

Abstract
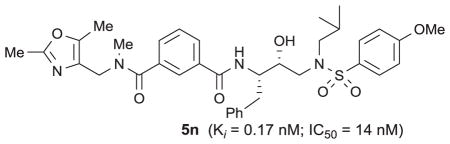
We describe the design, synthesis and biological evaluation of a series of novel HIV-1 protease inhibitors bearing isophthalamide derivatives as the P2–P3 ligands. We have investigated a range of acyclic and heterocyclic amides as the extended P2–P3 ligands. These inhibitors displayed good to excellent HIV-1 protease inhibitory activity. Also, a number of inhibitors showed very good antiviral activity in MT cells. Compound 5n has shown an enzyme Ki of 0.17 nM and antiviral IC50 of 14 nM. An X-ray crystal structure of inhibitor 5o-bound to HIV-1 protease was determined at 1.11 Å resolution. This structure revealed important molecular insight into the inhibitor-HIV-1 protease interactions in the active site.
Keywords: HIV-1 Protease, Inhibitors, Design, Synthesis, Antiviral, Isophthalamide
Graphical Abstract
HIV-1 protease inhibitors (PIs) are an important component of highly active antiretroviral therapy (HAART).1,2 This treatment regimen has had a major impact in improving the quality of life and extending life expectancy of HIV/AIDS patients.3,4 In fact, recent analyses have revealed that the mortality rates of HIV/AIDS patients have become closer to the general mortality rates due to HAART and first-line HAART with protease inhibitor-based regimens.5,6 Despite these advances, the development of potent and more effective HIV-1 protease inhibitor drugs is essential for successful long-term control of HIV-1 infection and AIDS.7,8 In our continuing efforts toward the design and synthesis of nonpeptide HIV-1 protease inhibitors with clinical potential, we reported a variety of exceptionally potent PIs including darunavir (1, Figure 1) that incorporated structurally novel ligands and scaffolds targeting the active site HIV-1 protease backbones.9–12 Our design of ligands has focused on non-peptidic cyclic/heterocyclic structures that mimic the biological mode of peptide bonds. In particular, we designed a variety of cyclic ether and poly-ether derived functionalities inherent to bioactive natural products.13,14 This led to the development of structurally intriguing bis-THF, Cp-THF, Tp-THF, and Tris-THF-derived exceptionally potent inhibitors with broad-spectrum activity against multidrug-resistant HIV-1 variants.9–12
Figure 1.
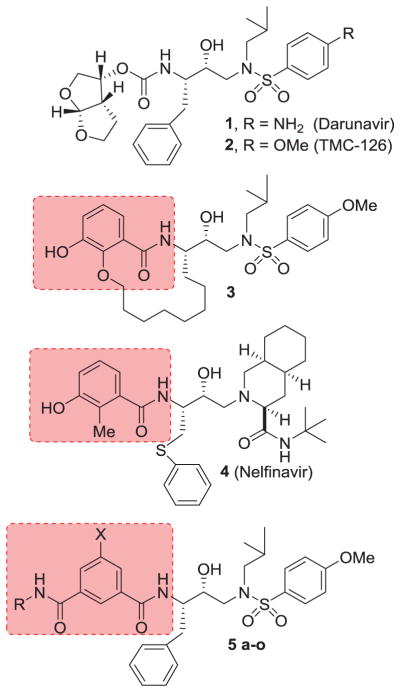
Structures of HIV-1 protease inhibitors 1–5.
Recently, we reported a series of HIV-1 protease inhibitors incorporating 3-hydroxy-2-alkyl or 3-hydroxy-2-alkoxy benzoic acid derivatives as the P2-ligands.15,16 A representative example is inhibitor 3. These inhibitors were designed by taking advantage of the large hydrophobic pocket in the HIV-1 protease S1–S2 subsites.16 The P2-ligands resemble 3-hydroxy-2-methyl benzamide inherent to nelfinavir (4), an FDA approved drug. One of the important features of inhibitor 3 is that the P2 ligand does not contain an asymmetric center. Based upon this result, we have now investigated isophthalamide-based P2-ligands in combination with hydroxyethylamine sulfonamide isosteres. Our preliminary models of such isophthalamide derivatives, based upon the X-ray structure of nelfinavir-bound HIV-1 protease, revealed that both carboxamide functionalities of the isophthalamide ligand can form hydrogen bonds with the Gly-27 backbone carbonyl as well as with Asp-29 backbone amide NHs.17 Also, we speculated that appropriately functionalized substituents can fill in the hydrophobic pocket in the S2–S3 subsites. Interestingly, isophthalamide derivatives were previously utilized in the design of BACE1 inhibitors, an aspartic acid protease implicated in Alzheimer’s disease.18–20 However, there is hardly any report of the use of isophthalamide-derived P2 ligands in the design of HIV-1 protease inhibitors. Herein, we report the design and synthesis of a series of potent HIV-1 protease inhibitors incorporating substituted isophthalamide as the P2–P3 ligands. These inhibitors displayed excellent enzyme inhibitory potency and a number of inhibitors exhibited excellent antiviral activity. A high resolution X-ray structure of an inhibitor-bound HIV-1 protease has been determined. The structure revealed important molecular insight into the ligand-binding site interactions.
