

Abstract
Purpose
Aurora A and B are oncogenic serine/threonine kinases that regulate mitosis. Over-expression of Auroras promotes resistance to microtubule targeted agents. We investigated mechanistic synergy by inhibiting the mitotic spindle apparatus in the presence of MLN8237 [M], an Aurora A inhibitor with either vincristine [MV] or docetaxel [MD] in aggressive B-NHL. The addition of rituximab [R] to MV or MD was evaluated for synthetic lethality.
Experimental Design
Aggressive B-NHL cell subtypes were evaluated in vitro and in vivo for target modulation and anti-NHL activity with single agents, doublets and triplets by analyzing cell proliferation, apoptosis, tumor growth, survival and mechanisms of response/relapse by gene expression profiling with protein validation.
Results
MV is synergistic while MD is additive for cell proliferation inhibition in B-NHL cell culture models. Addition of R to MV is superior to MD but both significantly induce apoptosis compared to doublet therapy. Mouse xenograft models of mantle cell lymphoma showed modest single agent activity for M, R, D and V with tumor growth inhibition (TGI) of ~10–15%. Of the doublets, MV caused tumor regression, while TGI was observed with MD (~55–60%) and MR (~25–50%) respectively. Although MV caused tumor regression, mice relapsed 20 days after stopping therapy. In contrast, MVR was curative, while MDR led to TGI of ~85%. PCNA, Aurora B, cyclin B1, cyclin D1 and Bcl-2 proteins of harvested tumors confirmed response and resistance to therapy.
Conclusions
Addition of R to MV is a novel therapeutic strategy for aggressive B-NHL and warrants clinical trial evaluation.
Introduction
Aggressive B-cell non-Hodgkin’s lymphomas (B-NHL) includes diffuse large B-cell lymphoma (DLBCL), mantle cell lymphoma (MCL), Burkitt’s lymphoma (BL) and transformed follicular lymphoma (TFL) that have disparate responses to chemo-immunotherapies. A significant number of patients (~50–60%) failing frontline therapies have few therapeutic options(1). Therefore, the development of novel safe and effective treatments based on biologically validated targets is urgently needed for these therapy resistant patients.
Aurora kinase A has received great attention in recent years as potential therapeutic target for a variety of hematologic and solid malignancies (2–6). Aurora A is a serine/ threonine kinase that plays a key role in mitotic initiation, progression and spindle assembly checkpoint (SAC) activity during the mammalian cell cycle. Aurora A localizes to centrosomes and functions in centrosome maturation and the proper formation of mitotic spindle (7–9). Suppression of its activity results in defects in centrosome maturation and separation, mitotic spindle formation and chromosome alignment (10–14). Aurora A is able to transform rodent cells leading to tumor formation in xenograft mice (15–17). In humans Aurora A is over-expressed in numerous solid (breast, colorectal, pancreas, ovary, gastric, prostate) and hematological (acute myeloid leukemia, B-NHL) malignancies (18–21). Knockdown of Aurora A protein in tumor cells delays mitotic entry and progression, resulting in the accumulation of cells in G2/M, spindle defects, polyploid cells and apoptosis (22–25). In addition, over-expression of Aurora A overrides the SAC and results in resistance to microtubule targeted agent (MTAs, e.g. taxanes, vinca alkaloids) treatment (26, 27). Indeed, inhibition of Aurora A has demonstrated broad therapeutic potential with chemotherapeutics and synergy with MTA in several human tumor models (28–32).
MLN8237 is a second-generation small molecule inhibitor of Aurora-A kinase. It is orally bioavailable and is a highly selective inhibitor of Aurora A with antineoplastic activity (33–35). MLN8237 binds to and inhibits Aurora A kinase, which may result in disruption of the assembly of the mitotic spindle apparatus, disruption of chromosome segregation, and inhibition of cell proliferation. Several studies show MLN8237 has significant activity in vitro and in vivo against numerous tumor models including multiple myeloma (36), T-cell leukemia (37), chronic myeloid leukemia (38), neuroblastoma and acute lymphoblastic leukemia (39). Recently, MLN8237 has entered Phase II clinical investigation in several hematologic malignancies.
Rituximab is a chimeric mouse anti-human CD20 monoclonal antibody used for the treatment of CD20+ B-NHLs. The overall response in FL patients is ~50% when it is used as a single agent, and the response rate is significantly increased when rituximab is used in combination with chemotherapy (40, 41). The mechanisms of antitumor effect of rituximab include apoptosis, complement dependent cytotoxicity (CDC), antibody dependent cellular cytotoxicity (ADCC) and antibody dependent cellular phagocytosis (ADCP) (42). Our previous study demonstrated that MLN8237 inhibited Aurora A kinase activity and induced apoptosis in aggressive B-NHL cell lines. Moreover, MLN8237 plus docetaxel demonstrated a significant tumor growth inhibition (TGI) with an associated improved overall survival in a mouse MCL xenograft model (32). Based on the efficacy of rituximab in inhibiting B-cell proliferation with chemotherapy, we hypothesized that addition of rituximab to an Aurora A inhibitor plus a MTA (e.g. docetaxel or vincristine) would enhance synergistic activity in B-NHL cells and mouse xenograft models. Here we show that MLN8237 plus vincristine plus rituximab (MVR) has superior anti-B-NHL activity and is curative in mice bearing MCL compared to MLN8237 plus docetaxel plus rituximab (MDR). These finding are highly correlated with harvested tumor analysis of markers of proliferation and cell cycle regulation.
Materials and Methods
Cells and reagents
B-NHL cell lines used in this study (RL, Granta-519 and SUDHL-4) were from Drs. S. Grant (Virginia Commonwealth University, VA) and C. Jordan (University of Rochester, NY) and maintained in RPMI 1640 medium (Mediatech, VA) supplemented with 10% fetal bovine serum, 2 mM sodium pyruvate and 100 units/ml penicillin/streptomycin at 37°C in a humidified atmosphere containing 5% CO2. MLN8237 was kindly provided by Millennium Pharmaceuticals Inc (Cambridge, MA). Rituximab, vincristine and docetaxel were a kind donation by the Arizona Cancer Center Clinic. The compounds were dissolved at 10 mM in DMSO as a stock solution, and then further diluted to desired concentrations for in vitro experiments. Anti-Aurora B (ab2254) antibody was purchased from Abcam (Cambridge, MA). Anti-PCNA and anti-Cyclin B1 (V152) antibodies were purchased from Cell Signaling Technology (Danvers, MA). Anti-Cyclin D1 (sc-718) and anti-β-actin antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA) and Sigma (St Louis, MO), respectively. Anti-Bcl2 antibody was purchased from (XXX, YY).
