

Abstract
Neuronal nicotinic acetylcholine receptors (nAChRs) are the primary binding sites for nicotine within the brain. Using alpha(α)2 nAChR subunit-null mutant mice, the current study evaluates whether the absence of this gene product during mecamylamine-precipitated nicotine withdrawal eliminates neuronal activity within selective midbrain and limbic brain regions, as determined by the expression of the immediate early gene, cfos. Our results demonstrate that nicotine withdrawal enhances neuronal activity within the interpeduncular nucleus and dorsal hippocampus, which is absent in mice null for α2-containing nAChRs. In contrast, we observe that α2-null mutant mice exhibit a suppression of neuronal activity in the dentate gyrus in mice undergoing nicotine withdrawal. Interestingly, α2-null mutant mice display potentiated neuronal activity specifically within the stratum lacunosum moleculare layer of the hippocampus, independent of nicotine withdrawal. Overall, our findings demonstrate that α2-null mutant mice have altered cfos expression in distinct populations of neurons within selective midbrain and limbic brain structures that mediate baseline and nicotine withdrawal-induced neuronal activity.
Keywords: Nicotine Withdrawal, Addiction, Hippocampus, Interpeduncular Nucleus, Chrna2, Neuronal Activity
Graphical abstract
1.0 Introduction
Chronic tobacco use is the leading cause of preventable disease and death in the United States [1]. An important mediator of continued tobacco use relates to the aversive withdrawal symptoms induced by smoking cessation [2–5]. Thus, by understanding the mechanisms mediating tobacco withdrawal symptoms, more effective therapeutic interventions could be provided.
Nicotine, a primary psychoactive component within tobacco, binds to neuronal nicotinic acetylcholine receptors (nAChRs) [4, 6–9]. Nicotinic receptors are pentameric, ligand-gated ion channels composed of various combinations of alpha(α) and beta(β) subunits [2, 10]. The α and β nAChR subunits are proteins that interact to give rise to distinct receptors with selective pharmacology and developmental expression patterns throughout the brain [11–13]. Depending on their subunit composition, neuronal nAChRs display varying sensitivities to a wide range of agonists and antagonists [14]. To better understand how these unique receptors contribute to nicotine-modulated behaviors, including withdrawal, genetic animal models have been developed [2, 5, 15, 16]. Findings illustrate that certain nicotinic receptor subunit null mutant mice, but not others, differentially regulate nicotine withdrawal (for review see [2]). While less is known about the mechanisms mediating withdrawal symptoms in humans, genome-wide association and candidate gene studies have found consistent trends for polymorphisms in a number of nicotinic receptor subunit genes predicting tobacco addiction [17–19]. Thus, both clinical and preclinical evidence suggest that nicotinic receptors are important contributors to addiction, with possible relationships to the mechanisms mediating nicotine withdrawal [5, 20]. Such studies are assisting in the development of novel therapeutic ligands specific for selective nicotinic receptors for the purpose of combatting the tobacco addiction pandemic [21–24].
The α2 nAChR subunit (encoded by the Chrna2 gene) is expressed in a unique set of neuronal structures associated with addiction and withdrawal [11, 12, 25, 26]. In rodents, these regions include limbic and midbrain structures, with highest mRNA expression for α2 nAChR subunits found within the interpeduncular nucleus (IPN, all sub-regions), and more restricted expression within other brain regions such as the hippocampus (Stratum Oriens/Alveus) and the entorhinal cortex (layer II) [11, 12, 25, 27]. While the α2 nAChR subunit has lower levels of expression within the rodent central nervous system when compared to other nAChR subunits, wider expression in the primate and human brain is observed, suggesting a more global influence [28, 29].
Indeed, recent evidence has shown that multiple nAChR subunits within the habenulo-IPN axis influence nicotine reward and withdrawal [4, 5, 30–33]. The IPN is a midline structure located within the midbrain that receives a vast majority of its input bilaterally from the medial habenula via a pathway known as the fasciculus retroflexus [34]. Local injection of the general nAChR antagonist, mecamylamine, within either the habenula (Hb) or the IPN after chronic nicotine treatment is sufficient to precipitate somatic withdrawal in a home cage habituated environment. Notably, these withdrawal symptoms are absent in mice that are mutant knockouts for the α2 nAChR subunit [31]. Subsequent studies have supported these findings, while also illustrating that withdrawal behavior is dependent on the context in which the animals are assessed [35]. Taken together, the studies implicate a putative role for α2-containing nAChRs located within the IPN in regulating nicotine withdrawal [31, 35–37].
The IPN is also known as an integrative center for the limbic system, with output projections to the hippocampus, entorhinal cortex and the septum [34, 38]. Thus, the IPN in parallel with the limbic system could potentiate the aversive effects of withdrawal via negative psychological states and drug-associated or context-specific memories [34, 39–42]. Prior studies have demonstrated that genetic deletion of the α2 nAChR subunit eliminates nicotinic facilitation and depression of long-term potentiation (LTP) within the dorsal hippocampal (DH) CA1 [43]. These effects are likely mediated via oriens lacunosum moleculare (OLM) GABAergic interneurons within the stratum oriens (SO) layer of the hippocampal CA1 region [43–46], which can influence distal dendrite inhibition to facilitate fear memory in mice [47]. Nicotine can also influence hippocampal synaptic plasticity within the dentate gyrus (DG) via the disinhibition of granule cells, which modify local GABAergic circuit inhibition [48]. These effects could be influenced, at least in part, through α2–containing nAChR expression in layer II of the entorhinal cortex [25], a layer known to project to the DG via the perforant pathway [49]. Reportedly, α2-containing nAChR expression is also present locally within the DG during early development (post-natal day 3–5) [13] and may contribute to the maturation of the circuit. Nicotine-induced modifications in hippocampal synaptic plasticity could then mediate drug-associated or context-specific memories via changes in GABAergic systems [48, 50], possibly through α2-containing nAChRs. Since nicotine withdrawal signs are dependent on the context where behavior is assessed [35], we investigate the effects of nicotine withdrawal-induced neuronal activity within hippocampal circuits.
The current study examines whether α2-null mutant mice have altered cFos expression in restricted midbrain and limbic brain regions during nicotine withdrawal [51]. We compare α2 nAChR subunit null mutant versus wild type mice to better understand how the genetic deletion of this specific receptor subunit can influence neuronal activation in adult male mice, as demonstrated by the expression of the immediate early gene, cfos. We hypothesize that α2-containing nAChRs mediate nicotine withdrawal via enhanced neuronal activity within restricted midbrain and limbic brain structures, which should be absent in α2-null mutant mice. We assess cfos expression within the Hb-IPN axis, known for its influence in mediating nicotine withdrawal [31, 33]. Within the hippocampus, we evaluate the dorsal and ventral regions separately, as they are known to have functionally and anatomically distinct structures, corresponding to cognitive functions such as encoding spatial memory (dorsal hippocampus), as well as stress, emotion and affect (ventral hippocampus (VH)) [52].