Our general strategy for the synthesis of HIV-1 protease inhibitors bearing isophthalamide derivatives as the P2–P3 ligands is shown in Scheme 1. As shown, the designed inhibitors would be synthesized by amide coupling of isophthalic acid derivative 6 with the amine of the hydoxyethylamine sulfonamide isosteres (7a and 7b)21,22 to provide HIV-1 protease inhibitors. Various isophthalic acid derivatives would be obtained by coupling of readily available isophthalic monoacids 8 with various amines 9a–g and 10. Among these, amines 9a–d are commercially available. Other amines were synthesized as follows.
Scheme 1.

General synthetic routes to inhibitors.
The synthesis of various proline derivatives 10a–d is shown in Scheme 2. The synthesis of all four optically active hydroxyl proline derivatives 11 was carried out as described by Baker and co-workers.23 Protection of the hydroxyl group as the benzyl ether followed by LiBH4 reduction of the ethyl ester provided respective primary alcohol 12a–d in excellent yields. These alcohols were converted to methyl ethers by treatment of alcohols with NaH in THF for 30 min at 23 °C followed by addition of methyl iodide and stirring for 12 h. Methyl ethers were obtained in good yield. Removal of the Boc-group by exposure of the Bocderivatives to trifluoroacetic acid (TFA) in CH2Cl2 at 0 °C for 2 h afforded proline derivatives 10a–d in excellent yields. These proline derivatives were then coupled with isophthalic acid methyl ester 8 (X = H) in the presence of EDC, HOBt, and i-Pr2NEt to provide the corresponding amide derivative. Saponification of the methyl ester with aqueous lithium hydroxide afforded acid derivatives 13a–d in very good yields.
Scheme 2.

Reagents and conditions: (a) BnBr, NaH, DMF, 23 °C, 1 h; (b) LiBH4, THF, 23 °C, 2 h, (80–85%); (c) Mel, NaH, THF, 23 °C, 12 h; (d) TFA, CH2Cl2, 0 °C, 2 h, (65–71%); (e) acid 8 (X = H), EDC, HOBT, i-Pr2NEt, CH2Cl2, 23 °c; (f) 1N LiOH, THF, 23 °C, 2 h (67–82%).
The synthesis of oxazole derivatives 9e and 9f is shown in Scheme 3. Commercially available oxazole 14 was treated with NaN3 in DMF and heated to 120 °C for 2 h to provide the corresponding azide in 59% yield. Catalytic hydrogenation of azide using 10% Pd-C in the presence of Boc2O afforded the corresponding Boc-derivative in near quantitative yield. Reduction of ester with calcium borohydride afforded alcohol 15 in excellent yield.24 Reaction of alcohol 15 with NaH and MeI in THF for 6 h at 23 °C resulted in the formation of methyl ether and N-methylation to provide 16 in moderate yield. Removal of the Boc-group by exposure to TFA in CH2Cl2 at 0°C for 3 h provided amine 9e in near quantitative yield. Alcohol 15 was converted to methyl oxazole 17 in a two-step sequence involving mesylation of alcohol 15 with mesyl chloride and pyridine at 0°C to 23 °C for 4 h followed by reduction of the resulting mesylate with NaBH4 in HMPA to provide 17. Removal of the Boc-group by exposure of 17 to TFA in CH2Cl2 provided amine 9f in excellent yield. Amine 9e was coupled with isophthalic acid methyl ester 8 (X = H) using EDC, HOBt and in the presence of i-Pr2NEt to furnish the corresponding amide derivative. Saponification of the methyl ester with aqueous lithium hydroxide afforded acid 18a in very good yield. Similarly, acid 8 was coupled with oxazolylmethyl amine 9f to provide the corresponding amide. The resulting amide was methylated using NaH and MeI in THF for 6 h to provide the corresponding amide. Saponification of the methyl ester with aqueous lithium hydroxide afforded acid 18b. For the synthesis of dimethyloxazole derivatives 18c,d, the corresponding known oxazolylmethyl amine 9g was coupled with acids 8 (X = H, Me).19 The resulting amides were methylated with NaH and MeI to provide the corresponding N-methyl derivatives. Saponification of the methyl ester afforded acids 18c and 18d (50–55% yield for the 3-steps).