Cell proliferation assay
Lymphoma cells (Granta-519 or RL) were seeded at 10,000 per well in 96-well culture plates and allowed to grow for 24 hr followed by the desired treatment with increasing concentrations of the indicated agents (MLN8237, vincristine, docetaxel) for 4 days. Viable cell densities were determined using a CellTiter 96 Cell Proliferation Assay (Promega). Absorbance readings at 490 nm were analyzed against the control group for each drug treatment to determine cell viability. The studies were performed in triplicates × 4 and IC 50 values were estimated by Calcusyn software (Biosoft, UK). For combination studies of MLN8237 plus Vincristine or docetaxel, an equipotent ratio was calculated to determine a combined graded combination treatment. The equipotent ratio is the ratio of the median effects resulting from the single dose treatments of MLN8237 and vincristine or docetaxel. A control group was established for each drug treatment in six replicates. The effects of the combined treatments were determined by the combination-index (CI) and isobologram methods derived from the median-effect principle of Chou and Talalay.
Apoptosis assay
Using Annexin V staining to detect apoptosis, treated cells were harvested and rinsed with cold PBS once. After centrifugation for 5 min, cells were resuspended in 500 μl of 1× Annexin V binding buffer (BioVision, Annexin V-FITC Reagent Kit, Cat.#1001–1000) and then added 5μl of Annexin V-FITC and 5μl of Propidium Iodide (BioVision, Annexin V-FITC Reagent Kit). After incubation for 5 min at room temperature in the dark, the samples were analyzed by flow cytometry.
Immunoblotting
The cells were lysed in NP-40 lysis buffer containing 50 mM Tris.Cl (pH 7.4), 0.15 M NaCl, 0.5% NP-40, 1 mM DTT, 50 mM Sodium Fluoride, and 2 μl/ml Protease inhibitor cocktail (Sigma, St. Louis, MO). Protein concentrations were determined using the BioRad protein assay kit (Hercules, CA) and 50 μg of protein was resolved by electrophoresis on a 10% SDS-PAGE gel. The proteins were then transferred onto a nitrocellulose membrane and non-specific binding was blocked by incubating with 5% nonfat milk in TBST buffer (0.01 M Tris-Cl, 0.15 M NaCl, 0.5% Tween-20, pH 8.0) at room temperature for 1 hr. The membrane was subjected to the indicated antibodies and the proteins were detected by a LI-COR Odyssey Infrared Imaging System.
Immunohistochemistry
Paraffin-embedded sections were deparaffinized and rehydrated to distilled water. Antigen unmasking was carried out by bringing slides to a boil in 1 mM EDTA pH 8.0 followed by 15 min at a sub-boiling temperature. After washing in dH2O three times, the slides were incubated in 3% hydrogen peroxide for 10 min and then blocked with blocking solution (0.2% BSA, 0.01% saponin and 1% normal rabbit serum in PBS) for 1 hr at room temperature. The slides were then immunostained using anti-PCNA antibody at the dilution 1:1000 in blocking solution. The reaction was incubated overnight at 4°C. After washing 3 times with PBS, the secondary antibody conjugated horseradish peroxidase (KPL, Gaithersburg, MD) was applied for 30 min at room temperature. The signal was checked using AEC chromogen kit (Sigma) following the manufacturer’s protocol. Primary or secondary antibody replacement with normal serum from the same animal species was used as the negative controls.
MCL mouse xenograft model
Animal care and treatment were performed at Arizona Cancer Center’s experimental mouse shared services (EMSS) core facility. SCID mice were injected with 1×107 Granta-519 MCL cells subcutaneously into the right hind flank. When tumors reached a volume of ~60–120 mm3, mice were divided randomly (pair-matched) into different groups with 12 mice per cohort. The mice were treated with MLN8237 [M], rituximab [R], vincristine [V] and docetaxel [D] alone at indicated dosages or different combinations [MR, MV, MD, MVR and MDR]. MLN8237 was given orally once a day for 3 weeks, while R, V and D intravenously once a week for 4 weeks. The length (L) and width (W) of the subcutaneous tumors were measured by calipers and the tumor volume (TV) was calculated as: TV = (L×W2)/2. Mice were sacrificed at the end of treatment (3 mice per cohort), end of study or if they reached >2000 mm3 at any time during the study. Excised tumors (end of the treatment) were either fixed in paraffin for immunohistochemistry (IHC) analysis or snap frozen for Western blotting and DNA microarray studies. Overall survival for each cohort was analyzed by Kaplan-Meier method.
RNA isolation and Gene expression profiling
Total RNA was extracted from harvested tumors at the end of treatment using the RNeasy Mini Kit (Qiagen, CA). After checking for RNA quality, DNA microarray was performed utilizing the human HG-U133A Affymetrix (Santa Clara, CA) genechip consisting of 22 277 ‘probe sets’ (Genomic Core Facility at Arizona Cancer Center). Data analysis was performed using BioConductor libraries (http://www.bioconductor.org) and R programming (http://www.r-project.org). A quality control analysis for each array was performed using Affymetrix console methods and the affyQCReport library. The array data was background corrected and quantile normalized using the affy Bioconductor library. Probe set values for the same gene were summarized by averaging. To compare conditions, log ratios of expression values were calculated. For each comparison, the mean ± 2 SD of the log fold changes were used as cut-offs to determine outlying differentially expressed genes. Each list of genes was tested for over-representation of KEGG and GO terms by counting the number of genes in the list with each pathway or term and using the Fisher exact test to determine over-representation. All terms with a P-value ≤ 0.05 were reported.
