

Summary
Purpose
Juvenile myoclonic epilepsy (JME) accounts for 3 to 12% of all epilepsies. In 2004, we identified a mutation-harboring Mendelian gene that encodes a protein with one EF-hand motif (EFHC1) in chromosome 6p12. We observed one doubly heterozygous and three heterozygous missense mutations in EFHC1 segregating as an autosomal dominant gene with 21 affected members of six Hispanic JME families from California and Mexico. In 2006, similar and three novel missense mutations were reported in sporadic and familial Caucasian JME from Italy and Austria. In this study, we asked if coding single nucleotide polymorphisms (SNPs) of EFHC1 also contribute as susceptibility alleles to JME with complex genetics.
Methods
We screened using denaturing high-performance liquid chromatography (DHPLC) and then directly sequenced the 11 exons of EFHC1 in 130 unrelated JME probands, their 352 family members, and seven exons of EFHC1 in 400–614 ethnically matched controls. We carried out case-control association studies between 124 unrelated Hispanic JME probands and 552–614 ethnically matched controls using four SNPs, rs3804506, rs3804505, rs1266787, and rs17851770. We also performed family-based association on SNPs rs3804506 and rs3804505 in 84 complete JME families using the Family-Based Association Test (FBAT) program.
Results
We found no statistically significant differences between JME probands and controls in case-control association and no genetic transmission disequilibria in family-based association for the tested SNPs. In addition, we identified four new DNA variants in the coding region of EFHC1.
Conclusion
The four coding SNPs, rs3804506, rs3804505, rs1266787, and rs17851770, of EFHC1 may not be susceptibility alleles for JME.
Keywords: Juvenile myoclonic epilepsy, EFHC1, DNA variants
Juvenile myoclonic epilepsy (JME) is the most common cause of primary grand mal seizures and accounts for at least 3 to 12% of all epilepsies. JME can be inherited as a Mendelian autosomal dominant or autosomal recessive trait or as a non-Mendelian complex genetic trait. Three mutation-harboring Mendelian genes for JME have been reported. Mutations in α1 subunit of γ-aminobutyric acid receptor subtype A on chromosome 5q34 segregated with nine affected individuals of a three-generation French Canadian family with JME (Cossette et al., 2002). Mutations in a chloride-channel gene, CLCN2 on chromosome 3q26 segregated with five affected members of a three-generation German family with JME (Haug et al., 2003). Our group first mapped a JME locus in chromosome 6p12 (Liu et al., 1995, 1996; Serratosa et al., 1996; Bai et al., 2002) and identified a mutation-harboring Mendelian gene that encodes a protein with one EF-hand motif. Hence, we called the gene EFHC1. We reported one doubly heterozygous and three heterozygous missense mutations segregated in 21 clinical and electroencephalography (EEG) affected members of six unrelated two- to four-generation JME Hispanic families from California (U.S.A.) and Mexico (Suzuki et al., 2004). The 6p12 JME locus was again mapped independently by two separate groups; first, by a genome-wide linkage study of JME families from various countries of Europe (Hempelmann et al., 2006) and second by a chromosome 6p12 replication genetic linkage study in a smaller cohort of 18 Dutch families (Pinto et al., 2004). Similar and novel missense mutations in EFHC1 were reported in Caucasian JME patients from Tennessee (U.S.A.), Italy, and Austria (Ma et al., 2006; Stogmann et al., 2006; Annesi et al., 2007). Therefore the causality of the gene in a Mendelian JME has been established, but the influence of common functional single nucleotide polymorphisms (SNPs) in JME with complex genetics has not been established. Pinto et al. (2006) reported three SNPs of EFHC1 that were not associated with JME in a case-control study of 112 unrelated patients and 180 controls. In our present study, we chose the most common four coding SNPs of EFHC1 and performed both case-control and family-based association studies to determine if they contribute to the complex genetics of JME.