Scheme 3.

Reagents and conditions: (a) NaN3, DMF, 120 °C; (b) H2, Pd/C, Boc2O, EtOAc; (c) NaBH4, CaCl2, EtOH/THF, (3:2), (58% 3-steps); (d) Mel, NaH, THF, (51%); (e) TFA, CH2Cl2, 0 °C, (99%); (f) MsCl, pyridine, CH2Cl2, (46%); (g) NaBH4, HMPA, (66%).
Synthesis of HIV-1 protease inhibitors 5a–o containing isophthalamide as the P2 ligands is shown in Scheme 4. Coupling of various carboxylic acids 19a–c with amine 9a using EDC and HOBt in the presence of i-Pr2NEt afforded the corresponding amide derivative 20a–c in good yields (50–60%). For the synthesis of N-methyl amine derivatives, methyl esters 20a–c were reacted with NaH and MeI in THF at 0 °C to 23 °C for 6 h to provide the corresponding N-methyl derivatives 21a–c. Saponification of the resulting methyl esters with aqueous lithium hydroxide afforded carboxylic acids 22a–c in good yields (90–95%). For the synthesis of inhibitor 5a, methyl ester 20a was saponified and the resulting acid was coupled with amine 7 using EDC and HOBt in the presence of i-Pr2NEt to afford amide 5a in 65% yield. In the synthesis of inhibitors 5b–d, amide derivatives 20a–c were methylated with NaH and MeI to provide the corresponding N-methyl amides. Saponification of the methyl ester provided acids 22a–c. Coupling of acids 22a,b with amine 7 provided inhibitors 5b and 5c. Synthesis of inhibitor 5d was accomplished by coupling of acid 22c with amine 7 followed by treatment of the resulting amide with TFA in CH2Cl2 for 6 h to provide 5d. For the synthesis of inhibitors 5e–g, isophthalic acid methyl ester 8 (X = H) was coupled with amines 9b–d. The resulting esters were saponified and the acids were coupled with amine 7. For the synthesis of inhibitors 5h–k, proline derivatives 13a–d were coupled with amine 7 and the resulting amide derivatives were hydrogenated using 10% Pd-C in a mixture of EtOAc and ethanol. Inhibitors 5l–o were synthesized by coupling of acids 18a–d with amine 7a or 7b. 21,22 Standard EDC and HOBt coupling afforded the various amides in 55–65% yield.
Scheme 4.

Reagents and conditions: (a) EDCl, HOBT, DlPEA, CH2Cl2; (b) Mel, NaH, DMF; (c) 1N LiOH, THF, 23 °C, 2 h; (d) TFA, CH2Cl2, (90%); (e) H2, 10% Pd-C, EtOAc/EtOH (1:1), 23 °C, 2 h (90–95%)
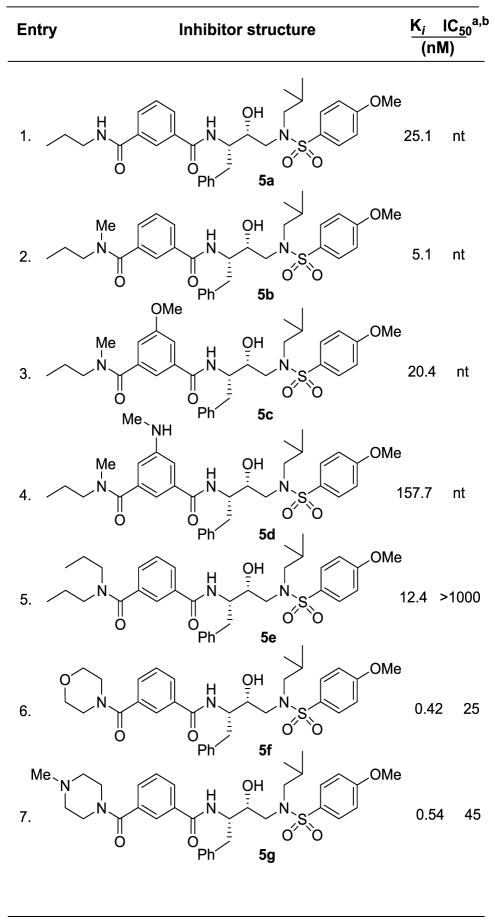
HIV-1 protease inhibitory potency of all inhibitors was first evaluated using assay protocol reported by Toth and Marshall.25 Compounds that showed potent enzyme inhibitory Ki values were then evaluated in an antiviral assay. The results of isophthalamide-derived inhibitors are shown in Table 1 and 2. As can be seen, N-alkylated amides are more potent than the secondary amide inhibitor 5a (entry 1). Interestingly, N-methyl amide derivative 5b showed 5-fold enhanced potency over 5a (entry 2). In inhibitors 5c and 5d, we examined the effect of polar substituents at the 3-position of isophthalamide (entries 3 and 4). As it turned out, neither the hydrogen bond donor nor the acceptor group improved potency over unsubstituted derivative 5b. Dipropyl amide 5e was less effective than N-methyl amide 5b. Compound 5e was evaluated in antiviral assay using MT-2 cells exposed to HIV-1 LAI.26 However, it showed no appreciable antiviral activity. Substitution of N-methyl-N-propyl amide in 5b with cyclic amines such as morpholine and N-methylpiperazine in inhibitors 5f and 5g resulted in over 10-fold improvement of protease inhibitory activity (entries 6 and 7). Both inhibitors 5f and 5g showed very good antiviral activity as well.