Statistical analysis
All in vitro experiments were performed in triplicate. The data were expressed as mean ± S.D. The difference between two mean values were evaluated using the Student’s t-test and considered to be statistically significant when p < 0.05. Statistical analysis of the mouse xenograft model data was performed by estimating the tumor growth for each mouse by fitting the least squares regression line of the tumor volume by day. The cube root of the observed tumor volumes was used to induce linearity in the raw data values. The slope of the regression line measures the tumor growth rate. Analysis of variance was used to test for the overall treatment effects on TGI. Tukey’s studentized range test was used to assess the significance of pair-wise differences between the groups adjusted for multiple comparisons. Survival of the mice was measured from the date of pair matching to sacrifice (event) or end of study (censored). The Kaplan-Meier method was used to estimate survival. The log rank test was used to compare survival between the respective treatment groups. Statistical adjustments were made for multiple comparisons. Analysis was performed using Prism (Graphpad, La Jolla, CA). All p-values ≤0.05 were considered statistically significant.
Results
A synthetic lethal interaction between spindle assembly dynamics and inhibition of Aurora A with MLN8237 in aggressive B-NHL cell lines
B-NHL cells (Granta-519 and RL) were treated in serial dilution with MLN8237 [M] or vincristine [V] or docetaxel [D]. The IC50 values for the B-NHL cells (Granta-519) determined for M, V and D are109.8 nM, 2.74 nM, and 4.53 nM respectively. For combination studies of M plus V, the dosage ratio was ~40:1 (M:V) for Granta-519 and 5.5:1 for RL cells. Both cell lines experienced a synergistic response with the combination of M plus V. The CI values for ED50 were determined to be 0.048 and 0.24 for Granta-519 and RL, respectively. Therefore, these interactions can be described as exhibiting very strong synergism. The combined dose median effect was 2.65 nM (in relation to M) and 0.067 nM (in relation to V) for Granta-519 cells (Figure 1) and this is consistent with tumor regression observed in the Granta-519 xenograft model (Figure 4A). These results indicate that the combination of M plus V is synergistic. Hence, V an agent that inhibits microtubule assembly plus Aurora A inhibitor [M] (inhibits centrosome assembly) is synthetic lethal.
Figure 1. Cell proliferation (MTS) assays of Granta-519 treated with MLN8237 or Vincristine or Both.

(A). The IC50 for MLN8237 was determined to be 109.8 nM and when combined with vincristine (V), decreased to 2.65 nM. (B). IC50 for V was determined to be 2.74 nM and when combined decreased to 0.067 nM with a CI value for ED50 to be 0.048.
Figure 4. MLN8237 plus Vincristine plus Rituximab is synthetic lethal and curative in a mouse xenograft model of MCL.
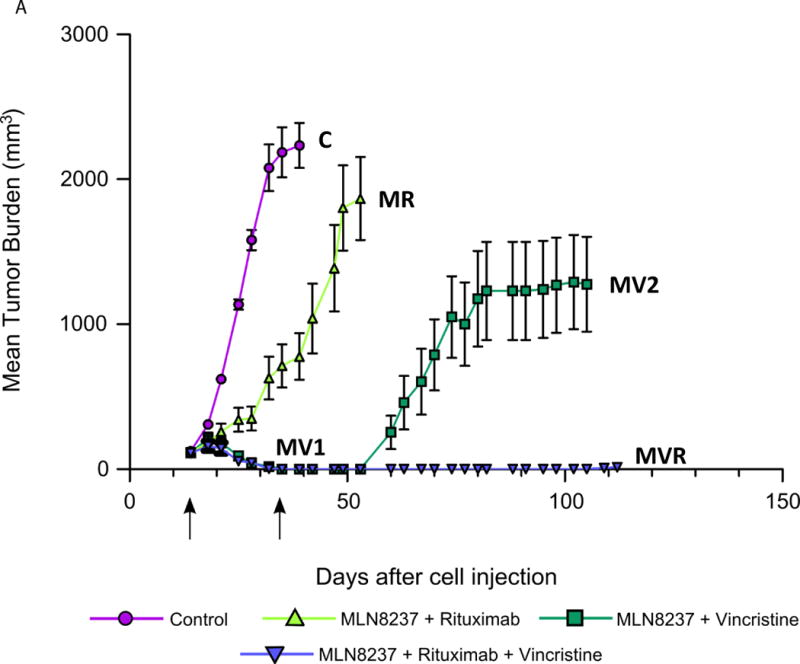

(A). Granta-519 xenograft mice (n=12 per cohort) were treated with saline (control), M 30 mg/kg + R 10 mg/kg, M 30 mg/kg + V 0.375 mg/kg and M 30 mg/kg + V 0.375 mg/kg + R 10 mg/kg. MLN8237 was given by PO Q1D × 3 weeks, vincristine and rituximab by IV Q1W × 4 weeks. MV1 represents the phase of tumor regression while MV2 represents the phase of lymphoma relapse. Tumor burdens were measured, graphed and represented as mean ± S.E.M. (B). Kaplan-Meier survival curves show overall survival differences between MVR in comparison to control, MR and MV.
Rituximab enhances apoptosis in B-cell NHL cells treated with MLN8237 plus microtubule targeting agents
MCL patients with high Aurora A expression demonstrated significantly worse survival than low expressers, suggesting Aurora A kinase is a potential target for aggressive B-cell NHL therapy (32). In a MCL (Granta-519) mouse xenograft model single agent MLN8237 [M] activity was modest with a TGI of ~10–15% (32). Recently, it was observed that MTAs such as docetaxel [D], paclitaxel and vincristine [V] had synergistic activity with an Aurora A kinase small molecular inhibitor or Aurora A transcriptional silencing in solid tumors (28–31). In a mouse MCL xenograft model, we demonstrated enhance TGI (~55–60%) when M was combined with D compared D alone (~10–15%) with an enhanced overall survival (32). Rituximab [R] is an effective treatment in B-NHL when combined with chemotherapy. To determine whether R enhances cytotoxicity of M, D or V and doublet combinations, we evaluated degree of apoptosis by flow cytometry in RL, Granta-519 and SUDHL-4 cell lines utilizing sub-lethal doses (D 5nM, R 10μg/ml, M 5nM). As shown in figure 2A, M, D or R alone had no significant effect on inducing apoptosis compared to control. However, the combination treatments of MR, MD, and MDR significantly induced apoptosis compared to control (p<0.05 and p<0.001) in all three B-NHL cell lines. Importantly, R significantly enhanced apoptosis of MD therapy (figure 2A). Interestingly, apoptosis was not increased by RD therapy. However, superior results were observed with V (0.1nM) and combinations MV and MVR at sub-lethal doses in Granta-519 cells (Figure 2B) that correlate well with the MTS cell viability data (Figure 1).
