

Abstract
Molecular analysis of proaerolysin selected glycosylphosphatidylinositol anchor (GPI-a) deficient isolates in the TK6 cell line was performed. Initial studies found that the expected X-linked PIGA mutations were rare among the spontaneous isolates but did increase modestly after ethyl methane sulfate (EMS) treatment (but to only 50% of isolates). To determine the molecular bases of the remaining GPI-a deficient isolates, real-time analysis for all the 25 autosomal GPI-a pathway genes was performed on the isolates without PIGA mutations, determining that PIGL mRNA was absent for many. Further analysis determined these isolates had several different homozygous deletions of the 5’ region of PIGL (17p12-p22) extending 5’ (telomeric) through NCOR1 and some into the TTC19 gene (total deletion >250,000bp). It was determined that the TK6 parent had a hemizygous deletion in 17p12-p22 (275,712bp) extending from PIGL intron 2 into TTC19 intron 7. Second hit deletions in the other allele in the GPI-a deficient isolates led to the detected homozygous deletions. Several of the deletion breakpoints including the original first hit deletion were sequenced. As strong support for TK6 having a deletion, a number of the isolates without PIGA mutations nor homozygous PIGL deletions had point mutations in the PIGL gene. These studies show that the GPI-a mutation studies using TK6 cell line could be a valuable assay detecting point and deletion mutations in two genes simultaneously.
Keywords: GPI-a, PIGA, PIGL, mutation, in vitro, TK6, glycosylphosphatidylinositol anchor
INTRODUCTION
The glycosylphosphatidylinositol anchors (GPI-a’s) are complex glycolipids that are covalently linked to the C-terminus of proteins as a posttranslational modification; they anchor the attached protein to the cell membrane and are essential for normal functioning of eukaryotic cells [Nishimura et al. 1999; Paulick and Bertozzi 2008]. Seven proteins (PIGA, PIGQ, PIGC, PIGN, PIGF, PIGH, PIGY) perform the first step of GPI-a biosynthesis, the transfer of N-acetylglucosamine (GlcNAc) from uridine 5-prime-diphospho-N-acetylglucosamide (UDP-GlcNAc) to phosphatydil-inositol (PI) to yield GlcNAc-PI. There are an additional 17 proteins that work in this GPI-a synthesis pathway (PIGP, PIGL, PIGM, PIGB, PIGO, PIGK, GPAA1, PIGS, PIGT, DPM1, DPM2, DPM3, SL15, PIGU, PIGV, PIGX, PIGW).
Currently, there is great interest in the development of flow cytometry-based assays to determine the loss of function of cell membrane glycosylphosphatidylinositols for mutagenicity testing of chemicals in rodents and monitoring of exposures in humans [Dertinger and Heflich 2011; Dertinger et al. 2011; Dobrovolsky et al. 2011; Sadiq et al. 2012; Dobrovolsky et al. 2013; Horibata et al. 2013; Kimoto et al. 2013; Dertinger et al. 2014; Kruger et al. 2014; Onami et al. 2014]. Loss of surface proteins maintained by GPI-a’s is conventionally assessed by flow cytometry or by resistance (lack of cell killing) to the toxin aerolysin. Fluorescent proaerolysin variant (FLAER), which binds with high affinity to the glycan portion of the GPI-a, can be used in flow cytometry to measure the frequency of GPI-a negative (GPI-a−) erythrocytes (RBCs) or lymphocytes. Resistance to aerolysin is the basis for selecting the putative PIGA mutant T-cells from peripheral blood by cloning assay [McDiarmid et al. 2011].
While the GPI-a pathway contains the 26 gene products discussed above, investigators developing the rodent and human assays have assumed that only the PIGA locus itself is the target for GPI-a inactivation because this is the only gene that is on the X-chromosome and, thus, loss of GPI-a function by PIGA mutation requires only a single hit. Confirmation of this assumption is provided by studies of patients with hematological disorder paroxysmal nocturnal hemoglobinuria (PNH), an acquired hematopoietic stem cell disease characterized by a loss of GPI-a protein on an affected stem cell and in all of its progeny [Boccuni et al. 2000; Okamoto et al. 2006], which have been shown to have PIGA mutations [Nafa et al. 1995; Mortazavi et al. 2003; Okamoto et al. 2006]. Rare GPI-a deficient cells with PIGA mutations are also found in healthy individuals [Nafa et al. 1998; Hu et al. 2005]. Loss of function in one of the 25 autosomal GPI-a genes would require two hits to inactivate both the maternal and paternal gene copies. However, in humans, genetic conditions are known which are the result of homozygous mutations in one of the GPI-a pathway genes (PIGL, PIGM, PIGN, PIGO, PIGT, PIGV, MPDUI, DPM1, DPM2, DPM3) [Kranz et al. 2001; Schenk et al. 2001; Garcia-Silva et al. 2004; Almeida et al. 2006; Lefeber et al. 2009; Krawitz et al. 2010; Maydan et al. 2011; Barone et al. 2012; Krawitz et al. 2012; Kvarnung et al. 2013], thus, individuals must exist in the population that are heterozygous for mutations in these genes, which is a point of concern.
In addition to mutations in a GPI-a synthesis pathway gene, inactivation of one or several of these genes by methylation or other epigenetic change has also been documented. Reports on the molecular basis of proaerolysin resistance in Burkitt lymphoma cell lines found that the mRNA for PIGL was reduced in some lines while PIGY was reduced in others [Hu et al. 2009]. Studies of the methylation status of the PIGL gene demonstrated that hypermethylation of these genes was associated with the low levels of mRNA. Their conclusion was that “GPI-anchored protein deficiency in Burkitt lymphoma cells is not due to a genetic mutation (e.g., PIGA); rather, the lack of GPI-anchored proteins results from transcriptional silencing of PIGL and PIGY”. Methylation silencing of the PIGA gene has also been suggested for loss of GPI-a on human T-lymphocytes following long-term culture [Gabdoulkhakova et al. 2007].
Initial molecular analyses for PIGA mutations among the GPI-a deficient isolates from experiments for development of an in vitro GPI-a mutation assay in the TK6 cell line (often used for HPRT mutation studies), found few PIGA mutations. Further real-time analysis for all the GPI-a pathway genes found that PIGL (17p12.1) mRNA was deficient; further molecular analyses of the PIGL gene region (multiplex PCR, cytogenetics, Long PCR, DNA sequencing) determined that the TK6 cell line was heterozygous for a chromosome 17p deletion including much of the TTC19 gene continuing through to past PIGL exon 2. GPI-a deficient isolates of TK6 thus consist of both PIGA and PIGL point and deletion mutations.
METHODS
TK6 cell line
The TK6 cell line used in these experiments was obtained from the laboratory of Dr. William Thilly approximately 20 years ago and has been maintained in the laboratory since that time. It is HPRT+ and TK+/−. TK6 cells were cultured in RPMI 1640 medium (Hyclone, Logan, UT) supplemented with 10% CBS at 37°C and 5.0% CO2.