2.0 Materials and Methods
2.1 Animals and Nicotine Withdrawal Drug Treatment
All studies described below conformed to the National Institute of Health Guidelines for the Care and Use of Laboratory Animals [35]. The creation of the α2 nAChR subunit null mutant mouse line, drug treatment conditions for the induction of nicotine withdrawal, behavioral testing and withdrawal assessment have been previously described [35]. Briefly, twenty male wild type mice and α2 nAChR subunit null mutant mice were chronically treated with either nicotine or vehicle at a dose of 24 mg/kg (expressed as free base) per day for 2 weeks using an Alzet mini-osmotic pump (model 1002). On day 13, animals were injected with mecamylamine at a dose of 3 mg/kg intraperitoneally (i.p.), to precipitate nicotine withdrawal in a habituated home cage environment. Two hours after assessment of mecamylamine-precipitated withdrawal, animals were perfused and brains were harvested for future assessment of cfos immunoreactivity. As previously reported, these mice exhibit enhanced mecamylamine-precipitated somatic withdrawal signs [35]. Nicotine and cotinine blood plasma concentrations were quantified on day 13 of osmotic minipump administration by Pura Tech UCSF-Clinical Pharmacology Laboratory [53]. Within a batch (n = 4) of the chronic nicotine-treated wild type male mice, nicotine (56.6 ng/ml ± 14.0 (S.E.M., Standard Error of the Mean)) and cotinine (91.5 ± 39.7 (S.E.M.) ng/ml) blood plasma levels were similar to predicted values based on prior studies [54].
2.2 Perfusion and Tissue Harvesting
Two hours after assessment of mecamylamine-precipitated withdrawal, animals were anesthetized in their home cage habituated environment with sodium pentobarbital (100 mg/kg, i.p.) prior to perfusion, similar to procedures used by [36, 37, 55, 56]. Anesthesia was confirmed with a sharp toe pinch. Mice were first pumped with 40 mL of 1×PBS solution (8.00 g/L NaCl, 0.20 g/L KCl, 1.44 g/L NaH2PO4, 1.76 g/L KH2PO4) until solutions exiting the heart ran clear. Mice were then perfused with 40 mL of fresh 4% paraformaldehyde solution, and fixation tremors were observed after 20–30 seconds for each animal. The brains were harvested and stored overnight in 4% paraformaldehyde solution. The brains were then stored in 30% sucrose solution for 48 hours until the brains sank, before being flash frozen with dry ice in 2-methyl butane. All brains were stored in the −80°C freezer until use.
2.3 Cryosectioning
Each brain was placed into a cryomold and coated with Tissue-Tek O.C.T. (Optimal Cutting Temperature) Compound, which was subsequently allowed to solidify in the −80°C freezer. The mold was then removed and the brain was mounted on the chuck within the cryostat. The brains were sliced into 40 μm sections and stored as free floating tissues in antifreeze solution (500 mL/L 0.1 M PBS, 300 g/L sucrose, 300 mL/L ethylene glycol and 10 g/L polyvinylpyrrolidone-40) in the −20° freezer until staining [51].
2.4 Immunohistochemistry
Methodology for immunohistochemistry was adapted from Okabe and Murphy, 2004 [51]. One to five slices per brain were chosen based on their location within the brain and the inclusion of the anatomical region of interest. Slices from individual brains were placed together in a single well of a 24-well plate. The slices were washed with 1×PBS for two consecutive 10 min washes. The slices were then soaked in 0.3% hydrogen peroxide solution for four 6 min periods and then washed with 1×PBS. The slices were soaked in blocking serum (1% BSA (2 mg/ml, Pierce #23209), 0.5% triton x-100, 5% donkey serum (Millipore, S30-100ML), 93.5% 1×PBS) for two hours and subsequently washed with 1×PBS and incubated in 1:2000 primary antibody solution overnight (cFos sc-50 Rabbit polyclonal IgG, Santa Cruz Biotech Inc, and 2% donkey serum). On the second day, the slices were washed with 1×PBS and incubated in 1:200 secondary antibody solution (donkey anti-rabbit IgG-B sc-2089 biotin conjugated antibody, Santa Cruz Biotech Inc, and 2% donkey serum) for two hours. The slices were then washed with 1×PBS and incubated in Vector Laboratories ABC reagent for one hour. The slices were washed with 1×PBS and finally incubated in peroxidase substrate solution (2 drops Buffer Stock Solution, 4 drops DAB sk-4100 reagent, 2 drops hydrogen peroxide solution, and 2 drops nickel solution in 5 ml of deionized distilled water, all obtained from Vector Laboratories DAB Peroxidase Substrate Kit) for two minutes and then washed with cold water in two 3-min periods. The slices were stored in 1XPBS solution in the 4°C fridge before being mounted on gelatin coated Fisher Superfrost slides. On the following day, the slides were taken through a dehydration scheme (consecutive 2 minute washes in the following order- 1× deionized distilled water, 1× 30% ethanol, 1× 50% ethanol, 1× 70% ethanol, 2× 90% ethanol, 2× 100% ethanol and 3× 5 minute xylenes washes) before the cover slips were mounted using Cytoseal.
2.5 Immunohistochemistry Imaging
For each brain slice, an image using an Olympus BX51 research microscope was taken with 1.25× magnification for location reference as well as an image using 10× magnification to conduct the cfos counts. From the 10× images, we examined a small pre-determined area within the anatomical region of interest. Experimenter was blind to both the genotype and drug treatment. All counts were performed using ImageJ64 software (National Institute of Health). A cell was determined to be cfos positive based on the darkness of its nuclear stain compared to background level cells within the brain regions expressing low to absent levels of cfos.
Fluorescent Tissue Plating and Imaging
To demonstrate the localization of α2 nAChR subunit-containing neurons using a fluorescent tag, we mated CHRNA2-Cre mice (OE25-cre, Gensat) with Ai9 mice (007909, Jackson Laboratory). The Ai9 mice harbor a lox-p flanked stop cassette inhibiting the constitutive promoter expression of a red fluorescent protein, tdTomato. In the presence of Cre-mediated recombination, the stop cassette is deleted, thus allowing for constitutive expression of tdTomato in α2-containing neurons. Brains were harvested from these animals during adulthood using the methodology for perfusion, cryosectioning and storage described above. Slices 40 μm in thickness that contained brain regions of interest were collected and plated onto gelatin coated Fisher Superfrost slides. The following day, the slides were taken through a dehydration scheme followed by a xylene wash before the cover slips were mounted using Cytoseal. Images were taken on a microscope with fluorescent capabilities.
2.6 Statistical Analysis
Data were analyzed using JMP statistical software (SAS Institute). We used ANOVA and repeated-measures ANOVA to identify differences between cfos counts within groups. Post hoc analysis was performed using Dunnett’s, or Student’s t test and Bonferroni corrected for multiple comparisons, when applicable. In particular, post-hoc analyses were performed for results that demonstrated significant and non-significant trends, as highlighted in the text. Data were analyzed from 20 perfused brains. No exclusion criteria was applied to remove mice from the reported cfos analysis based on, for example, whether withdrawal signs were or were not observed. Tissue sections that did not contain the entirety of the brain region being quantified or were lost and/or damaged during cryosectioning were excluded from analysis by a blind observer. From the IPI data of the IPN, one mouse was excluded for having data that was 3 standard deviations ± the mean.