Methods
Subjects and families
All JME probands and their family members were recruited through the Americas branch of our International Genetic Epilepsies Study (GENESS) consortium. The consortium includes adult neurologists and epileptologists, as well as child neurologists/epileptologists from the U.S.A. (Los Angeles, CA), Mexico, and Honduras. Each participating individual signed a consent form approved by human research committees from UCLA and participating institutes or university from Mexico and Honduras. The criteria for inclusion and exclusion of JME probands and affected family members have been published previously (Liu et al., 1996; Bai et al., 2002). We studied 130 JME families including 111 from Mexico City, Mexico, 11 from California, U.S.A. and 8 from Honduras. Thirty of these families were reported by Bai et al. in 2002 and Suzuki et al. in 2004. One large family was reported by Serratosa et al. in 1996 and was part of the 31 family studies by Suzuki and coworkers in 2004. They are all of Hispanic culture and were of American Indians mixture with Spanish ancestry.
We screened 130 JME probands and their 352 available family members (totally 482 members) for all the 11 exons of EFHC1 and 7 exons with DNA variants detected from the 130 JME families in 400–614 population controls. All control samples belonged to persons who did not have a history of epileptic seizures. These persons donated blood to the National Institute of Neurology & Neurosurgery, Mexico City, Mexico, were randomly selected, and matched to the case samples based on sex, age, and ancestral origin.
For case-control study, we used 124 probands and 552 to 614 controls. Six probands who had originally shown mutations in EFHC1 were excluded from the case-control study.
For Family-Based Association Test (FBAT), we used 84 complete families, which included 73 two-generation families and 11 three-generation families. These families had 187 affected members and 237 unaffected members. We excluded 46 families from the FBAT study, because 34 families only had the probands and no parents, while the other 12 families only had the probands and one parent.
Primer design and polymerase chain reaction amplification
Genomic DNA was extracted from peripheral venous blood of each subject using the QIAamp Blood Kit (Qiagen, Valencia, CA, U.S.A.). EFHC1 reference sequence was used from National Center for Biotechnology Information (NCBI; http://www.ncbi.com). Eleven primer pairs (Supplementary Material Table 1) were designed with the software program (primer 3, version) to amplify the 11 EFHC1 exons and their adjacent exon/intron boundary regions. Polymerase chain reaction (PCR) was performed using 50 ng DNA, 1.0 mM dNTPs, 1.5 mM MgSO4, and 1 μl Taq DNA polymerase (Transgenomic, Omaha, NE, U.S.A.) in 25-μl reaction volumes. Samples were amplified using ABI 9700 (Applied Biosystems, Foster City, CA, U.S.A.) with a program of 95°C for 5 min, 30 cycles of 95°C for 30 s, annealing temperature 57°–61°C for 30 s, and 72°C for 30 s, followed by 72°C for 7 min at the end. The correct melting temperature (Tm) was established experimentally using the recommendation of Primer 3 program and WAVE program (Transgenomic, Omaha, NE, U.S.A.).
Mutation detection and SNP genotyping
We used heteroduplex formation and detection by denaturing high-performance liquid chromatography (DHPLC) analysis and direct DNA sequencing for DNA variant detection and SNP genotyping. We first screened each sample with DHPLC and then confirmed the DNA variants by sequencing.
DHPLC
PCR products were denatured for 5 min at 95°C and then cooled to 25°C over 40 min to allow for heteroduplex formation. Denatured products were analyzed with WAVE DNA Fragment Analysis System (Transgenomic, Omaha, NE, U.S.A.). The WAVEMAKER software predicted the melting domains of DNA fragments. Column temperatures for screening each DNA fragment were selected on the basis of its melting domains, whereby the domains were each 75%–90% in helical conformation at the given temperatures. Consequently, each PCR product was analyzed at one to three column temperatures.
To identify and characterize SNPs, each PCR product was analyzed individually using the above DHPLC conditions, but only at a single column temperature at which the multipeak elution profile was most distinctive. Known homozygotes were mixed with a reference homozygous sample in equal volumes and reanalyzed again using the same conditions. With the second analysis, the sample had the same genotype as the control homozygote if a single peak was observed and was homozygous for the other allele if multiple peaks were seen. Similarly, samples showing multiple peaks (heterozygotes) at the first analysis were also mixed with a reference heterozygous sample and reanalyzed. Identical multiple peaks as in the first analysis confirmed that sample had the same heterozygous genotype as the control heterozygote. A distorted multi-peak elution profile indicated the presence of additional SNP in the fragment and would be further investigated.