GPI-a deficient isolate generation
TK6 cells were resuspended to a concentration of 0.5 × 106 cell/ml in RPMI-10% CBS, and a treatment fraction was then incubated with 30μg/ml EMS (ethyl methanesulphonate, Sigma-Aldrich, St. Louis, MO) overnight. For this day 1, and on the following days 3, 6, 8, and 10, the cells were counted and the GPI-a cloning assay was performed as described [McDiarmid et al. 2011]. On each of the five days, 10 × 106 cells at 0.5 × 106 cells/ml were maintained in RPMI-10% CBS for the next day of analysis. Surviving colonies from selection plates were identified and expanded in culture for subsequent molecular analysis. In experiment #1, 20 control (spontaneous) and 80 EMS treated cells were obtained. In experiment #2, 40 GPI-a deficient spontaneous isolates were expanded for a second round of PA-selection to confirm the PA resistant phenotype.
Chromosomal locations
All chromosomal locations are based on reference GRCh38 Primary Assembly annotation 106.
PIGA mutation analysis
The six-exon PIGA coding sequence is larger than can be sequenced in one run (1455bp) and it has several alternate splice products in the very large exon 2 making analysis more difficult. Therefore, PIGA mutants were analyzed in a two-step manner. RT-PCR was performed for the 3’ part of the gene (mid ex2 to ex6) and if no mutation was discovered in the 3’ region then genomic sequencing was performed on exon 2 (exon 1 is very small and non-coding) or vice versa. If an exon was missing from the cDNA, genomic sequencing for that exon was performed. If no exon could be amplified or no cDNA was produced, then a multiplex(es) of all six PIGA exons was performed. For most mutants with no mutation, the 26bp non-coding exon 1 was sequenced to look for splice mutations (none were found).
Mutants are described using the Human Genome Variation Society recommendations with several exceptions. First, the IVS nomenclature is used for mutations in the introns rather than the nnn+ or nnn- nomenclature (where nnn is the number of the last or first base of the exon) because it is more informative to individuals who are not familiar with the number of the first and last bases of each exon. Second, because the large deletion nomenclature is not intuitively obvious for mutants lacking one or more exons (e.g., 28-?_318+?del for a deletion of exons 2-3), these are simply referred to as del ex2-3, del ex4 etc. Also, the base sequence change is also included after the amino acid change description, i.e. cys66>his (tgt>tat). Lastly, since all mutation descriptions are based on the cDNA numbering (A of ATG is 1, last A of TAA is 657), c. does not preface all of the descriptions for simplicity. The suggested but not yet recommended designation “los” was used to identify mutations with a deletion of one or more repeat units in a repeated sequence.
Primer Design
New primers were designed using the Primer3 from the Whitehead Institute (MIT) (http://frodo.wi.mit.edu/primer3/) or the NCBI Primer-Blast website (http://www.ncbi.nlm.nih.gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome). The PIGA primers used are shown in Supplement 1, the real-time primers in Supplement 2, and the PIGL region primers in Supplement 3. All primers were purchased from Integrated DNA Technologies (Coralville, IA).
mRNA extraction
mRNA was isolated from frozen 50,000 cell pellets using Qiagen RNeasy mini kits or Qiagen RNeasy 96 kits (Valencia, CA) following the manufacturer’s directions. The mRNA was snap frozen in liquid N2 and kept at −70°C. This mRNA was also used as a source of genomic DNA for multiplex and exon specific amplifications.
RT-PCR for exons 3-6
The High Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Life Technologies, Grand Island, NY) was used following manufacturer’s directions. For the PCR, a 20ul reaction PCR was performed consisting of 2.0ul Qiagen 10X buffer, 1.6ul 2.5mM (each) dNTPs, 12.7ul HPLC water, 0.4ul 20mM MIDF primer, 0.4ul 20mM PIGAEx6R primer, 0.1ul HotStar Taq (Qiagen) plus 2ul of the cDNA preparation. PCR cycles were 95°C 15min then 40X of 94°C-30s, 58°C-30s, 72°C-90s then 72°C-10min and a 4°C hold.
PIGA specific exon PCR
A 20ul reaction PCR was performed consisting of 2.0ul Qiagen 10X buffer, 1.6ul 2.5mM (each) dNTPs, 13.5ul HPLC water, 0.4ul 20mM forward primer, 0.4ul 20mM reverse primer, 0.1ul HotStar Taq plus 2ul of mRNA preparation. PCR cycles were 95°C 15min then 40X of 94°C-30s, 58°C-30s, 72°C-90s then 72°C-10min and a 4°C hold. The primers PIGAEx1F and PIGAEx1R3 were used for exon1 (sequence with PIGAEx1R3), PIGAEx2F and PIGAEx2R for exon 2 (sequence with PIGAEx2R), PIGAEx3F and PIGAEx3R for exon 3 (sequence with PIGAEx3F), PIGAEx4-5F and PIGAEx4-5R for exons 4 and 5 together (sequence with PIGAEx4-5F) and PIGAEx6F and PIGAEx6R for exon 6 (sequence with PIGAEx6F).
PIGA Genomic Multiplex
Initially a multiplex of the six PIGA exons in five amplicons was performed. Later a separate exon 1, 3’UTR and dystrophin (DYS, positive control) multiplex was created. The original PIGA multiplex (20ul reaction) consisted of 2.0ul Qiagen 10X buffer, 1.6ul 2.5mM (each) dNTPs, 1.6ul of 25mM MgCl2, 8.5ul HPLC water, 3.2ul multiplex primer mix (final 200nM exon 1, exon 3 and exon 6 primers; 400nM exon 2 and exon 4-5 primers), 0.1ul HotStar Taq plus 3ul of the mRNA preparation. PCR cycles were 95°C 15min then 36X of 94°C-30s, 58°C-30s, 72°C-2min then 72°C-7min and a 10°C hold. Products were run on a 2% agarose gel. The PIGA Ex1-Ex6-Ex6+-Dystrophin multiplex was a 20ul reaction consisting of 2.0ul Qiagen 10X buffer, 1.6ul 2.5mM (each) dNTPs, 9.3ul HPLC water, 1.6ul of 25mM MgCl2. 0.3ul of each 20uM primer [PIGAEx1F, PIGAEx1R3, PIGAEx6F, PIGAEx6R, PIGAEx6+F, PIGAEx6+R, DYSF, DYSR], 0.12ul HotStar Taq plus 2ul of the mRNA preparation. PCR cycles were the same as for the PIGA Genomic Multiplex. Products were run on a 1.5% agarose gel.
Real-time PCR for GPI-a mRNAs
The real-time primers either came from the literature, were found in qPrimerDepot (http://primerdepot.nci.nih.gov/) or Primer Bank (http://pga.mgh.harvard.edu/primerbank/) or were designed on the NCBI Primer-Blast website (see Supplement 2). Real-time amplification was performed using 4.4ul ddH20, 10ul 2X Qiagen Quantitect SYBR Green PCR mix, 0.3ul of 20uM forward primer, 0.3ul of 20uM reverse primer, plus 5ul cDNA. Real-time amplification was performed in a Stratagene MX3005 with the parameters: 15 min 95°C, then 45X of 94°C 15sec, 55°C 30sec, 72°C 30sec.
