

Abstract
Increasingly mechanistic virology studies require dependable and sensitive methods for isolating purified organelles containing functional cellular sub-domains. The mitochondrial network is, in part, closely apposed to the endoplasmic reticulum (ER). The mitochondria-associated membrane (MAM) fraction provides direct physical contact between the ER and mitochondria. Characterization of the dual localization and trafficking of human cytomegalovirus (HCMV) UL37 proteins required establishing protocols in which the ER and mitochondria could be reliably separated. Because of its documented role in lipid and ceramide transfer from the ER to mitochondria, a method to purify MAM from infected cells was also developed. Two robust procedures were developed to efficiently isolate mitochondria, ER, and MAM fractions while providing substantial protein yields from HCMV-infected primary fibroblasts and from transfected HeLa cells. Furthermore, this unit includes protocols for isolation of detergent resistant membranes from subcellular fractions as well as techniques that allow visualization of the mitochondria network disruption that occurs in permissively infected cells by their optimal resolution in Percoll gradients.
Keywords: subcellular fractionation, human fibroblasts, ER, mitochondria, MAM, HCMV, protein localization, sucrose gradient, Percoll gradient, differential centrifugation
INTRODUCTION
Fractionation, the mechanical separation and purification of subcellular compartments, is an invaluable tool for studying protein localization, trafficking, processing, and functions. Subcellular fractionation is also useful for the concentration of relatively low-abundance proteins, isolation of enzymatic complexes, or proteomic identification of organelle components. Increasingly mechanistic analyses of cellular biology and virology require dependable and sensitive methods for isolating purified organelles and tethered organelles from which functional subcellular domains, such as translocation complexes, lipid rafts, and viral envelopment sites, can be studied. Moreover, functional contacts between distinct organelles have been identified and fractionated. For example, the contact sites between endoplasmic reticulum (ER) and mitochondria are being characterized as sites for exchange of calcium (Bononi et al., 2012; Marchi et al., 2014; Rizzuto et al., 2004) and lipids (Vance, 2014) between these organelles, as well as defining locations for mitochondrial division (Friedman et al., 2011). In all, ~5% to 20% of the mitochondrial network surface within a cell is in close apposition to the ER (Rizzuto et al., 1998). The mitochondria-associated membrane (MAM) fraction, a subdomain of the ER, which consists of membrane tubules that provide direct physical contact between the ER and mitochondria, has been fractionated to high purity (Vance, 1990). Importantly, the purified MAM fraction is enriched in lipid synthetic enzymes, which produce phosphatidylserine and ceramide, and transport these products from the ER into mitochondria (Ardail et al., 2003; Bionda et al., 2004; Stone and Vance, 2000).
The authors’ laboratory investigates human cytomegalovirus (HCMV) UL37 proteins, which dually target the ER and mitochondria of transfected and of HCMV-infected human foreskin fibroblasts (HFFs) (Colberg-Poley et al., 2000; Colberg-Poley and Williamson, 2013; Goldmacher et al., 1999; Hayajneh et al., 2001; Mavinakere and Colberg-Poley, 2004a; Mavinakere and Colberg-Poley, 2004b; Williamson and Colberg-Poley, 2010). The authors found that HCMV UL37 proteins traffic sequentially from the ER into the mitochondrial outer membrane (Bhuvanendran et al., 2014; Colberg-Poley et al., 2015; Mavinakere et al., 2006). Characterization of the subcellular localization and trafficking of HCMV UL37 proteins required establishing a protocol in which ER and mitochondria could be reliably separated from one another, despite their prevalent interconnection. Because of the documented role of MAM in lipid and ceramide transfer from the ER to mitochondria, the authors also sought to highly purify the MAM from transfected and from infected cells, adapting an established protocol from Vance (1990). The Vance protocol was originally used for the isolation of microsomes, mitochondria, and MAM from rat liver tissue (Vance, 1990). The following protocols were developed to maximally isolate mitochondria, ER, and MAM fractions while providing the best protein recoveries from HCMV-infected HFFs and from transfected, non-permissive HeLa cells. Basic Protocol 2 has proved especially valuable because it allows visualization of the mitochondria network disruption that occurs in permissively infected cells (Bozidis et al., 2010; McCormick et al., 2003; Zhang et al., 2011) by their resolution in Percoll gradients. Basic Protocol 3 allows for the isolation of detergent resistant membrane (DRM) fractions from crude ER or crude mitochondrial fractions. This procedure can also act as an alternative method for enriching MAM detergent resistant membranes, without the need for Percoll gradients.
BASIC PROTOCOL 1: DIFFERENTIAL SUCROSE GRADIENT ISOLATION OF ER AND MITOCHONDRIA
This protocol utilizes discontinuous sucrose gradients to band purified ER and mitochondrial organelles. Initially, cells are lysed mechanically with sonication and, then, a low-speed centrifugation (700 × g) is used to remove large cellular debris. Supernatant from this step is collected as a total lysed protein fraction. A subsequent 15,000 × g centrifugation crudely pellets mitochondria and separates it from ER and other organelles. The supernatant is loaded onto a three-layered sucrose gradient and purified ER is banded by centrifugation at 152,000 × g. Separately, the pellet is washed and loaded onto a two-layer sucrose gradient and purified mitochondria are banded using a 40,000 × g centrifugation. The high protein yields and considerable purity of banded organelles makes this fractionation of great utility for studies involving ER- or mitochondrial-resident proteins. The critical steps are shown in Figure 3.27.1.
Figure 3.27.1.
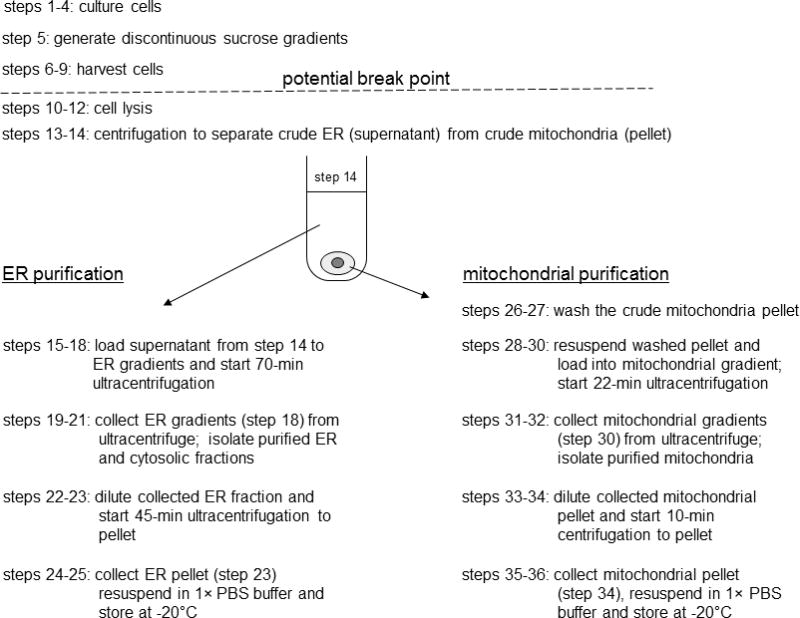
A flow chart for Basic Protocol 1 is shown. Basic Protocol 1, step 14, separates crude ER (supernatant) from crude mitochondria (pellet). Subsequent steps are grouped by the organelle which is to be purified for clarity and to provide a sense of continuity. To streamline the timing of the procedure and to reduce protein degradation; however, ER and mitochondrial purification steps should be carried out simultaneously.
Materials
Human foreskin fibroblasts (HFFs; Viromed SF cells)
HeLa cells (ATCC CCL-2)
HCMV (desired strain) or DNA for transfection
2% and 10% (v/v) FBS
Lipofectamine 2000 (Invitrogen; UNIT 20.6)
Opti-MEM I (Invitrogen)
1.0, 1.3, 1.5, 1.7, and 2.0 M sucrose solutions (see recipes), sterile
1× PBS, pH 7.4 (APPENDIX 2A)
0.25% trypsin/EDTA (Life Technologies, cat. no. 25200114)
1× mannitol/Tris/EDTA (1× MTE) buffer (see recipe)
100 mM PMSF stock (see recipe)
Ultrapure water
70% ethanol
175-cm2 flasks (~0.8–2 × 107 cells/flask)
37°C CO2 incubator
Pre-sterilized (autoclaved) Beckman polyallomer centrifuge tubes: 14 × 89–mm (cat. no. 331372) or 11 × 60–mm (cat. no. 328874)
5-ml serological pipets
Aspirator
Sterile 15-ml conical tubes
Beckman GS-6R centrifuge with GH-3.8 swinging-bucket rotor
Analog sonicator with 1/8-in. microtip (Branson Ultrasonics model 250)
250-ml glass beakers
1.5-ml microcentrifuge tubes
14-ml polypropylene, round-bottom, snap-cap tubes (17 × 100–mm; Falcon cat. no. 2059)
Beckman J2-MI centrifuge with JA20.1 rotor
Beckman XL-90 ultracentrifuge with SW60 Ti and SW41 Ti rotors
1-ml syringes and 20-G needles
Parafilm
Beckman GS-15R centrifuge with F2402 rotor
Culture cells
-
1
For HCMV infection, seed two 175-cm2 flasks of 60% to 70% density, actively growing primary HFFs. For transfection, seed two 175-cm2 flasks of HeLa cells, 25% to 30% density and actively growing cells.
For best results, use HFFs at or less than passage 18 as primary HFFs undergo senescence following 50 cell doublings or ~25 passages.
Transfer viral DNA to cells
To infect HFF cells with HCMV
-
2a
Twenty-four hr after seeding, infect permissive HFFs with HCMV (strain AD169 or desired strain), for 1 hr at 37°C in low-serum (2% FBS) medium, at a multiplicity of three plaque forming units (pfu) per cell (Mavinakere and Colberg-Poley, 2004a).
A cell density of ~80% the day of infection is best for maximal virus infectivity, as HCMV grows better in actively growing cells. -
3a
After 1 hr, remove virus inocula by suction and overlay cells with complete medium (10% FBS). Proceed to step 4.
To transfect HeLa cells
-
2b
Transfect HeLa cells with Lipofectamine 2000 (UNIT 20.6) according to manufacturer’s protocol. For each 175-cm2 flask, use 20 μg DNA, 35 μl Lipofectamine 2000, and a total of 8.8 ml Opti-MEM I.
-
3b
Proceed to step 4.
A cell density of 80% to 90% is critical for maximal transfection efficiency using Lipofectamine 2000. If it is necessary to attain the correct cell density, incubate cultures for an extra day after seeding before transfecting.Lipofectamine 2000 was found to give reliably higher transfection efficiencies in HeLa cells and HFFs than either Oligofectamine or Lipofectamine. -
4
Incubate HCMV-infected cells according to the temporal class of examined viral protein. Allow transfected cells to incubate 24 hr at 37°C.
Depending on the abundance of the protein of interest or the kinetics of its movement through the cell, the incubation time may be adjusted to anywhere between 8 to 120 hr.
Prepare discontinuous sucrose gradients
-
5a
For mitochondria gradients. At room temperature, dispense 1 ml of 1.7 M sucrose into a sterile 11 × 60–mm Beckman polyallomer ultracentrifuge tube. Mark the top of sucrose layer on the outside of the tube with an indelible felt-tip marker. Using a 5-ml serological pipet, carefully overlay with 1.6 ml of 1.0 M sucrose.
Hold the tube at a slight angle and slowly add top layer, to prevent sucrose layers from mixing. -
5b
For ER gradients. At room temperature, dispense 2 ml of 2.0 M sucrose to the bottom of a sterile 14 × 89–mm Beckman polyallomer ultracentrifuge tube. Using a 5-ml serological pipet, slowly layer 3 ml of 1.5 M sucrose onto the 2.0 M sucrose. Overlay with 3 ml of 1.3 M sucrose on top of the gradient.
The 14 × 89–mm polyallomer tubes used for ER gradients fit snugly into a polypropylene holder/dryer rack with 102 drying pins (made for 10- to 13-mm tubes).Use the discontinuous sucrose gradients within 10 hr of preparation, for best results. The fine demarcation between sucrose layers may fade rapidly, but gradients remain stable at room temperature for several hours.
Harvest cells
-
6
Remove medium from cells by aspiration and wash the monolayers with 10 ml of 1× PBS, pH 7.4. Add 2 ml of 0.25% trypsin/EDTA and incubate 5 min at 37°C.
-
7
Inactivate the trypsin by adding 8 ml complete medium (10% FBS) to each flask and resuspend cells by vigorous pipetting. Transfer the resuspended cells from each flask into separate, sterile 15-ml conical tubes.
-
8
Pellet cells by centrifuging 5 min at 200 × g (1,000 rpm in tabletop Beckman GS-6R centrifuge), 4°C. Aspirate the supernatant and resuspend the cell pellet in 10 ml of 1× PBS, pH 7.4.
-
9
Centrifuge cell suspension 5 min at 1,400 × g (2,500 rpm in tabletop Beckman GS-6R centrifuge), 4°C. Remove the supernatant by aspiration and store the cell pellet on ice (average pellet size is ~0.145 g).
Cell pellets are stable on ice for up to 2 hr. Alternatively, the cell pellets can be frozen up to 1 month at −80°C at this stage and saved for later processing.