Figure 2. Rituximab and microtubule targeting agents enhance MLN8237-induced apoptosis in B-cell NHL cells.



(A) RL, Granta-519 and SUDHL-4 cells were treated with docetaxel at 5 nM, rituximab at 10 μg/ml and MLN8237 at 5 nM alone or the combinations as indicated at same doses for 72 hr. (B) Granta-519 cells were treated as same as (A) except docetaxel was substituted for vincristine at the dose of 0.1 nM. Apoptosis was analyzed by flow cytometry after annexin V and PI staining. The graph represents the mean percentage of apoptosis ± S.D. (n=3). * p<0.05 and ** p<0.001. (C) Granta-519 cells treated with AT9283 (5nM) (pan-Aurora inhibitor) and vincristine (0.1nM) ± rituximab (10 μg/ml). Apoptosis was analyzed by flow cytometry after annexin V and PI staining. The graph represents the mean percentage of apoptosis ± S.D. (n=3).
Rituximab increases in vivo anti-NHL activity of MLN8237 and MLN8237 plus docetaxel
Based on our in vitro data that targeting Aurora A in combination with rituximab [R] was more effective in inducing apoptosis, we evaluated this combination in a SCID mouse xenograft model of MCL (Granta-519). There were 6 cohorts of 12 mice: vehicle control, M at 10 mg/kg and 30 mg/kg PO once a day for 3 weeks, R at 10 mg/kg IV once/week × 4, M at 10 mg/kg or 30 mg/kg for 3 weeks + R 10 mg/kg IV once/week × 4. The dose of R was based on a clinically relevant dose used in mouse xenograft tumor models. Treatments with M or R alone showed a ~10–15% TGI compared to vehicle control. However, M (10 mg/kg or 30mg/kg) plus R showed significant TGI of ~50% compared to vehicle control (p < 0.05) (Figure 3A) indicating R enhances anti-NHL activity of M. Next, we evaluated the effect of M plus D or M plus R versus MDR triple combination to increase anti-NHL activity in a MCL xenograft mouse model. SCID mice bearing Granta-519 were treated with vehicle, M 30 mg/kg + R 10 mg/kg, M 30 mg/kg + D 10 mg/kg and M 30 mg/kg + R 10 mg/kg + D 10 mg/kg, respectively. As shown in figure 3B, TGI was superior for MDR (~85%) compared to MD (~55–60%) and MR (~25%). Statistical analysis showed that tumor growth rate for MR, MD and MDR was significantly less than that for control with p=0.004, p<0.0001 and p<0.0001 respectively. MD and MDR also significantly inhibited tumor growth compared to MR with p=0.0024 and p<0.0001 respectively. Together, these data indicate that D increased anti-NHL activity of M and MR significantly. The overall survival for mice treated with MDR was >45 days compared control versus MD (~30 days) and MR (~15 days) (p=0.01). No weight loss >10% was observed in any of the treatment arms.
Figure 3. Rituximab increases in vivo anti-NHL activity of MLN8237 and MLN8237 plus docetaxel in a Granta-519 MCL xenograft mouse model.


(A). Evaluation of in vivo therapeutic activity of MLN8237 [M], rituximab [R] and MR in a MCL xenograft mouse model. SCID mice bearing Granta-519 tumors (n = 12) were treated with saline (control), M at 10 mg/kg or 30 mg/kg, R at 10 mg/kg, M 10 mg/kg + R 10 mg/kg and M 30 mg/kg + R 10 mg/kg. Tumor burden were measured three times a week and graphed. All values are presented as mean ± S.E.M. (B). Evaluation of tumor growth inhibition of M + R versus M + docetaxel [D] versus M + D + R. Granta-519 xenograft mice were treated with M 30 mg/kg plus R 10 mg/kg, M 30 mg/kg plus D 10 mg/kg and M 30 mg/kg + D 10 mg/kg + R 10 mg/kg. Tumor volume versus time is presented as mean ± S.E.M. The arrows represent the start and the end of treatment.
MLN8237 plus Vincristine plus Rituximab is curative in a mouse xenograft model of Mantle Cell Lymphoma
Since V is used commonly in B-NHL as part of standard chemotherapy (e.g. CHOP), we replaced D with V, the former targets microtubule polymerization while the latter microtubule depolymerization. We treated xenograft mice (n=4 per treatment arm) bearing Granta-519 with V at four different doses (0.0375 mg/kg, 0.07 mg/kg, 0.14 mg/kg and 0.375 mg/kg) IV once per week ×4. The results showed V alone had a modest TGI of ~10–15% compared to control (p>0.05) (data not shown). However, M (30 mg/kg PO, once a day for 3 weeks) plus V (0.375mg/kg IV, once a week ×4) [MV] and M (30 mg/kg PO, once a day for 3 weeks) plus V (0.375mg/kg IV, once a week ×4) plus R (10 mg/kg IV, once a week x4) [MVR] led to tumor regression [TR] compared to control or MR (p=0.0072 and p<0.0001, respectively) (Figure 4A). Interestingly tumors treated with MV relapsed 20 days after stopping treatment, while those treated with MVR did not relapse >100 days after stopping treatment (curative) (Figure 4A). Kaplan-Meier analysis of overall survival showed that the mice treated with MV and MVR had a statistically significant improvement in overall survival when compared with the control (p<0.0001) or MR (p=0.0043) (Figure 4B). The mice in all cohorts tolerated treatment well with no weight loss >10%. In fact mice in the MVR were healthy and some were separated due to in-fighting.