Deletion Breakpoints on chromosome 17p
Multiplex PIGL PCR had determined that PIGL exons 1 and 2 were absent in many isolates (with PIGL exon 3 always present) so PCR primer pairs within PIGL intron 2 were designed (NCBI primer-blast) to pinpoint the 3’ deletion. For the 5’ deletion end, primer pairs were created at intervals through NCOR1 into TTC19 until the total deletion ends were identified. Numerous multiplex PCRs were performed to narrow the breakpoint region for the various deletions. PCR reactions contained three to four sets of primers including DYS exon 45 as a positive control for amplification. Care was taken so that adjacent amplicons were of different sizes and not 550bp (size of DYS positive control) for ease of interpreting screening results. A 20ul reaction PCR was performed consisting of 2.0ul Qiagen 10X buffer, 1.6ul 2.5mM (each) dNTPs, 13.5ul HPLC water, 0.4ul 20mM forward primer, 0.4ul 20mM reverse primer, 0.1ul HotStar Taq plus 2ul of mRNA preparation. PCR cycles were 95°C 15min then 32X of 94°C-30s, 58°C-30s, and 72°C-90s, then 72°C-10min and a 4°C hold. Products were run on a 1.5 to 2% agarose gel.
PCR across deletion breakpoints
Once the total deletion ends were determined in any isolate, primer pairs were chosen that flanked the deletion ends to perform an amplification across the breakpoint (see Supplement 3). When no breakpoints were obtained using conventional PCR, long PCR was performed using the TaKaRa LA PCR Kit Ver 2.1 (Clontech, Mountain View, CA) following manufacturer’s directions.
PIGL mutation analysis
Genomic primers were designed (NCBI Primer-Blast website) for each of the seven PIGL exons to create a multiplex PCR (Fig. 1) (see Supplement 3). Genomic PCR was performed as described for PIGA with an exception of alternative primer concentrations of 200nM. RT-PCR and sequencing were performed as described for PIGA except using the PIGLseq primers.
Fig 1.
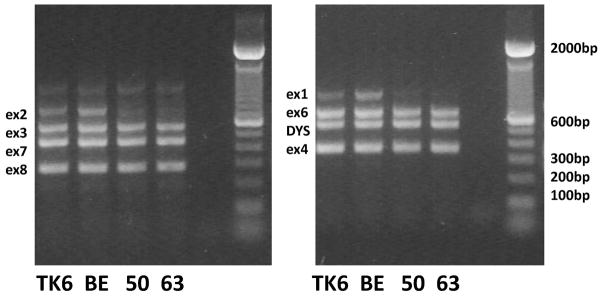
PIGL genomic multiplex in two amplifications. TK6 is the parental cell line, BE has no PIGA or PIGL mutation, 50 and 63 both have the PIGL exon 1/exon 2 deletion. Lane 5 is the blank negative control.
DNA sequencing
PCR products were ExoSAP-IT (USB Corporation, Cleveland, OH) treated according to manufacturer’s recommendations. DNA sequencing was performed at the University of Vermont, Vermont Cancer Center DNA Analysis Facility utilizing an Applied Biosystems 3130XL.
Cytogenetics
Cytogenetics was performed in the University of Vermont Medical Center Cytogenetics Laboratory, Department of Pathology and Laboratory Medicine. The cells were analyzed using G-banded karyotyping and interphase and metaphase FISH for the SMS region at chromosome 17p11.2 and RARA region at 17q21 (Abbott Molecular, Inc. Des Plaines, IL). This probe is 146kb long and includes the LLGL1, FLII, TOP3A, and SMHT1 genes (~18,225kb to 18,363kb).
Epigenetic Studies
DNA was extracted from the frozen pellets and bisulfite treated using the Qiagen EpiTect Plus Bisulfite Kit according to manufacturer’s directions. For Pyrosequencing, PCR of the PIGA promoter was performed using the primers from the Qiagen PyroMark CpG Assays for the human PIGA gene (PM00133700 and PM00133707) and NCOR1/PIGL gene (PM00065513) using the Qiagen PyroMark PCR Kit according to manufacturer’s directions. Pyrosequencing was performed on a Qiagen PyroMark Q24 using PyroMark Gold Q24 Reagents following manufacturer’s protocols at the University of Vermont COBRE Cell and Molecular Biology Core Facility.
RESULTS
In experiment #1, the EMS treated cells showed a gradual increase in GPI-a mutant frequency (MF) with values rising from approximately 20×10−6 (background) to 470×10−6 (day 10 post-treatment). However, initial molecular analysis of 74 control and treated (x - day 1 to × - day 8) GPI-a deficient isolates found that only 21 (28.4%) had determinable mutations in the PIGA gene. This was not due to methodologic issues causing mutations to be missed as PIGA mutations were found in >80% of GPI-a deficient isolates from in vivo studies of eight control individuals performed concurrently (data not shown).
To confirm that the isolated aerolysin-resistant isolates from TK6 were in fact GPI-a deficient isolates, experiment #2 (MF 4.4 to 28.1 x10−6) was performed in which 40 spontaneous isolates were isolated and re-tested a second time confirming aerolysin resistance. Molecular analysis was then performed on these isolates as well as the Day 10 isolates from experiment #1. None of the isolates from experiment #2 (all spontaneous isolates) had mutations in the PIGA gene. However, a greater percentage of the 24 isolates from the experiment #1-Day 10 isolates had PIGA mutations (13, 54%) indicating that PIGA mutations were being induced by the EMS treatment.
Since the isolates seemed to be stably aerolysin-resistant, real-time PCR was performed for each of the other twenty-five GPI-a pathway genes in the hope that at least some isolates would show mRNA alterations/loss in the other pathway genes. For PIGL, two real-time primer sets were used and many of the isolates showed total loss of amplification of one or both amplicons (Fig. 2).
Fig. 2.


Real-time analysis of PIGL cDNA (A) and actin (ACTB) (B) positive control for amplification. CC, CX and DA have the PIGL type A deletion. TK6 is the parental line. DB and DC do not make PIGL mRNA for unknown reasons. CJ and CW have point mutations in PIGA.
Genomic primers were then designed for all the PIGL exons (17p11.2, 16,217kb to 16,327kb, seven exons) and a two-amplification multiplex PCR designed (Fig. 1). This multiplex PCR showed that the isolates that failed real-time amplification exhibited deletions of PIGL exons 1 and 2 but not loss of exon 3. The 188Kb NCOR1 gene is very closely linked on the telomeric side of PIGL in a head to head (5’ to 5’) orientation. Therefore, primer pairs were designed for NCOR1 for use in multiplex amplifications to determine if the deletion(s) extended into NCOR1 and, if so, how far. In all the isolates with PIGL deletions, the deletion extended into NCOR1 but to different extents. Some deletions extended through NCOR1 into the adjacent TTC19 gene. Primer pairs were also made for PIGL intron 2 for use in other multiplex amplifications to determine where the deletions ended in the PIGL gene. Fig. 3 shows the homozygous deleted areas on a graph of the TTC19/NCOR1/PIGL region and with the deletion endpoint ranges and deletion size ranges.