Lyse cells
-
10
Dispense 15 ml of 1× MTE and add fresh PMSF to a final concentration of 1 mM. Keep solution on ice. Add 2 ml of this solution to each 15-ml conical tube (step 9), and resuspend cell pellets.
-
11
Wash the sonicator microtip with ultrapure water and then with 70% ethanol. Blot dry with a Kimwipe before use. Set the sonicator for continuous pulse on a power setting of 3.5.
-
12
Place the 15-ml conical tube with resuspended cells into a 250-ml glass beaker partially filled with a slurry of ice and cold water. Submerge the cleaned, dry sonicator tip into the cell suspension to just above the bottom of the tube, avoiding touching the bottom. Sonicate the cell suspension on ice three times for 10 sec each, separated by 10-sec rest intervals. Keep the lysed cells on ice.
Cell pellets from each flask should be kept separately up to this point to maximize the lysis efficiency. In step 14, however, combine the lysed cells from two 175-cm2 flasks to maximize protein yields during fractionation.The 10-sec lysis condition had the best combination of high protein yields and fraction purity. If one wishes to sacrifice protein yield to obtain maximal purity of subcellular fractions, Basic Protocol 2 is recommended using homogenization lysis. -
13
Centrifuge the lysed cells in 15-ml conical tubes 10 min at 1,400 × g (2,500 rpm in a tabletop Beckman GS-6R centrifuge), 4°C. Collect 100 μl of supernatant from each 15-ml conical tube, pool together duplicate samples into a single 1.5-ml microcentrifuge tube and label as “total protein.” Store immediately at –20°C.
Depending upon the cell lysis procedure, a pellet may not be seen at this step (corresponding to large cellular debris). A large pellet can be seen after gentle Dounce homogenization, while no pellet is usually visualized after long sonication times (see Table 3.27.1). -
14
Decant remaining supernatant into a sterile, pre-chilled 14-ml polypropylene round-bottom snap-cap tube. Combine material from both 175-cm2 flasks into a common chilled 14-ml tube. Centrifuge tubes without a snap-cap in the inner row of a JA20.1 rotor 10 min at 15,000 × g, 4°C.
Use only polypropylene tubes for this procedure, as polystyrene tubes will crack and leak because of centrifugal stress.This centrifugation separates crude ER (supernatant) from crude mitochondria (pellet). Subsequent steps are grouped by the organelle, which is to be purified for clarity and to provide a sense of continuity. To streamline the timing of the procedure and to reduce protein degradation; however, ER and mitochondrial purification steps should be carried out simultaneously.
Table 3.27.1.
Mitochondrial and ER Yields and Purity from Differentially Lysed HeLa Cells
| Average
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Lysis method | Yield samples | Initial cell pellet (g) | Total protein fraction (mg) | Average Mitochondria yield (μg) | ER yield (μg) | Yield | MAM detected in | Purity |
| Sonication | ||||||||
| Low (3 × 5 sec) | 1, 2 | 0.153 | 9.87 | 415.86 | 842.6 | High | ER + mitochondria | Good |
| Medium (3 × 10 sec) | 1, 2 | 0.148 | 9.584 | 350.34 | 824.4 | High | ER | Good |
| High (3 × 15 sec) | 1, 2 | 0.146 | 8.778 | 163.14 | 820.5 | Medium | ER | Good |
| Freeze/thaw | 1, 2 | 7.634 | ND | 195.2 | Medium | ER + mitochondria | Good | |
| Homogenization | 1, 2 | 0.146 | 1.484 | NDa | NDa | Low | MAM/mitochondria | Excellent |
| 3, 4 | 0.147 | 1.392 | NDa | NDa | Requires large scale-up | Mitochondria | ||
ND, not determined as extract protein concentrations were below detection level of the BCA assay.
Load ER gradient for ER purification
-
15
After centrifugation, transfer tubes to ice.
There should be a large yellowish-brown pellet containing mitochondrial proteins. -
16
Using a micropipet, carefully withdraw 1.7 ml of the supernatant (containing crude ER) and layer it onto the top of the ER sucrose gradient, creating a new layer. Pipet slowly to keep from mixing the top layer of sucrose with the sample. Decant and discard any excess supernatant remaining in the 14-ml tubes containing the protein pellet, and then return the pellets to ice.
-
17
Transfer an additional 1.3 ml of ice-cold 1× MTE plus PMSF onto the top of the ER gradient.
-
18
Ultracentrifuge ER gradients 70 min at 152,000 × g (35,000 rpm in an SW41 rotor), 4°C. Set acceleration and deceleration profiles to 1 (transition speed of 170 rpm for 2 min).
It is important to add the extra 1× MTE plus PMSF buffer on top of the samples to keep the polyallomer tubes from collapsing during ultracentrifugation.
Isolate ER fractions
-
19
Collect the ER gradient tubes from the ultracentrifuge. Withdraw the upper 1 ml of solution from the tube, using a micropipet, and transfer into a sterile 1.5-ml microcentrifuge tube. Label as “cytosol” and store immediately at –20°C.
-
20
Extract 0.4 to 0.6 ml volume of the large band at the interface of the 1.3 M sucrose gradient layer, using a 20-G needle and 1-ml syringe (Fig. 3.27.2).
The samples can be stored for 1 to 2 months at –20°C prior to their use for western analysis. -
21
Remove the needle from the syringe, and transfer the extracted band to a sterile 11 × 60–mm Beckman polyallomer tube. Add an additional 3.6 to 3.8 ml of ice-cold 1× MTE plus PMSF buffer to dilute out the sucrose.
It is important to remove the needle from the syringe before dispensing liquid into the new ultracentrifuge tube, to avoid shear forces that would damage the sample. -
22
Cover the top of the tube with Parafilm and mix by inversion until the suspension looks homogeneous (until the highly viscous sucrose swirling within the tube is no longer distinguishable).
-
23
Ultracentrifuge 45 min at 126,000 × g (35,000 rpm in an SW60 Ti rotor), 4°C.
-
24
Collect tube from ultracentrifuge. Decant and discard the supernatant.
There will be a large, translucent pellet at the bottom of the tube. -
25
Allow tubes to dry, inverted, for a few minutes. Resuspend the pellet in 100 μl of 1× PBS, pH 7.4, and label as “ER.” Store immediately at –20°C.
The samples can be stored for 1 to 2 months at –20°C prior to their use for western analysis.
Figure 3.27.2.

Representative pictures of visible bands seen upon ultracentrifugation as described in Basic Protocol 1. The ER and mitochondria bands are indicated on their respective gradients following ultracentrifugation.
Load mitochondria gradient for mitochondrial purification
-
26
For the mitochondrial pellet wash 1, return to the 14-ml tube on ice containing the protein pellets (step 16). Gently wash the inside sides of the tube with 0.5 ml of ice-cold 1× MTE plus PMSF, being careful not to disrupt the protein pellet. Wash the sides three to five times using the same aliquot of buffer. Decant and discard wash solution.
-
27
For mitochondrial pellet wash 2, use a fresh aliquot of 0.5 ml of ice-cold 1× MTE plus PMSF buffer to carefully wash the periphery of the protein pellet, which contains ER contaminant proteins. Wash three to five times using the same aliquot of buffer. Decant and discard the wash.
The ER contaminant proteins make a large, loose ring around the more stable mitochondrial pellet. The mitochondrial pellet will appear more yellow in color, while the surrounding contamination appears whiter in color. A large amount of contaminant protein towards the bottom of the pellet will be seen. -
28
Resuspend the washed mitochondrial pellet in 0.8 ml of ice-cold 1× MTE plus PMSF. Load slowly on top of the mitochondrial sucrose gradient so that it forms a new layer.
-
29
Top off gradient with an additional 0.8 ml of ice-cold 1× MTE plus PMSF buffer.
-
30
Ultracentrifuge the mitochondrial gradient 22 min at 40,000 × g (19,500 rpm in an SW60 Ti rotor), 4°C. Set acceleration and deceleration profiles to 1 (transition speed of 170 rpm for 2 min).
Isolate mitochondrial fractions
-
31
Collect mitochondrial gradients from the ultracentrifuge (step 30). Using a 1-ml syringe with a 20-G needle, extract a volume of 0.4 ml from the band at the interface of the 1.7 M and 1.0 M sucrose layers (Fig. 3.27.2).
The mitochondrial band can vary in visibility and is often very thin and hard to see, which is why marking the location of the interface with an indelible felt tip marker before centrifugation is important. -
32
Remove the needle and dispense the extracted volume into a sterile 1.5-ml microcentrifuge tube.
-
33
Add 1.1 ml of ice-cold 1× MTE plus PMSF, cap microcentrifuge tube, and mix well by inversion until suspension is homogeneous.
-
34
Centrifuge 10 min at 15,000 × g, 4°C (in tabletop Beckman GS-15R centrifuge).
-
35
Decant and discard supernatant (a small, stable yellowish-brown pellet should be visible).
The mitochondrial pellet can sometimes move slightly as you withdraw the supernatant. Do not aspirate the supernatant at this step, as you risk losing some of the pellet. -
36
Allow tube to dry, inverted, for a few minutes. Resuspend pellet in 30 μl of 1× PBS, pH 7.4, and label as “mitochondria.” Store immediately at –20°C.
The samples can be stored for 1 to 2 months at –20°C prior to their use for western analysis.
BASIC PROTOCOL 2: SEPARATION OF MITOCHONDRIA AND MITOCHONDRIA-ASSOCIATED MEMBRANE FRACTION
This procedure combines differential and Percoll gradient centrifugations. Its critical steps are underscored in Figure 3.27.3. During the first steps, the post-nuclear supernatant (PNS) is separated from nuclei and cellular debris by differential centrifugation at low g forces. The post-nuclear supernatant is then subjected to centrifugation at 10,300 × g during which the crude mitochondrial fraction is separated from the total microsomal fraction. The total microsomal fraction consists mainly of vesicles derived from rough and smooth ER and membranes from the Golgi apparatus and plasma membrane. The microsomal fraction is then recovered as a pellet after centrifugation of the total microsomal fraction at 100,000 × g. The post-microsomal (high-speed) supernatant is generally used as “cytosol” in downstream applications. The crude mitochondrial fraction is subjected to a density gradient fractionation through a self-generating Percoll gradient (Pertoft et al., 1978). Because both mitochondria and mitochondria-associated membrane (MAM) have similar densities, a two step purification follows that includes a centrifugation at 6,300 × g and collection of “crude MAM”. Finally, the supernatant containing purified MAM is subjected to centrifugation at 100,000 × g and the MAM is isolated as a pellet above the tight Percoll pellet.
Figure 3.27.3.

Schematic representation of Basic Protocol 2. Critical steps in the procedure, which improve purity of the microsomes, mitochondria, and MAM, are emphasized.
NOTE: All solutions, glassware, centrifuge tubes, and equipment should be pre-cooled to 0°C to 4°C and kept on ice throughout the procedure.
Materials
Untransfected or transfected HeLa cells (3 × 107 cells, ten 100 × 20–mm tissue culture dishes, 100% confluent) or uninfected or HCMV-infected HFF cells (5 × 107 cells, four 850-cm2 roller bottles, 90% confluent; or twenty 100 × 20-mm tissue culture dishes, 70–90% confluent)
Phosphate-buffered saline (PBS; see recipe)
Sucrose homogenization medium (SHM; see recipe), ice cold
Protease inhibitors (optional but recommended, see recipe)
Mannitol buffer A (see recipe), ice cold
30% (v/v) Percoll suspension in mannitol buffer B (see recipe), ice cold
Mannitol buffer B (see recipe), ice cold
Beckman GS-15R tabletop centrifuge with swinging-bucket rotor (e.g., S4180)
14-ml polypropylene round-bottom centrifuge tubes (e.g., Falcon)
2-ml Potter-Elvehjem plastic coated tissue grinder with PTFE pestle (e.g., Wheaton safe-grind type, cat. no. 358003)
Overhead stirrer for tissue grinder (Wheaton, cat no. 903475)
Phase-contrast microscope
1.5-ml microcentrifuge tubes
Beckman GS-15R centrifuge with fixed-angle rotor (e.g., F2402)
Centrifuge tubes, 1.5-ml microcentrifuge tubes
Millipore Amicon Ultra-15 Centrifugal Filter Units with Ultracel-3 membrane (24-pk: Millipore cat. no. 900324)
Beckman XL-90 ultracentrifuge with swinging-bucket rotor (e.g., SW 41)
Ultracentrifuge SW41 tubes (ultraclear tubes are highly recommended, e.g., 14 × 89–mm, cat. no. 344059)
1-ml syringes and 20-G needles
Harvest cells
-
1
Remove medium from cells by aspiration and wash the cell monolayers with 1× PBS, pH 7.4. Use approximately 5 ml PBS per 100-mm dish, or 12 ml per roller bottle. Remove PBS wash by aspiration.
-
2
Harvest cells using either a cell lifter, or by trypsinization, and pool together experimental sets of ten 100-mm dishes of HeLa cells, or twenty 100-mm dishes of HFF cells (can alternatively use 4 roller bottles of HFF cells).
Cell Lifter: Add 5 ml 1× PBS, pH 7.4 to each 100-mm dish and harvest cells with a sterile cell lifter. Combine cells into sterile 50-ml conical tubes.