DNA sequencing
The DNA sequences of representative samples that showed distinct elution patterns were determined by direct sequencing. After amplification and analyses using DPHLC, samples that represented distinct patterns were chosen for direct sequencing. The same samples were cleaned or purified using Edge Gel Filtration Cartridge 750 G or Sephadex G-50 following analyses with DPHLC. Cycle sequencing was performed on the purified products using Big Dye Terminator Cycle Sequencing Ready reaction kit (Applied Biosystems). The sequencing products were analyzed in ABI 3700 Capillary DNA Analyzer (Applied Biosystems).
Statistical analysis
Case-control association study
Allele and genotype frequencies were calculated using the gene counting method. Allele and genotype association between JME probands and controls were tested using Pearson’s chi-square (χ2) test with Yates’ correction for continuity (Preacher, 2001). As stated above, we only include 124 unrelated probands and excluded the six probands with EFHC1 mutations reported by Suzuki et al. (2004).
Family-based association study
FBAT was performed for 84 complete JME families with two or more generation family members both under null hypothesis of no association and no linkage and under no association in the presence of linkage with FBAT program (Laird et al., 2000; Lake et al., 2000; Rabinowitz & Laird, 2000).
Statistical power calculations for the samples used in the association analyses
Assuming a high-risk allele frequency of 0.05, a disease prevalence of 0.001 in general population, and a type I error rate of 0.05, the power to detect a dominant genotype effect of a relative risk (RR) of 3 for rs3804506 and rs3804505 is over 0.80, both for a case-control and FBAT study. The power for rs1266787 is 0.78 for a case-control study. However, none of the sample sizes can reach a statistical power of 0.80 when we performed power calculations assuming an RR of 2. The results of statistical power calculations for the samples used in each association analysis were presented in Table 2 of the Supplementary Material. All power calculations were performed using Genetic Power Calculator (Purcell et al., 2003).
Results
DNA variants in coding region of EFHC1 and their characteristics
We screened by DHPLC and then directly sequenced the 11 exons and their immediate flanking regions of EFHC1 in 130 JME families and identified 13 DNA variants from seven exons in the coding region of EFHC1. Table 1 shows the assignment of DNA variants, their nucleotide substitution, amino acid changes, and genotype counts in our cohort of JME probands and 400–614 ethically matched population controls. Nucleotide position was based on the genomic sequence GenBank accession number NM1800, GI: 8922435 and protein accession NP060570. We confirmed all five mutations identified by Suzuki et al. (2004) in the same probands with JME, including one proband who not only had the 685T>C mutation but also had the SNP 545G>A. In addition, we found four new DNA variants: 91G>A in exon 2, 661T>C in exon 4, 755C>A in exon 5, and 1821C>T in exon 10. The variants 755C>A and 1821C>T are found in two JME index cases and were not present in normal population controls. Variant 755C>A is a missense mutation causing a threonine to lysine substitution in 252 amino acid positions. Variant 1821C>T is a silent mutation and does not change the structure of protein. Variants 91G>A and 661T>C are both found in population controls, and 661T>C is also present in one JME proband.
Table 1.
DNA variants of EFHC1 coding region in JME probands and controls
| No. | Location | NCBI dbSNP ID | Nucleotide substitution | Amino acid changes | Comment and functionb | Genotype counts (no. of subjects)
|
|
|---|---|---|---|---|---|---|---|
| With JME | Normal controls | ||||||
| 1 | Exon 2 | New variantc | 91G>A | T30T | Synonymous | 0/130 | 1/401 |
| 2 | Exon 2 | Mutationa | 229C>A | P77T | Missense | 1/130 | 0/401 |
| 3 | Exon 3 | rs3804506ad | 475 C>T | R159W | Missense | 24/130 | 95/604 |
| 475 T/T | R159W | Missense | 0/130 | 3/604 | |||
| 4 | Exon 3 | rs3804505ad | 545G>A | R182H | Missense | 21/130 | 65/604 |
| 5 | Exon 4 | Mutationa | 628G>A | D210N | Missense | 1/130 | 0/400 |
| 6 | Exon 4 | New variantc | 661T>C | R221C | Missense | 1/130 | 1/400 |
| 7 | Exon 4 | Mutationa | 662G>A | R221H | Missense | 1/130 | 0/401 |
| 8 | Exon 4 | Mutationa | 685T>C | F229L | Missense | 2/130 | 0/401 |
| 9 | Exon 5 | New variantc | 755C>A | T252K | Missense | 1/130 | 0/402 |
| 10 | Exon 5 | Mutationa | 757G>T | D253Y | Missense | 1/130 | 0/402 |
| 11 | Exon 8 | rs1266787cd | 1343T>C | M448T | Missense | 6/130 | 36/614 |
| 12 | Exon 10 | new variantc | 1821C>T | N607N | Synonymous | 1/130 | 0/401 |
| 13 | Exon 11 | rs17851770ad | 1855A>C | I619L | Missense | 5/130 | 16/552 |
Reported in Suzuki et al., 2004.