Fig. 3.

Extent of the homozygous PIGL deletions on chromosome 17p. Numbering is based on Genbank GRCh38 Primary Assembly annotation 106. Adapted from the NCBI “Genomic regions, transcripts, and products” diagram for PIGL (http://www.ncbi.nlm.nih.gov/gene/9487).
The fact that these GPI-a deficient isolates had homozygous deletions for an autosomal region suggested that the original TK6 cell line had a hemizygous deletion on chromosome 17p (first hit) and that these isolates had a second hit which resulted in a homozygous deletion for part of 17p. The homozygous deletion detected was the overlap between the first and second hit deletions; however the breakpoint ends of either individual heterozygous deletion were unclear.
That at least part if not all of PIGL was likely hemizygous implied that point mutations (as the second hit) in PIGL could also be occurring in the GPI-a isolates. All the isolates without a PIGA mutation or a PIGL deletion were then sequenced using RT-PCR for the PIGL gene. Seventeen of these isolates had point mutations in the PIGL gene which was confirmation that part or all of PIGL is hemizygous in TK6. The point mutations occurred in exon 1 up to exon 7 (up to base pair 685 of the cDNA (A of ATG is 1)). Five isolates had a loss of the 32bp exon 5 in the cDNA but no exon 5 mutation was found in the genomic DNA; two additional isolates failed to produce any PIGL RT-PCR product. While the actual mutations were not found, all of these have an implied mutation in PIGL.
Promoter methylation studies were performed by pyrosequencing for PIGA and PIGL on the isolates without defined mutations [AI, DB and DC (no PIGL mRNA), DI (no PIGA mRNA) and BE, EH, EP as well as several controls). No methylation was found on either the PIGA or PIGL promoter for any of the isolates (data not shown).
Table I summarizes all the results for PIGA and PIGL isolates and mutations in the spontaneous and EMS treated isolates. The complete list of mutations is given in Supplement 4.
Table I.
Summary of TK6 mutation type results
| Type of point mutation | Spontaneous mutations | EMS mutations | ||
|---|---|---|---|---|
| PIGA Mutations | PIGL Mutations | PIGA Mutations | PIGL Mutations | |
| G>A | 0 | 0 | 19 | 9 |
| C>T | 0 | 0 | 11 | 7 |
| G>T | 0 | 0 | 3 | 0 |
| C>A | 0 | 0 | 2 | 0 |
| T>A | 0 | 0 | 1 | 0 |
| A>T | 0 | 0 | 0 | 0 |
| G>C | 0 | 0 | 0 | 0 |
| C>G | 0 | 0 | 0 | 0 |
| A>G | 0 | 0 | 0 | 1 |
| T>C | 0 | 0 | 0 | 0 |
| A>C | 0 | 0 | 0 | 0 |
| T>G | 0 | 0 | 0 | 0 |
| frameshift | 0 | 0 | 1 | 0 |
| no mRNA | 0 | 2 | 1 | 0 |
| genomic deletion | 0 | 57 | 0 | 17 |
| exon loss, no mutation found | 0 | 0 | 0 | 5 |
| Total | 0 | 59 | 38 | 39 |
| None in PIGA or PIGL | 1 | 3 | ||
| TOTAL | 60 | 80 | ||
To attempt to determine the size of the original hemizygous TK6 17p11-p12 region deletion, karyotypic analysis of the TK6 cell line was performed. No obvious changes in the 17p region were found although several other abnormalities were found including three copies of chromosome 13 and added material on chromosomes 14 and 21 (Fig. 4A and 4B). This result conforms to a previous reported karyotype for TK6 [Kodama et al. 1989]. FISH was performed using a probe for the Smith-Magenis Syndrome deletion region (17p11) which is approximately 2×106 basepairs 3’ (centromeric) to the PIGL gene. This probe was found in two copies in the TK6 cells indicating that the hemizygous deletion does not extend that far centromeric (Fig. 4C).
Fig. 4.

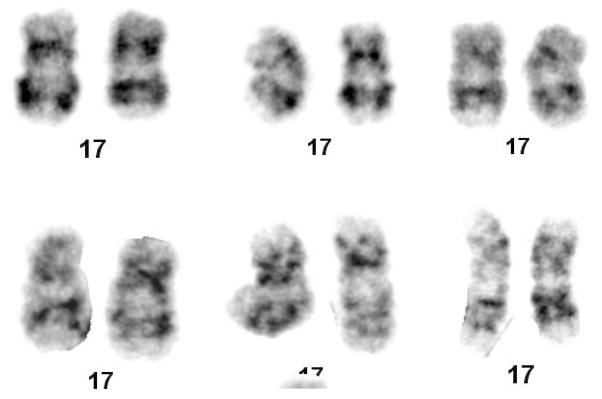

Cytogenetic Analysis of the TK6 cell line. A. Karyotype, B. Closeups of chromosome 17, C. Fish results with a Smith-Magenis probe.
The fact that none of the homozygous deletions extended past PIGL exon 3 suggested either that the original hemizygous deletion in TK6 extends only that far or that a homozygous deletion past PIGL exon 3 might be non-viable because of a required gene product. There is a micro RNA (MIR1288) mapping in the large PIGL intron 2 (65,817bp); however, it is included in several of the deletions, thus, this is not the reason for the limited deletion.
The multiplex amplifications narrowed the placement of the ends of possible breakpoints in the different deletion groups to within 1–2kb on each end. Attempts were then made to sequence across the breakpoints of both the original heterozygous deletion in TK6 and the second hit deletions in the GPI-a deficient isolates. Each end of the homozygous deletion in any mutant could represent an end of the original TK6 deletion or an end of the new deletion in the mutant. Primers facing the homozygous deletion in each isolate were used in Long PCR amplification reactions for both that isolate and TK6 in the hope that one combination would amplify across a deletion breakpoint in either that isolate or TK6 (Table II).
Table II.
Primers Used to Sequence Across Breakpoints and Products Generated
| Cells/Deletion | primer closest to5’ end of deletion | primer closest to3’ end of deletion | expected maximum size of PCR product | actual observed size of PCR product |
|---|---|---|---|---|
| TK6 | TTC19int7eF | PIGLex3R | 2469 bp | ~2000 bp |
| A | TTC19int7eF | PIGLex3R | 2469 bp | ~2000 bp |
| B | NCOR1 int15R | PIGLint2kR | 1526 bp | no product |
| C | NCOR1int1hR | PIGL int2bR | 5798 bp | ~4000 bp |
| D | NCOR1 int15dR | PIGL int2bR | 3826 bp | ~1500 bp |
| E | NCOR1 int14R | PIGLex3R | 3720 bp | no product |
| F | NCOR1 int19R | PIGLex3R | 5512 bp | no product |
| G | NCOR1int18bR | PIGL int2bR | 5366 bp | ~3500 bp |
based on distance of primers from breakpoint
Deletion breakpoints found from sequencing of the generated amplification products are given in Supplement 5. The ~2000bp product representing the group A deletion was sequenced and found to be the result of an 275,712bp deletion extending from 16023577 in TTC19 intron 7 to 16299286 of PIGL intron 2 (near exon 3 at 16299888 to 16299978). Primers were designed flanking this breakpoint (614bp amplicon). Representative GPI-a deficient TK6 isolates were tested as well as the TK6 parent; all had this deletion indicating this deletion was the original hemizygous deletion in the TK6 parent (control normal DNA did not give the 614bp product). In addition, three samples of the TK6 cell line obtained in 2009 from Drs. Peter Wogan, William Thilly, and Howard Liber, respectively, all had the PIGL deletion while Jurkat and WTK1 (also from Dr. Liber in 2009) did not.