Trypsinization: Add 1 ml of 0.25% trypsin/EDTA per 100-mm dish, or 5 ml per roller bottle, and incubate 5 min at 37°C. After 5 min, check to see that cells have been adequately lifted off of the culture surface using a brightfield microscope. If not, you may need to agitate cells by firmly hitting the side of the dish/roller bottle, or incubate an additional 5 min at 37°C. Inactivate the trypsin by adding 3 ml of complete medium to each 100-mm dish, or 20 ml to each roller bottle. Combine harvested cells into sterile 50-ml conical tubes, and pellet cells by centrifuging 5 min at 200 × g (1,000 rpm in tabletop Beckman GS-6R centrifuge), 4°C. To ensure all active trypsin is removed from cells wash cell pellet once with 20 ml complete medium and once with 20 ml 1× PBS, pH 7.4, centrifuging as previously described after each wash.
-
3
Resuspend final cell pellet in 2 ml of ice-cold sucrose homogenization medium (SHM) containing protease inhibitors, and keep on ice.
Cellular Homogenization
-
4
Attach the pre-cooled pestle of a 2-ml Potter-Elvehjem tissue grinder set to a motorized overhead stirrer. Set the stirrer to a medium speed (e.g., ~500 rpm). While pestle is turning, wash pestle and glass mortar with 70% ethanol followed by ultrapure water. Dry both briefly with paper towel.
-
5
Transfer the first 1 ml of harvested cells to the glass mortar. Homogenize the cells for 1 min with quick up-and-down motions (45 – 50 pumps of the pestle into the mortar). After 1 min, transfer the homogenized suspension into a pre-cooled 12 × 75-mm polypropylene tube. Repeat homogenization procedure with the second 1 ml of harvested cells, and collect into the same 12 × 75 mm polypropylene tube as before. Repeat this homogenization process for each set of harvested cells in your experiment, to keep all samples at the same stage.
Between experimental samples you will want to clean the pestle and mortar with 70% ethanol followed by ultrapure water. Keep pestle running to save time.Keep mortar on ice between homogenization steps. -
6
Centifuge homogenate at 600 × g for 5 min, 4°C, in capped 12 × 75-mm polypropylene tubes (e.g., in Sorvall Legend RT centrifuge). A large pellet will form. Transfer the supernatant into a 15-ml conical tube labeled “HOMOGENIZED”; this contains fully homogenized cell material.
-
7
To increase yields, the large pellet from step 6 should be subjected to further homogenization. Resuspend the pellet in 2 ml of sucrose homogenization medium. As in step 5, homogenize 1 ml at a time for 1 min and collect in a 12 × 75-mm polypropylene tube labeled “IN PROCESS.”
-
8
Centrifuge homogenate at 600 × g for 5 min, 4°C. Again, you will see a substantial pellet. Transfer supernatant into the “HOMOGENIZED” 15-ml conical tube from step 6. Resuspend the remaining pellet in 2 ml sucrose homogenization medium and repeat cycle (steps 5 and 6).
Although time consuming, repeat homogenization steps on the 600 × g pellets dramatically increases final yields of ER, MAM, and mitochondrial fractions. The authors recommend repeating this homogenization cycle 4–7 times (or until a pellet no longer forms after centrifugation), always collecting the supernatant into a common 15-mL collection tube.After 2–3 homogenization cycles, use 1 ml sucrose homogenization medium to resuspend the 600 × g pellet (instead of 2 ml). This will increase the viscosity of the cell suspension, increase the efficiency of homogenization, and save time.To check the efficiency of the homogenization, pipet 2 to 3 μl of the homogenized suspension onto a glass slide, overlay with coverslip, and observe using a microscope. A shiny ring around the nuclei indicates that cells are still intact. If >90% of the nuclei do not have the shiny ring, proceed to the next step. Otherwise, repeat the homogenization. -
9
After all homogenization is complete, remove 300 μl of sample from the common “HOMOGENIZED” 15-ml conical tube to serve as a “total” fraction. Freeze sample immediately at −80°C.
-
10
Using the graduations on the side of the 15-ml conical tube, note the final volume of your homogenized supernatant. If necessary, add sucrose homogenization buffer to bring to a final volume of at least 8 ml.
If the final volume of homogenized cell material is less than 8 ml, we find that the purity of the microsomal fraction suffers.Optional: It is possible to stop at this step by storing the capped 15-ml conical tubes at 4°C overnight without significant reduction of yield or purity of final fractions.
Fractionation: Pelleting MAM and Mitochondria out of homogenized cells
-
11
Divide cell homogenates from step 10 into 1.5-ml microcentrifuge tubes (you will need at least 6 tubes per homogenate). Centrifuge 10 min at 10,300 × g, 4°C, using a tabletop centrifuge (e.g., Beckman GS-15R) with a fixed-angle rotor (e.g., F2402).
-
12
Transfer the supernatant from step 11 to a new 1.5-ml microcentrifuge tube and repeat 10,300 × g centrifugation (10 min, 4°C). Keep remaining pellet on ice. Repeat this step until no pellet is formed from the 10,300 × g centrifugation (this typically requires 4 or 5 rounds of centrifugation).
During these steps remember to keep all tubes on ice, and do not throw away any material. Here you are pelleting the dense mitochondria and connected MAM membranes out of the cell homogenate. By repeating the centrifugation until no pellet is seen, you will increase the purity of your final microsomal fraction.On the last two centrifugations leave plenty of supernatant behind in the microcentrifuge tube. The pellet is very soft, and you don’t want to accidentally transfer it out with the collected supernatant. -
13
Combine all supernatants from microcentrifuge tubes in step 12 into a common 16 × 76-mm Ultraclear ultracentrifuge tube (Beckman, #344085). From these supernatants you will isolate purified microsomes and cytosolic fractions (steps 15–19).
-
14
Resuspend pellets from the first three centrifugations in steps 11 and 12 (as these will be the biggest pellets) in a total of 1 ml sucrose homogenization medium, and combine in a common 1.5-ml microcentrifuge tube. Keep tubes on ice. From these pellets you will isolate MAM and mitochondrial fractions (steps 20–26).
To reduce protein loss, resuspend pellets initially in a single 700 μl aliquot of sucrose homogenization medium, then wash tubes from which the pellets were resuspended three times with 100 μl fresh sucrose homogenization medium (to a final volume of 1 ml).
Fractionation: Microsome purification
-
15
Using a small (e.g. 50-ml) beaker to hold tubes, balance the microsome/cytosol-containing ultracentrifuge tubes (from step 13) from different experimental samples. Add sucrose homogenization medium as needed to balance tubes.
-
16
Ultracentrifuge mixture at 100,000 × g for 60 min, 4°C (33,000 rpm in a pre-chilled Beckman XL-90 Ultracentrifuge with a Ti70 fixed-angle rotor). The supernatant from this step will be the cytosol fraction, while the pellet will contain purified microsomes.
The fixed-angle Ti70 rotor gives a tighter microsome pellet that sticks well to the ultracentrifuge tube, and better facilitates getting a clean separation of cytosol and microsomal fractions. You can also use a swinging bucket rotor here, such as the SW41 Ti rotor (24,000 rpm for 60 min), but the microsome pellet will be loose and more care should be taken when separating it from the supernatant containing cytosol.To save time, skip ahead to complete steps 20–23 during this hour-long ultracentrifugation. -
17
Collect 4 ml of the supernatant from step 16 and transfer to Millipore Amicon centrifugal fiulter unit with Ultracel-3 membrane (3 kDa MW cutoff). Centrifuge 60 min at 3,000 × g, 4°C (e.g., in Sorvall Legend RT centrifuge).
If necessary, the 4 ml supernatant can be loaded into filter devices and stored overnight at 4°C before concentrating the sample in the centrifuge. -
18
After centrifugation, mix retained sample volume in the concentrator well, being sure to pipette up and down over the filter surface. Transfer 300 μl of this concentrated sample to a sterile 1.5-ml microcentrifuge tube and label as “cytosol.” Store immediately at −80°C.
-
19
From ultracentrifuge tube in step 16, decant off remaining supernatant and resuspend the pellet in 200 μl 1× PBS, pH 7.4. Transfer to sterile 1.5-ml microcentrifuge tube, label as microsomes, and store immediately at −80°C.
Fractionation: Percoll separation of MAM and mitochondria
-
20
Centrifuge combined (1 ml) mixtures of MAM and mitochondria (from step 14) at 10,300 × g for 10 min, 4°C (Beckman GS-15R centrifuge with F2402 rotor).
-
21
Discard supernatant. Add 600 μl of Mannitol Buffer A and pipette up and down ~45 times with a P1000 micropipettor to resuspend the pellet. Using a P200 micropipettor, pipette up and down an additional 35 times. Transfer mixture to glass mortar and homogenize using 30–40 pumps of the motorized pestle (set at medium speed, as before, or slightly lower).
-
22
Dispense 9.5 ml of 30% Percoll suspension into a 14 × 89 Beckman Ultraclear ultracentrifuge tube. Carefully layer homogenized sample from step 21 on top of the 30% Percoll suspension, adding liquid in a dropwise manner to the side of the tube to avoid mixing the two layers. You should see a well-defined interface between these two layers.
-
23
Using a small (e.g. 50-ml) beaker to support the ultracentrifuge tubes, weigh tubes and add Mannitol Buffer A as necessary to balance.
-
24
Ultracentrifuge at 95,000 × g for 65 min, 4°C (23,600 rpm using an SW41 Ti swinging-bucket rotor in a Beckman XL-90 Ultracentrifuge). Set acceleration profile to 3 (transition speed of 500 rpm for 3 min) and deceleration profile to 9 (transition speed of 500 rpm for 6 min).
A total of 1 to 5 mg of protein (~0.5 ml) on 10 ml of gradient material results in satisfactory separation of banded material without overloading the gradient. -
25
After ultracentrifugation, use 20 gauge needles to extract banded MAM and mitochondrial fractions from the Percoll gradient (Figure 3.27.4). To maximize separation of the two fractions, insert one needle into the ultracentrifuge tube just above the banded MAM (to collect this fraction from the top, down) and one just below the banded mitochondria (to collect this fraction from the bottom, up). Withdraw 200 μl of banded MAM, and 2 ml of banded mitochondria.
To avoid leaking, add a small amount of Vaseline or vacuum grease to the shaft of the needle, approximately ¼ inch back from the needle’s tip. While holding the ultracentrifuge tube with your index finger at the top and your thumb at the bottom, gently “drill” the needle into the tube by slowly adding pressure and rotating the needle back and forth. This will minimize agitation of the Percoll gradient and mixing of the banded fractions when the needle finally penetrates through the wall of the tube.Start at the top. Insert the first needle above the compact, white band labeled as fraction 2 in Figure 3.27.4. Extract the indicated volume of banded MAM into an appropriately sized syringe, being careful not to extract material too close to the interface of banded MAM and mitochondria. Leave the syringe and needle in place (stuck in the tube) while you then proceed to insert a second needle below the multi-band mitochondrial fraction (fraction 3 in Figure 3.27.4), and extract the indicated volume of mitochondria being careful not to take material too close to the MAM/mitochondria interface. Once you remove the needles or syringes from the tube, material will immediately start leaking out. Only remove needles and syringes after all fractions have been collected.To avoid excessive shear stress on collected samples, remove the needle from the end of the syringe before dispensing sample into a new sterile collection tube. -
26
Percoll must now be removed from the collected samples, as it will interfere with subsequent analysis of proteins. This requires diluting the collected samples in at least 5 volumes of buffer, and pelleting purified membrane fractions away from the Percoll.
MAM fraction: Dispense the 200 μl MAM fraction, collected in step 25, into a sterile 1.5-ml microcentrifuge tube. Dilute sample in ice-cold 1× PBS, pH 7.4 + 1 mM PMSF to a final volume of 1.2 ml. Mix well by inversion, or with gentle vortexing. Centrifuge at 6,300 × g for 10 min, 4°C (Beckman GS-15R centrifuge with F2402 rotor). Proceed to step 30.
Mitochondria fraction: Dispense the 2 ml mitochondria fraction, collected in step 25, into a sterile 14 × 89-mm Ultraclear ultracentrifuge tube. Dilute sample in ice-cold 1× PBS, pH 7.4 + 1 mM PMSF to a final volume of 10 ml. Seal tube with parafilm and mix well by inversion. Remove parafilm and balance tubes as necessary with dilution buffer. Centrifuge at 6,300 × g for 20 min, 4°C (6,000 rpm in Beckman XL-90 centrifuge with SW41 rotor). Proceed to step 27.
Figure 3.27.4.

Separation of MAM and mitochondrial fractions using self-generating Percoll (30%) gradients. Crude mitochondrial extracts from uninfected or HCMV-infected HFFs were subjected to ultracentrifugation in a Percoll gradient (see Basic Protocol 2). The diagram on the left illustrates the distinct features of banded organelles within the Percoll gradient. Fractions 2 and 3 were collected to isolate purified MAM and mitochondria, respectively. Gradients on the right demonstrate proper banding of MAM and mitochondria in Percoll gradients, as well as highlight the alterations in mitochondrial banding observed during HCMV infection.
