followed by 40 cycles at 95°C for 10 s and 60°C for 32 s.
Table 1.
Sequence of RT-PCR primers of inflammatory genes.
| Prime | Sequence | Product (bp) |
|---|---|---|
| β-Actin | Forward primer: AGAGCTACGAGCTGCCTGAC | 184 |
| Reverse primer: AGCACTGTGTTGGCGTACAG | ||
| TNF-α | Forward primer: CCTGTGAGGAGGACGAACAT | 240 |
| Reverse primer: AGGCCCCAGTTTGAATTCTT | ||
| TLR4 | Forward primer: CCATAAAAGCCGAAAGGTGA | 159 |
| Reverse primer: CTGAGCAGGGTCTTCTCCAC | ||
| NF-κB1 | Forward primer: TCGTTTCCGTTATGTATGT | 227 |
| Reverse primer: CCTTGGGTCCAGCAGTTA | ||
| IL-6 | Forward primer: AGGAGACTTGCCTGGTGAAA | 180 |
| Reverse primer: CAGGGGTGGTTATTGCATCT | ||
| STAT3 | Forward primer: TGTGCGTATGGGAACACCTA | 170 |
| Reverse primer: AGAAGGTCGTCTCCCCCTTA | ||
| HIF1A | Forward primer: GAAAACTTGGCAACCTTGGA | 194 |
| Reverse primer: ATCTCCGTCCCTCAACCTCT | ||
| i-NOS | Forward primer: CTCTATGTTTGCGGGGATGT | 179 |
| Reverse primer: TTCTTCGCCTCGTAAGGAAA |
RT-PCR: real-time polymerase chain reaction.
Table 2.
Sequence of RT-PCR primers of cell cycle–related genes.
| Prime | Sequence | Product (bp) |
|---|---|---|
| β-Actin | Forward primer: AGAGCTACGAGCTGCCTGAC | 184 |
| Reverse primer: AGCACTGTGTTGGCGTACAG | ||
| p21 | Forward primer: TTAGCAGCGGAACAAGGAGT | 225 |
| Reverse primer: GCCGAGAGAAAACAGTCCAG | ||
| P27 | Forward primer: CGCTTTGTTTTGTTCGGTTT | 221 |
| Reverse primer: TCTCTGCAGTGCTTCTCCAA | ||
| CDK2 | Forward primer: GCCCTAATCTCACCCTCTCC | 211 |
| Reverse primer: AAGGGTGGTGGAGGCTAACT | ||
| CDK6 | Forward primer: AGCCCAAGATGACCAACATC | 181 |
| Reverse primer: AGGTCAAGTTGGGAGTGGTG | ||
| Cyclin Dl | Forward primer: GAGGAAGAGGAGGAGGAGGA | 231 |
| Reverse primer: AGAGATGGAAGGGGGAAAGA |
RT-PCR: real-time polymerase chain reaction.
Analysis of mRNA expression using the 2−ΔΔCT method
The analysis of mRNA expression was previously described using the 2−ΔΔCT.14 The mean mRNA expression level for the three measurements was calculated. The 2−ΔΔCT method was used to calculate relative changes in mRNA expression determined from RT-PCR experiments. In this study, the data are presented as the fold change in target gene expression in breast carcinoma cells treated with BetA normalized to the internal control gene (β-actin) and relative to the untreated breast carcinoma cells. The results of the RT-PCR data were represented as CT values, where CT was defined as the threshold cycle number of PCRs at which amplified product was first detected. The average CT was calculated for both the target genes and β-actin and the ΔCT was determined as the mean of the triplicate CT values for the target gene − the mean of the triplicate CT values for β-actin. The ΔΔCT represented the difference between the paired tissue samples, as calculated by the formula ΔΔCT = (ΔCT of treated cells − ΔCT of untreated cells). The fold differential expression in the target gene of treated cells compared to the untreated cells was expressed as 2−ΔΔCT. In this study, increased mRNA expression was defined as fold ≥2.0, normal expression was a fold ranging from 0.51 to 1.99 and decreased mRNA expression was fold ≤0.5.
Cell cycle kinetics
Breast carcinoma MDA-MB-231 and MDA-MB-468 cell lines were seeded at a density of 1 × 104 cells/well in flat-bottomed 12-well culture plate in the FBS-free L15 culture medium. The upper chambers of the transwell plate coated with Matrigel were inoculated with stromal cells. The plate was then incubated at 37°C for 24 h in a humidified atmosphere to allow the cells to attach. Treatments of cells with BetA were applied in triplicates. Followed by incubating the plate under normal growth conditions for 48 h (80% confluence), the cells were simultaneously digested with 0.25% trypsin and 0.02% EDTA. Then, the cell suspension was centrifuged for 5 min at 1000 r/min. The supernatant was then discarded, followed by resuspending the cell pellet with D-Hank’s solution and centrifuged for 5 min at 1000 r/min. The supernatant was discarded leaving 0.5 mL and passed through a 200-mesh. The cells were then fixed in ice-cold 70% ethanol for 24 h at 4°C. The cells were centrifuged for 5 min at 1000 r/min, washed with PBS and resuspended in 1 mL PBS. A volume of 5 µL of RNAse (10 mg/mL) was added, followed after gentle mixing and 1 h of incubation at 37°C by the addition of 1 mL propidium iodide (PI; 100 µg/mL) solution. The mixed cells were incubated in the dark at room temperature for 30 min. Progression of cells through the cell cycle and cell apoptosis were examined by flow cytometry where 1 × 104 cells were counted with the instrument set on cell apoptosis analysis.