Synonymous substitution or silent mutation, one single base change in DNA, but no amino acid change in protein; missense mutation or nonsynonymous substitution, one single base change in DNA lead to an amino acid change in protein.
Newly found DNA variants in this report.
DNA variants with association analysis in this paper.
Case-control association analysis
We chose four coding SNPs, rs3804506, rs3804505, rs1266787, and rs17851770, for case-control association analysis, because their minor allele frequencies in control population were over 0.01. The heteroduplex chromatogram and sequence chromatogram for the four SNPs are shown in Figures 1–4 in the Supplementary Material. The genotype and allele frequency of the four SNPs from 124 unrelated JME probands and ethnically matched controls are shown in Table 2. None of the observed genotypes and allele frequencies in either group showed statistical significance. There is no statistical deviation from Hardy-Weinberg equilibrium for the four tested SNPs in population controls.
Table 2.
Comparisons of the genotype and allele frequency of the four EFHC1 SNPs between JME probands and controls
| SNPs | n | Genotype, n (percent) | Allele, n (percent) | p-valuea | ||
|---|---|---|---|---|---|---|
| rs3804506 | C/C | C/T | C | T | ||
| JME | 124 | 100 (0.81) | 24 (0.19) | 224 (0.90) | 24 (0.10) | 0.58 |
| Controlsb | 604 | 506 (0.84) | 95 (0.16) | 1107 (0.92) | 101 (0.08) | |
| rs3804505 | G/G | G/A | G | A | ||
| JME | 124 | 105 (0.85) | 19 (0.15) | 229 (0.92) | 19 (0.08) | 0.21 |
| Controls | 604 | 539 (0.89) | 65 (0.11) | 1143 (0.95) | 65 (0.05) | |
| rs1266787 | T/T | T/C | T | C | ||
| JME | 124 | 118 (0.95) | 6 (0.05) | 242 (0.98) | 6 (0.02) | 0.82 |
| Controls | 614 | 578 (0.94) | 36 (0.06) | 1192 (0.97) | 36 (0.03) | |
| rs17851770 | A/A | A/C | A | C | ||
| JME | 124 | 119 (0.96) | 5 (0.04) | 243 (0.98) | 5 (0.02) | 0.71 |
| Controls | 552 | 536 (0.97) | 16 (0.03) | 1088 (0.99) | 16 (0.01) | |
p-values refer to comparisons of allele frequencies.
There were three controls with T/T genotype of rs3804506.
Family-based association analysis
Table 3 presents FBAT results for rs3804506 and rs3804505 in 84 complete JME families with two or more generations. No association between the two SNPs and JME is present both under null hypothesis of no association and no linkage and under null hypothesis of no association in the presence of linkage (Laird et al., 2000; Lake et al., 2000; Rabinowitz & Laird, 2000). The number of families with minor allele is too small for SNPs rs1266787 and rs17851770 to be included in FBAT.
Table 3.