Long PCR products were also generated for deletion types C, D and G and the breakpoints sequenced (Supplement 5). However, no specific Long PCR products were generated for groups B, E and F. Since there were seven isolates which appeared to have the same homozygous deletion D, they were tested with primers flanking the deletion breakpoint found in the first mutant. All were found to give the same 784bp product as the first mutant suggesting this was an in vitro clone.
The breakpoints were examined for repeats that might be sites for recombination and for VDJ recombinase sites as a possible mechanism for the deletion. The original TK6 deletion had an unspecified DNA element at the 5’ breakpoint and an Alu sequence at the right breakpoint. Both deletion C and deletion D had Alu’s at the left breakpoint while deletion F had no repeat. Deletions C, D and F all had an ERV_ClassI element at the 3’ breakpoint which is not surprising given their proximity (within 1424 bp).
DISCUSSION
The 275,712bp TTC19 intron 7 to PIGL intron 2 deletion is clearly the original hemizygous deletion in TK6; it is present not only in our stock of TK6 but also in TK6 stocks in other laboratories as well. The TK6 parental line, WI-L2, was originally isolated in 1968 [Levy et al. 1968]. TK6 (has the PIGL deletion) and WTK1 (does not have the PIGL deletion) were both derived from WI-L2 by separate ICR-191 mutagenesis to first a thymidine kinase (TK) −/− cell line and then another round of ICR-191 to a TK+/− state [Skopek et al. 1978; Benjamin et al. 1991; Amundson et al. 1993]. Of interest, the TK gene is also on chromosome 17; however, TK is on the distal long arm (17q23.2-q25.3; 78,174kb to 78,187kb) while PIGL is on the p arm (17p11.2). Thus, it appears unlikely that the chromosome 17 events causing the PIGL deletion and the TK mutation(s) are related.
The endpoints of the original (first hit) PIGL deletion were defined as those of the deletion in group A only because the second hit deletion(s) in group A are larger than the original TK6 deletion. The deletion A group is undoubtedly made of a number of different deletions of different extents, their only commonality is that they extend farther on both ends than the first hit TK6 hemizygous deletion. Similarly, the deletions in groups B, E and F are undoubtedly longer on the 3’ PIGL end than the TK6 original hemizygous deletion, hence it was not possible to PCR across their breakpoints despite their 5’ breakpoints being undoubtedly correctly defined as within NCOR1. Deletion B has both breakpoint ends within the bounds of the first hit deletion, so it would be expected that PCR across the breakpoints would be successful; that it was not despite several attempts suggests that it may involve a translocation or other rearrangement as well as a deletion.
Of the seven isolates with no identifiable mutation in PIGA or PIGL, AI, DB and DI clearly have mutations/methylation in PIGA or PIGL, respectively, since they demonstrate no mRNA expression. While no promoter methylation was found in these three isolates, we have found one example of PIGA promoter methylation in a different study of in vivo GPI-a deficient lymphocytes from Veterans exposed to depleted uranium [manuscript in preparation].
The genomic basis for GPI-a deficiency in the remaining three isolates (BE, EH, EP) is unclear. They could be a mixed well with two isolates with different mutations, one a deletion in PIGL and one a deletion in PIGA, then each mutant would hide the presence of the other. There were eight isolates with heterozygous mutations (Day 6 – 13%, Day 8 – 4.2% and Day 10 – 17%). Two isolates (AQ, DH) had two different PIGA mutations each at less than 50%. For the other six, five had a PIGA mutation (BH, EG, EI, EM, ER) and one had a PIGL mutation (BO) at less than 50%. Since homozygous PIGL deletions are common among the isolates, it would not be unexpected in a well with two isolates that one isolate would have a homozygous PIGL deletion and a second isolate have a PIGA mutation. The Day 6, Day 8 and Day 10 experiments had high percentages of positive wells (40%, 54%, 91%) so the P2 class from the Poisson would suggest that 16%, 21% and 31%, respectively, of the wells would have two isolates which is consistent with our observation. The four isolates without mutations could also have some cryptic mutation or a mutation in another gene in the pathway; however, these are overall a small percentage of the isolates studied (3/140 = 2.1%).
Fig. 5 graphs the percentage of the three major types of mutations seen: PIGA mutations, PIGL deletions and PIGL point mutations in controls and after EMS treatment. PIGL deletions are the major mutation type for the spontaneous mutations on Day 1 and Day 3 while PIGA and PIGL point mutations increase at Day 6, Day 8 and Day 10. These mutations are of the type expected for EMS treatment For the 33 independent mutations with basepair changes, 16 (48.5%) are G>A and 11 (33.3%) are C>T. Previous studies of EMS treatment in TK6 cells found 65% G>A and 14% C>T mutations [Branda et al. 2007]. This is expected because EMS reacts with the O6 and N7 positions of guanine. O6-ethylguanine pairs with thymine during replication, while N7-alkylation leads to apurinic sites [Laval et al. 1981; Margison and Pegg 1981; Singer and Brent 1981; Beranek 1990; Vogel and Nivard 1994].
Fig. 5.

Frequency of PIGA mutations, PIGL deletions and PIGL point mutations at different days of selection for EMS-treated TK6 cells.
Deletion Type A is the most frequent deletion occurring in 32 of 40 (80%) spontaneous isolates in experiment #2 and 14 of 20 (70%) isolates in the spontaneous isolates for experiment #1. All but one of the other deletions (C) also occur in spontaneous isolates. Deletion A also occurs in 17 of the 78 (%) EMS treated isolates. However, as discussed above Deletion type A is probably made up of a number of different deletions.
The conventional assumption of an obligatory PIGA mutation as the causative genetic event in GPI-a deficiency ignores the possibility of constitutional heterozygosity for any of the 25 autosomal genes in the pathway, rendering the remaining wild-type allele hemizygous and similar to PIGA in terms of vulnerability to single-hit inactivation. There are however, rare examples of PNH with a normal, wild-type PIGA gene. In a well described case, an individual with a constitutional (inherited) deletion mutation in the autosomal PIGT gene subsequently acquired a somatic mutation in the remaining PIGT wild-type allele and developed a clinically typical PNH [Krawitz et al. 2013]. Also, obviously, parents and other relatives of individuals homozygous for the various described GPI-a gene diseases would be expected to be heterozygous.