3.0 Results
Based on expression patterns of α2–containing nAChRs and brain regions involved in nicotine withdrawal, we conducted an analysis of cfos stained cells within the interpeduncular nucleus, the habenula (medial (MHb) and lateral (LHb)), the CA1 region of both the dorsal and ventral hippocampus and the dorsal dentate gyrus [25, 27]. The CA1 was further subdivided into four layers (in dorsal to ventral order): the stratum oriens layer, pyramidal layer (PL), stratum lacunosum moleculare layer (SLM) and the stratum radiatum (SR) layer. The DG was also subdivided into the outermost molecular layer (ML), the middle granular layer (GL) and the innermost polymorphic layer (iPL).
3.1 Interpeduncular Nucleus
Fluorescent imaging confirms the presence of α2-positive neurons within the IPN, which associates with high levels of α2 nAChR subunit mRNA expression in the same region (Figure 1A [11, 25]. Using a two-way ANOVA to assess cfos expression within the IPN, we found a non-significant trend for an interactive effect for withdrawal condition × genotype (F(3,19) = 3.64, p = 0.075). Dunnett’s post-hoc analysis reveals that nicotine treatment significantly enhances cfos expression in the IPN in wild type nicotine withdrawal versus vehicle treated mice (p < 0.05) (Figure 2A–D, 3A). This effect of nicotine withdrawal enhancement of cfos activity in the IPN is absent in the α2 nAChR subunit null mutant mice. These results parallel previously published mecamylamine-precipitated withdrawal behavior that demonstrate an enhancement of somatic withdrawal signs in wild type mice, which is absent in the α2-null mutant mouse in a habituated home cage environment [31, 35]. Assessing a more selective subdivision of the IPN, i.e. the interpeduncular intermediate region, as performed by others [56], also highlighted enhanced cfos counts during nicotine withdrawal, but this effect was not dependent on the genotype of α2 mutant mice (data not shown).
Figure 1. Fluorescent images demonstrate expression of α2-positive neurons.
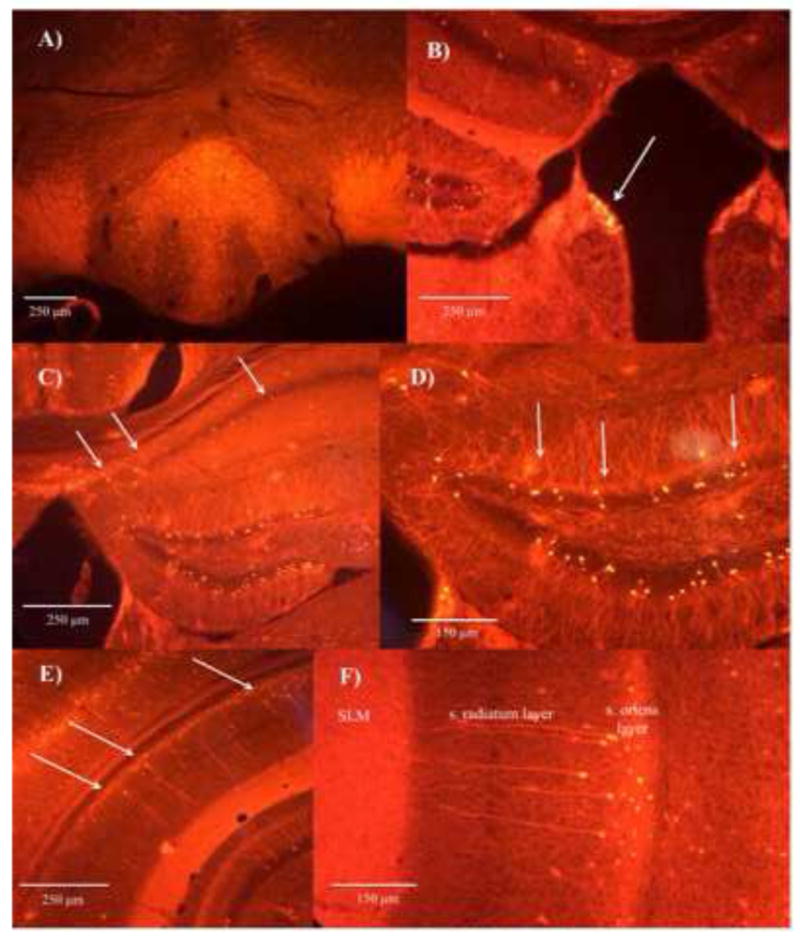
within the IPN (A), habenula region (B), oriens layer of the dorsal CA1 hippocampal region (C), granular layer of the dentate gyrus (D), and oriens layer of the ventral CA1 hippocampal region (E). The α2-positive neurons within the oriens layer of the CA1 hippocampal region send projections directly into the s. lacunosum moleculare layer (F).
Figure 2. Representative photomicrographs demonstrating cfos staining within midbrain and limbic brain structures.
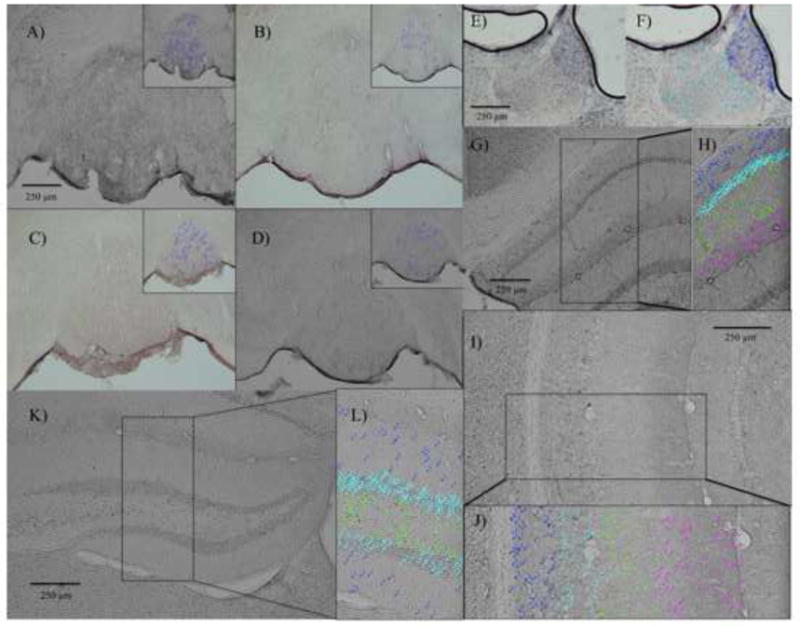
Cfos staining within the IPN, with inset images showing nuclei that were counted as cfos-positive (wild type, nicotine withdrawal (A), wild type, vehicle-treated (B), knockout, nicotine withdrawal (C) and knockout, vehicle-treated (D)); cfos staining within the MH and LH (E) and the nuclei that were marked as cfos-positive for the MH (dark blue markers) and LH (light blue markers) (F); cfos staining within the CA1 region of the DH (G), and the magnified segment of the same brain section showing the nuclei marked as cfos-positive across all four layers (H); cfos staining within the CA1 region of the VH (I), and the magnified segment of the same brain section showing the nuclei marked as cfos-positive within the four layers (J); cfos staining within the dentate gyrus (K), and the magnified segment of the same brain section showing the nuclei marked as cfos-positive within the three layers (K).