Tube formation assay
HMMECs, breast carcinoma cell lines and stromal cells were resuspended in ECM made from 5% FBS, 1% P/S and 1% ECGS and transferred into the coated flasks at 7.5 × 103 cells/cm2. The morphology and number of tube-like formations of HMMEC cells were assessed using an inverted phase-contrast microscope (DC300F; Leica, Germany) coupled to a digital camera. Three groups were set based on the cells tested; HMMECs alone, HMMECs in co-culture with stromal cells and HMMECs in co-culture with breast carcinoma cell lines. When HMMECs were treated alone, 1 × 105 cells/well were seeded in a 24-well plate pre-coated with 300 µL/well Matrigel for 30 min. When in co-culture with breast carcinoma MDA-MB-231 or MDA-MB-468 cell lines or stromal cells, 5 × 105 cells/well HMMECs were seeded in the lower chamber of 12-hole transwell plate also coated with Matrigel and 5 × 105 cells/well MDA-MB-231, MDA-MB-468 or stromal cells were seeded in the upper chamber of transwell plate; 1 µg/mL BetA was added to triplicate wells. Preliminary experiments using concentrations of 1, 5 and 10 µg/mL showed that 5 and 10 µg/mL induced great cell death (results not shown). The number of tube-like structures formed was observed after 6, 12, 24 and 36 h.
Statistical analysis
Each experiment was repeated at least three times and presented as mean ± standard deviation (SD). Statistical comparisons were carried out by analysis of variance (ANOVA) using SPSS 13.0 software; p < 0.05 was considered statistically significant.
Results
The effect of BetA on the morphology of MDA-MB-231 and MDA-MB-468 cell lines
To determine whether BetA, structure shown in Figure 1, had anti-tumour activity against breast cancer, breast carcinoma MDA-MB-231 and MDA-MB-468 cell lines and HMMECs were treated with BetA for 24 h. Microscopic examination of the treated cells using an inverted phase-contrast microscope revealed that the lower concentrations of BetA had no significant effects on cell number or morphology when compared to the untreated control cells. In fact, the proliferation of HMMECs was found to be enhanced following treatment with the low 0.625 µg/mL concentration of BetA. However, this cell line showed sensitivity to Betulinic with a marked degree of inhibition of cell proliferation observed with treatments greater than 2.5 µg/mL. HMMECs appeared to be more susceptible to the treatment with BetA with cell kill occurring at much lower concentrations compared to breast carcinoma MDA-MB-231 and MDA-MB-468 cell lines. BetA at concentrations of 10 and 20 µg/mL and higher caused marked degree of cell kill and produced significant reduction in cell number and morphological changes in breast carcinoma MDA-MB-231 and MDA-MB-468 cell lines, respectively. Compared to the control, cells treated with these concentrations appeared to lose their characteristic spindle shape and necrotic cells, and cell debris were also observed (Figure 2).
Figure 2.

The effect of increasing concentrations of BetA on the cellular morphology of breast carcinoma MDA-MB-231 and MDA-MB-468 cell lines. Images were taken using an inverted phase-contrast microscope set at 40× magnification.
BetA: betulinic acid.
The cytotoxicity of BetA against HMMECs and MDA-MB-231 and MDA-MB-468 cell lines
The cytotoxic effect of increasing concentrations of BetA in HMMECs and the two breast carcinoma cell lines MDA-MB-231 and MDA-MB-468 shown in Figure 3 was used for the determination of its IC50 values, that is, the concentration required for 50% cell kill. Table 3 gives the IC50 values of BetA for the three cell lines. HMMECs found earlier to be more sensitive to BetA had a much lower IC50 (4.6 µM) compared to MDA-MB-231 and MDA-MB-468 cell lines. BetA was twice more active in MDA-MB-231 (21.9 µM) compared to MDA-MB-468 cell line. BetA was least active against MDA-MB-468 with an IC50 value of 46.0 µM.
Figure 3.