FBAT results for rs3804506 and rs3804505 showing no evidence for association in 84 complete JME familiesa
| SNPs | Allele | Allele frequency | Under no association and no linkage
|
Under no association in the presence of linkage
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Family no. | Sb | E(S)c | p-value | Family no. | Sb | E(S)c | p-value | |||
| rs3804506 | C | 0.903 | 27 | 56.00 | 56.00 | 1.00 | 20 | 63.00 | 63.00 | 1.00 |
| rs3804506 | T | 0.097 | 27 | 24.00 | 24.00 | 1.00 | 20 | 21.00 | 21.00 | 1.00 |
| rs3804505 | G | 0.917 | 28 | 66.00 | 67.50 | 0.68 | 20 | 83.00 | 84.50 | 0.77 |
| rs3804505 | A | 0.083 | 28 | 28.00 | 26.50 | 0.68 | 20 | 27.00 | 25.50 | 0.77 |
The number of families with minor allele is too small for SNPS-rs1266787 and rs17851770 to be included in FBAT.
S, tested statistic.
E(S), estimated statistic.
Discussion
In this study, we screened 11 exons and their immediately adjacent intron in EFHC1 in 130 JME probands and their 352 family members. We also screened seven exons with DNA variants detected in our JME families in 400–614 population controls. We provide information on frequencies of both alleles and genotypes in JME probands and control population of Hispanic ethnic background. We confirmed the five mutations and three SNPs previously reported by Suzuki et al. (2004). We found four new DNA variants in the coding region of EFHC1. 755C>A and 1821C>T were found only in JME families and not in 402 and 401 separate control populations suggesting they could be functional mutations; however, 1821C>T is a silent mutation with no change in protein structure. Both 91G>A and 661T>C exist in control populations suggesting they are rare polymorphisms. EFHC1 mutations were reported in two other published studies aside from our original studies of six JME families from California (U.S.A.) and Mexico (Suzuki et al., 2004). Ma et al. (2006) reported one Caucasian family with R221H mutation, and Stogmann et al. (2006) reported one sporadic Caucasian JME patient with 2014T>C mutation. Here we identified two more novel EFHC1 mutations, 755C>A and 1821C>T, in Hispanic JME patients. These mutation reports on index cases from Tennessee (U.S.A.) and Austria and our new present data supporting EFHC1 as a JME Mendelian gene question whether EFHC1 SNP alleles could also contribute to the complex genetics of JME. Using case-control and family-based association methods, we were unable to find any relationship of common SNP polymorphisms and JME. A lack of an association has also been found in a cohort of Dutch JME population (Pinto et al., 2006). Functional studies have failed to show any difference in cell-death effects between the SNPs of rs3804506, rs3804505, and rs17851770 and wild-type EFHC1 (Suzuki et al., 2004). In contrast, human JME mutation in EFHC1 that are inherited as an autosomal dominant trait reversed apoptosis induced by wild-type myoclonin/EFHC1 (Suzuki et al., 2004). Our present sample sizes have enough statistical power (>0.80) to detect a dominant genotype effect of a RR of 3 for rs3804506 and rs3804505, both for a case-control and FBAT study, and nearly enough power (0.78) for rs1266787 for a case-control study (see Table 2 in the Supplementary Material). We would miss a low-risk susceptibility allele (RR = 2 or RR < 2) giving the present sample sizes. Although larger sample size or more replication studies are needed for further verification, combining all above evidences, we conclude that the four SNPs of EFHC1 are not JME-associated alleles and may not contribute to the complex genetics of JME as major genetic susceptibility alleles.
Of the 18 genes in the 3.5 cM EJM1 region (MIM254770) in chromosome 6p12 identified by linkage, Suzuki and colleagues excluded 17 genes with the exceptional EFHC1 (Fig. 1). Five coding missense mutations (P77T, D210N, R221H, F229L and D253Y) were identified in Hispanic JME families (Suzuki et al., 2002, 2004, 2006). Stogmann et al. (2006) found four EFHC1 coding mutations (I174V, C259Y, A394S, and 2014T>C) in three Caucasian patients or families with idiopathic generalized epilepsy (IGE) and one with cryptogenic temporal lobe epilepsy, respectively. They also found that F229L exists both in patients and study populations with equal frequency, suggesting it is unlikely to be pathogenic. Among the two new EFHC1 mutations (T252K and N607N) identified in this study N607N, which does not produce any change in protein structure, is unlikely to be functional. Of all the nine SNPs identified from both JME or IGE and control population (Suzuki et al., 2004; Ma et al., 2006; Pinto et al., 2006; Stogmann et al., 2006), we provide evidences that the four most frequent coding SNPs in EFHC1 (Table 2) may not contribute to susceptibility of genetically complex JME. This is consistent with the association studies reported in a cohort of Dutch JME population (Pinto et al., 2006). These results suggest a further search for novel coding EFHC1 mutations and for mutations in its promoter regions and splice sites. We should also carefully sequence other candidate genes in the neighboring region of EJM1 among JME families that were previously linked to 6p12, but do not contain EFHC1 mutations. Undoubtedly, we also need to search for more JME genes outside of 6p12. A whole genome scan of 101 JME families who do not link to 6p12 is now underway in our laboratories to find more new JME genes.