Recently, Kruger et al. [2014] have reported a flow cytometry-based GPI deficiency assay in TK6 cells that they proposed as an “in vitro PIG-A mutation assay”. Although acknowledging the existence of autosomal genes in the GPI-a synthesis pathway, it was assumed that most or all of the deficiency isolates in the reported study resulted from mutations in the PIGA gene. Our results presented here demonstrate that this may not necessarily be so. The status of the autosomal genes in the TK6 cells used by Kruger et al. [2014] cannot be known and will not necessarily be the same as in the cells in our laboratory. In continuously growing cell lines and in outbred species such as humans, it is inevitable that hemizygosity for one or more genes in the GPI anchor pathway will have occurred as demonstrated by our observations in the TK6 cell line.
While this might be considered an impediment to using GPI-a deficiency as an end-point in mutagenicity studies, it could present an advantage over currently used in vitro selection schemes, i.e. 6-thioguanine resistance for HPRT mutations or trifluorothymidine for TK mutations. Mutations of various types (in two genes) can be selected in a single assay using GPI-a deficiency. Flow cytometry can be used in quantitative studies as proposed by Kruger et al. [2014], thereby greatly increasing throughput and reducing costs. For an assessment of an agent’s overall mutagenicity, a simple quantitative result will suffice. This may be all that is necessary for hazard identification and to eliminate false negatives due to a mutagen inducing primarily a clastogenic response when an assay that detects primarily point mutations is used. For example, an in vitro mutagenicity study in cells that are hemizygous for one or more genes in the GPI-a synthesis genetic pathway and therefore able to detect large deletion mutations could serve as check on in vivo Piga animal studies that give negative results for clastogens due to mutations arising only in Piga. However, this requires confidence that there is no such hemizygosity in the animals.
When further characterization of mutagenic action is required, a differentiation between a point mutagenic versus a clastogenic response can be made if two positive controls are used in an in vitro mutagenicity study – a primarily point mutagen such as EMS in the current study and an agent that primarily acts through chromosomal level effects. However, for any of these advantages to be realized for any target cell line used for in vitro studies, the GPI-a synthesis genetic pathway should first be characterized, as done here for our TK6 cells.
It should be obvious that comparing GPI-a deficiency mutant frequencies in different cells lines to infer genetic instability in one or more of the lines, as has been done for various malignant cell lines, requires that the cells be comparable in terms of potential genetic targets for the deficiency [Araten et al. 2010].
The existence of multiple potential genetic targets for GPI-a deficiency makes mutagenicity monitoring of humans also more problematic when exposed and unexposed populations consisting of different individuals are compared to infer the mutagenicity of an agent of concern. Heterozygosity of any of the autosomal genes in this pathway may give one or more additional targets for GPI-a deficiency in some individuals. This will not be the case however, if before and after exposure studies are done in the same individuals. These considerations also apply to animal studies, although inbred or closely related animals are usually used to eliminate or minimize genetic heterogenicity. Eventually, characterization of genes in the GPI-a synthesis pathway as begun here should allow development of knock-out rodent models for more precise in vivo mutagenicity studies.
Supplementary Material
Acknowledgments
The authors acknowledge that this work was supported by the U.S. Department of Veterans Affairs. The automated DNA sequencing was performed in the VT Cancer Center DNA Analysis Facility (special thanks to Mary Lou Shane and Jessica Hoffman) and was supported in part by the Vermont Cancer Center, the Lake Champlain Cancer Research Organization, and the UVM College of Medicine. Methylation studies were performed on a Qiagen Pyrosequencer Q24 in the University of Vermont COBRE Cell and Molecular Biology Core Facility (special thanks to Thomm Buttolph) which is supported by NIH Grant Numbers 5 P30 RR032135 from the COBRE Program of the National Center for Research Resources and 8 P30 GM 103498 from the National Institute of General Medical Sciences. Special thanks to Terri Messier for comments on the manuscript.
Footnotes
AUTHOR CONTRIBUTIONS
J.A.N. performed all the molecular studies. E.W.C performed the GPI-a cell cloning and mutagenesis experiments. R.J.A. was the principal investigator and helped design experiments and write the manuscript. All authors provided critical review and approved the final manuscript. None of the authors have a conflict of interest in regard to this manuscript.
References
- 1.Almeida AM, Murakami Y, Layton DM, Hillmen P, Sellick GS, Maeda Y, Richards S, Patterson S, Kotsianidis I, Mollica L, Crawford DH, Baker A, Ferguson M, Roberts I, Houlston R, Kinoshita T, Karadimitris A. Hypomorphic promoter mutation in PIGM causes inherited glycosylphosphatidylinositol deficiency. Nat Med. 2006;12(7):846–851. doi: 10.1038/nm1410. [DOI] [PubMed] [Google Scholar]
- 2.Amundson SA, Xia F, Wolfson K, Liber HL. Different cytotoxic and mutagenic responses induced by X-rays in two human lymphoblastoid cell lines derived from a single donor. Mutat Res. 1993;286(2):233–241. doi: 10.1016/0027-5107(93)90188-l. [DOI] [PubMed] [Google Scholar]
- 3.Araten DJ, Martinez-Climent JA, Perle MA, Holm E, Zamechek L, DiTata K, Sanders KJ. A quantitative analysis of genomic instability in lymphoid and plasma cell neoplasms based on the PIG-A gene. Mutat Res. 2010;686(1–2):1–8. doi: 10.1016/j.mrfmmm.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barone R, Aiello C, Race V, Morava E, Foulquier F, Riemersma M, Passarelli C, Concolino D, Carella M, Santorelli F, Vleugels W, Mercuri E, Garozzo D, Sturiale L, Messina S, Jaeken J, Fiumara A, Wevers RA, Bertini E, Matthijs G, Lefeber DJ. DPM2-CDG: a muscular dystrophy-dystroglycanopathy syndrome with severe epilepsy. Ann Neurol. 2012;72(4):550–558. doi: 10.1002/ana.23632. [DOI] [PubMed] [Google Scholar]