Figure 3. Alterations in nicotine withdrawal-induced neuronal activity as measured by cfos expression across selective midbrain and limbic brain regions.
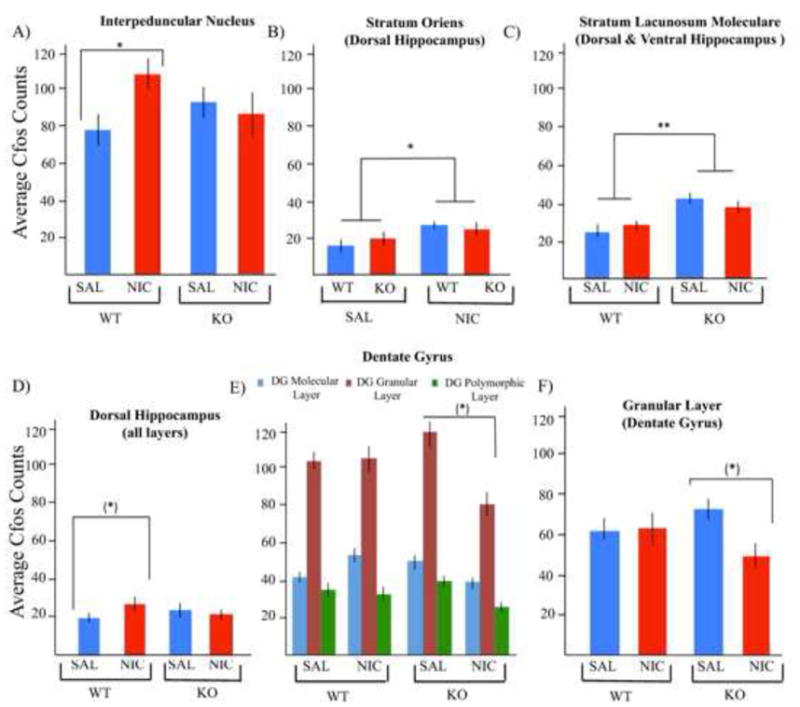
Nicotine withdrawal increases cfos expression within the IPN of wild type mice, an effect that is absent in α2 nAChR subunit null mutant mice (*p < 0.05 vs. wild type vehicle-treated mice, n=3–6/group) (A). Nicotine withdrawal increases cfos activity within the oriens layer of the dorsal hippocampus independent of genotype; asterisk represents treatment effect (*p = 0.05, nicotine withdrawal vs. vehicle-treated groups, n=9–11/group) (B). The absence of the α2 nAChR subunit increases cfos activity within the SLM layer of both the dorsal and ventral hippocampus independent of nicotine withdrawal condition; asterisk represents genotype effect (**p < 0.01, wild type vs. mutant mice, n=7–12/group) (C). Nicotine withdrawal results in an overall increase in cfos activity across all four layers of the dorsal hippocampus in wild type, but not α2 nAChR subunit null mutant mice; asterisk represents treatment effect within genotype ((*)p = 0.03, wild type, vehicle-treated vs. wild type, nicotine withdrawal mice, not corrected for multiple comparisons testing, n=3–6/group) (D). Distribution of cfos staining across individual layers of the DG, with an overall effect driven by the granular cell layer ((*)p < 0.05, α2 KO, vehicle-treated vs. α2 KO, nicotine withdrawal mice, not corrected for multiple comparisons testing) as opposed to the molecular layer (blue) or the polymorphic layer (green), n=3–5/group) (E). Nicotine withdrawal suppresses cfos activity across all layers of the dentate gyrus within α2 nAChR subunit null mutant mice; this effect is absent in wild type mice ((*)p < 0.05, α2 KO, vehicle vs. α2 KO, nicotine withdrawal mice, not corrected for multiple comparisons testing, n=3–5/group) (F).
3.2 Habenula
Fluorescent imaging demonstrates the localization of α2-positive neurons to very dorsal regions of the MHb (Figure 1B), a finding that is supported by in situ hybridization mRNA expression within the brain region, particularly early in development (postnatal day 3–5) [13]. We did not find any significant results for cfos expression with regards to withdrawal condition or genotype within the MHb. We did see an interactive effect for genotype × withdrawal condition for cfos counts within the LHb (F(3,17) = 4.60, p = 0.05), but post-hoc analysis revealed a lack of significance (Figure 2E–F).
3.3 CA1 Region of the Hippocampus
Fluorescent imaging demonstrates α2-positive neurons within the CA1 stratum oriens layer of both the DH and the VH (Figure 2C–F); these findings associate with α2 nAChR subunit mRNA expression patterns within the hippocampus [11, 13, 25, 27]. When assessing cfos neuronal activity within the SO layer of the DH, results using a two-way ANOVA demonstrate a main effect of withdrawal condition (F(3,19) = 4.68, p < 0.05), but not genotype or genotype × withdrawal condition interaction. Post-hoc analysis reveals that nicotine withdrawal significantly increased neuronal activity within the SO layer of the DH independent of genotype (Figure 2G–H, 3B). On the other hand, when examining the SO neuronal projection site of the SLM layer, we observe a main effect of genotype (F(3,18) = 7.23, p < 0.05) within the SLM of the VH, and a non-significant trend for genotype (F(3,19) = 3.82, p = 0.09) within the SLM of the DH. Combined scores for both the DH and VH SLM layer demonstrated a main effect of genotype (F(3,18) = 11.46, p < 0.01), but not withdrawal condition or genotype × withdrawal condition interaction. Post-hoc analysis reveals that α2 nAChR subunit null mutant mice express potentiated neuronal activity within the SLM layers of both the DH and VH compared to wild type mice independent of nicotine withdrawal condition, with the overall effect driven primarily by the VH (Figure 2I–J, 3C). Finally, when we averaged cfos counts for all four different layers across the DH, we found a non-significant trend for an interactive effect for genotype × withdrawal condition (F(3,19) = 2.79, p = 0.11); this effect would likely be significant with a larger number of mice in the study. Post-hoc analysis demonstrates a significant enhancement of neuronal activity during nicotine withdrawal within wild type mice that is absent in the α2 nAChR subunit null mutant mice (p = 0.03), but the effect does not remain significant after Bonferroni correction of multiple comparisons (Figure 2G–J, 3D). The results suggest that dorsal hippocampal neuronal activity during nicotine withdrawal is likely mediated by α2-containing nAChRs. No significant main or interactive effects were observed when assessing the combined four layers across the VH.