Figure 1.
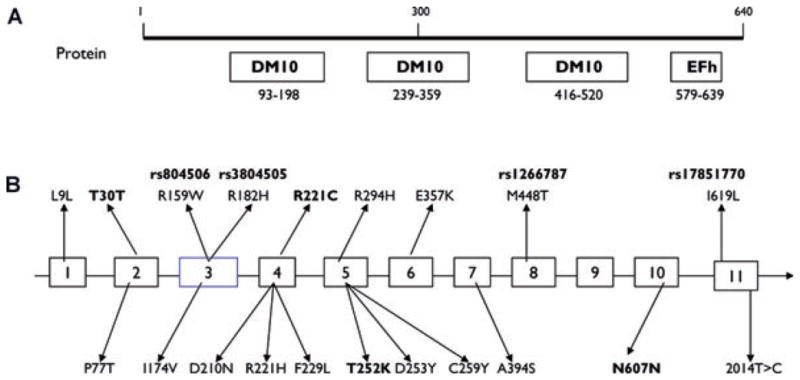
The diagram of genomic organization of the EFHC1 gene. (A) Translated protein with functional domain and motif. (B) Structure of the EFHC1 gene and the positions of the variants identified. The upper row is the reported SNPs, and the lower row is the mutations. Those with bold letters are reported in this paper, and the four with NCBI SNP names are used for association study.
Epilepsia © ILAE
Supplementary Material
Acknowledgments
We would like to thank all of the families who participated in this study. We wish to gratefully acknowledge our colleagues in the International Consortium GENESS (Genetic Epilepsies Studies) who had helped us with this and other studies of the genetic epilepsies. Study supported by the National Institute of Neurological Disorders and Stroke (NINDS) grant no. 5R01NS042376-03.
Footnotes
Conflict of interest: We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. None of the authors have any conflicts of interest to disclose.
Electronic Database Information
dbSNP Home Page, http://www.ncbi.nlm.nih.gov/SNP/
GenBank, http://www.ncbi.nlm.nih.gov/GenBank/ (EF-HC1, Gene ID 114327, and genomic clone containing EFHC1 accession NM018100 GI: 8922435 and accession number AB001328)
Primer3, http://www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi
Chi-square test, http://www.quantpsy.org
Human genome working draft, http://genome.ucsc.edu/cgi-bin/hgGateway
FBAT program, http://www.biostat.harvard.edu/fbat/fbat.htm
Genetic power calculator, http://pngu.mgh.harvard.edu/purcell/gpc/
HWE test, http://www.kursus.kvl.dk/shares/vetgen/_Popgen/genetics/2/2.htm
Additional Supporting Information may be found in the online version of this article:
Figure S1. (A) rs3804506 heteroduplex chromatogram: C/T. (B) rs3804506 sequence chromatogram: C/T.
Figure S2. (A) rs3804505 heteroduplex chromatogram: G/A. (B) rs3804505 sequence chromatogram: G/A.
Figure S3. (A) rs1266787 heteroduplex chromatogram: T/C. (B) rs1266787 sequence chromatogram: T/C.
Figure S4. (A) rs17851770 heteroduplex chromatogram: A/C. (B) rs17851770 sequence chromatogram: A/C.