- 5.Benjamin MB, Potter H, Yandell DW, Little JB. A system for assaying homologous recombination at the endogenous human thymidine kinase gene. Proc Natl Acad Sci U S A. 1991;88(15):6652–6656. doi: 10.1073/pnas.88.15.6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beranek DT. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat Res. 1990;231(1):11–30. doi: 10.1016/0027-5107(90)90173-2. [DOI] [PubMed] [Google Scholar]
- 7.Boccuni P, Del Vecchio L, Di Noto R, Rotoli B. Glycosyl phosphatidylinositol (GPI)-anchored molecules and the pathogenesis of paroxysmal nocturnal hemoglobinuria. Crit Rev Oncol Hematol. 2000;33(1):25–43. doi: 10.1016/s1040-8428(99)00052-9. [DOI] [PubMed] [Google Scholar]
- 8.Branda RF, O'Neill JP, Brooks EM, Powden C, Naud SJ, Nicklas JA. The effect of dietary folic acid deficiency on the cytotoxic and mutagenic responses to methyl methanesulfonate in wild-type and in 3-methyladenine DNA glycosylase-deficient Aag null mice. Mutat Res. 2007;615(1–2):12–17. doi: 10.1016/j.mrfmmm.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Dertinger SD, Heflich RH. In vivo assessment of Pig-a gene mutation-recent developments and assay validation. Environ Mol Mutagen. 2011;52(9):681–684. doi: 10.1002/em.20685. [DOI] [PubMed] [Google Scholar]
- 10.Dertinger SD, Phonethepswath S, Avlasevich SL, Torous DK, Mereness J, Cottom J, Bemis JC, Macgregor JT. Pig-a gene mutation and micronucleated reticulocyte induction in rats exposed to tumorigenic doses of the leukemogenic agents chlorambucil, thiotepa, melphalan, and 1,3-propane sultone. Environ Mol Mutagen. 2014;55(4):299–308. doi: 10.1002/em.21846. [DOI] [PubMed] [Google Scholar]
- 11.Dertinger SD, Phonethepswath S, Weller P, Avlasevich S, Torous DK, Mereness JA, Bryce SM, Bemis JC, Bell S, Portugal S, Aylott M, MacGregor JT. Interlaboratory Pig-a gene mutation assay trial: Studies of 1,3-propane sultone with immunomagnetic enrichment of mutant erythrocytes. Environ Mol Mutagen. 2011;52(9):748–755. doi: 10.1002/em.20671. [DOI] [PubMed] [Google Scholar]
- 12.Dobrovolsky VN, Elespuru RK, Bigger CA, Robison TW, Heflich RH. Monitoring humans for somatic mutation in the endogenous PIG-a gene using red blood cells. Environ Mol Mutagen. 2011;52(9):784–794. doi: 10.1002/em.20667. [DOI] [PubMed] [Google Scholar]
- 13.Dobrovolsky VN, Shaddock JG, Mittelstaedt RA, Miura D, Heflich RH. Detection of in vivo mutation in the Hprt and Pig-a genes of rat lymphocytes. Methods Mol Biol. 2013;1044:79–95. doi: 10.1007/978-1-62703-529-3_4. [DOI] [PubMed] [Google Scholar]
- 14.Gabdoulkhakova A, Henriksson G, Avkhacheva N, Sofin A, Bredberg A. High rate of mutation reporter gene inactivation during human T cell proliferation. Immunogenetics. 2007;59(2):135–143. doi: 10.1007/s00251-006-0180-8. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Silva MT, Matthijs G, Schollen E, Cabrera JC, Sanchez del Pozo J, Marti Herreros M, Simon R, Maties M, Martin Hernandez E, Hennet T, Briones P. Congenital disorder of glycosylation (CDG) type Ie. A new patient. J Inherit Metab Dis. 2004;27(5):591–600. doi: 10.1023/b:boli.0000042984.42433.d8. [DOI] [PubMed] [Google Scholar]
- 16.Horibata K, Ukai A, Kimoto T, Suzuki T, Kamoshita N, Masumura K, Nohmi T, Honma M. Evaluation of in vivo genotoxicity induced by N-ethyl-N-nitrosourea, benzo[a]pyrene, and 4-nitroquinoline-1-oxide in the Pig-a and gpt assays. Environ Mol Mutagen. 2013;54(9):747–754. doi: 10.1002/em.21818. [DOI] [PubMed] [Google Scholar]
- 17.Hu R, Mukhina GL, Lee SH, Jones RJ, Englund PT, Brown P, Sharkis SJ, Buckley JT, Brodsky RA. Silencing of genes required for glycosylphosphatidylinositol anchor biosynthesis in Burkitt lymphoma. Exp Hematol. 2009;37(4):423–434. e422. doi: 10.1016/j.exphem.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu R, Mukhina GL, Piantadosi S, Barber JP, Jones RJ, Brodsky RA. PIG-A mutations in normal hematopoiesis. Blood. 2005;105(10):3848–3854. doi: 10.1182/blood-2004-04-1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimoto T, Horibata K, Chikura S, Hashimoto K, Itoh S, Sanada H, Muto S, Uno Y, Yamada M, Honma M. Interlaboratory trial of the rat Pig-a mutation assay using an erythroid marker HIS49 antibody. Mutat Res. 2013;755(2):126–134. doi: 10.1016/j.mrgentox.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 20.Kodama Y, Boreiko CJ, Skopek TR, Recio L. Cytogenetic analysis of spontaneous and 2-cyanoethylene oxide-induced tk-/- mutants in TK6 human lymphoblastoid cultures. Environ Mol Mutagen. 1989;14(3):149–154. doi: 10.1002/em.2850140304. [DOI] [PubMed] [Google Scholar]
- 21.Kranz C, Denecke J, Lehrman MA, Ray S, Kienz P, Kreissel G, Sagi D, Peter-Katalinic J, Freeze HH, Schmid T, Jackowski-Dohrmann S, Harms E, Marquardt T. A mutation in the human MPDU1 gene causes congenital disorder of glycosylation type If (CDG-If) J Clin Invest. 2001;108(11):1613–1619. doi: 10.1172/JCI13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krawitz PM, Hochsmann B, Murakami Y, Teubner B, Kruger U, Klopocki E, Neitzel H, Hoellein A, Schneider C, Parkhomchuk D, Hecht J, Robinson PN, Mundlos S, Kinoshita T, Schrezenmeier H. A case of paroxysmal nocturnal hemoglobinuria caused by a germline mutation and a somatic mutation in PIGT. Blood. 2013;122(7):1312–1315. doi: 10.1182/blood-2013-01-481499. [DOI] [PubMed] [Google Scholar]
- 23.Krawitz PM, Murakami Y, Hecht J, Kruger U, Holder SE, Mortier GR, Delle Chiaie B, De Baere E, Thompson MD, Roscioli T, Kielbasa S, Kinoshita T, Mundlos S, Robinson PN, Horn D. Mutations in PIGO, a member of the GPI-anchor-synthesis pathway, cause hyperphosphatasia with mental retardation. Am J Hum Genet. 2012;91(1):146–151. doi: 10.1016/j.ajhg.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krawitz PM, Schweiger MR, Rodelsperger C, Marcelis C, Kolsch U, Meisel C, Stephani F, Kinoshita T, Murakami Y, Bauer S, Isau M, Fischer A, Dahl A, Kerick M, Hecht J, Kohler S, Jager M, Grunhagen J, de Condor BJ, Doelken S, Brunner HG, Meinecke P, Passarge E, Thompson MD, Cole DE, Horn D, Roscioli T, Mundlos S, Robinson PN. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010;42(10):827–829. doi: 10.1038/ng.653. [DOI] [PubMed] [Google Scholar]