Re-activation of cell cycle regulators is a mechanism of resistance to MV abrogated by MVR therapy
In order to gain mechanistic insights for MV or MVR driven tumor regression, tumors (n=3) were harvested at end of treatment (3 weeks) for each cohort. Figure 5A shows the relative sizes of control versus MR versus MV versus MVR. These tumors were evaluated for up- and down-regulated genes by expression profiling and validated by Western blotting for selected cell cycle regulators and biomarkers of proliferation. Unique and common Gene Ontologies (GO) were repressed by individual treatments (MR Versus MV Versus MVR) consistent with modulation of cell cycle regulators (Figure 5B) that provides mechanistic insight of the effectiveness of MV or MVR therapy. Over-expression cyclin D1 associated with G1 and S phase of the cell cycle (a hallmark of MCL) and over-expression of Bcl-2, continues to be elevated with MR but is suppressed by MV1 (tumor regression phase) and is re-expressed in MV2 (tumor relapse phase), but remains completely suppressed with MVR therapy (Figure 5C). Cyclin B1 associated with G2/M phase of the cell cycle follows a similar expression pattern as cyclin D1 implicating an active cell cycle is inhibited by MVR therapy. Over-expression of Aurora B (mitotic phase and SAC) is inhibited by MV1 (regression phase) but is re-activated on relapse (MV2). However, MVR therapy continues to inhibit Aurora B by repressing mitotic sister chromatid segregation by repressed gene expression analyzed by GO. Inhibition of mitotic sister chromatid segregation is unique to MVR therapy and is most likely due to R mediated inhibition of cell proliferation. This effect is highlighted by proliferation cell nuclear antigen (PCNA) by IHC and Western blotting analysis which demonstrated proliferation to be completely repressed by MVR therapy and correlates with down regulation of cell cycle activators identified by gene expression profiling (Figure 5D).
Figure 5. Re-activation of cell cycle regulators and anti-apoptosis is a mechanism of resistance to MVabrogated by MVR therapy.

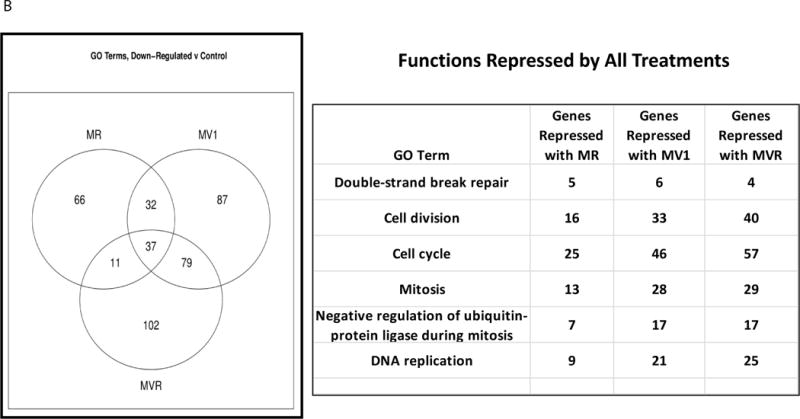


(A). Tumor sizes of 3 mice per cohort sacrificed 3h after end of the last treatment are shown for control, MR, MV and MVR therapy. (B). Gene expression profiling identified unique and common gene ontologies repressed by individual drug treatments compared to control and each other (Venn diagram and list of repressed functionalities). (C). Protein was isolated from the tumors 3h after the end of last treatment as indicated and at relapse (MV2). Western blotting analysis demonstrated that Proliferating cell nuclear antigen (PCNA), Aurora B, cyclin B1 cyclin D1 and Bcl-2 were inhibited by MV1 (tumor regression phase) and MVR but not MR and MV2 (tumor relapse phase). (D). PCNA was evaluated by immunohistochemistry from tumors 3h after the end of last treatment for control, MR, MV1, MVR and MV2 (at relapse).
Discussion
Lymphoma is a malignant transformation of B- or T- lymphocytes and is the most common type of hematologic malignancy in the United States which represents ~5% of all cancers and ~55% of blood cancers. Although modern treatment options such as R-Hyper-CVAD for MCL, R-CHOP for DLBCL and TFL have improved clinical outcomes (43–45), there are no curative therapies for >50% patients. Here, we demonstrate that pre-clinical in vitro and in vivo mechanistic investigations with MLN8237 [M], an Aurora A selective small molecule inhibitor (SMI) combined with vincristine [V] is potently synergistic. However, B-NHL treated with MV eventually acquires resistance and relapse. In contrast, when rituximab [R] is combined with MV there is enhanced apoptosis and complete responses (curative) in a mouse xenograft model of MCL. Hence, MVR is a promising novel therapy and an early phase clinical trial has been initiated with this combination in relapsed/refractory aggressive B-NHL.
Studies using RNAi and selective SMIs have shown that Aurora A inhibition is characterized by a SAC-induced mitotic arrest with formation of unipolar spindles, a tetraploid phenotype and biphasic apoptosis. Thus, inhibiting its enzyme activity with specific SMIs to the catalytic domain ATP-binding site is regarded as feasible for targeted cancer therapy, and numerous inhibitors including M have been developed and are now in clinical trials (2). Recently, we demonstrated Aurora A to be over-expressed in a number of B-NHL cell lines and in MCL patient tissues increased expression correlated with decreased survival (32). Hence, we hypothesized that Aurora A is an attractive therapeutic target for the treatment of aggressive B-NHL. MLN8237 had potent activity against Aurora A kinase (32) and induced apoptosis in cultured cells (Figure 2). However, as a single agent TGI is modest (Figure 3A), suggesting that inhibition of Aurora A kinase alone is insufficient as an anti-NHL therapy. We (32) and others (26, 27) have shown that Aurora A amplification overrides the SAC leading to paclitaxel resistance. Hence, inhibition of Aurora A abrogates the mitotic delay induced by paclitaxel (27) at sub-lethal doses. Therefore, Aurora SMIs in combination with microtubule targeting agents (MTAs) such as the taxanes (32, 46) and vincristine [V] show synergy in vitro cell culture models of apoptosis and in vivo anti-tumor activity. Further, we demonstrate that the combination of M plus V (targets microtubule depolymerization and hence inhibits spindle assembly) to have superior synergistic activity (Figure 1 and 4A) than M plus D (targets microtubule polymerization and inhibits spindle disassembly). The molecular mechanism(s) that determines synergy of Aurora inhibition plus vincristine is due to complete inhibition to form centrosome driven mitotic spindle formation.