Table S1. Primer pairs used to amplify the EFHC1 coding sequence and the sizes of PCR products
Table S2. The statistical power for the samples used in each association analysis*
Please note: Wiley-Blackwell is not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Annesi F, Gambardella A, Michelucci R, Bianchi A, Marini C, Canevini MP, Capovilla G, Elia M, Buti D, Chifari R, Striano P, Rocca FE, Castellotti B, Cali F, Labate A, Lepiane E, Besana D, Sofia V, Tabiadon G, Tortorella G, Vigliano P, Vignoli A, Beccaria F, Annesi G, Striano S, Aguglia U, Guerrini R, Quattrone A. Mutational analysis of EFHC1 gene in Italian families with juvenile myoclonic epilepsy. Epilepsia. 2007;48:1686–1690. doi: 10.1111/j.1528-1167.2007.01173.x. [DOI] [PubMed] [Google Scholar]
- Bai D, Alonso ME, Medina MT, Bailey JN, Morita R, Cordova S, Rasmussen A, Ramos-Peek J, Ochoa A, Jara A, Donnadieu FR, Cadena G, Yamakawa K, Delgado-Escueta AV. Juvenile myoclonic epilepsy: linkage to chromosome 6p12 in Mexico families. Am J Med Genet. 2002;113:268–274. doi: 10.1002/ajmg.10724. [DOI] [PubMed] [Google Scholar]
- Cossette P, Liu L, Brisebois K, Dong H, Lortie A, Vanasse M, Saint-Hilaire JM, Carmant L, Verner A, Lu WY, Wang YT, Rouleau GA. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nature Genet. 2002;31:184–189. doi: 10.1038/ng885. [DOI] [PubMed] [Google Scholar]
- Haug K, Warnstedt M, Alekov AK, Sander T, Ramirez A, Poser B, Maljevic S, Hebeisen S, Kubisch C, Rebstock J, Horvath S, Hallmann K, Dullinger JS, Rau B, Haverkamp F, Beyenburg S, Schulz H, Janz D, Giese B, Muller-Newen G, Propping P, Elger CE, Fahlke C, Lerche H, Heils A. Mutations in CLCN2 encoding a voltage-gated chloride channel are associated with idiopathic generalized epilepsies. Nat Genet. 2003;33:527–532. doi: 10.1038/ng1121. [DOI] [PubMed] [Google Scholar]
- Hempelmann A, Taylor KP, Heils A, Lorenz S, Prud’homme JF, Nabbout R, Dulac O, Rudolf G, Zara F, Bianchi A, Robinson R, Gardiner RM, Covanis A, Lindhout D, Stephani U, Elger CE, Weber YG, Lerche H, Nurnberg P, Kron KL, Scheffer IE, Mulley JC, Berkovic SF, Sander T. Exploration of the genetic architecture of idiopathic generalized epilepsies. Epilepsia. 2006;47:1682–1690. doi: 10.1111/j.1528-1167.2006.00677.x. [DOI] [PubMed] [Google Scholar]
- Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epi. 2000;19(Suppl 1):S36–S42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Lake SL, Blacker D, Laird NM. Family-based tests of association in the presence of linkage. Am J Hum Genet. 2000;67:1515–1525. doi: 10.1086/316895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu AW, Delgado-Escueta AV, Gee MN, Serratosa JM, Zhang W, Alonso ME, Medina MT, Cordova S, Zhao HZ, Spellman JM, Rubio Donnadieu F, Ramos Peek J, Treiman LJ. Juvenile epilepsy locus in chromosome 6p21.2-p11: linkage to convulsions and electroencephalograph trait. Am J Hum Genet. 1995;57:368–381. [PMC free article] [PubMed] [Google Scholar]