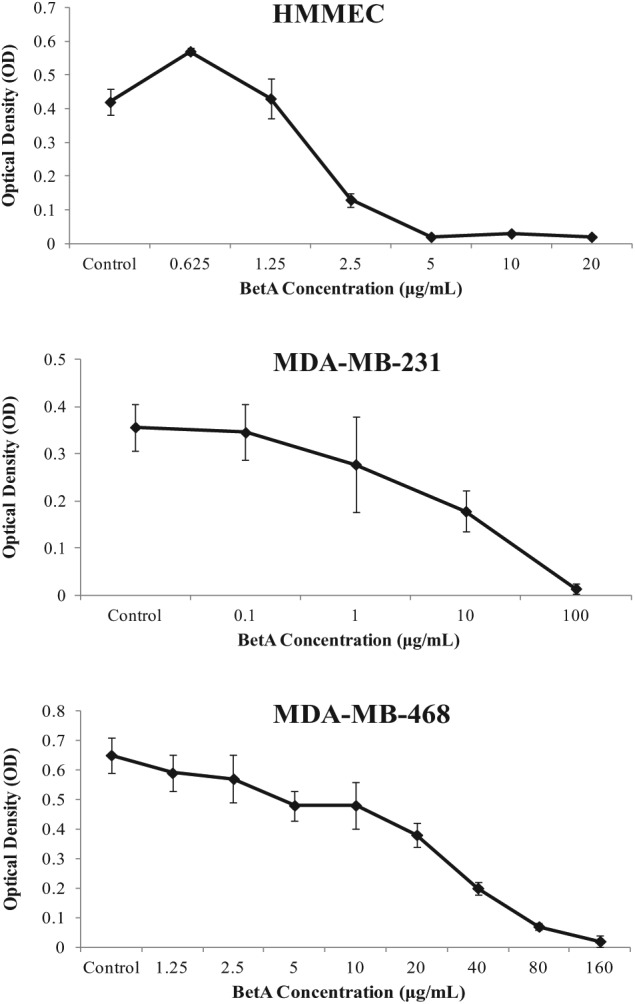
The effect of BetA on the proliferation of HMMECs, MDA-MB-231 and MDAMB- 468 cell lines. Cell proliferation was determined in hexaplicates after 24 h, using the MTT assay. Each point represents the mean of independent experiments with vertical bars representing the standard deviation. *p < 0.05 for comparison of optical density for that given concentration compared to the control indicates statistical significance.
HMMEC: human mammary microvascular endothelial cell; BetA: betulinic acid; MTT: 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
Table 3.
IC50 values (µM) of BetA against HMMECs and breast carcinoma MDA-MB-231 and MDA-MB-468 cell lines.
| Cell line | IC50 |
|---|---|
| HMMEC | 4.6 |
| MDA-MB-231 | 21.9 |
| MDA-MB-468 | 46.0 |
HMMEC: human mammary microvascular endothelial cell.
The effect of BetA on the mRNA expression of inflammatory factors in breast carcinoma cell lines
The involvement of inflammatory factors in cancer prompted the investigation of the inflammatory response to BetA in the breast carcinoma cell lines. The effect of 1, 10 and 20 µg/mL BetA on the mRNA expression of TNF-α, TLR4, NF-κB1, HIF1A, IL-6, STAT3 and i-NOS was determined in breast carcinoma MDA-MB-231 and MDA-MB-468 cell lines cultured alone and in co-culture with breast stromal cells (Table 4). The treatment of MDA-MB-231 cells with 1 µg/mL BetA lowered the expression of all inflammatory factors except for the expression of IL-6 remaining at a normal level. The 10 µg/mL BetA concentration found to inhibit 50% of MDA-MB-231 cells lowered the expression of TLR4, STAT3 and i-NOS and had no effect on the expression of TNF-α, NF-kB1 and HIF1A but increased the expression of IL-6. The highest concentration of BetA had generally no effect on the expression of TNF-α, NF-kB1, HIF1A, STAT3 and i-NOS, though further lowered the expression of TLR4, while increasing IL-6 expression. The treatment of MDA-MB-231 co-cultured with breast stromal cells with 5 µg/mL BetA lowered the expression of IL-6, whereas the expression of the other inflammatory factors remained normal. The results indicate that BetA was effective in blocking inflammation by the down-regulation of the expression of inflammatory factors such as TNF-α, i-NOS and NF-κB1. Interestingly, it appears that the lowest concentration of BetA (1 µg/mL) was the most effective at lowering the expression of the inflammatory factors tested.
Table 4.
The effect of BetA on mRNA expression levels of inflammatory factors in breast cancer MDA-MB-231 and MDA-MB-468 cell lines and their co-culture with breast stromal cellsa.
| Cells | BetA concentration (µg/mL) | TNF-α | TLR4 | NF-κB1 | HIF1A | IL-6 | STAT3 | i-NOS |
|---|---|---|---|---|---|---|---|---|
| MDA-MB-231 | 1 | 0.03 | 0.13 | 0.31 | 0.40 | 1.67 | 0.20 | 0.34 |
| 10 | 1.25 | 0.32 | 0.52 | 0.56 | 5.20 | 0.25 | 0.48 | |
| 20 | 0.69 | 0.15 | 0.79 | 0.92 | 10.84 | 0.53 | 0.88 | |
| 5b | 1.24 | 1.10 | 1.04 | 0.81 | 0.38 | 0.95 | 0.71 | |
| MDA-MB-468 | 5 | 6.71 | 2.09 | 1.61 | 1.81 | 1.45 | 3.09 | 1.05 |
| 10 | 31.44 | 3.70 | 3.16 | 1.68 | 3.75 | 2.56 | 0.49 | |
| 20 | 167.65 | 6.47 | 5.47 | 2.66 | 16.97 | 9.96 | 0.68 | |
| 5b | 0.43 | 1.04 | 0.91 | 1.34 | 0.75 | 2.21 | 1.05 | |
| 10b | 0.14 | 0.72 | 0.90 | 1.35 | 1.02 | 2.07 | 1.09 |
TNF: tumour necrosis factor; IL-6: interleukin-6.