Since FDA approval in 1997, rituximab [R] has become one of the most widely prescribed therapeutic agents for the B-NHL patients due to the ubiquitous expression of the target protein CD20 on the surface of B lymphocytes and the fact that the vast majority of NHLs are B-cell malignancies (47). CD20 is a B-cell marker of differentiation and proliferation. The mechanisms of anti-lymphoma effect of R from experimental evidence are apoptosis, complement dependent cytotoxicity (CDC), antibody dependent cellular cytotoxicity (ADCC), antibody dependent cellular phagocytosis (ADCP) and vaccinal effect (48). In combination with chemotherapy, R significantly improves survival outcomes for patients with B-NHLs (49, 50). Here, we demonstrate that R induced apoptosis in different subtypes of B-NHL cells in vitro and importantly, when combined with M or M plus a MTA (particularly vincristine) at sub-lethal doses induced significantly higher percentage of apoptosis (decreased Bcl-2) (Figure 5C) than the each agent alone (Figure 2). Although MV is a synergistic in mice bearing MCL tumors, despite an initial complete response there is eventual tumor relapse (Figure 4A). However, addition of R to MV completely abrogated this relapse. In order to gain mechanistic insight gene expression profiling (GEP) and confirmatory Western blotting showed re-emergence of expression of Aurora B, cyclin B1, cyclin D1 and Bcl-2 with MV therapy but not with MVR therapy. Comparisons of GEP of MR versus MV and MVR indicate that the latter combination directly and indirectly interferes with all aspects of the cell cycle including inhibition of sister chromatid segregation (Figure 5B). In contrast, addition of R to MD led to superior TGI (~85%) but not to tumor regression (Figure 3B), as was observed with MV and MVR. Gene expression profiling showed several B-cell surface markers including CD19 and CD24 to be down-regulated with MVR compared to MV, which could contribute to complete responses observed with the former therapy. We hypothesize that lymphoma progenitor or stem cells to be extremely sensitive to MVR therapy and this concept is under investigation.
In conclusion, collectively our findings indicate that Aurora A is an excellent therapeutic target for aggressive B-NHLs. The potential of a synergistic interaction between Aurora inhibition (MLN8237) and microtubule spindle assembly (vincristine) is identified as a potent therapy for aggressive B-NHL. However, the MV combination is not curative in mice bearing B-NHL. The addition of R to MV prevents relapse and is synthetic lethal with curative potential, most likely by interfering with cellular components of proliferation not affected by MV therapy alone. Therefore, MLN8237 plus vincristine plus rituximab represents a novel therapeutic strategy in aggressive B-NHL and warrants early phase clinical trial evaluation.
Translational Relevance.
Targeting synergistic mechanisms within the context of the hallmarks of cancer may yield novel effective therapies with minimal toxicity for B-NHL. Auroras (A and B) are a family of mitotic serine/threonine kinases that are involved in high fidelity cell division. Aberrant over-expression of Auroras is oncogenic leading to genetic instability, tumor initiation and progression. Over-expression of Auroras has been shown to promote resistance to microtubule targeting agents (MTAs) such as taxanes and vinca alkaloids. Inhibition of Auroras with siRNA knockdown or pharmacologic intervention with a small molecule inhibitor [MLN8237] leads to enhanced sensitivity of cancer cells to MTAs. Here, we demonstrated that MLN8237 plus vincristine (MV) is synergistic and addition of rituximab to MV (MVR) is synthetic lethal and curative in established B-NHL xenograft mice. These findings suggest that MVR may represent a novel therapeutic strategy in aggressive B-NHL and a phase I/II trial is enrolling patients based on our data.
Acknowledgments
The authors thank the Lymphoma SPORE (1 P5O CA B080501A1) from the NIH/NCI for funding this project, Dr. Richard I. Fisher, Principal Investigator (University of Rochester), Dr. David Mount (AZCC Bioinformatics Core) for providing analysis of gene expression profiling of harvested tumors. The mouse xenograft studies were conducted by the AZCC experimental mouse shared services (EMSS) and statistical analysis by Dr. H. Cui (AZCC, Biometry and Lymphoma SPORE bio-statistic core). The authors also thank Larry and Marcia Greene; Bill and Ginny Noyes; Drs. June and Cedric Dempsey for providing donations to fund research conducted on Mantle Cell Lymphoma as well as Millennium Pharmaceuticals and Astex Pharmaceuticals for providing Aurora kinase inhibitors for this study.
Footnotes
Contribution: D.M. and W.Q. designed and performed research, analyzed the data, and wrote the manuscript; A.S. and C.M. performed MTS assay; L.S.C. and A.M. analyzed the data. D.O.P., R.I.F. and T.P.M. reviewed and edited the manuscript.
Conflict-of-interest disclosure
The authors declare no competing financial interests.