3.4 Dentate Gyrus
Fluorescent imaging demonstrates the presence of α2-containing neurons within the GL of the DG (Figure 1D). Using a two-way ANOVA, we examined cfos immunoreactivity within the three different layers of the DG (iPL, GL, and ML) and saw a non-significant trend for a main effect for withdrawal condition (F(3,17) = 3.90, p = 0.07), and a non-significant trend for an interaction for withdrawal condition × genotype (F(3,17) = 3.71, p = 0.08) within the GL (Figure 2K–L, 3E). Post-hoc analysis demonstrates mice undergoing withdrawal have reduced neuronal activity in the GL, and this effect is only present in the α2 nAChR subunit null mutant mice. Subsequent analysis of neuronal activity across all layers of the DG revealed a non-significant trend for an interactive effect between withdrawal condition × genotype (F(3,17) = 4.14, p = 0.06). Post-hoc analysis revealed that nicotine withdrawal condition verses vehicle treatment suppressed neuronal activity within the DG of the α2 nAChR subunit null mutant mice (Figure 2K–L, 3F). This effect was absent in wild type mice. Thus, the results of cfos immunoreactivity within the GL may be driving the overall main effect across all layers the DG. However, none of the effects within the DG remained significant after Bonferroni correction of multiple comparisons.
4.0 Discussion
Our results demonstrate that nicotine withdrawal enhances neuronal activity within the interpeduncular nucleus and dorsal hippocampus, which is absent in mice null for α2-containing nAChRs (Figure 4). In contrast, we observe that the absence of α2-containing nAChRs results in a suppression of neuronal activity in the dentate gyrus in mice undergoing nicotine withdrawal (Figure 4). The interpeduncular nucleus has been accepted both for its role in mediating nicotine withdrawal in conjunction with the Hb-IPN axis, and as an overall integrative center for limbic brain regions [5, 31, 34–36, 38, 56]. The region also has the highest levels of α2 nAChR subunit expression within the rodent brain [11, 12, 25, 26] and α2-positive neuronal fluorescence (Figure 1). Our present study demonstrates that α2-containing nAChRs influence nicotine withdrawal-induced neuronal activity within the IPN, as assessed by the immediate early gene cfos. Such findings are supported by published reports illustrating that (i) nicotinic receptor antagonist activity within the IPN precipitates nicotine withdrawal, (ii) optogenetic activation of GABAergic neurons within the IPN induce nicotine withdrawal signs, and (iii) the genetic deletion of α2 nAChR subunits ablate nicotine withdrawal in a habituated home cage environment [31, 36, 37, 56]. Thus, α2-containing nAChRs within the IPN are likely important for mediating the mechanisms influencing nicotine withdrawal symptoms.
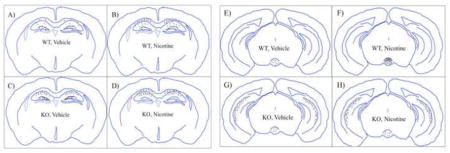
Figure 4. Summary diagram of baseline and nicotine withdrawal-induced neuronal activity across selective midbrain and limbic brain regions.
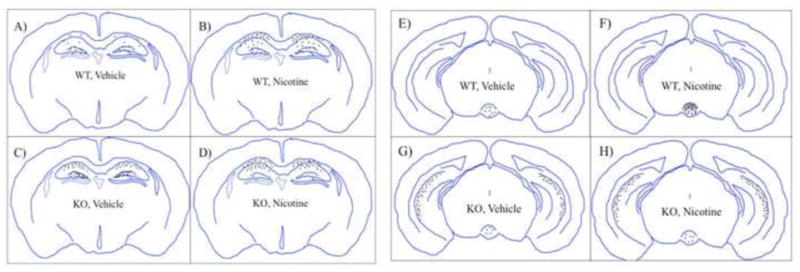
Representations of cfos activity across the four different groups (A & E. wild type, vehicle; B & F. wild type, nicotine withdrawal; C & G. α2 KO, vehicle; D & H. α2 KO, nicotine withdrawal) within the dorsal hippocampus and dentate gyrus regions (A–D) and the ventral hippocampus and interpeduncular nucleus (E–H).
The lack of a main effect for nicotine withdrawal-induced neuronal activity within habenula regions was surprising due to the fact that the IPN receives a vast majority of its excitatory cholinergic and glutamatergic input from the medial habenula structure via the fasciculus retroflexus [38, 57]. However, other investigators have reported selective enhancement of IPN neuronal activity during mecamylamine-precipitated nicotine withdrawal [36, 37]. These results suggest that the habenula input is not necessary to potentiate nicotine withdrawal-induced neuronal activity within the IPN.
Our fluorescent images demonstrate that α2-positive neurons are restricted to very dorsal regions of the medial habenula. Due to this selected localization, we predicted and observed no significant genotype effect within the medial and lateral habenula regions. Interestingly, prior immunohistochemistry experiments demonstrated that expression of other α-type nAChR subunits is restricted to the ventral regions of the medial habenula, while β2 and β4 nAChR subunits are located across both ventral and dorsal regions of the medial habenula [37]. Thus, the α2 subunit could combine with either the β2 or β4 nAChR subunits to form functional channels within both the Hb and IPN, a hypothesis that is supported by prior immunoprecipitation experiments within the IPN [58]. Nevertheless, our overall findings provide further evidence that α2-containing nAChRs within the IPN, but not habenula, are important contributors of neuronal activity during nicotine withdrawal.
Limbic regions such as the hippocampus have also been implicated in nicotine reward and withdrawal due to the roles that learning and memory play in reinforcing drug-associated behaviors [40, 59]. Learning, memory and withdrawal behaviors involve many of the same neural areas and cellular processes, and changes in pathways underlying plasticity may contribute to addiction. For example, the development of drug-context associations can facilitate drug cravings upon re-exposure to a specific context during withdrawal [40, 50]. Interestingly, nicotine gates long-term potentiation within the hippocampus CA1 likely via specific activation of α2-containing nAChRs and can influence cue and context dependent nicotine withdrawal behavior [35, 43]. We therefore were interested in examining neuronal activity across various regions of the hippocampus during nicotine withdrawal, as well as how the absence of the α2 nAChR subunit could influence this activity using α2-null mutant mice.
Most noteworthy within the dorsal hippocampus, our findings suggest that α2-null mutant mice exhibit an absence of nicotine withdrawal-induced enhancement of neuronal activity within all layers. These findings are similar to those discovered within the IPN, although the hippocampal effect is much more subtle. It is important to note that the α2 nAChR subunit has much lower expression levels within the hippocampus when compared to the IPN. This may account for the more subtle interactive effect within the DH in α2-null mutant mice. Nicotinic receptor antagonism within more ventral hippocampal targets of chronic nicotine treated mice does not precipitate somatic withdrawal signs, demonstrating that hippocampal modulation of nicotine withdrawal is likely region dependent [31]. Overall, the results indicate that α2-null mutant mice exhibit a coordinated reduction of cellular activity during nicotine withdrawal within both the IPN and DH. Thus, we speculate that using a pharmacological intervention to reduce α2-containing nAChR activity during nicotine withdrawal may help alleviate negative behaviors associated with drug relapse. These effects may be dependent on a habituated environmental context. Further experimental exploration is needed to determine whether opposing results would be observed in novel environmental contexts using the α2-null mutant mice [35].