The values represent fold change in expression of the target gene relative to the internal control gene (β-actin) and untreated control cells.
Breast carcinoma cell line co-cultured with breast stromal cells.
The expression levels of the inflammatory factors in MDA-MB-468 cell line were found to remain normal or increase following the treatment with BetA. Generally, increasing concentrations of BetA increased the expression of the inflammatory factors especially the expression of TNF-α and IL-6. Although emerging evidence has shown TNF-α is one of the major mediators of cancer-related inflammation and acts as a tumour-promoting factor, paradoxically, high doses of human recombinant TNF-α induced haemorrhagic tumour necrosis in tumours when injected locally and repeatedly.15 Interestingly, the expression of TNF-α in MDA-MB-468 cell line markedly increased with increasing concentrations of BetA indicating that apoptosis due to BetA may be mediated by this inflammatory cytokine. Therefore, as TNF-α is part of the extrinsic apoptotic pathway, apoptosis may be triggered through the association and activation of caspases. Further experiments outlining the involvement of caspases are needed to confirm this hypothesis. On the other hand, when MDA-MB-468 cell line is co-cultured with stromal cells, the expression of TNF-α following the treatment with BetA decreases. It appears that BetA is influencing the expression of TNF-α either by decreasing its expression and thus blocking the inflammatory response or, on the contrary, by markedly increasing its expression causing apoptosis.
Effect of BetA treatment on the cell cycle in breast carcinoma cell lines
To examine the possible molecular mechanisms by which BetA induces cell cycle arrest in breast carcinoma MDA-MB-231 and MDA-MB-468 cell lines, BetA-treated cells were analysed for their DNA content by PI staining. Flow cytometry analysis of MDA-MB-231 cells co-cultured with stromal cells detected an increase in G1 DNA content in comparison with the control group. The percentage of cells in the S-phase decreased, whereas the percentage of cells in the G2-phase doubled following BetA treatment. The data suggest that BetA treatment of MDA-MB-231 cell line could lead to the inhibition of DNA synthesis causing the arrest of cells in the G2-phase, eventually restraining the proliferation of cells. However, BetA at the concentrations tested had no significant effect on the cell cycle of MDA-MB-468 co-cultured with stromal cells and in fact may have led to a higher percentage of cells in the S-phase (Table 5 and Figure 4). To elucidate the exact mechanism by which BetA causes cell cycle arrest in MDA-MB-468 cell line, further experiments with higher concentrations of BetA need to be conducted.
Table 5.
The effect of BetA on the cell cycle of breast carcinoma MDA-MB-231 and MDA-MB-468 cell lines co-cultured with breast stromal cells.
| BetA concentration | %G1 | %G2 | %S | |
|---|---|---|---|---|
| MDA-MB-231 | Control | 37.20 ± 3.54 | 11.15 ± 0.50 | 51.70 ± 4.10 |
| 5 µg/mL | 41.10 ± 7.50 | 23.10 ± 16.83 | 35.83 ± 9.40 | |
| MDA-MB-468 | Control | 70.27 ± 4.31 | 23.39 ± 1.32 | 8.76 ± 0.42 |
| 5 µg/mL | 61.91 ± 2.86 | 20.89 ± 4.60 | 15.52 ± 0.65 | |
| 10 µg/mL | 68.50 ± 1.17 | 19.87 ± 2.72 | 11.33 ± 2.02 |
Figure 4.
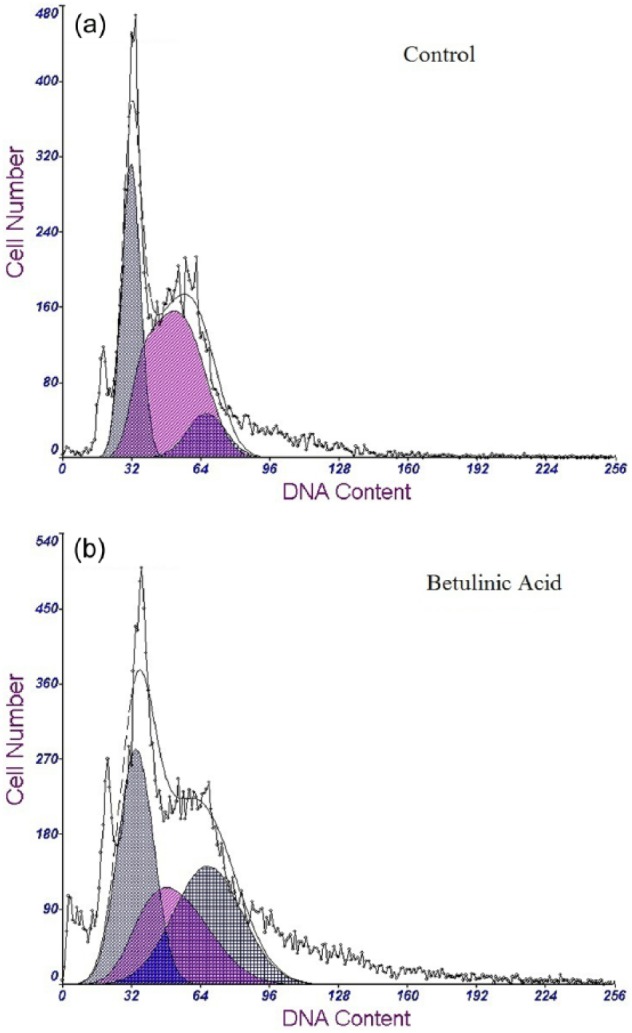
Influence of BetA on cell cycle progression of MDA-MB-231 and MDA-MB-468 cell lines in co-culture with breast stromal cells analysed using flow cytometry. (a) Cell cycle of untreated cells and (b) cell cycle is altered after the treatment of MDA-MB-231 with 5 µg/mL BetA.