- Liu AW, Delgado-Escueta AV, Gee MN, Serratosa JM, Zhang W, Alonso ME, Medina MT, Cordova S, Zhao HZ, Spellman JM, Rubio Donnadieu F, Ramos Peek J, Treiman LJ. Juvenile myoclonic epilepsy in chromosome 6p12-p11: locus heterogeneity and recombination. Am J Med Genet. 1996;63:438–446. doi: 10.1002/(SICI)1096-8628(19960614)63:3<438::AID-AJMG5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Ma S, Blair MA, Abou-Khalil B, Lagrange AH, Gurnett CA, Hedera P. Mutations in the GABRa1 and EFHC1 gene in familial juvenile myoclonic epilepsy. Epilepsy Res. 2006;71:129–134. doi: 10.1016/j.eplepsyres.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Pinto D, de Haan GJ, Janssen GA, Boezeman EH, van Erp MG, Westland B, Witte J, Bader A, Halley DJ, Kasteleijn-Nolst Trenite DG, Lindhout D, Koeleman BP. Evidence for linkage between juvenile myoclonic epilepsy-related idiopathic generalized epilepsy and 6p11-12 in Dutch families. Epilepsia. 2004;45:211–217. doi: 10.1111/j.0013-9580.2004.36003.x. [DOI] [PubMed] [Google Scholar]
- Pinto D, Louwaars S, Westland B, Volkers L, de Haan GJ, Trenite DG, Lindhout D, Koeleman BP. Heterogeneity at the JME 6p11-12 locus: absence of mutations in the EFHC1 gene in linked Dutch families. Epilepsia. 2006;47:1743–1736. doi: 10.1111/j.1528-1167.2006.00676.x. [DOI] [PubMed] [Google Scholar]
- Preacher KJ. Calculation for the chi-square test: an interactive calculation tool for chi-square tests of goodness of fit and independence [computer software] 2001 [Available from http://www.quantpsy.org]
- Purcell S, Cherny SS, Sham PC. Genetic power calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics. 2003;19:149–150. doi: 10.1093/bioinformatics/19.1.149. [DOI] [PubMed] [Google Scholar]
- Rabinowitz D, Laird NM. A unified approach to adjusting association tests for population admixture with arbitrary pedigree structure and arbitrary missing marker information. Hum Hered. 2000;50:211–223. doi: 10.1159/000022918. [DOI] [PubMed] [Google Scholar]
- Serratosa JM, Delgado-Escueta AV, Medina MT, Zhang Q, Iranmanesh R, Sparkes RS. Clinical and genetic analysis of a large pedigree with juvenile myoclonic epilepsy. Ann Neurol. 1996;39:187–195. doi: 10.1002/ana.410390208. [DOI] [PubMed] [Google Scholar]
- Stogmann E, Lichtner P, Baumgartner C, Bonelli S, Assem-Hilger E, Leutmezer F, Schmied M, Hotzy C, Strom TM, Meitinger T, Zimprich F, Zimprich A. Idiopathic generalized epilepsy phenotypes associated with different EFHC1 mutations. Neurology. 2006;67:2029–2031. doi: 10.1212/01.wnl.0000250254.67042.1b. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Morita R, Sugimoto Y, Sugawara T, Bai DS, Alonso ME, Medina MT, Bailey JN, Rasmussen A, Ramos-Peek J, Cordova S, Rubio-Donnadieu F, Ochoa A, Jara-Prado A, Inazawa J, Delgado-Escueta AV, Yamakawa K. Identification and mutational analysis of candidate genes for juvenile myoclonic epilepsy on 6p11-p12: LRRC1, GCLC, KIAA0057 and CLIC5. Epilepsy Res. 2002;50:265–275. doi: 10.1016/s0920-1211(02)00052-9. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Delgado-Escueta AV, Aguan K, Alonso ME, Shi J, Hara Y, Nishida M, Numata T, Medina MT, Morita R, Bai D, Ganesh S, Sugimoto Y, Inazawa J, Bailey JN, Ochoa A, Jara-Prado A, Rasmussen A, Ramos-Peek J, Cordova S, Rubio-Donnadieu F, Inoue Y, Osawa M, Kaneko S, Oguni H, Mori Y, Yamakawa K. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet. 2004;36:842–849. doi: 10.1038/ng1393. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Delgado-Escueta AV, Alonso ME, Morita R, Okamura N, Sugimoto Y, Bai D, Medina MT, Bailey JN, Rasmussen A, Ramos-Peek J, Cordova S, Rubio-Donnadieu F, Ochoa A, Jara-Prado A, Inazawa J, Yamakawa K. Mutation analyses of genes on 6p12-p11 in patients with juvenile myoclonic epilepsy. Neurosci Lett. 2006;405:126–131. doi: 10.1016/j.neulet.2006.06.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.