Final collection: purified mitochondria
-
27
After centrifugation of the mitochondria fraction from step 26 is complete, pipette off the bulk of the supernatant (you will eventually discard this, but store it on ice temporarily until you have successfully completed the next step), leaving approximately 1 ml of supernatant in the bottom of the ultracentrifuge tube. You may already see a cloudy accumulation of mitochondria as well as a Percoll pellet from this centrifugation. Alternatively, it may take a second dilution before these fractions resolve. Even if you can already distinguish cloudy mitochondria material and a Percoll pellet, a subsequent dilution and centrifugation step increases separation and yield.
-
28
Add 8 ml of ice-cold 1× PBS, pH 7.4 + 1 mM PMSF dilution buffer, to a final volume of 9 ml. Mix well. Resuspend any Percoll pellet and mix with the cloudy mitochondria, if necessary. Centrifuge as before, at 6,300 × g for 20 min, 4°C (6,000 rpm in Beckman XL-90 centrifuge with SW41 rotor).
-
29
After this second centrifugation, you should observe a small, dense Percoll pellet at the bottom of the tube with a cloudy accumulation of mitochondria floating above it. Capture as much of this cloudy accumulation as possible by extracting 50 – 200 μl with a micropipettor. Transfer into a sterile 1.5-ml microcentrifuge tube and label “mitochondria.” Pipette up and down to create a homogenous mixture, and store immediately at −80°C. Dilute final sample in ice-cold 1× PBS, pH 7.4 + 1 mM PMSF dilution buffer, if necessary.
Final collection: purified MAM
-
30
Add 8 ml of ice-cold 1× PBS, pH 7.4 + 1 mM PMSF dilution buffer to a 16 × 76-mm Beckman Ultraclear ultracentrifuge tube, and transfer the MAM fraction supernantant into this tube. Seal tube with parafilm and mix well by inversion. Remove parafilm and balance tubes as necessary with dilution buffer. Proceed to step 32.
-
31
Resuspend the MAM fraction pellet, from the centrifugation in step 26, in 100–200 μl ice-cold 1× PBS, pH 7.4 + 1 mM PMSF dilution buffer and transfer into a sterile 1.5-ml microcentrifuge tube. Label as “6,300 × g MAM”, and store immediately at −80°C.
Depending upon your experimental needs, you may wish to stop the procedure here and just analyze the 6,300 × g MAM. This material contains integral MAM membrane proteins as well as a number of peripherally-associated proteins (eg. Grp75). This pellet is often referred to as “crude MAM” to distinguish it from the more stringently processed MAM sample collected in step 33, but it is by all means a purified, Percoll-banded MAM fraction. This fraction has a much higher protein yield, if that is a consideration for your particular analysis. Moving forward to pellet membranes from the remaining supernatant (as in step 32) will allow you to purify integral membrane MAM constituents away from peripherally-associated proteins. -
32
Centrifuge supernatant from step 30 at 100,000 × g for 30 min, 4°C (33,000 rpm in Beckman XL-90 centrifuge with Ti70 rotor). A loose pellet of MAM will form just above a dense Percoll pellet at the bottom of the ultracentrifuge tube.
-
33
Capture as much of this loose MAM pellet as possible by extracting 50 – 200 μl with a micropipettor. Transfer into a sterile 1.5-ml microcentrifuge tube and label “100,000 × g MAM.” Pipette up and down to create a homogenous mixture, and store immediately at −80°C. Dilute final sample in ice-cold 1× PBS, pH 7.4 + 1 mM PMSF dilution buffer, if necessary.
These fractions may be stored up to 6 months at –80°C.
BASIC PROTOCOL 3: FLOTATION OF DETERGENT-RESISTANT MAM MEMBRANES FROM CRUDE ER OR MITOCHONDRIA FRACTIONS
The MAM subdomains of the ER are enriched in detergent-resistant membrane (DRM)-forming lipids such as cholesterol, shingolipids, and ceramide (Hayashi and Fujimoto, 2010). Consistent with this finding, a number of constituent MAM proteins have been characterized as organized into DRMs in the ER (Browman et al., 2006; Hayashi and Su, 2003; Williamson et al., 2011). Critically, cholesterol appears to regulate MAM-mitochondrial connectivity (Fujimoto et al., 2012), with MAM cholesterol enrichment potentially favoring transient, restrained, and carefully controlled organelle interactions.
The procedure outlined here exploits the ability to easily isolate DRMs from bulk membranes, by flotation through dense sucrose, to purify MAM domains from bulk ER or mitochondria. This represents a powerful technique to quickly probe and analyze proteins within the MAM while avoiding the experimentally demanding Percoll fractionation outlined in Basic Protocol 2. Modified from flotation procedures that use whole cell lysates as starting material (Carman et al., 1999; Song et al., 1996; Williamson et al., 2011), our flotation protocol isolates DRMs from sub-fractionated crude ER/MAM or MAM/mitochondria membranes to help ensure specificity of purified fractions. Recent proteomic profiling of DRMs isolated from crude MAM/mitochondria fractions identified 74% of proteins collected in this way to match known MAM-resident and MAM-associated proteins (Poston et al., 2011).
Materials
Human foreskin fibroblasts (HFFs; Viromed SF cells)
HeLa cells (ATCC CCL-2)
HCMV (desired strain) or DNA for transfection
ER/MAM pellet or MAM/mitochondria pellet from Basic Protocols 1 or 2, respectively
2% and 10% (v/v) FBS
Lipofectamine 2000 (Invitrogen; UNIT 20.6)
Opti-MEM I (Invitrogen)
100 mM PMSF stock (see recipe)
MBS buffer, ice cold (see recipe)
MBST buffer, ice cold (see recipe)
5%, 30%, and 90% sucrose + MBS buffer, ice cold (see recipe)
Ultrapure water
175-cm2 flasks (~0.8–2 × 107 cells/flask)
37°C CO2 incubator
5-ml serological pipets
Pre-sterilized (autoclaved) Beckman polyallomer centrifuge tubes: 11 × 60–mm (cat. no. 328874)
Analog sonicator with 1/8-in. microtip (Branson Ultrasonics model 250)
1.5-ml microcentrifuge tubes
Beckman XL-90 ultracentrifuge with SW60 Ti rotor
Sub-fractionate samples
-
1
Follow fractionation protocols listed above to isolate either an ER/MAM pellet (from Basic Protocol 1, step 24) or a MAM/mitochondria pellet (from Basic protocol 2, step 20).
Solubilize detergent-sensitive membranes
-
2
Thoroughly resuspend above pellet in 0.5 ml pre-chilled MBST buffer (see recipe) by pipetting up and down 10 to 15 times. Incubate 30 min in a slurry of ice and cold water to solubilize detergent-sensitive membranes.
An alternative to 1% Triton X-100 in the MBST buffer is to use 1% Triton X-114, which is better suited to isolate intracellular DRMs and may give higher yields of MAM DRMs (Hayashi and Fujimoto, 2010).When working with infected cells, the authors find that slightly modifying this step aids in experimental reproducibility. Resuspend the pellet in 0.3 ml of MBST and sonicate the suspension three times for 10 sec each. Be very careful not to introduce bubbles at this point. Use 0.2 ml of MBST to wash sonicator tip, bringing the final sample volume to 0.5 ml, then incubate in a slurry of ice and cold water for 30 min. -
3
Dilute solubilized membranes 1:1 in MBS + 90% sucrose by adding 0.5 ml of buffer to resuspended sample, and mix well. Transfer solution to the bottom of a pre-chilled polypropylene 11 × 60 mm SW60 ultracentrifuge tube.
-
4
Flotation gradient. Using a 5-ml serological pipet, carefully overlay with 2 ml of MBS + 30% sucrose. Next, overlay with 1 ml of MBS + 5% sucrose. Balance tubes with MBS + 5% sucrose solution prior to ultracentrifugation.
Hold the tube at a slight angle and slowly add top layer, to prevent mixing of the sucrose layers.The 2 ml 30% sucrose layer is a buffer layer to help separate DRMs, which float through it, from detergent-sensitive membranes which cannot float through it. If you do not get adequate separation of DRMs from detergent-sensitive membranes with the 30% sucrose layer, use 2 ml of a 20% sucrose solution here instead. In our hands, intracellular lipid rafts band in solutions with sucrose composition between 20% – 15% (see Figure 3.27.7A). -
5
Ultracentrifuge gradients 16 hr at 187,813 × g, 4°C, in a swinging bucket rotor (39,000 rpm in an SW60 Ti rotor). Set acceleration profile to 9 (transition speed of 500 rpm for 6 min) and deceleration profile to 1 (transition speed of 170 rpm for 2 min).
Pre-chill the ultracentrifuge and rotor to 4°C prior to use. -
6
Collect 12 fractions, 0.33 ml each. Store immediately at −20°C.
If more than 0.33 ml remains in the last fraction, mix well to homogenize sample and collect all remaining solution and label as fraction 12. It is important to have a set number of fractions for comparison of results between experiments. DRMs would be expected to appear concentrated between fractions 3 to 7 (see Figure 3.27.7B and C).The samples can be stored for 1 to 2 months at −20°C prior to their use for western analysis.
Figure 3.27.7.

Flotation of detergent-resistant MAM membranes from crude ER or mitochondrial fractions (Protocol 3). A. Crude ER or mitochondria were solubilized with MBST buffer and floated through sucrose gradients (as in Basic Protocol 3). Banding of DRMs in discontinuous sucrose gradients is demonstrated on the right. DRMs band in sucrose between 20–15%. To maximize separation from bulk membranes, it is preferable to band DRMs near the top of the sucrose gradient. Basic protocol 3 utilizes a 30% sucrose buffer cushion, as shown in the diagram to the left. A 20% cushion also allows DRMs to band similarly, while increasing the separation from bulk membranes.
B. DRMs from ER/MAM and mitochondria from HCMV infected cells. HFFs were HCMV (strain BADwt)-infected (multiplicity of infection of 3) and ER/MAM and mitochondria were isolated at 48 hrs. Lipid rafts were then isolated from the indicated purified subcellular fractions by cold 1% Triton X-100 extraction and flotation in 5%, 35% and 45% sucrose gradients. Each gradient fraction (20 μl) was resolved by SDS-PAGE and analyzed by Western blots using anti-vMIA/UL37x1 antibody.
C. DRMs from ER/MAM and mitochondria from transfected cells. HeLa cells were transfected with a vector expressing untagged vMIA/pUL37x1 and harvested 36 hr after transfection and ER/MAM and mitochondria were isolated. DRMs were then isolated by extraction with 1% Triton X-100 on ice and flotation in discontinuous 5%, 35% and 45% sucrose gradients. Twelve fractions (330μl) were collected from top of the gradient and aliquots (30 μl) of each were subjected to SDS-PAGE and Western analyses with anti-Grp75 (1:1000, Stressgen), Calnexin (1:500; Santa Cruz); HSP60, Mfn2 (1:200; Cell Signal), Prohibitin (1:500, GeneTEX), VDAC (1:1000, Cell Signal), Erlin2 (1:500, a gift from Dr. Stephen Robbins).
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see APPENDIX 2A; for suppliers, see SUPPLIERS APPENDIX.
Mannitol buffer A
To 100 ml ultrapure H2O, add:
9.13 g mannitol (0.25 M final)
38 mg EGTA (0.5 mM final)
10 ml 100 mM HEPES (5 mM final)
Adjust pH to 7.4 with 1 N NaOH
Add ultrapure H2O to 200 ml
Sterilize by autoclaving for 20 min using a liquid cycle
Store up to 2 or 3 months at 4°C
Mannitol buffer B
To 100 ml ultrapure H2O, add:
8.22 g mannitol (0.225 M final)
76 mg EGTA (1 mM final)
50 ml 100 mM HEPES (25 mM final)
Adjust pH to 7.4 with 1 N NaOH
Add ultrapure H2O to 200 ml
Sterilize by autoclaving for 20 min using a liquid cycle
Store up to 2 or 3 months at 4°C
MTE solution, 1×
In 60 ml ultrapure H2O, dissolve:
4.914 g D-mannitol (270 mM final)
121.1 mg Tris-base (10 mM final)
3.72 mg EDTA (0.1 mM final)
Adjust pH to 7.4 with 6 M HCl
Add ultrapure H2O to 100 ml
Sterilize through a 0.22-μm vacuum filter
Store 3 to 6 months at room temperature
MBS buffer
In 300 ml ultrapure H20, dissolve:
2.44 g 4-morpholineethanesulfonic acid (25 mM final)
4.4 g NaCl (150 mM final)
Adjust pH to 6.5
Add ultrapure H20 to 500 ml
Sterilize by autoclaving for 20 min using a liquid cycle
Store 2 or 3 months at room temperature
MBS + 5% sucrose
In 300 ml ultrapure H2O, dissolve:
2.44 g 4-morpholineethanesulfonic acid (25 mM final)
4.4 g NaCl (150 mM final)
25 g sucrose (5% w/v final)
Adjust pH to 6.5
Add ultrapure H2O to 500 ml
Sterilize by autoclaving for 20 min using a liquid cycle
Store 2 or 3 months at room temperature
MBS + 30% sucrose
In 300 ml ultrapure H2O, dissolve:
2.44 g 4-morpholineethanesulfonic acid (25 mM final)
4.4 g NaCl (150 mM final)
150 g sucrose (30% w/v final)
Adjust pH to 6.5
Add ultrapure H2O to 500 ml
Sterilize by autoclaving for 20 min using a liquid cycle
Store 2 or 3 months at room temperature
MBS + 90% sucrose
In 300 ml ultrapure H2O, dissolve:
2.44 g 4-morpholineethanesulfonic acid (25 mM final)
4.4 g NaCl (150 mM final)
Adjust pH to 6.5
Add 450 g sucrose (90% w/v final)
Add ultrapure H2O to 500 ml
Sterilize by autoclaving for 20 min using a liquid cycle
Store 2 or 3 months at room temperature
MBST
50 ml MBS buffer (see recipe)
0.5 ml Triton X-100 (1% final)
500 μL 100 mM PMSF (1 mM final)
Make fresh for each experiment
Store on ice
Percoll solution, 30% (v/v)
1 vol 90% (v/v) stock isotonic Percoll (see recipe)
2 vol mannitol buffer B (see recipe)
Sterilize by autoclaving for 20 min using a liquid cycle
Store 2 or 3 months at 4°C
Phosphate-buffered saline, pH 7.4
3 liters distilled water
32 g NaCl (140 mM final)
0.8 g KH2PO4 (1.5 mM final)
8.7 g Na2HPO4 ·7H2O (8.1 mM final)
0.8 g KCl (2.7 mM final)
Adjust pH to 7.4 with 1 N NaOH
Add H2O to 4 liters
Sterilize by autoclaving for 20 min using a liquid cycle
Store 2 or 3 months at 4°C
PMSF, 100 mM (Protease Inhibitor)
In 50 ml absolute ethanol, dissolve:
0.871 g PMSF
-
Store up to 1 year at −20°C
Add fresh to all solutions as required so that the final concentrations are 1 mM.