Independent of nicotine withdrawal condition, we also found that α2-null mutant mice exhibit potentiated cfos activity within the SLM layer of both the dorsal and ventral CA1 hippocampal regions. The α2-containing nAChRs within the hippocampus CA1 are particularly localized to GABAergic interneurons within the OLM layer [11, 13, 27, 46]. This subset of GABAgeric interneurons of the OLM have been shown to send their axonal projections to synapse onto the distil dendrites of the pyramidal neurons, which are located in the SLM layer of the hippocampus CA1 [43]. Thus, it is possible that removal of α2-containing nAChRs, in α2-null mutant mice, results in a lack of or decreased inhibition of the SLM region via reduced activity of a specific subset of the GABAergic OLM neurons. This reduction of inhibition would subsequently yield higher levels of neuronal activity overall in the SLM. These effects appear to be mediated solely by a genotype effect, and not by nicotine withdrawal condition or interactive genotype by nicotine withdrawal condition. Whether such a genotype effect could influence stress, learning, memory, emotion and affect needs further exploration. This is particularly important, as prior studies have demonstrated that hippocampal dendritic inhibition can facilitate learning and memory behavior [47].
Within the dentate gyrus, α2-null mutant mice exhibit an entirely different effect for nicotine withdrawal-induced changes in neuronal activity. Nicotine withdrawal induced a reduction of cfos activity across all three layers of the dentate gyrus specifically in α2-null mutant mice, an effect that was absent in wild type mouse brains. Further studies are needed to clarify whether such effects may influence, at least in part, mechanisms mediating context dependent nicotine withdrawal and/or the learning and memory deficits in adult α2-null mutant mice [35]. Our results indicate that there may be a mechanism dependent on α2-containing nAChRs within the mouse brain which functions to prevent nicotine-induced suppression of neuronal activity within the dentate gyrus. Furthermore, this effect appears to be highly driven by neurons within granular cell layer, which has interesting implications given their role in modulating synaptic plasticity, pattern separation and pattern completion [48, 60].
Examination of our fluorescent images containing the dentate gyrus area shows high levels of α2-positive neurons within the granular cell layer that project their axons outwards into the molecular layer. The α2 nAChR subunit is developmentally expressed within the dentate gyrus (highest expression at postnatal day 3–5), and while mRNA hybridization show no α2-containing nAChR subunits within the dentate gyrus in adult mice, the presence of the fluorescent marker supports the idea that there is expression within the dentate gyrus early in development [13]. It is therefore possible that the absence of α2-containing nAChRs dysregulates circuitry within the dentate gyrus during early development, altering subsequent neuronal activity levels within the region during nicotine withdrawal. Indeed, we should highlight the caveat that we are not reporting α2 expression directly, but rather Chrna2-promoter driven Cre expression. There are limitations of Cre driver mouse lines, which can include ectopic expression, lack of expression in bona fide α2 expressing cells, and lack of reporter expression in cell types where Chrna2 promoter activity is weak. These limitations should be taken into account when interpreting the results of our current paper.
Overall, α2-null mutant mice have altered neuronal activity in specific brain regions during nicotine withdrawal. Importantly, cfos activity within the interpeduncular nucleus and the dorsal hippocampus mimic, at least in part, the context dependent nicotine withdrawal behavioral findings previously found in α2-null mutant mice [35]. Thus, our studies suggest that α2-containing nAChRs are important in regulating nicotine withdrawal likely through changes in neuronal activity within distinct midbrain and limbic brain structures. Our results also show that α2-null mutant mice have changes in neuronal hyperactivity, independent of nicotine withdrawal, across both the dorsal and ventral hippocampus. Further studies are needed to determine the behavioral consequences of such effects. Our findings present a possible molecular-genetic target for therapeutic interventions to help negate the side effects associated with nicotine withdrawal.
Acknowledgments
The study was funded by Tobacco-Related Disease Research Program Fellowship 20FT-0072 and project grant 22RT-0103 (S.L.), UCLA Friends of the Semel Institute (S.L.), T32 National Institute of Mental Health postdoctoral fellowship MH17140 (S.L.), and the National Alliance for Research on Schizophrenia and Depression Young Investigator Grants from the Brain & Behavior Research Foundation (S.L.). The authors would like to thank Dr. Jim Boulter, Dr. Celina Mojica, Dr. Amynah Pradhan, Dr. Chris Evans, Sarah Isaac Nessah and Kevin Lee with their assistance on the current study.
Abbreviations
- CA1
field CA1 of the hippocampus
- DG
dentate gyrus
- DH
dorsal hippocampus
- GL
granular layer of the dentate gyrus
- IPN
interpeduncular nucleus
- LHb
lateral habenula
- MHb
medial habenula
- ML
molecular layer of the dentate gyrus
- OLM
oriens lacunosum moleculare of the stratum oriens
- iPL
polymorphic layer of the dentate gyrus
- PL
pyramidal layer
- SLM
stratum lacunosum moleculare
- SO
stratum oriens layer of the hippocampus
- SR
stratum radiatum layer of the hippocampus
- VH
ventral hippocampus
Footnotes
Author contributions: M.U. and S.L. designed research, M.U. and S.L. performed research; S.L contributed unpublished reagents/analytic tools; M.U. and S.L. analyzed data; M.U. and S.L. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7.0 References
- 1.Jamal A, Agaku IT, O’Connor E, King BA, Kenemer JB, Neff L. Current cigarette smoking among adults--United States, 2005–2013. MMWR Morb Mortal Wkly Rep. 2014;63:1108–12. [PMC free article] [PubMed] [Google Scholar]
- 2.Changeux JP. Nicotine addiction and nicotinic receptors: lessons from genetically modified mice. Nat Rev Neurosci. 2010;11:389–401. doi: 10.1038/nrn2849. [DOI] [PubMed] [Google Scholar]
- 3.West RJ, Hajek P, Belcher M. Severity of withdrawal symptoms as a predictor of outcome of an attempt to quit smoking. Psychol Med. 1989;19:981–5. doi: 10.1017/s0033291700005705. [DOI] [PubMed] [Google Scholar]