References
- 1.Mahadevan D, Fisher RI. Novel Therapeutics for Aggressive Non-Hodgkin’s Lymphoma. J Clin Oncol. 2011;29(14):1876–84. doi: 10.1200/JCO.2010.32.7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Green MR, Woolery JE, Mahadevan D. Update on Aurora Kinase Targeted Therapeutics in Oncology. Expert Opin Drug Discov. 2011;6(3):291–307. doi: 10.1517/17460441.2011.555395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009;8:547–66. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- 4.Marumoto T, Zhang D, Saya H. Aurora-A - a guardian of poles. Nat Rev Cancer. 2005;5:42–50. doi: 10.1038/nrc1526. [DOI] [PubMed] [Google Scholar]
- 5.Arbitrario JP, Belmont BJ, Evanchik MJ, Flanagan WM, Fucini RV, Hansen SK, et al. SNS-314, a pan-Aurora kinase inhibitor, shows potent anti-tumor activity and dosing flexibility in vivo. Cancer Chemother Pharmacol. 65:707–17. doi: 10.1007/s00280-009-1076-8. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka R, Squires MS, Kimura S, Yokota A, Nagao R, Yamauchi T, et al. Activity of the multitargeted kinase inhibitor, AT9283, in imatinib-resistant BCR-ABL-positive leukemic cells. Blood. 116:2089–95. doi: 10.1182/blood-2009-03-211466. [DOI] [PubMed] [Google Scholar]
- 7.Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, et al. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol. 2002;22:874–85. doi: 10.1128/MCB.22.3.874-885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kufer TA, Nigg EA, Sillje HH. Regulation of Aurora-A kinase on the mitotic spindle. Chromosoma. 2003;112:159–63. doi: 10.1007/s00412-003-0265-1. [DOI] [PubMed] [Google Scholar]
- 9.Terada Y, Uetake Y, Kuriyama R. Interaction of Aurora-A and centrosomin at the microtubule-nucleating site in Drosophila and mammalian cells. J Cell Biol. 2003;162:757–63. doi: 10.1083/jcb.200305048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glover DM, Leibowitz MH, McLean DA, Parry H. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995;81:95–105. doi: 10.1016/0092-8674(95)90374-7. [DOI] [PubMed] [Google Scholar]
- 11.Giet R, McLean D, Descamps S, Lee MJ, Raff Jw, Prigent C, et al. Drosophila Aurora A kinase is required to localize D-TACC to centrosomes and to regulate astral microtubules. J Cell Biol. 2002;156:437–51. doi: 10.1083/jcb.200108135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roghi C, Giet R, Uzbekov R, et al. The Xenopus protein kinase pEg2 associates with the centrosome in a cell cycle-dependent manner, binds to the spindle microtubules and is involved in bipolar mitotic spindle assembly. J Cell Sci. 1998;111(Pt 5):557–72. doi: 10.1242/jcs.111.5.557. [DOI] [PubMed] [Google Scholar]
- 13.Hannak E, Kirkham M, Hyman AA, Oegema K. Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. J Cell Biol. 2001;155:1109–16. doi: 10.1083/jcb.200108051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marumoto T, Honda S, Hara T, Nitta M, Hirota T, Kohmura E, et al. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem. 2003;278:51786–95. doi: 10.1074/jbc.M306275200. [DOI] [PubMed] [Google Scholar]
- 15.Bischoff JR, Anderson L, Zhu Y, Mossie K, Nf L, Sousa B, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17:3052–65. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–93. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- 17.Giet R, Petretti C, Prigent C. Aurora kinases, aneuploidy and cancer, a coincidence or a real link? Trends Cell Biol. 2005;15:241–50. doi: 10.1016/j.tcb.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Hamada M, Yakushijin Y, Ohtsuka M, Kakimoto M, Yasukawa M, Fujita S. Aurora2/BTAK/STK15 is involved in cell cycle checkpoint and cell survival of aggressive non-Hodgkin’s lymphoma. Br J Haematol. 2003;121:439–47. doi: 10.1046/j.1365-2141.2003.04311.x. [DOI] [PubMed] [Google Scholar]
- 19.Mortlock AA, Keen NJ, Jung FH, Heron NM, Foote KM, Wilinson RW, et al. Progress in the development of selective inhibitors of aurora kinases. Curr Top Med Chem. 2005;5:807–21. doi: 10.2174/1568026054637719. [DOI] [PubMed] [Google Scholar]
- 20.Mahadevan D, Spier C, Della Croce K, Miller S, George B, Riley C, et al. Transcript profiling in peripheral T-cell lymphoma, not otherwise specified, and diffuse large B-cell lymphoma identifies distinct tumor profile signatures. Mol Cancer Ther. 2005;4:1867–79. doi: 10.1158/1535-7163.MCT-05-0146. [DOI] [PubMed] [Google Scholar]
- 21.Camacho E, Bea S, Salaverria I, Lopez-Fuillerma A, Puig X, Benavente Y, et al. Analysis of Aurora-A and hMPS1 mitotic kinases in mantle cell lymphoma. Int J Cancer. 2006;118:357–63. doi: 10.1002/ijc.21370. [DOI] [PubMed] [Google Scholar]
- 22.Hata T, Furukawa T, Sunamura M, Egawa S, Motoi F, Ohmura N, et al. RNA interference targeting aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res. 2005;65:2899–905. doi: 10.1158/0008-5472.CAN-04-3981. [DOI] [PubMed] [Google Scholar]
- 23.Hirota T, Kunitoku N, Sasayama T, Marumoto T, Zhang D, Nitta M, et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114:585–98. doi: 10.1016/s0092-8674(03)00642-1. [DOI] [PubMed] [Google Scholar]
- 24.Marumoto T, Hirota T, Morisaki T, Kunitoku N, Zhang D, Ichikawa, et al. Roles of aurora-A kinase in mitotic entry and G2 checkpoint in mammalian cells. Genes Cells. 2002;7:1173–82. doi: 10.1046/j.1365-2443.2002.00592.x. [DOI] [PubMed] [Google Scholar]
- 25.Liu Q, Ruderman JV. Aurora A, mitotic entry, and spindle bipolarity. Proc Natl Acad Sci U S A. 2006;103:5811–6. doi: 10.1073/pnas.0601425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anand S, Penrhyn-Lowe S, Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 2003;3:51–62. doi: 10.1016/s1535-6108(02)00235-0. [DOI] [PubMed] [Google Scholar]
- 27.Wysong DR, Chakravarty A, Hoar K, Ecsedy JA. The inhibition of Aurora A abrogates the mitotic delay induced by microtubule perturbing agents. Cell Cycle. 2009;8:876–88. doi: 10.4161/cc.8.6.7897. [DOI] [PubMed] [Google Scholar]