- 25.Kruger CT, Hofmann M, Hartwig A. The in vitro PIG-A gene mutation assay: mutagenicity testing via flow cytometry based on the glycosylphosphatidylinositol (GPI) status of TK6 cells. Arch Toxicol. 2014 doi: 10.1007/s00204-014-1413-5. [DOI] [PubMed] [Google Scholar]
- 26.Kvarnung M, Nilsson D, Lindstrand A, Korenke GC, Chiang SC, Blennow E, Bergmann M, Stodberg T, Makitie O, Anderlid BM, Bryceson YT, Nordenskjold M, Nordgren A. A novel intellectual disability syndrome caused by GPI anchor deficiency due to homozygous mutations in PIGT. J Med Genet. 2013;50(8):521–528. doi: 10.1136/jmedgenet-2013-101654. [DOI] [PubMed] [Google Scholar]
- 27.Laval J, Pierre J, Laval F. Release of 7-methylguanine residues from alkylated DNA by extracts of Micrococcus luteus and Escherichia coli. Proc Natl Acad Sci U S A. 1981;78(2):852–855. doi: 10.1073/pnas.78.2.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lefeber DJ, Schonberger J, Morava E, Guillard M, Huyben KM, Verrijp K, Grafakou O, Evangeliou A, Preijers FW, Manta P, Yildiz J, Grunewald S, Spilioti M, van den Elzen C, Klein D, Hess D, Ashida H, Hofsteenge J, Maeda Y, van den Heuvel L, Lammens M, Lehle L, Wevers RA. Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am J Hum Genet. 2009;85(1):76–86. doi: 10.1016/j.ajhg.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levy JA, Virolainen M, Defendi V. Human lymphoblastoid lines from lymph node and spleen. Cancer. 1968;22(3):517–524. doi: 10.1002/1097-0142(196809)22:3<517::aid-cncr2820220305>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 30.Margison GP, Pegg AE. Enzymatic release of 7-methylguanine from methylated DNA by rodent liver extracts. Proc Natl Acad Sci U S A. 1981;78(2):861–865. doi: 10.1073/pnas.78.2.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maydan G, Noyman I, Har-Zahav A, Neriah ZB, Pasmanik-Chor M, Yeheskel A, Albin-Kaplanski A, Maya I, Magal N, Birk E, Simon AJ, Halevy A, Rechavi G, Shohat M, Straussberg R, Basel-Vanagaite L. Multiple congenital anomalies-hypotonia-seizures syndrome is caused by a mutation in PIGN. J Med Genet. 2011;48(6):383–389. doi: 10.1136/jmg.2010.087114. [DOI] [PubMed] [Google Scholar]
- 32.McDiarmid MA, Albertini RJ, Tucker JD, Vacek PM, Carter EW, Bakhmutsky MV, Oliver MS, Engelhardt SM, Squibb KS. Measures of genotoxicity in Gulf war I veterans exposed to depleted uranium. Environ Mol Mutagen. 2011;52(7):569–581. doi: 10.1002/em.20658. [DOI] [PubMed] [Google Scholar]
- 33.Mortazavi Y, Merk B, McIntosh J, Marsh JC, Schrezenmeier H, Rutherford TR. The spectrum of PIG-A gene mutations in aplastic anemia/paroxysmal nocturnal hemoglobinuria (AA/PNH): a high incidence of multiple mutations and evidence of a mutational hot spot. Blood. 2003;101(7):2833–2841. doi: 10.1182/blood-2002-07-2095. [DOI] [PubMed] [Google Scholar]
- 34.Nafa K, Bessler M, Castro-Malaspina H, Jhanwar S, Luzzatto L. The spectrum of somatic mutations in the PIG-A gene in paroxysmal nocturnal hemoglobinuria includes large deletions and small duplications. Blood Cells Mol Dis. 1998;24(3):370–384. doi: 10.1006/bcmd.1998.0203. [DOI] [PubMed] [Google Scholar]
- 35.Nafa K, Mason PJ, Hillmen P, Luzzatto L, Bessler M. Mutations in the PIG-A gene causing paroxysmal nocturnal hemoglobinuria are mainly of the frameshift type. Blood. 1995;86(12):4650–4655. [PubMed] [Google Scholar]
- 36.Nishimura J, Murakami Y, Kinoshita T. Paroxysmal nocturnal hemoglobinuria: An acquired genetic disease. Am J Hematol. 1999;62(3):175–182. doi: 10.1002/(sici)1096-8652(199911)62:3<175::aid-ajh7>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 37.Okamoto M, Shichishima T, Noji H, Ikeda K, Nakamura A, Akutsu K, Maruyama Y. High frequency of several PIG-A mutations in patients with aplastic anemia and myelodysplastic syndrome. Leukemia. 2006;20(4):627–634. doi: 10.1038/sj.leu.2404135. [DOI] [PubMed] [Google Scholar]
- 38.Onami S, Cho YM, Toyoda T, Horibata K, Ishii Y, Umemura T, Honma M, Nohmi T, Nishikawa A, Ogawa K. Absence of in vivo genotoxicity of 3-monochloropropane-1,2-diol and associated fatty acid esters in a 4-week comprehensive toxicity study using F344 gpt delta rats. Mutagenesis. 2014;29(4):295–302. doi: 10.1093/mutage/geu018. [DOI] [PubMed] [Google Scholar]
- 39.Paulick MG, Bertozzi CR. The glycosylphosphatidylinositol anchor: a complex membrane-anchoring structure for proteins. Biochemistry. 2008;47(27):6991–7000. doi: 10.1021/bi8006324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sadiq R, Bhalli JA, Yan J, Woodruff RS, Pearce MG, Li Y, Mustafa T, Watanabe F, Pack LM, Biris AS, Khan QM, Chen T. Genotoxicity of TiO(2) anatase nanoparticles in B6C3F1 male mice evaluated using Pig-a and flow cytometric micronucleus assays. Mutat Res. 2012;745(1–2):65–72. doi: 10.1016/j.mrgentox.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 41.Schenk B, Imbach T, Frank CG, Grubenmann CE, Raymond GV, Hurvitz H, Korn-Lubetzki I, Revel-Vik S, Raas-Rotschild A, Luder AS, Jaeken J, Berger EG, Matthijs G, Hennet T, Aebi M. MPDU1 mutations underlie a novel human congenital disorder of glycosylation, designated type If. J Clin Invest. 2001;108(11):1687–1695. doi: 10.1172/JCI13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singer B, Brent TP. Human lymphoblasts contain DNA glycosylase activity excising N-3 and N-7 methyl and ethyl purines but not O6-alkylguanines or 1-alkyladenines. Proc Natl Acad Sci U S A. 1981;78(2):856–860. doi: 10.1073/pnas.78.2.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skopek TR, Liber HL, Penman BW, Thilly WG. Isolation of a human lymphoblastoid line heterozygous at the thymidine kinase locus: possibility for a rapid human cell mutation assay. Biochem Biophys Res Commun. 1978;84(2):411–416. doi: 10.1016/0006-291x(78)90185-7. [DOI] [PubMed] [Google Scholar]
- 44.Vogel EW, Nivard MJ. International Commission for Protection Against Environmental Mutagens and Carcinogens. The subtlety of alkylating agents in reactions with biological macromolecules. Mutat Res. 1994;305(1):13–32. doi: 10.1016/0027-5107(94)90123-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.