Stock isotonic Percoll, 90% (v/v)
9 vol Percoll (Pharmacia cat. no. P1644)
1 vol 2.5 M sucrose (0.25 M final)
Sterilize by autoclaving for 20 min using a liquid cycle
Store 2 or 3 months at 4°C
Sucrose, 1.0 M
In 60 ml ultrapure H2O, dissolve:
34.23 g sucrose
121.1 mg Tris-base (10 mM final)
3.72 mg EDTA (0.1 mM final)
Adjust pH to 7.6 with 6 M HCl
Add ultrapure H2O to 100 ml
Sterilize by autoclaving 20 min using a liquid cycle
Store 3 to 6 months at room temperature
Open only in a laminar flow hood to avoid contamination
Sucrose, 1.3 M
In 60 ml ultrapure H2O, dissolve:
44.50 g sucrose
121.1 mg Tris-base (10 mM final)
3.72 mg EDTA (0.1 mM final)
Adjust pH to 7.6 with 6 M HCl
Add ultrapure H2O to 100 ml
Sterilize by autoclaving 20 min using a liquid cycle
Store 3 to 6 months at room temperature
Sucrose, 1.5 M
In 60 ml ultrapure H2O, dissolve:
51.35 g sucrose
121.1 mg Tris-base (10 mM final)
3.72 mg EDTA (0.1 mM final)
Adjust pH to 7.6 with 6 M HCl
Add ultrapure H2O to 100 ml
Sterilize by autoclaving 20 min using a liquid cycle
Store 3 to 6 months at room temperature
Sucrose, 1.7 M
In 60 ml ultrapure H2O, dissolve:
58.19 g sucrose
121.1 mg Tris-base (10 mM final)
3.72 mg EDTA (0.1 mM final)
Adjust pH to 7.6 with HCl
Add ultrapure H2O to 100 ml
Sterilize by autoclaving 20 min using a liquid cycle
Store 3 to 6 months at room temperature
Sucrose, 2.0 M
In 60 ml ultrapure H2O, dissolve:
68.46 g sucrose
121.1 mg Tris-base (10 mM final)
3.72 mg EDTA (0.1 mM final)
Adjust pH to 7.6 with HCl
Add ultrapure H2O to 100 ml
Sterilize by autoclaving 20 min using a liquid cycle
Store 3 to 6 months at room temperature
Sucrose homogenization medium (SHM)
To 100 ml ultrapure H2O, add:
17.1 g sucrose (0.25 M final)
20 ml 100 mM HEPES (10 mM final)
Adjust pH to 7.4 with 1 N NaOH
Add ultrapure H2O to 200 ml
Sterilize by autoclaving for 20 min using a liquid cycle
Store 2 or 3 months at 4°C
COMMENTARY
Background Information
Highly purified mitochondria are very difficult to obtain in appreciable amounts. The discontinuous sucrose gradient protocol (see Basic Protocol 1) is valuable for experiments that require higher yields of mitochondrial samples. It is often imperative to use the same pool of purified mitochondria to observe fraction purity, probe for the presence of mitochondrially localized viral proteins, and examine the post-translational modifications of those proteins. Obtaining enough purified mitochondria for all of these experiments by the Percoll protocol (see Basic Protocol 2) presented here would not be feasible. The discontinuous sucrose gradient protocol takes slightly less time, is more user-friendly, requires less starting material (number of cells), and produces higher yields of mitochondrial fractions. These mitochondrial fractions are well-purified and practical for almost any application. Also, the utility of starting with fewer cells is that one can compare more experimental conditions and controls within a single fractionation experiment. However, when one is willing to sacrifice protein yield for even higher purity mitochondrial fractions, or when one needs to isolate MAM fractions to compare to either ER or mitochondria, then the Percoll protocol (see Basic Protocol 2) is the preferred method.
Points of contact between the ER and mitochondria make up 5% to 20% of the total mitochondrial network (Rizzuto et al., 1998). These connections, as well as the ER and mitochondria organelles themselves, are quite variable and can undergo rapid changes in overall morphology, size, and composition (Bereiter-Hahn and Voth, 1994; Collins et al., 2002). Factors such as calcium homeostasis, cell metabolism, and perceived stress can have dramatic impacts on both the form and function of these organelles. The fractionation techniques presented here facilitate not only a sub-cellular localization analysis of proteins of interest, but when combined with techniques such as proteinase digestion or lipid raft isolation can further inform investigators on the sub-organellar localization (or sub-domain enrichment) of these proteins. More recent applications of the methods outlined in this chapter have begun to probe biologically relevant changes in ER, MAM, or mitochondria protein composition during cellular conditions such as the innate anti-viral immune response (Goswami et al., 2013; Horner et al., 2011), apoptosis (Zhang et al., 2013), and autophagy (Bartolome et al., 2012). Furthermore, proteomic profiling of purified MAM in uninfected and virally infected human fibroblasts has identified a number of resident MAM proteins, as well as highlighted a potential role for the MAM in metabolism (Zhang et al., 2011).
Transfection reagents are continually being modified and improved. While Lipofectamine 2000 remains the authors’ preferred choice for use with HeLa and HFF cells, you may wish to test multiple reagents to optimize transfection conditions in your cells and for your plasmids. Xtremegene9 (Roche) and Fugene HD (Promega) have also given reliable transfections in our hands. Following transfection, it is important to check your cells often. Lipofectamine 2000 was the least toxic, in the authors’ hands, to both HeLa and HFF cells, but cell viability can still change significantly in response to overexpression of a transfected plasmid. When using control plasmids encoding innocuous or beneficial (anti-apoptotic) proteins, Lipofectamine 2000 can be left in the culture medium for up to 24 hr. However, for transfection of most plasmids, it is prudent to change Lipofectamine 2000 transfection medium after 4 hr, replacing it with complete cell medium so as to minimize the stress or apoptotic signals on the cells. Furthermore, overexpression of some plasmids may be more toxic to cells, in which case, one must wash out the lipofection medium after 4 hr (as before) and, moreover, may have to decrease the total incubation time before cell harvesting. Prior to harvesting, monitor the cells for viability as cells that are heavily stressed or apoptotic are not reliable for fractionation experiments.
The need to obtain intact and highly purified subcellular fractions, including MAM, imposes the additional use of density gradient centrifugation to remove contamination by broken membranes or organelles of similar size as in Basic Protocol 2. Percoll is considered to be one of the best density gradient medium available for the separation of cells, organelles, viruses, and subcellular particles. Because of its low osmolarity (<25 mOs/kg water), Percoll forms a density gradient without generating an osmolarity gradient (Pertoft et al., 1978). Osmolarity of the gradient medium must be taken into consideration since cellular membrane-bound organelles such as mitochondria and MAM act as osmometers. A high external osmolarity will result in shrinkage of membrane-bound organelles while low osmolarities will result in swelling of organelles, hence altering their buoyant densities. Differences in the osmolarity of the gradient medium can explain differences in the size and apparent buoyant densities of subcellular compartments when different gradient media are used. Buoyant densities of organelles in sucrose rise as water is removed from their enclosed spaces. In contrast, organelles in Percoll gradients in physiological range (280 to 320 mOs/kg) have lower apparent buoyant densities than in sucrose (Pertoft et al., 1978).
Another unique feature of Percoll is that it can form self-generated gradients by centrifugation at moderate g forces. Fixed-angle rotors are most commonly used for Percoll gradients. The advantage of using fixed-angle or vertical rotors is that the path-length for formation of the gradient is shorter and the gradient forms more rapidly. Both Vance (1990) and Hovius et al. (1990) used a fixed-angle rotor to isolate purified mitochondria from a self-generated Percoll (30% v/v) gradient. Conversely, the authors used a swinging-bucket rotor for the formation of the Percoll gradient. There are two main reasons for this choice: first, because both fractions (mitochondria and MAM) have similar densities and they are banded within the density range of ~1.039 to 1.051 g/ml (Fig. 3.27.4), a better separation of this density range can be achieved by using a rotor with a longer path-length. Second, a good spatial separation between resolved mitochondria subpopulations, which differ slightly in density and size from one another, can be achieved (Collins et al., 2002). The latter feature is especially desirable in the case of HCMV-infected cells in which disruption of mitochondria networks (McCormick et al., 2003) can be visualized by density banding of mitochondrial species in these Percoll gradients (Fig. 3.27.4) (Bozidis et al., 2010).
Based upon the authors’ experience, the transfection protocol does not affect the pertinent physical properties of the organelles and their constituents during fractionation. Thus, no difference should be expected between fractions that are isolated from either untransfected or transfected cells. However, HCMV infection markedly alters the fractionation protocol. A change in the banding patterns of mitochondria (fraction 3) was observed after HCMV infection (Fig. 3.27.4). As well, an increase in the presence of mitochondrial proteins in the 6,300 × g MAM pellet (see Basic Protocol 2, step 31) was also observed after HCMV infection. Usually, only background levels of the mitochondrial markers can be detected in this pellet. It is possible that HCMV infection causes a shift of mitochondrial densities such that some mitochondria band in the density range of MAM, which could be related to the documented disruption of mitochondria networks by HCMV. Alternatively, HCMV infection may increase the connectivity of MAM-mitochondrial contacts, as has been observed in response to ER stress (Simmen et al., 2005), and protein complexes tethering the two organelles together could be isolated with the 6,300 × g centrifugation. Indeed, HCMV infection increased the abundance of most MAM-resident proteins two- to five-fold by 72 hours post-infection, consistent with an increase in connectivity of the two organelles (Zhang et al., 2011). Additionally, the mitochondrial markers Grp75 and VDAC were increasingly detected in the 6300 × g MAM fraction from HCMV-infected cells (Bozidis et al., 2010; Zhang et al., 2011), but rather than representing mitochondrial contamination, they appeared to reflect the formation of a macromolecular complex known to be involved in calcium signaling (Bozidis et al., 2010; Szabadkai et al., 2006).
The analysis of both the 6300 × g and 100,000 × g MAM fractions is highly recommended. A conservative interpretation of results is best based upon the more rigorous fraction that is isolated after the final ultracentrifugation step at 100,000 × g, as it is described in Basic Protocol 2, step 33. This fraction is consistently devoid of mitochondrial markers in both uninfected and HCMV-infected cells. However, our experience has clearly demonstrated that unusual proteins identified in the 6300 × g MAM fraction should not be written off as merely contaminants, but warrant further investigation. It is imperative to monitor fraction purity for each experiment by analyzing total, ER, mitochondria, and MAM pools by utilizing suitable organelle markers (Table 3.27.2). Not only will this ensure that the identity and purity of the fractions are sufficient for unequivocal conclusions, but appropriate controls can provide additional information about cell status at the time of fractionation.
Table 3.27.2.