- 4.Fowler CD, Kenny PJ. Nicotine aversion: Neurobiological mechanisms and relevance to tobacco dependence vulnerability. Neuropharmacology. 2014;76(Pt B):533–44. doi: 10.1016/j.neuropharm.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLaughlin I, Dani JA, De Biasi M. Nicotine Withdrawal. In: Balfour DJK, Munafo MR, editors. The Neuropharmacology of Nicotine Dependence. Springer International Publishing; 2015. pp. 99–123. [Google Scholar]
- 6.Brody AL, Mandelkern MA, London ED, Olmstead RE, Farahi J, Scheibal D, et al. Cigarette smoking saturates brain alpha 4 beta 2 nicotinic acetylcholine receptors. Arch Gen Psychiatry. 2006;63:907–15. doi: 10.1001/archpsyc.63.8.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Benowitz NL. Pharmacology of nicotine: addiction and therapeutics. Annu Rev Pharmacol Toxicol. 1996;36:597–613. doi: 10.1146/annurev.pa.36.040196.003121. [DOI] [PubMed] [Google Scholar]
- 8.Brody AL, Mandelkern MA, Olmstead RE, Allen-Martinez Z, Scheibal D, Abrams AL, et al. Ventral striatal dopamine release in response to smoking a regular vs a denicotinized cigarette. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2009;34:282–9. doi: 10.1038/npp.2008.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–4. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- 10.Vizi ES, Lendvai B. Modulatory role of presynaptic nicotinic receptors in synaptic and non-synaptic chemical communication in the central nervous system. Brain Res Brain Res Rev. 1999;30:219–35. doi: 10.1016/s0165-0173(99)00016-8. [DOI] [PubMed] [Google Scholar]
- 11.Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, et al. Distribution of alpha 2, alpha 3, alpha 4, and beta 2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol. 1989;284:314–35. doi: 10.1002/cne.902840212. [DOI] [PubMed] [Google Scholar]
- 12.Wada K, Ballivet M, Boulter J, Connolly J, Wada E, Deneris ES, et al. Functional expression of a new pharmacological subtype of brain nicotinic acetylcholine receptor. Science. 1988;240:330–4. doi: 10.1126/science.2832952. [DOI] [PubMed] [Google Scholar]
- 13.Son JH, Winzer-Serhan UH. Postnatal expression of alpha2 nicotinic acetylcholine receptor subunit mRNA in developing cortex and hippocampus. J Chem Neuroanat. 2006;32:179–90. doi: 10.1016/j.jchemneu.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chavez-Noriega LE, Crona JH, Washburn MS, Urrutia A, Elliott KJ, Johnson EC. Pharmacological characterization of recombinant human neuronal nicotinic acetylcholine receptors h alpha 2 beta 2, h alpha 2 beta 4, h alpha 3 beta 2, h alpha 3 beta 4, h alpha 4 beta 2, h alpha 4 beta 4 and h alpha 7 expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1997;280:346–56. [PubMed] [Google Scholar]
- 15.De Biasi M, Salas R. Influence of neuronal nicotinic receptors over nicotine addiction and withdrawal. Exp Biol Med (Maywood) 2008;233:917–29. doi: 10.3181/0712-MR-355. [DOI] [PubMed] [Google Scholar]
- 16.Jackson KJ, Muldoon PP, De Biasi M, Damaj MI. New mechanisms and perspectives in nicotine withdrawal. Neuropharmacology. 2014 doi: 10.1016/j.neuropharm.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bierut LJ. Nicotine dependence and genetic variation in the nicotinic receptors. Drug Alcohol Depend. 2009;104 (Suppl 1):S64–9. doi: 10.1016/j.drugalcdep.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sullivan PF, Neale BM, van den Oord E, Miles MF, Neale MC, Bulik CM, et al. Candidate genes for nicotine dependence via linkage, epistasis, and bioinformatics. Am J Med Genet B Neuropsychiatr Genet. 2004;126B:23–36. doi: 10.1002/ajmg.b.20138. [DOI] [PubMed] [Google Scholar]
- 19.Li MD, Xu Q, Lou XY, Payne TJ, Niu T, Ma JZ. Association and interaction analysis of variants in CHRNA5/CHRNA3/CHRNB4 gene cluster with nicotine dependence in African and European Americans. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:745–56. doi: 10.1002/ajmg.b.31043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brunzell DH, Stafford AM, Dixon CI. Nicotinic Receptor Contributions to Smoking: Insights from Human Studies and Animal Models. Current Addicion Reports. 2014;2:33–46. doi: 10.1007/s40429-015-0042-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cahill K, Stead LF, Lancaster T. Nicotine receptor partial agonists for smoking cessation. Cochrane Database Syst Rev. 2012;4:CD006103. doi: 10.1002/14651858.CD006103.pub2. [DOI] [PubMed] [Google Scholar]
- 22.Jin Z, Khan P, Shin Y, Wang J, Lin L, Cameron MD, et al. Synthesis and activity of substituted heteroaromatics as positive allosteric modulators for alpha4beta2alpha5 nicotinic acetylcholine receptors. Bioorg Med Chem Lett. 2014;24:674–8. doi: 10.1016/j.bmcl.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshimura RF, Hogenkamp DJ, Li WY, Tran MB, Belluzzi JD, Whittemore ER, et al. Negative allosteric modulation of nicotinic acetylcholine receptors blocks nicotine self-administration in rats. J Pharmacol Exp Ther. 2007;323:907–15. doi: 10.1124/jpet.107.128751. [DOI] [PubMed] [Google Scholar]
- 24.Toll L, Zaveri NT, Polgar WE, Jiang F, Khroyan TV, Zhou W, et al. AT-1001: a high affinity and selective alpha3beta4 nicotinic acetylcholine receptor antagonist blocks nicotine self-administration in rats. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2012;37:1367–76. doi: 10.1038/npp.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishii K, Wong JK, Sumikawa K. Comparison of alpha2 nicotinic acetylcholine receptor subunit mRNA expression in the central nervous system of rats and mice. J Comp Neurol. 2005;493:241–60. doi: 10.1002/cne.20762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, et al. Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci. 1992;12:2765–84. doi: 10.1523/JNEUROSCI.12-07-02765.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Son JH, Winzer-Serhan UH. Expression of neuronal nicotinic acetylcholine receptor subunit mRNAs in rat hippocampal GABAergic interneurons. J Comp Neurol. 2008;511:286–99. doi: 10.1002/cne.21828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han ZY, Le Novere N, Zoli M, Hill JA, Jr, Champtiaux N, Changeux JP. Localization of nAChR subunit mRNAs in the brain of Macaca mulatta. Eur J Neurosci. 2000;12:3664–74. doi: 10.1046/j.1460-9568.2000.00262.x. [DOI] [PubMed] [Google Scholar]
- 29.Aridon P, Marini C, Di Resta C, Brilli E, De Fusco M, Politi F, et al. Increased sensitivity of the neuronal nicotinic receptor alpha 2 subunit causes familial epilepsy with nocturnal wandering and ictal fear. Am J Hum Genet. 2006;79:342–50. doi: 10.1086/506459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leslie FM, Mojica CY, Reynaga DD. Nicotinic receptors in addiction pathways. Mol Pharmacol. 2013;83:753–8. doi: 10.1124/mol.112.083659. [DOI] [PubMed] [Google Scholar]