BetA: betulinic acid.
The effect of BetA on the mRNA expression of cell cycle–related genes in breast carcinoma cell lines
To further determine the effect of BetA on the cell cycle progression, the mRNA expression pattern of cell cycle genes of breast carcinoma MDA-MB-231 and MDA-MB-468 cells was examined. The treatment of MDA-MB-231 cell line with 1 and 10 µg/mL BetA lowered the expression of P21, P27, CDK2, CDK6 and Cyclin Dl, whereas 20 µg/mL BetA lowered the expression of p27 and CDK6 only. BetA treatment of MDA-MB-231 cell line co-cultured with stromal cells lowered the expression of p21, p27 and Cyclin D1. BetA treatment of MDA-MB-468 cell line alone and in co-culture with stromal cells generally appeared to increase the expression of the genes with increasing concentration (Table 6).
Table 6.
The effect of BetA on the mRNA expression levels of cell cycle genes in breast carcinoma MDA-MB-231 and MDA-MB-468 cell lines and their co-culture with breast stromal cellsa.
| BetA concentration (µg/mL) | p21 | P27 | CDK2 | CDK6 | Cyclin Dl | |
|---|---|---|---|---|---|---|
| MDA-MB-231 | 1 | 0.26 | 0.10 | 0.32 | 0.12 | 0.36 |
| 10 | 0.42 | 0.14 | 0.42 | 0.12 | 0.40 | |
| 20 | 0.74 | 0.29 | 0.69 | 0.25 | 1.12 | |
| 5b | 0.19 | 0.32 | 2.22 | 1.07 | 0.23 | |
| MDA-MB-468 | 5 | 1.67 | 7.60 | 2.32 | 3.54 | 1.85 |
| 10 | 2.49 | 4.68 | 0.73 | 5.16 | 2.59 | |
| 20 | 3.45 | 10.21 | 4.13 | 5.06 | 2.99 | |
| 5b | 0.55 | 2.44 | 1.21 | 3.15 | 1.91 | |
| 10b | 6.27 | 6.70 | 1.07 | 2.80 | 0.95 |
The values represent fold change in expression of the target gene relative to the internal control gene (β-actin) and untreated control cells.
Breast carcinoma cell line co-cultured with breast stromal cells.
The effect of BetA on angiogenesis and the mRNA expression of VEGF and bFGF
When plated on Matrigel, HMMECs alone and its co-culture with the breast carcinoma cells or breast stromal cells underwent rapid reorganization and formed tube-like structures. The treatment of HMMEC alone and its co-culture with the breast carcinoma cell lines with 1 µg/mL BetA had no significant inhibitory effect on the Matrigel-induced network formations over the course of treatment (only images of HMMECs co-cultured with MBA-MB-231 cells are shown in Figure 5(b)). In contrast, 1 µg/mL BetA treatment of HMMECs co-cultured with breast stromal cells caused significant inhibition of networks formed following 6, 12, 24 and 36 h of incubation (Figure 6). These findings further confirm that BetA can modify HMMEC functions using a low BetA concentration and suggest that it might indeed prevent the process of angiogenesis.
Figure 5.

The effect of BetA on the formation of tube-like structures in (a) HMMECs, (b) HMMECs co-cultured with MDA-MB-231 breast carcinoma cell line and (c) HMMECs co-cultured with breast stromal cells. Images were taken using an inverted phase-contrast microscope set at 40× magnifications. The effect of BetA was determined following 6, 12, 24 and 36 h of incubation.
HMMEC: human mammary microvascular endothelial cell; BetA: betulinic acid.
Figure 6.

The effect of BetA on the inhibition of tube-like structure formation in HMMEC co-cultured with breast stromal cells. *p < 0.05 for the comparison of the tubes formed at that time point compared to the control indicates statistical significance.
BetA: betulinic acid.
To further determine the mechanism behind the anti-angiogenic activity of BetA in HMMECs, the mRNA expression levels of VEGF and bFGF were determined using RT-PCR. Compared to the control group, the mRNA expression levels of VEGF and bFGF following 1 µg/mL BetA treatment were 98.7% and 4.8%, respectively. The results indicate that the anti-angiogenic activity of BetA may be mediated via the inhibition of bFGF rather than VEGF.