Cellular Markers for Verification and Assessment of ER, Mitochondria, and MAM Subcelluar Fractions
| Subcellular compartment | Marker | Antibody | Vendor | Reference |
|---|---|---|---|---|
| ER (microsomes) | DPM1 | I-20 | Santa Cruz Biotechnology cat. no. sc-15836 | Maeda et al., 1998 |
| Calreticulin | Affinity Bioreagents cat. no. PA3-900 | Johnson et al., 2001; Gelebart et al., 2005 | ||
| Mitochondria | SDH | MitoSciences | Lynes et al., 2012 | |
| ATPase-α | Invitrogen no.459240 | Guardia-Laguarta et al., 2014 | ||
| COXII | K-20 | Santa Cruz Biotechnology cat. no. sc-23984 | Scheffler, 2001 | |
| Grp75 | Stressgen cat. no. SPS-825 | Manning-Krieg et al., 1991 | ||
| MAM | mEGFP-huPSS-1 | B-2 (anti-GFP) | Santa Cruz Biotechnology cat. no. sc-9996 | Bozidis et al., 2008 |
| huPSS-1 | Y-19 | Santa Cruz Biotechnology cat. no. sc-51410 | Morita et al., 2012 | |
| FACL4 | Abgent cat. no. AP2536b | Simmen et al., 2005 | ||
| Sig-1R | Santa Cruz Biotechnology | Williamson et al., 2011 | ||
| Erlin-2 | Cell Signaling Technology cat. No. 2959 | Guardia-Laguarta et al., 2014 | ||
| Mfn2 | Abcam | Zhang et al., 2011 | ||
| ACAT1 | Affinity Bioreagents | Myhill et al., 2008 | ||
| TMX1 | Sigma | Lynes et al., 2012 | ||
| TMX2 | Lifespan Biosciences | Lynes et al., 2012 | ||
| α-synuclein | Cell Signaling Technology cat. No. 2628 | Guardia-Laguarta et al., 2014 | ||
| p66Shc | Abcam | Wieckowski et al., 2009 |
Critical Parameters and Troubleshooting
Sucrose gradient fractionation
Sometimes a mitochondrial band will not be seen after centrifugation, so make sure to mark the gradient interface between the 1.7 M and 1.6 M sucrose layers when preparing the gradients. If a band is not seen after centrifugation, insert the collecting needle into the side of the 11 × 60–mm Beckman tube at the marked gradient interface and carefully withdraw ~0.4 ml of liquid.
When collecting protein fractions with needles, the use of small-bore needles should be avoided as they increase the shear force on the extracted samples. Extract the protein band slowly into the syringe, then remove the needle before dispensing the protein fraction into a microcentrifuge tube for storage. This saves the sample from the shear force of flowing a second time through the needle.
Proteins can be degraded extremely rapidly. Once the cells have been lysed, work as quickly as possible. Also make sure to keep samples on ice at all times. Pre-cooling buffers to 4°C can help. While the sucrose gradients are made at room temperature to facilitate marking interfaces on the tubes with sharpie markers, they can be chilled to 4°C after they are created or rotors can be chilled to 4°C to help ensure protein samples stay cold throughout the procedure. Furthermore, PMSF is a serine protease inhibitor. It does not inhibit other proteases, and does not even inhibit all serine proteases. If protein degradation appears to be a problem, it may be necessary to add a commercially available protease inhibitor cocktail (e.g., Roche Complete Protease Inhibitor Cocktail tablets) to buffers in addition to PMSF. Freeze fractionated samples immediately upon collection to avoid unwarranted degradation.
Differential pelleting by centrifugation
A lot of care must be taken in the centrifugal separation of the mitochondrial/MAM fraction from the microsomal fraction (steps 11–14). Extra time must be allotted in the procedure to remove all traces of unpelleted mitochondrial/MAM. It is therefore recommended to use a fixed-angle rotor and 4–5 centrifugations under the same conditions (Fig. 3.27.3). In the authors’ experience, if the above conditions are not followed, the supernatant microsomal fraction is still contaminated with detectable mitochondrial/MAM fraction.
Percoll density gradients
To generate Percoll gradients, stock isotonic Percoll (SIP) solution is prepared at the desired density in 0.25 M sucrose. To accurately measure buoyant density, samples are premixed with the gradient material. However, if samples are layered onto the top of the Percoll gradient, better resolution of subcellular particles from soluble proteins is attained. Soluble proteins tend to remain above the Percoll gradient and subcellular particles will sediment into the gradient, thus achieving better separation.
Percoll gradients continuously change during high-speed centrifugation. Therefore, depending on the centrifugation conditions (type of rotor, RCF, time of centrifugation), the density profile of the gradient and, consequently, the separation of the organelles will be altered. Thus, using density marker beads (Amersham Biosciences cat. no. 17–0459-01) to monitor densities in a duplicate gradient is recommended. Their use verifies the consistency of density gradient formation and greatly facilitates identification of the desired fractions.
In spite of their many advantages, Percoll particles are difficult to remove from purified fractions. A simple approach is to separate subcellular particles from Percoll-coated silica particles by high-speed centrifugation in a swinging-bucket rotor or fixed-angle rotor (Calaminus et al., 1979). Percoll is in a tight pellet; whereas, the biological material remains loosely packed and can be gently resuspended.
Cellular responses to stress, metabolic demands, and signaling may alter the composition, morphology, or connectivity of ER and mitochondria. This can impact the banding pattern for these organelles, as well as the separation between banded MAM and mitochondria. The authors recommend the practice of photographying the gradients to document the banding patterns during experiments.
Anticipated Results
Different lysis methods
The effects of various lysis conditions were evaluated using untransfected HeLa cells fractionated by discontinuous sucrose gradients (Table 3.27.1). Five conditions were surveyed: low sonication (three, 5-sec pulses), medium sonication (three, 10-sec pulses), high sonication (three, 15-sec pulses), homogenization (10 strokes with Dounce homogenizer), or freeze/thaw cycles (three cycles of 1-hr incubation at −80°C followed by a rapid thaw in a 37°C water bath).
Homogenization, the gentlest lysis procedure, had the lowest ER and mitochondrial yields of the lysis protocols tested. The low-speed spin after lysis, to remove large cellular debris, produced large pellets of intact cells after homogenization, which decreased the material available for subsequent banding on ER and mitochondrial gradients. Basic Protocol 2 utilizes large amounts of cell culture starting material to ensure that all final fractions can be collected in appreciable amounts. Starting amounts of cellular material can be decreased, however, as needed if you do not require high yields of all three purified organelles (microsomes, MAM, and mitochondria).
To determine the purity of the fractionated mitochondria and ER from the various lysis procedures, an examination of the presence of ER (DPM1), MAM (FACL4), and mitochondrial (Grp75, COXII) markers in the banded ER and mitochondria fractions (Fig. 3.27.5) was done. Sonication resulted in good separation of ER and mitochondria although the anti-DPM1 antibody detected mitochondrial DPM (Gasnier et al., 1992) in this experiment. However, the other markers tested (FACL4, Grp75, and COXII) showed good separation of ER and mitochondria. Medium or high sonication disrupted the association between the MAM and mitochondrial compartments, as the MAM marker (FACL4) was detected in the ER fraction and was barely detected in the mitochondrial fraction. Conversely, the freeze/thaw lysis showed an equal distribution of MAM in both ER and mitochondrial fractions. Of the lysis procedures tested, homogenization was the most effective at preserving the association of the MAM with mitochondria. Nonetheless, the highest yields of ER and mitochondrial fractions were obtained from low and medium sonication procedures. Medium sonication was therefore chosen as the preferred lysis condition for discontinuous sucrose gradient fractionation (see Basic Protocol 1). This procedure produces superior yields of well-purified ER and mitochondrial fractions, with MAM membranes predominantly appearing in the ER fraction.
Purification of mitochondria and mitochondria-associated membranes
The ultracentrifugation of the crude mitochondrial extract in a self-generating Percoll (30%) gradient using a swinging-bucket rotor produces three distinct fractions (Fig. 3.27.4). Using density marker beads (Pharmacia), the authors found that MAM and mitochondria from cultured human cells span a range of densities between 1.039 and 1.051 g/ml (data not shown). The higher density beads (> 1.069 g/ml) sediment near the bottom of the Percoll gradient.
Fraction 1 appears as a faint, diffuse layer just above fraction 2; whereas, fraction 2 is the more compact white in color band, less dense than mitochondria. Fraction 3 consists of multiple denser mitochondrial bands. Because the borders of the fractions usually are partially overlapping, care must be taken during the collection of the bands to minimize mixing of the samples. If a needle and syringe is used for this purpose, it is recommended that the collection should start from the top in the case of fraction 1 and from the bottom for fraction 3. In every case, the interface between the fractions should be carefully avoided.
The isolated fractions were verified by western blot analysis using known cellular protein markers for cytosol (Hsp70), microsomes (DPM1, calreticulin), MAM (mEGFP-huPSS-1), and mitochondria (COXII, Grp75) (Fig. 3.27.6 and Table 3.27.2). The MAM is physically associated and pelleted with mitochondria. Upon resolution in Percoll gradients, its position is consistent with previously reported relative densities of MAM (Vance, 1990). Most compellingly, the high relative abundance of mEGFP-huPSS-1 fusion protein in fraction 2 as well as the presence of the other ER markers (DPM1, calreticulin) indicate that it is an ER subcompartment, enriched for phosphatidylserine synthetic enzymes as previously documented in rat liver tissue (Stone and Vance, 2000). Therefore, it has been concluded that fraction 2 contains the purified mitochondria-associated membranes from cultured human cells.
Figure 3.27.6.

Western analyses of fractionated HeLa cells that stably express mEGFP-huPSS-1 fusion protein. Stably transfected cells were fractionated according to the procedure described in Basic Protocol 2 and subcellular fractions were isolated. Fractionated proteins (10 μg) were separated by 10% SDS PAGE and transferred onto nitrocellulose membranes using a semi-dry protein transfer apparatus (BioRad). Blotted proteins were probed against markers for microsomes (anti-DPM1, 1:100 or anti-calreticulin, 1:1000), MAM (mEGFP-huPSS-1 using anti-GFP, 1:100), and mitochondria (anti-COX, 1:100 or anti-Grp75, 1:2500) and with the corresponding horseradish peroxidase-conjugated secondary Ab (1:2000). Reactivity was detected using the chemiluminescent method (Amersham, GE Healthcare).
Western analyses identified fraction 3 as the purified mitochondria (Fig. 3.27.6). The parallel bands that can be observed within this fraction reflect mitochondria of slightly different sizes and densities. Mitochondria in the cell differ in size and morphology (Bereiter-Hahn and Voth, 1994; Collins et al., 2002). It should be noted that the position of the mitochondria in the gradient, exactly below fraction 2, is another indication that fraction 2 represents the mitochondria-associated membranes.
The identity of fraction 1 is less clear. Western analyses clearly indicate that it is ER-related as it shares markers (Fig. 3.27.6, calreticulin) with the ER and MAM. Nonetheless, fraction 1 notably differs in the presence of an alternative DPM species detected by the anti-DPM1 antibody. Moreover, fraction 1 has markedly less mEGFP-huPSS-1 than the MAM fraction.
Cellular markers for verification of subcellular compartment identity and purity
To verify the purity of the isolated microsomal and mitochondria fractions, established organelle-specific markers (Colberg-Poley et al., 2000; Mavinakere and Colberg-Poley, 2004a; Mavinakere and Colberg-Poley, 2004b; Mavinakere et al., 2006) were used. To unequivocally identify the MAM fraction (fraction 2) from Percoll gradients, phosphatidylserine synthase 1 (PSS-1), which has been shown to be enriched in the rat liver MAM (Stone and Vance, 2000), was chosen. However, neither commercial antibodies nor human PSS-1 cDNA clones were originially available when the authors adapted this protocol for viral studies in human cells. Therefore, the complete human PSS-1 cDNA was cloned from a HeLa cDNA library and its open reading frame was tagged with EGFP (Bozidis et al., 2008). A HeLa transfectant cell line (HeLa-PSS-120) stably expresses the mEGFP-huPSS-1 fusion protein. The fractionated subcellular compartments from HeLa-PSS-120 cells verified the identity of fraction 2 as the MAM by virtue of the selective presence of PSS-1 in fraction 2 but not in fraction 3 (Fig. 3.27.6).
Recent studies have uncovered a number of new MAM-constituent proteins, as well identified biologically-relevant circumstances under which MAM composition changes. As well, different laboratories use different methodologies to purify MAM fractions. Thus, picking the right markers to verify organelle purity of contiguous ER and MAM membranes, or closely related MAM and mitochondria membranes, for your studies will not be a trivial task. We have chosen to focus here on organelle markers that have been used in the field, which also have commercially available antibodies compatible with Western analyses of human cell fractionations. Table 3.27.2 summarizes these cellular makers, with relevant antibodies, commercial sources, and original references. The authors refer readers to (Schon and Area-Gomez, 2013) for a more comprehensive list of published MAM markers gathered from studies in rat, mouse, primate, as well as human cells.
Microsomes
Dolichol phosphate-mannose synthase 1 (DPM1)
DPM synthase 1, a transmembrane protein, which catalyzes mannosyl transfer from GDP-mannose hydrophobic long-chain acceptor dolichol-phosphate, is present in the rough ER (Maeda et al., 1998). In human cells, DPM1, which is located at the cytosolic side of the ER, associates with DPM2 and DPM3 to form the complete multicomponent enzyme of human DPM synthase (Maeda et al., 2000). In addition, a mitochondrial DPM synthase is located on the cytosolic face of the outer membrane of mitochondria (Gasnier et al., 1992). On occasion, the anti-DPM1 (I-20, Santa Cruz Biotechnology) antibody used detected these different DPM species in the ER or in mitochondrial fractions (Bozidis et al., 2010; Mavinakere et al., 2006).
Calreticulin
Calreticulin is a Ca2+-binding chaperone that contains an N-terminal amino acid signal sequence and a C-terminal KDEL ER retrieval sequence, which are responsible for its localization in the ER lumen (Gelebart et al., 2005). However, calreticulin has also been detected in cellular compartments, other than ER and including the cell surface (Johnson et al., 2001). Anti-calreticulin (Affinity Bioreagents) antibody detected calreticulin in the cytosol; whereas, other markers for the ER (anti-DPM1), MAM (anti-mEGFP-PSS-1), or mitochondria (anti-Grp75 and anti-COXII) did not.