- 28.VanderPorten EC, Taverna P, Hogan JN, Ballinger MD, Flanagan WM, Fucini RV. The Aurora kinase inhibitor SNS-314 shows broad therapeutic potential with chemotherapeutics and synergy with microtubule-targeted agents in a colon carcinoma model. Mol Cancer Ther. 2009;8:930–9. doi: 10.1158/1535-7163.MCT-08-0754. [DOI] [PubMed] [Google Scholar]
- 29.Lentini L, Amato A, Schillaci T, Insalaco L, Di Leonardo A. Aurora-A transcriptional silencing and vincristine treatment show a synergistic effect in human tumor cells. Oncol Res. 2008;17:115–25. doi: 10.3727/096504008785055521. [DOI] [PubMed] [Google Scholar]
- 30.Scharer CD, Laycock N, Osunkoya AO, Logani S, McDonald JF, Benigno BB, et al. Aurora kinase inhibitors synergize with paclitaxel to induce apoptosis in ovarian cancer cells. J Transl Med. 2008;6:79. doi: 10.1186/1479-5876-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazumdar A, Henderson YC, El-Naggar AK, Sen S, Clayman GL. Aurora kinase A inhibition and paclitaxel as targeted combination therapy for head and neck squamous cell carcinoma. Head Neck. 2009;31:625–34. doi: 10.1002/hed.21007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qi W, Cooke LS, Liu X, Rimsa L, Roe DJ, Manziello A, et al. Aurora inhibitor MLN8237 in combination with docetaxel enhances apoptosis and anti-tumor activity in mantle cell lymphoma. Biochem Pharmacol. 81:881–90. doi: 10.1016/j.bcp.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carol H, Boehm I, Reynolds CP, Kang MH, Maris JM, Morton CL, et al. Efficacy and pharmacokinetic/pharmacodynamic evaluation of the Aurora kinase A inhibitor MLN8237 against preclinical models of pediatric cancer. Cancer Chemother Pharmacol. 2011;68(5):1291–304. doi: 10.1007/s00280-011-1618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sloane DA, Trikic MZ, Chu ML, Lamers MB, Mason CS, Mueller I, et al. Drug-resistant aurora A mutants for cellular target validation of the small molecule kinase inhibitors MLN8054 and MLN8237. ACS Chem Biol. 5:563–76. doi: 10.1021/cb100053q. [DOI] [PubMed] [Google Scholar]
- 35.Lipsitz E, Moorthy G, Mosse Y, Fox E, Adamson PC. A sensitive and selective liquid chromatography/tandem mass spectrometry method for determination of MLN8237 in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 878:2369–73. doi: 10.1016/j.jchromb.2010.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gorgun G, Calabrese E, Hideshima T, Ecsedy J, Perrone G, Mani M, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. 115:5202–13. doi: 10.1182/blood-2009-12-259523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomita M, Mori N. Aurora A selective inhibitor MLN8237 suppresses the growth and survival of HTLV-1-infected T-cells in vitro. Cancer Sci. 101:1204–11. doi: 10.1111/j.1349-7006.2010.01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelly KR, Ecsedy J, Medina E, Mahalingam D, Padmanabhan S, Nawrocki ST, et al. The novel Aurora A kinase inhibitor MLN8237 is active in resistant chronic myeloid leukemia and significantly increases the efficacy of nilotinib. J Cell Mol Med. 2011;15(10):2057–70. doi: 10.1111/j.1582-4934.2010.01218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, et al. Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP) Pediatr Blood Cancer. 55:26–34. doi: 10.1002/pbc.22430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vega MI, Huerta-Yepez S, Martinez-Paniagua M, Martinez-Miguel B, Hernandez-Pando R, Gonzalez-Bonilla CR, et al. Rituximab-mediated cell signaling and chemo/immuno-sensitization of drug-resistant B-NHL is independent of its Fc functions. Clin Cancer Res. 2009;15:6582–94. doi: 10.1158/1078-0432.CCR-09-1234. [DOI] [PubMed] [Google Scholar]
- 41.Hu W, Ge X, You T, Xu T, Zhang J, Wu G, et al. Human CD59 Inhibitor Sensitizes Rituximab-Resistant Lymphoma Cells to Complement-Mediated Cytolysis. Cancer Res. 71:2298–307. doi: 10.1158/0008-5472.CAN-10-3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li M, Liu L, Xi N, Wang Y, Dong Z, Tabata O, et al. Imaging and measuring the rituximab-induced changes of mechanical properties in B-lymphoma cells using atomic force microscopy. Biochem Biophys Res Commun. 404:689–94. doi: 10.1016/j.bbrc.2010.12.043. [DOI] [PubMed] [Google Scholar]
- 43.Flowers CR, Sinha R, Vose JM. Improving outcomes for patients with diffuse large B-cell lymphoma. CA Cancer J Clin. 60:393–408. doi: 10.3322/caac.20087. [DOI] [PubMed] [Google Scholar]
- 44.Goy A, Kahl B. Mantle cell lymphoma: The promise of new treatment options. Crit Rev Oncol Hematol. 2010;75(2):110–21. doi: 10.1016/j.critrevonc.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 45.Ghielmini M. Follicular lymphoma. Ann Oncol. 21(Suppl 7):vii151–3. doi: 10.1093/annonc/mdq287. [DOI] [PubMed] [Google Scholar]
- 46.Shimomura T, Hasako S, Nakatsuru Y, Mita T, Ichikawa, Kodera T, et al. MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol Cancer Ther. 9:157–66. doi: 10.1158/1535-7163.MCT-09-0609. [DOI] [PubMed] [Google Scholar]
- 47.Zwick C, Murawski N, Pfreundschuh M. Rituximab in high-grade lymphoma. Semin Hematol. 47:148–55. doi: 10.1053/j.seminhematol.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 48.Cartron G, Trappe RU, Solal-Celigny P, Hallek M. Interindividual variability of response to rituximab: from biological origins to individualized therapies. Clin Cancer Res. 17:19–30. doi: 10.1158/1078-0432.CCR-10-1292. [DOI] [PubMed] [Google Scholar]
- 49.Marcus R, Imrie K, Solal-Celigny P, Catalano JV, Dmoszynska A, Raposo JC, et al. Phase III study of R-CVP compared with cyclophosphamide, vincristine, and prednisone alone in patients with previously untreated advanced follicular lymphoma. J Clin Oncol. 2008;26:4579–86. doi: 10.1200/JCO.2007.13.5376. [DOI] [PubMed] [Google Scholar]
- 50.Friedberg JW, Vose JM, Kelly JL, Young F, Bernstein SH, Peterson D, et al. The combination of bendamustine, bortezomib, and rituximab for patients with relapsed/refractory indolent and mantle cell non-Hodgkin lymphoma. Blood. 117:2807–12. doi: 10.1182/blood-2010-11-314708. [DOI] [PMC free article] [PubMed] [Google Scholar]