Discussion
In this study, the efficacy of BetA in HMMEC and triple-negative breast carcinoma MDA-MB-231 and MDA-MB-468 cell lines was investigated. The rationale behind this idea is that plant products have been used for the treatment of human diseases including cancer for thousands of years due to their wide range of biological properties. The interplay between the induction of cell cycle arrest, inhibition of angiogenesis, overcoming multidrug resistance (MDR) and boosting the immune system is a pivotal approach in the fight against cancer.16 Initially identified for its anti-melanoma activity, BetA has recently been reported to be a promising agent for the treatment of multiple forms of cancer including those of the lung, colorectum, prostate, cervix and brain.10,11
Emerging studies also demonstrate the effectiveness of BetA in the treatment of breast cancer. Treatment of ErbB2-overexpressing BT474 and MDA-MB-453 breast cancer cells with 1–10 mmol/L BetA was found to inhibit cell growth, induce apoptosis, down-regulate specificity protein (Sp) transcription factors Sp1, Sp3 and Sp4 and decrease expression of ErbB2.17 BetA decreased proliferation and induced apoptosis of MDA-MB-231 cells through Sp1, Sp3 and Sp4 down-regulation accompanied by increased zinc finger ZBTB10 expression, a putative Sp-repressor and decreased microRNA-27a levels, a microRNA involved in the regulation of ZBTB10.18 In MCF-7 cells, microencapsulated BetA extracted from jujube fruits was able to induce apoptosis via the mitochondria transduction pathway in a dose-dependent manner and arresting cell cycle in the G2/M-phase.19
The current results further show the efficiency of BetA in the treatment of TNBC MDA-MB-231 and MDA-MB-468 cell lines. BetA also proved to be potent against HMMECs, microvascular epithelial cell of breast cancer tissue, cells that are responsible for the formation of new blood vessels. Increasing concentrations of BetA induced cell kill in MDA-MB-231, MDA-MB-468 and HMMEC cell lines in a concentration-dependent manner. BetA has been reported to suppress inflammation and modulate the immune response by interfering with NF-κB activation triggered by inflammation.20 Indeed, BetA was found to suppress the inflammatory mechanisms of MDA-MB-231 and MDA-MB-468 cell lines. At the concentrations tested, it was most effective in blocking inflammation in MDA-MB-231 cell line through down-regulating the expression of NF-κB1, TNF-α, TLR4, STAT3 and i-NOS. Interestingly, it appears that the lowest concentration of BetA (1 µg/mL) was the most effective at lowering the expression of the inflammatory factors tested. The efficacy of BetA in lowering the inflammatory response of a MDA-MB-468 culture alone was not significant. BetA was found to cause marked overexpression of TNF-α in MDA-MB-468 cells cultured alone, whereas the expression of this inflammatory cytokine was down-regulated in MDA-MB-468 cells co-cultured with breast stromal cells. This supports the idea that the role of TNF-α in cancer is complex with both pro-tumourigenic and anti-tumourigenic roles.21,22 Here, we propose that BetA is directly influencing the expression of TNF-α either by decreasing its expression and thus blocking the inflammatory response or, on the contrary, by markedly increasing its expression causing apoptosis.
Besides blocking the inflammatory response in these cell lines, the mechanism of BetA-induced cell cycle arrest was investigated. BetA has been reported to down-regulate cyclin A2 resulting in cell cycle G2/M arrest in lung cancer.23 BetA caused G2/M cell cycle arrest leading to apoptosis in AGS cells in vitro.24 On the other hand, G1 cell cycle inhibition due to treatment with BetA has been reported in cultured vascular smooth muscle cells.25 In this study, BetA was found to lower the percentage of S-phase cells and increase the percentage of G2-phase cells thus causing G2-phase cell cycle arrest in MDA-MB-231 cell line co-cultured with breast cancer stromal cells. Although the mRNA expression of cell cycle genes in BetA-treated MDA-MB-231 cell line was determined, the results were not conclusive. BetA lowered the expression of both the G1 cell cycle inhibitors p21 and p27 and the G1 cell cycle activators CDK2, CDK6 and cyclin D1 in MDA-MB-231 cell line. Although CDK2 is expressed in G1-, S- and G2-phases, it is difficult to predict precisely the phase in which the arrest of the cell cycle has occurred. Other cyclins and CDKs prominent in S- and G2-phases such as cyclins A and E may be explored to confirm that the activity of BetA predominately affects cells in the S- and G2-phases of the cell cycle. Thus, determining the expression of other cyclins may help elucidate the exact mechanism of BetA-induced cell cycle arrest in MDA-MB-231 cells. In the MDA-MB-468 cell line, the concentrations of BetA used in this experiment appeared to increase the mRNA expression of G1-phase inhibitors p21 and p27 and activators CDK2, CDK6 and cyclin D1. Flow cytometric analysis did not show significant arrest of the cell cycle. This may be partly due to the use of concentrations that are lower than the IC50 of BetA against this cell line, thus higher concentrations may be necessary to provide a better understanding of the mechanism of BetA-induced cell cycle arrest in MDA-MB-468 cell line.