Mitochondria
SDH (succinate dehydrogenase, or mitochondrial complex II)
SDH is a protein complex of the inner mitochondrial membrane composed of four nuclear-encoded subunits, two of which are integral membrane proteins of the inner mitochondrial membrane (Rutter et al., 2010). SDH is both an enzyme of the tricarboxylic acid cycle and a component of the electron transport chain. Moreover, its distribution across subcellular fractions using multiple MAM purification techniques in human cells has been published (Gilady et al., 2010).
ATPase-α (ATP synthase α-subunit, or mitochondrial complex V)
The ATP synthase enzyme is composed of 17 subunits, and predominantly localizes to the inner mitochondrial membrane where it is responsible for ATP production in oxidative phosphorylation (Ruhle and Leister, 2015). It has also been identified on the plasma membrane in several cell types including endothelial cells and cancer cells (Kenan and Wahl, 2005). Anti-bovine ATPase-α antibody (Invitrogen) recognizes the human ATP synthase complex, and acts as a strong mitochondrial marker by western analysis (Guardia-Laguarta et al., 2014).
COXII (cytochrome c oxidase subunit II)
COXII is one of three mitochondrial DNA–encoded subunits (MTCO1–3) of respiratory complex IV. Complex IV localizes to the mitochondrial inner membrane and is the terminal enzyme of the electron transport chain (Scheffler, 2001). Although COXII is mainly localized in the mitochondria, recently, it was shown that the protein is present at extra-mitochondrial sites (Sadacharan et al., 2005).
Grp75 (glucose regulated protein 75 kDa)
Grp75 is a molecular chaperone that is predominantly localized in the mitochondrial matrix, where, in concert with Hsp60, it is thought to participate in the refolding of proteins translocated into this organelle (Manning-Krieg et al., 1991). It has been shown that Grp75 is also present in a macromolecular complex with VDAC and IP3Rs at the ER-mitochondria interface (Szabadkai et al., 2006), and that its presence in MAM fractions can dramatically increase in response to viral infection (Bozidis et al., 2010).
Mitochondria-associated membranes
PSS-1 (phosphatidylserine synthase 1)
Both the PSS-1 and PSS-2 enzymes are involved in phosphatidylserine biosynthesis in mammalian cells. It has been shown that PSS-1 is highly enriched in MAM of rat liver and is largely excluded from the bulk of the ER (Stone and Vance, 2000). The fusion protein mEGFP-huPSS-1, mentioned above, localizes similarly to endogenous protein in human cells, and can be used as an exogenous MAM marker. More recently, commercial antibodies raised to human PSS-1 have become available. One such antibody from Santa Cruz, PSS-1 (Y-19), was published to successfully recognize both exogenous, myc-tagged, human PSS-1 as well as endogenous PSS-1 from human embryonic kidney cells by western blot analysis (Morita et al., 2012).
FACL4 (fatty acid CoA ligase, long chain 4)
FACL4 converts long-chain fatty acids into fatty acyl-CoA esters for use in synthesizing complex lipids (Piccini et al., 1998). Similar to PSS-1, it is enriched in MAM compartments and is a marker for MAM fractions in western blot analyses (Simmen et al., 2005).
Sig-1R (sigma-1 receptor)
Sig-1R is a nonopioid ER protein that regulates the compartmentalization of lipids in the ER, as well as their export (Hayashi and Su, 2003). Sig-1R also acts as a calcium-sensitive chaperone in the ER, and is enriched in the MAM where it can interact with and stabilize IP3R3 proteins to prolong calcium signaling from ER into mitochondria (Hayashi and Su, 2007).
Erlin-2
Erlin-2 is a novel member of the prohibitin family of proteins. It localizes to lipid raft enriched MAM subdomains of the ER (Browman et al., 2006) and forms large, heteromeric complexes that bind IP3Rs and regulate their ER-associated degradation (Pearce et al., 2007). Erlin-2 can be detected in purified microsomes and MAM using Percoll gradient fractionation techniques (Zhang et al., 2011) or in detergent-resistant MAM domains purified from crude mitochondria (Guardia-Laguarta et al., 2014).
Mfn2 (mitofusin 2)
Mfn2 is a dynamin-like GTPase required for mitochondrial outer membrane fusion. It is enriched in MAM and helps to tether ER and mitochondrial membranes together (de Brito and Scorrano, 2008). Mutations in Mfn2 cause Charcot-Marie-Tooth type IIa neuropathy.
ACAT1 (acetyl-CoA:cholesterol acyltransferase 1)
ACAT1 is a lipid biosynthetic enzyme involved in the synthesis of cholesteryl esters. The MAM is highly enriched, compared to bulk ER, in both ACAT1 activity (Rusiñol et al., 1994) as well as protein abundance (Gilady et al., 2010).
TMX1 and TMX2 (thioredoxin-related transmembrane proteins 1 and 2)
TMX1 is a thioredoxin-related transmembrane protein with disulfide reductase activity (Matsuo et al., 2001). TMX1 is enriched in MAM fractions, along with related TMX2. Both TMX proteins require palmitoylation of two membrane-proximal cytosolic cytseines for MAM enrichment (Lynes et al., 2012).
α-synuclein
α-synuclein is a predominantly cytoplasmic protein that is abundantly found in the presynaptic terminals of neurons. While its exact function is not known, it is thought to play a role in regulating synaptic vesicle turnover in these presynaptic terminals (Kim et al., 2014). α-synuclein is linked to a number of neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease, whose characteristic feature is the presence of α-synuclein aggregates. Wild-type α-synuclein was demonstrated to localize to MAM in human brain tissue and cultured HeLa cells, and to modulate mitochondrial morphology (Guardia-Laguarta et al., 2014).
p66Shc (66-kDa isoform of src-homology 2 domain-containing transforming protein 1)
p66Shc is a growth factor adaptor protein which is involved in signal transduction, can modulate mitochondrial metabolism, and participates in reactive oxygen species production (Suski et al., 2011). It has been shown that p66Shc can localize to plasma membrane-associated membranes as well as MAM (Wieckowski et al., 2009) in an age-related manner, with p66Shc increasing in these locaizations with increasing age (Suski et al., 2011).
Time Considerations
Basic Protocol 1 can easily be accomplished within 1 full working day. The time required heavily depends on the number of transfection or infection conditions being tested. It is best to limit the scale of the experiment to ultracentrifuge availability (i.e., each ultracentrifuge rotor can only hold six tubes). For a point of reference, twelve 175-cm2 flasks (six duplicate sets of experimental conditions) can be processed at once using a single rotor for each ultracentrifugation set, taking a total of ~6 to 7 hr. Preparation and sterilization of buffers is done prior to the day of fractionation. PMSF is added fresh to 1× MTE buffer just before use. Creation of sucrose gradients and harvesting of cell pellets are done first. Cell pellets are stable on ice for up to 2 hr, providing a convenient resting point. Alternatively, cell pellets can be frozen, providing a stopping point in the protocol. After sonication of cells, there is no other stopping point in the protocol, and one should work as quickly as possible. The ultracentrifugation for the ER fraction is relatively long (70 min), and this should be started first. After beginning this ultracentrifugation, go back and perform the pellet washes and load the gradients for the mitochondrial fractions. Both centrifugations will probably finish around the same time. Again, work with the ER fractions first, as they require an additional 45 min ultracentrifugation. Then, go back and collect banded mitochondria from the other tubes.
Basic Protocol 2 is a more time consuming protocol, and can be difficult to finish in a single day particularly due to the numerous homogenization and centrifugation steps early in the procedure to maximize yield and ensure robust separation of mitochondria and MAM organelles from microsomal fractions. A stopping point can be included after step 10. Also, depending on your desired final yields of each organelle, the laborious early steps may be modified to speed up the procedure. For example, if you wish to throw away more of the pellet from steps 6–8, this can decrease the time spent homogenizing and centrifuging. Alternatively, if you are not interested in collecting purified microsomes, you can modify step 12 to expedite the procedure. The latter steps in the protocol are grouped procedurally by organelle being purified, for simplicity. Some of these steps can be done simultaneously to save time, so that you are processing one set of samples while others are centrifuging. The availability of ultracentrifuges in your lab will also affect the time required for Basic Protocol 2.
Basic Protocol 3 is rather simple, and quick to set up. It does, however, require fractions collected from Basic Protocol 1 or Basic Protocol 2, and utilizes an overnight centrifugation before final samples can be collected. Starting material obtained from Basic Protocol 1 or Basic Protocol 2 may be frozen at −80°C, if needed, and stored overnight prior to the start of this procedure.
Figure 3.27.5.

Western analyses of ER (DPM1), mitochondrial (Grp75, COXII), and MAM (FACL4) markers in HeLa cells lysed by different methods. HeLa cells were fractionated using discontinuous sucrose gradients (see Basic Protocol 1) as detailed in Table 3.27.1. Briefly, cells were lysed using sonication (three times, 5, 10, or 15 sec each), Dounce homogenization (ten strokes), or freeze/thaw cycles (three times 1 hr at −80°C, followed by rapid thawing). Twenty micrograms of total, ER, and mitochondrial protein fractions from each sonication condition were subjected to SDS-PAGE in 10% Bis-Tris NuPage gels (Invitrogen). For homogenization and freeze/thaw lysis conditions, 40 μl of each fraction were used (due to low protein concentrations). Proteins were then transferred to nitrocellulose membranes and probed for DPM1 (1:100 goat anti-DPM1, 1:2000 donkey anti-goat HRP). Membranes were stripped and reprobed for Grp75 (1:2000 mouse anti-Grp75, 1:2500 goat anti-mouse). Similarly, membranes were then stripped and reprobed sequentially for FACL4 (1:250 rabbit anti-FACL4, 1:3000 goat anti-rabbit HRP) then COX II (1:200 goat anti-COXII, 1:2500 donkey anti-goat HRP).
Acknowledgments
The authors thank Drs. Eric P. Hoffman and Kanneboyina Nagaraju for their unwavering support. The studies were supported by Public Health Service grants R01 AI057906 and R21 AI081957 from the National Institute for Allergy and Infectious Diseases, the National Science Foundation grant MCB 1244509 and Children’s Research Institute funds to ACP.