- 31.Salas R, Sturm R, Boulter J, De Biasi M. Nicotinic receptors in the habenulo-interpeduncular system are necessary for nicotine withdrawal in mice. J Neurosci. 2009;29:3014–8. doi: 10.1523/JNEUROSCI.4934-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dao DQ, Salas R, De Biasi M. Nicotinic Acetylcholine Receptors Along the Habenulo-Interpeduncular Pathway: Roles in Nicotine Withdrawal and Other Aversive Aspects. In: Lester RAJ, editor. Nicotinic Receptors. New York Springer; New York: 2014. pp. 363–82. [Google Scholar]
- 33.Antolin-Fontes B, Ables JL, Gorlich A, Ibanez-Tallon I. The habenulo-interpeduncular pathway in nicotine aversion and withdrawal. Neuropharmacology. 2014 doi: 10.1016/j.neuropharm.2014.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morley BJ. The interpeduncular nucleus. Int Rev Neurobiol. 1986;28:157–82. doi: 10.1016/s0074-7742(08)60108-7. [DOI] [PubMed] [Google Scholar]
- 35.Lotfipour S, Byun JS, Leach P, Fowler CD, Murphy NP, Kenny PJ, et al. Targeted deletion of the mouse alpha2 nicotinic acetylcholine receptor subunit gene (Chrna2) potentiates nicotine-modulated behaviors. J Neurosci. 2013;33:7728–41. doi: 10.1523/JNEUROSCI.4731-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao-Shea R, Liu L, Pang X, Gardner PD, Tapper AR. Activation of GABAergic neurons in the interpeduncular nucleus triggers physical nicotine withdrawal symptoms. Curr Biol. 2013;23:2327–35. doi: 10.1016/j.cub.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shih PY, Engle SE, Oh G, Deshpande P, Puskar NL, Lester HA, et al. Differential expression and function of nicotinic acetylcholine receptors in subdivisions of medial habenula. J Neurosci. 2014;34:9789–802. doi: 10.1523/JNEUROSCI.0476-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klemm WR. Habenular and interpeduncularis nuclei: shared components in multiple-function networks. Med Sci Monit. 2004;10:RA261–73. [PubMed] [Google Scholar]
- 39.Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–79. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 40.Gould TJ. The Effects of Nicotine on Learning and Memory. In: Lester RAJ, editor. Nicotinic Receptors. New York: Springer New York; 2014. [Google Scholar]
- 41.Simmons SJ, Gould TJ. Involvement of neuronal beta2 subunit-containing nicotinic acetylcholine receptors in nicotine reward and withdrawal: implications for pharmacotherapies. J Clin Pharm Ther. 2014;39:457–67. doi: 10.1111/jcpt.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis JA, Gould TJ. Hippocampal nAChRs mediate nicotine withdrawal-related learning deficits. Eur Neuropsychopharmacol. 2009;19:551–61. doi: 10.1016/j.euroneuro.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakauchi S, Brennan RJ, Boulter J, Sumikawa K. Nicotine gates long-term potentiation in the hippocampal CA1 region via the activation of alpha2* nicotinic ACh receptors. Eur J Neurosci. 2007;25:2666–81. doi: 10.1111/j.1460-9568.2007.05513.x. [DOI] [PubMed] [Google Scholar]
- 44.Leao RN, Mikulovic S, Leao KE, Munguba H, Gezelius H, Enjin A, et al. OLM interneurons differentially modulate CA3 and entorhinal inputs to hippocampal CA1 neurons. Nat Neurosci. 2012;15:1524–30. doi: 10.1038/nn.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maccaferri G. Stratum oriens horizontal interneurone diversity and hippocampal network dynamics. J Physiol. 2005;562:73–80. doi: 10.1113/jphysiol.2004.077081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jia Y, Yamazaki Y, Nakauchi S, Ito K, Sumikawa K. Nicotine facilitates long-term potentiation induction in oriens-lacunosum moleculare cells via Ca2+ entry through non-alpha7 nicotinic acetylcholine receptors. Eur J Neurosci. 2010;31:463–76. doi: 10.1111/j.1460-9568.2009.07058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lovett-Barron M, Kaifosh P, Kheirbek MA, Danielson N, Zaremba JD, Reardon TR, et al. Dendritic inhibition in the hippocampus supports fear learning. Science. 2014;343:857–63. doi: 10.1126/science.1247485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang TA, Tang J, Pidoplichko VI, Dani JA. Addictive nicotine alters local circuit inhibition during the induction of in vivo hippocampal synaptic potentiation. J Neurosci. 2010;30:6443–53. doi: 10.1523/JNEUROSCI.0458-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 50.Gould TJ. Nicotine and hippocampus-dependent learning: implications for addiction. Mol Neurobiol. 2006;34:93–107. doi: 10.1385/MN:34:2:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Okabe C, Murphy NP. Short-term effects of the nociceptin receptor antagonist Compound B on the development of methamphetamine sensitization in mice: a behavioral and c-fos expression mapping study. Brain Res. 2004;1017:1–12. doi: 10.1016/j.brainres.2004.04.076. [DOI] [PubMed] [Google Scholar]
- 52.Fanselow MS, Dong HW. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65:7–19. doi: 10.1016/j.neuron.2009.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lotfipour S, Mandelkern M, Alvarez-Estrada M, Brody AL. A single administration of low-dose varenicline saturates alpha4beta2* nicotinic acetylcholine receptors in the human brain. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2012;37:1738–48. doi: 10.1038/npp.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology. 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- 55.Fowler CD, Lu Q, Johnson PM, Marks MJ, Kenny PJ. Habenular alpha5 nicotinic receptor subunit signalling controls nicotine intake. Nature. 2011;471:597–601. doi: 10.1038/nature09797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao-Shea R, DeGroot SR, Liu L, Vallaster M, Pang X, Su Q, et al. Increased CRF signalling in a ventral tegmental area-interpeduncular nucleus-medial habenula circuit induces anxiety during nicotine withdrawal. Nat Commun. 2015;6:6770. doi: 10.1038/ncomms7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qin C, Luo M. Neurochemical phenotypes of the afferent and efferent projections of the mouse medial habenula. Neuroscience. 2009;161:827–37. doi: 10.1016/j.neuroscience.2009.03.085. [DOI] [PubMed] [Google Scholar]
- 58.Whiteaker P, Wilking JA, Brown RW, Brennan RJ, Collins AC, Lindstrom JM, et al. Pharmacological and immunochemical characterization of alpha2* nicotinic acetylcholine receptors (nAChRs) in mouse brain. Acta Pharmacol Sin. 2009;30:795–804. doi: 10.1038/aps.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dani JA, Ji D, Zhou FM. Synaptic plasticity and nicotine addiction. Neuron. 2001;31:349–52. doi: 10.1016/s0896-6273(01)00379-8. [DOI] [PubMed] [Google Scholar]
- 60.Nakashiba T, Cushman JD, Pelkey KA, Renaudineau S, Buhl DL, McHugh TJ, et al. Young dentate granule cells mediate pattern separation, whereas old granule cells facilitate pattern completion. Cell. 2012;149:188–201. doi: 10.1016/j.cell.2012.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]