The anti-tumour activity of BetA is multifactorial, with possible anti-angiogenic properties.26,27 While in this study BetA had no significant anti-angiogenic effect in HMMECs cultured alone, BetA inhibited angiogenesis in HMMECs co-cultured with breast stromal cells. Two factors associated with angiogenesis are bFGF and VEGF.28,29 Interestingly, the mRNA expression levels of VEGF and bFGF in this study indicated that the BetA inhibited angiogenesis through the inhibition of bFGF rather than VEGF. The expression of bFGF was lowered to 4.8% of the control group, whereas the expression of VEGF remained unaffected. This is in contrary to previous reports suggesting BetA causes the down-regulation of VEGF in MDA-MB-231 cell line.18 In support of the present finding, increased level of bFGF in serum is associated with the presence of breast cancer.30 The down-regulation of bFGF may inhibit the formation of new blood vessels, therefore reducing the blood supply to the tumour microenvironment. Thus, the inhibition of bFGF may further enhance the effectiveness of BetA in breast cancer. Finally, the selective cytotoxicity of BetA against tumour cell lines but not normal cells makes it a great candidate for combination therapies.31, 32
In vitro experiment results cannot fully represent in vivo efficacy; in order to verify if it has in vivo anti-tumour effect and whether it is toxic to normal tissue cells, further animal experiments will be carried out in the following article. Experiments in animals revealed that BetA also has an anti-tumour effect in vivo.33 Normal cells seem to tolerate relatively high concentrations of BetA. In vivo studies on the acute toxicity profile of BetA in Kunming mice bearing melanoma showed that 500 mg/kg BetA had no acute or chronic signs of toxicity in mice.8 The lack of toxicity was also apparent in a Hippocratic screen of rats treated with 200 and 400 mg/kg BetA.34 Local injection can be taken into account if it injures normal cells; however, the evidence to date is extremely positive regarding BetA as a novel anti-proliferation agent in breast cancer cells.
Conclusion
In summary, BetA proved to be a potent anti-inflammatory, anti-proliferative and anti-angiogenic agent against breast carcinoma MDA-MB-231 and MDA-MB-468 cell lines. Given the fact that BetA treatment in vitro has been effective against TNBC, further studies are clearly warranted to determine its effect in vivo. The effectiveness of BetA shows that it may be highly effective and has great potential in combination with conventional chemotherapeutic regimes, therefore possibly enhancing the inhibition of tumour proliferation, inflammation and angiogenesis.
Acknowledgments
D.W. and M.Z. contributed equally to this article.
Footnotes
Declaration of conflicting interests: The authors declare that there is no conflict of interest.
Funding: This project was funded by a grant from CHM/RD P.L. sponsored by the Peoples Republic of China, serial no. ‘10JCZDJC21300’.
References
- 1. Zhang H, Cohen A, Krishnakumar S, et al. Patient-derived xenografts of triple-negative breast cancer reproduce molecular features of patient tumors and respond to mTOR inhibition. Breast Cancer Res 2014; 16(2): R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carey LA, Dees EC, Sawyer L, et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res 2007; 13(8): 2329–2334. [DOI] [PubMed] [Google Scholar]
- 3. Ueno NT, Zhang D. Targeting EGFR in triple negative breast cancer. J Cancer 2011; 2: 324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Oakman C, Viale G, Di Leo A. Management of triple negative breast cancer. Breast 2010; 19(5): 312–321. [DOI] [PubMed] [Google Scholar]
- 5. Weber D, Wheat JM, Currie GM. Inflammation and cancer: tumor initiation, progression and metastasis, and Chinese botanical medicines. J Chinese Integr Med 2010; 8(11): 1006–1013. [DOI] [PubMed] [Google Scholar]
- 6. Pierce BL, Ballard-Barbash R, Bernstein L, et al. Elevated biomarkers of inflammation are associated with reduced survival among breast cancer patients. J Clin Oncol 2009; 27(21): 3437–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res 2009; 15(2): 425–430. [DOI] [PubMed] [Google Scholar]
- 8. Pisha E, Chai H, Lee I-S, et al. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nat Med 1995; 1(10): 1046–1051. [DOI] [PubMed] [Google Scholar]
- 9. Moghaddam MG, Ahmad FBH, Samzadeh-Kermani A. Biological activity of betulinic acid: a review. Pharmacol Pharm 2012; 3: 119–123. [Google Scholar]
- 10. Kessler JH, Mullauer FB, de Roo GM, et al. Broad in vitro efficacy of plant-derived betulinic acid against cell lines derived from the most prevalent human cancer types. Cancer Lett 2007; 251(1): 132–145. [DOI] [PubMed] [Google Scholar]