Literature Cited
- Ardail D, Popa I, Bodennec J, Louisot P, Schmitt D, Portoukalian J. The mitochondria-associated endoplasmic-reticulum subcompartment (MAM fraction) of rat liver contains highly active sphingolipid-specific glycosyltransferases. Biochem J. 2003;371:1013–1019. doi: 10.1042/BJ20021834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolome A, Guillen C, Benito M. Autophagy plays a protective role in endoplasmic reticulum stress-mediated pancreatic beta cell death. Autophagy. 2012;8:1757–1768. doi: 10.4161/auto.21994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereiter-Hahn J, Voth M. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microscopy research and technique. 1994;27:198–219. doi: 10.1002/jemt.1070270303. [DOI] [PubMed] [Google Scholar]
- Bhuvanendran S, Salka K, Rainey K, Sreetama SC, Williams E, Leeker M, Prasad V, Boyd J, Patterson GH, Jaiswal JK, Colberg-Poley AM. Superresolution imaging of human cytomegalovirus vMIA localization in sub-mitochondrial compartments. Viruses. 2014;6:1612–1636. doi: 10.3390/v6041612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bionda C, Portoukalian J, Schmitt D, Rodriguez-Lafrasse C, Ardail D. Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria-associated membrane) and/or mitochondria? Biochem J. 2004;382:527–533. doi: 10.1042/BJ20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bononi A, Missiroli S, Poletti F, Suski JM, Agnoletto C, Bonora M, De Marchi E, Giorgi C, Marchi S, Patergnani S, Rimessi A, Wieckowski MR, Pinton P. Mitochondria-associated membranes (MAMs) as hotspot Ca(2+) signaling units. Advances in experimental medicine and biology. 2012;740:411–437. doi: 10.1007/978-94-007-2888-2_17. [DOI] [PubMed] [Google Scholar]
- Bozidis P, Williamson CD, Colberg-Poley AM. Mitochondrial and secretory human cytomegalovirus UL37 proteins traffic into mitochondrion-associated membranes of human cells. J Virol. 2008;82:2715–2726. doi: 10.1128/JVI.02456-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozidis P, Williamson CD, Wong DS, Colberg-Poley AM. Trafficking of UL37 proteins into mitochondrion-associated membranes during permissive human cytomegalovirus infection. J Virol. 2010;84:7898–7903. doi: 10.1128/JVI.00885-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browman DT, Resek ME, Zajchowski LD, Robbins SM. Erlin-1 and erlin-2 are novel members of the prohibitin family of proteins that define lipid-raft-like domains of the ER. J Cell Sci. 2006;119:3149–3160. doi: 10.1242/jcs.03060. [DOI] [PubMed] [Google Scholar]
- Calaminus JM, Bruggen J, Sorg C. Isolation, characterization and cultivation of human trophoblastic cells. Immunobiology. 1979;156:287. [Google Scholar]
- Carman CV, Lisanti MP, Benovic JL. Regulation of G protein-coupled receptor kinases by caveolin. J Biol Chem. 1999;274:8858–8864. doi: 10.1074/jbc.274.13.8858. [DOI] [PubMed] [Google Scholar]
- Colberg-Poley AM, Patel MB, Erezo DP, Slater JE. Human cytomegalovirus UL37 immediate-early regulatory proteins traffic through the secretory apparatus and to mitochondria. J Gen Virol. 2000;81:1779–1789. doi: 10.1099/0022-1317-81-7-1779. [DOI] [PubMed] [Google Scholar]
- Colberg-Poley AM, Patterson GH, Salka K, Bhuvanendran S, Yang D, Jaiswal JK. Superresolution imaging of viral protein trafficking. Medical microbiology and immunology. 2015 doi: 10.1007/s00430-015-0395-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colberg-Poley AM, Williamson CD. Intracellular sorting and trafficking of cytomegalovirus proteins during permissive infection. In: Reddehase MJ, editor. Cytomegaloviruses: From Molecular Pathogenesis to Intervention. 2. I. Caister Academic Press/Horizon; Norwich, U.K: 2013. pp. 196–229. [Google Scholar]
- Collins TJ, Berridge MJ, Lipp P, Bootman MD. Mitochondria are morphologically and functionally heterogeneous within cells. Embo J. 2002;21:1616–1627. doi: 10.1093/emboj/21.7.1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Hayashi T, Su TP. The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem Biophys Res Commun. 2012;417:635–639. doi: 10.1016/j.bbrc.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasnier F, Rousson R, Lerme F, Vaganay E, Louisot P, Gateau-Roesch O. Mitochondrial dolichyl-phosphate mannose synthase. Purification and immunogold localization by electron microscopy. European journal of biochemistry / FEBS. 1992;206:853–858. doi: 10.1111/j.1432-1033.1992.tb16993.x. [DOI] [PubMed] [Google Scholar]
- Gelebart P, Opas M, Michalak M. Calreticulin, a Ca2+-binding chaperone of the endoplasmic reticulum. The international journal of biochemistry & cell biology. 2005;37:260–266. doi: 10.1016/j.biocel.2004.02.030. [DOI] [PubMed] [Google Scholar]
- Gilady SY, Bui M, Lynes EM, Benson MD, Watts R, Vance JE, Simmen T. Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM) Cell Stress Chaperones. 2010;15:619–629. doi: 10.1007/s12192-010-0174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmacher VS, Bartle LM, Skaletskaya A, Dionne CA, Kedersha NL, Vater CA, Han JW, Lutz RJ, Watanabe S, Cahir McFarland ED, Kieff ED, Mocarski ES, Chittenden T. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc Natl Acad Sci U S A. 1999;96:12536–12541. doi: 10.1073/pnas.96.22.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami R, Majumdar T, Dhar J, Chattopadhyay S, Bandyopadhyay SK, Verbovetskaya V, Sen GC, Barik S. Viral degradasome hijacks mitochondria to suppress innate immunity. Cell research. 2013;23:1025–1042. doi: 10.1038/cr.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardia-Laguarta C, Area-Gomez E, Rub C, Liu Y, Magrane J, Becker D, Voos W, Schon EA, Przedborski S. alpha-Synuclein is localized to mitochondria-associated ER membranes. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:249–259. doi: 10.1523/JNEUROSCI.2507-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayajneh WA, Colberg-Poley AM, Skaletskaya A, Bartle LM, Lesperance MM, Contopoulos-Ioannidis DG, Kedersha NL, Goldmacher VS. The sequence and antiapoptotic functional domains of the human cytomegalovirus UL37 exon 1 immediate early protein are conserved in multiple primary strains. Virology. 2001;279:233–240. doi: 10.1006/viro.2000.0726. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Fujimoto M. Detergent-resistant microdomains determine the localization of sigma-1 receptors to the endoplasmic reticulum-mitochondria junction. Mol Pharmacol. 2010;77:517–528. doi: 10.1124/mol.109.062539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Su TP. Sigma-1 receptors (sigma(1) binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: roles in endoplasmic reticulum lipid compartmentalization and export. J Pharmacol Exp Ther. 2003;306:718–725. doi: 10.1124/jpet.103.051284. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Horner SM, Liu HM, Park HS, Briley J, Gale M., Jr Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A. 2011;108:14590–14595. doi: 10.1073/pnas.1110133108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S, Michalak M, Opas M, Eggleton P. The ins and outs of calreticulin: from the ER lumen to the extracellular space. Trends in cell biology. 2001;11:122–129. doi: 10.1016/s0962-8924(01)01926-2. [DOI] [PubMed] [Google Scholar]
- Kenan DJ, Wahl ML. Ectopic localization of mitochondrial ATP synthase: a target for anti-angiogenesis intervention? Journal of bioenergetics and biomembranes. 2005;37:461–465. doi: 10.1007/s10863-005-9492-x. [DOI] [PubMed] [Google Scholar]
- Kim WS, Kagedal K, Halliday GM. Alpha-synuclein biology in Lewy body diseases. Alzheimer’s research & therapy. 2014;6:73. doi: 10.1186/s13195-014-0073-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynes EM, Bui M, Yap MC, Benson MD, Schneider B, Ellgaard L, Berthiaume LG, Simmen T. Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. The EMBO journal. 2012;31:457–470. doi: 10.1038/emboj.2011.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y, Tanaka S, Hino J, Kangawa K, Kinoshita T. Human dolichol-phosphate-mannose synthase consists of three subunits, DPM1, DPM2 and DPM3. The EMBO journal. 2000;19:2475–2482. doi: 10.1093/emboj/19.11.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y, Tomita S, Watanabe R, Ohishi K, Kinoshita T. DPM2 regulates biosynthesis of dolichol phosphate-mannose in mammalian cells: correct subcellular localization and stabilization of DPM1, and binding of dolichol phosphate. The EMBO journal. 1998;17:4920–4929. doi: 10.1093/emboj/17.17.4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning-Krieg UC, Scherer PE, Schatz G. Sequential action of mitochondrial chaperones in protein import into the matrix. The EMBO journal. 1991;10:3273–3280. doi: 10.1002/j.1460-2075.1991.tb04891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta. 2014;1837:461–469. doi: 10.1016/j.bbabio.2013.10.015. [DOI] [PubMed] [Google Scholar]
- Matsuo Y, Akiyama N, Nakamura H, Yodoi J, Noda M, Kizaka-Kondoh S. Identification of a novel thioredoxin-related transmembrane protein. J Biol Chem. 2001;276:10032–10038. doi: 10.1074/jbc.M011037200. [DOI] [PubMed] [Google Scholar]
- Mavinakere MS, Colberg-Poley AM. Dual targeting of the human cytomegalovirus UL37 exon 1 protein during permissive infection. J Gen Virol. 2004a;85:323–329. doi: 10.1099/vir.0.19589-0. [DOI] [PubMed] [Google Scholar]
- Mavinakere MS, Colberg-Poley AM. Internal cleavage of the human cytomegalovirus UL37 immediate-early glycoprotein and divergent trafficking of its proteolytic fragments. J Gen Virol. 2004b;85:1989–1994. doi: 10.1099/vir.0.80094-0. [DOI] [PubMed] [Google Scholar]
- Mavinakere MS, Williamson CD, Goldmacher VS, Colberg-Poley AM. Processing of human cytomegalovirus UL37 mutant glycoproteins in the endoplasmic reticulum lumen prior to mitochondrial importation. Journal of virology. 2006;80:6771–6783. doi: 10.1128/JVI.00492-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick AL, Smith VL, Chow D, Mocarski ES. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J Virol. 2003;77:631–641. doi: 10.1128/JVI.77.1.631-641.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita SY, Shirakawa S, Kobayashi Y, Nakamura K, Teraoka R, Kitagawa S, Terada T. Enzymatic measurement of phosphatidylserine in cultured cells. J Lipid Res. 2012;53:325–330. doi: 10.1194/jlr.D021808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce MM, Wang Y, Kelley GG, Wojcikiewicz RJ. SPFH2 mediates the endoplasmic reticulum-associated degradation of inositol 1,4,5-trisphosphate receptors and other substrates in mammalian cells. J Biol Chem. 2007;282:20104–20115. doi: 10.1074/jbc.M701862200. [DOI] [PubMed] [Google Scholar]
- Pertoft H, Laurent TC, Laas T, Kagedal L. Density gradients prepared from colloidal silica particles coated by polyvinylpyrrolidone (Percoll) Analytical biochemistry. 1978;88:271–282. doi: 10.1016/0003-2697(78)90419-0. [DOI] [PubMed] [Google Scholar]
- Piccini M, Vitelli F, Bruttini M, Pober BR, Jonsson JJ, Villanova M, Zollo M, Borsani G, Ballabio A, Renieri A. FACL4, a new gene encoding long-chain acyl-CoA synthetase 4, is deleted in a family with Alport syndrome, elliptocytosis, and mental retardation. Genomics. 1998;47:350–358. doi: 10.1006/geno.1997.5104. [DOI] [PubMed] [Google Scholar]
- Poston CN, Duong E, Cao Y, Bazemore-Walker CR. Proteomic analysis of lipid raft-enriched membranes isolated from internal organelles. Biochem Biophys Res Commun. 2011;415:355–360. doi: 10.1016/j.bbrc.2011.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Duchen MR, Pozzan T. Flirting in little space: the ER/mitochondria Ca2+ liaison. Sci STKE. 2004;2004:re1. doi: 10.1126/stke.2152004re1. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- Ruhle T, Leister D. Assembly of FF-ATP synthases. Biochimica et biophysica acta. 2015 doi: 10.1016/j.bbabio.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Rusiñol AE, Cui Z, Chen MH, Vance JE. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J Biol Chem. 1994;269:27494–27502. [PubMed] [Google Scholar]
- Rutter J, Winge DR, Schiffman JD. Succinate dehydrogenase - Assembly, regulation and role in human disease. Mitochondrion. 2010;10:393–401. doi: 10.1016/j.mito.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadacharan SK, Singh B, Bowes T, Gupta RS. Localization of mitochondrial DNA encoded cytochrome c oxidase subunits I and II in rat pancreatic zymogen granules and pituitary growth hormone granules. Histochemistry and cell biology. 2005;124:409–421. doi: 10.1007/s00418-005-0056-2. [DOI] [PubMed] [Google Scholar]
- Scheffler IE. A century of mitochondrial research: achievements and perspectives. Mitochondrion. 2001;1:3–31. doi: 10.1016/s1567-7249(00)00002-7. [DOI] [PubMed] [Google Scholar]
- Schon EA, Area-Gomez E. Mitochondria-associated ER membranes in Alzheimer disease. Molecular and cellular neurosciences. 2013;55:26–36. doi: 10.1016/j.mcn.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. Embo J. 2005;24:717–729. doi: 10.1038/sj.emboj.7600559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song KS, Scherer PE, Tang Z, Okamoto T, Li S, Chafel M, Chu C, Kohtz DS, Lisanti MP. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem. 1996;271:15160–15165. doi: 10.1074/jbc.271.25.15160. [DOI] [PubMed] [Google Scholar]
- Stone SJ, Vance JE. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J Biol Chem. 2000;275:34534–34540. doi: 10.1074/jbc.M002865200. [DOI] [PubMed] [Google Scholar]
- Suski JM, Karkucinska-Wieckowska A, Lebiedzinska M, Giorgi C, Szczepanowska J, Szabadkai G, Duszynski J, Pronicki M, Pinton P, Wieckowski MR. p66Shc aging protein in control of fibroblasts cell fate. International journal of molecular sciences. 2011;12:5373–5389. doi: 10.3390/ijms12085373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem. 1990;265:7248–7256. [PubMed] [Google Scholar]
- Vance JE. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim Biophys Acta. 2014;1841:595–609. doi: 10.1016/j.bbalip.2013.11.014. [DOI] [PubMed] [Google Scholar]
- Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nature protocols. 2009;4:1582–1590. doi: 10.1038/nprot.2009.151. [DOI] [PubMed] [Google Scholar]
- Williamson CD, Colberg-Poley AM. Intracellular sorting signals for sequential trafficking of human cytomegalovirus UL37 proteins to the endoplasmic reticulum and mitochondria. Journal of virology. 2010;84:6400–6409. doi: 10.1128/JVI.00556-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson CD, Zhang A, Colberg-Poley AM. The human cytomegalovirus protein UL37 exon 1 associates with internal lipid rafts. J Virol. 2011;85:2100–2111. doi: 10.1128/JVI.01830-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A, Hildreth RL, Colberg-Poley AM. Human cytomegalovirus inhibits apoptosis by proteasome-mediated degradation of Bax at endoplasmic reticulum-mitochondrion contacts. J Virol. 2013;87:5657–5668. doi: 10.1128/JVI.00145-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A, Williamson CD, Wong DS, Bullough MD, Brown KJ, Hathout Y, Colberg-Poley AM. Quantitative proteomic analyses of human cytomegalovirus-induced restructuring of endoplasmic reticulum-mitochondrial contacts at late times of infection. Molecular & cellular proteomics : MCP. 2011;10:M111009936. doi: 10.1074/mcp.M111.009936. [DOI] [PMC free article] [PubMed] [Google Scholar]