- 11. Fulda S, Jeremias I, Steiner HH, et al. Betulinic acid: a new cytotoxic agent against malignant brain-tumor cells. Int J Cancer 1999; 82(3): 435–441. [DOI] [PubMed] [Google Scholar]
- 12. Damle AA, Pawar YP, Narkar AA. Anticancer activity of betulinic acid on MCF-7 tumors in nude mice. Indian J Exp Biol 2013; 51(7): 485–491. [PubMed] [Google Scholar]
- 13. Cailleau R, Olivé M, Cruciger QJ. Long-term human breast carcinoma cell lines of metastatic origin: preliminary characterization. In Vitro 1978; 14(11): 911–915. [DOI] [PubMed] [Google Scholar]
- 14. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001; 25(4): 402–408. [DOI] [PubMed] [Google Scholar]
- 15. Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer 2009; 9(5): 361–371. [DOI] [PubMed] [Google Scholar]
- 16. Parekh H, Liu G, Wei M. A new dawn for the use of traditional Chinese medicine in cancer therapy. Mol Cancer 2009; 8(1): 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu X, Jutooru I, Lei P, et al. Betulinic acid targets YY1 and ErbB2 through cannabinoid receptor-dependent disruption of microRNA-27a:ZBTB10 in breast cancer. Mol Cancer Ther 2012; 11(7): 1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mertens-Talcott SU, Noratto GD, Li X, et al. Betulinic acid decreases ER-negative breast cancer cell growth in vitro and in vivo: role of Sp transcription factors and microRNA-27a:ZBTB10. Mol Carcinog 2013; 52(8): 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun Y-F, Song C-K, Viernstein H, et al. Apoptosis of human breast cancer cells induced by microencapsulated betulinic acid from sour jujube fruits through the mitochondria transduction pathway. Food Chem 2013; 138(2–3): 1998–2007. [DOI] [PubMed] [Google Scholar]
- 20. Takada Y, Aggarwal BB. Betulinic acid suppresses carcinogen-induced NF-κB activation through inhibition of IκBα kinase and p65 phosphorylation: abrogation of cyclooxygenase-2 and matrix metalloprotease-9. J Immunol 2003; 171(6): 3278–3286. [DOI] [PubMed] [Google Scholar]
- 21. Mocellin S, Rossi CR, Pilati P, et al. Tumor necrosis factor, cancer and anticancer therapy. Cytokine Growth Factor Rev 2005; 16(1): 35–53. [DOI] [PubMed] [Google Scholar]
- 22. Lejeune FJ, Liénard D, Matter M, et al. Efficiency of recombinant human TNF in human cancer therapy. Cancer Immun 2006; 6: 6. [PubMed] [Google Scholar]
- 23. Hsu T-I, Wang M-C, Chen S-Y, et al. Betulinic acid decreases specificity protein 1 (Sp1) level via increasing the sumoylation of sp1 to inhibit lung cancer growth. Mol Pharmacol 2012; 82(6): 1115–1128. [DOI] [PubMed] [Google Scholar]
- 24. Yuan J, Lovejoy DB, Richardson DR. Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity: in vitro and in vivo assessment. Blood 2004; 104(5): 1450–1458. [DOI] [PubMed] [Google Scholar]
- 25. Vadivelu RK, Yeap SK, Ali AM, et al. Betulinic acid inhibits growth of cultured vascular smooth muscle cells in vitro by inducing arrest and apoptosis. Evid Based Complement Alternat Med 2012; 251362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dehelean CA, Feflea S, Ganta SMA. Anti-angiogenic effects of betulinic acid administered in nanoemulsion formulation using chorioallantoic membrane assay. J Biomed Nanotechnol 2011; 7(2): 317–324. [DOI] [PubMed] [Google Scholar]
- 27. Mukherjee R, Jaggi M, Rajendran P, et al. Betulinic acid and its derivatives as anti-angiogenic agents. Bioorg Med Chem Lett 2004; 14(9): 2181–2184. [DOI] [PubMed] [Google Scholar]
- 28. Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev 2004; 25(4): 581–611. [DOI] [PubMed] [Google Scholar]
- 29. Presta M, Dell’Era P, Mitola S, et al. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev 2005; 16(2): 159–178. [DOI] [PubMed] [Google Scholar]
- 30. Granato A, Nanni O, Falcini F, et al. Basic fibroblast growth factor and vascular endothelial growth factor serum levels in breast cancer patients and healthy women: useful as diagnostic tools? Breast Cancer Res 2004; 6(1): R38–R45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zuco V, Supino R, Righetti SC, et al. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett 2002; 175(1): 17–25. [DOI] [PubMed] [Google Scholar]
- 32. Cichewicz RH, Kouzi SA. Chemistry, biological activity, and chemotherapeutic potential of betulinic acid for the prevention and treatment of cancer and HIV infection. Med Res Rev 2004; 24(1): 90–114. [DOI] [PubMed] [Google Scholar]
- 33. Bache M, Zschornak MP, Passin S, et al. Increased betulinic acid induced cytotoxicity and radiosensitivity in glioma cells under hypoxic conditions. Radiat Oncol 2011; 6: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sandberg F, Dutschewska H, Christov V, et al. Spondianthus preussii var. glaber Engler. Pharmacological screening and occurrence of triterpenes. Acta Pharm Suec 1987; 24(5): 253–256. [PubMed] [Google Scholar]