Table 1.
Enzyme inhibitory and antiviral activity of inhibitors 5a–g
|
Darunavir (1) exhibited KI = 16 pM, antiviral IC50 = 3 nM;
nt = not tested
Table 2.
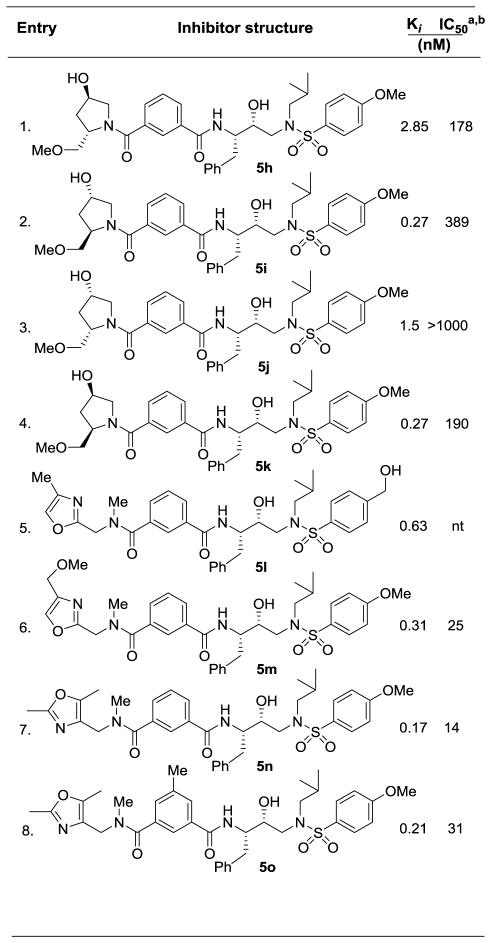
Enzyme inhibitory and antiviral activity of inhibitors 5h–o
|
Darunavir (1) exhibited KI = 16 pM, antiviral IC50 = 3 nM;
nt = not tested
We then examined various proline-derived isophthalamide derivatives. These results are shown in Table 2. Both donor and acceptor functionalities were introduced to form hydrogen bonds in the S2–S3 subsites. We explored all four possible diastereomers of hydroxyproline ligand. As shown, the 2-(R)-methoxymethyl configuration is preferred (entries 2 and 4) over 2-(S)-diastereomers in inhibitors 5h and 5j (entries 1 and 3). The effect of the C-4 hydroxy stereochemistry was not very pronounced. Inhibitors 5h, 5i and 5k showed moderate antiviral activity. We then examined various oxazolylmethyl-derived P3-ligands in inhibitors 5l–o. Inhibitors 5l and 5m (entries 5 and 6) exhibited comparable HIV-1 protease inhibitory activity, indicating minor contribution of the methoxy functionality in inhibitor 5m. Inhibitor 5m displayed an antiviral IC50 value of 25 nM. Inhibitors 5n and 5o containing dimethyloxazole derivatives showed enhanced potency over methyloxazole derivative in inhibitor 5l. However, incorporation of 3-methyl on the isophthalic phenyl ring (entry 8) resulted in reduction of enzyme affinity and antiviral activity. Inhibitor 5n displayed very potent antiviral activity (IC50 = 14 nM). To obtain molecular insight into the inhibitor-HIV-1 protease interactions, we have determined the X-ray crystal structure of the related inhibitor 5o and HIV-1 protease complex.
The crystal structure of 5o-bound HIV-1 was refined using X-ray data at 1.11 Å resolution. The overall protease structure showed similarity to that of the complex with darunavir27 with an RMSD of 0.21 Å for Cα atoms. The largest differences between corresponding Cα atoms are 0.9 Å for residue 78′ to 79′, 0.6 Å for flap residues 48′ and 51′ and 0.9 Å for the N-terminal residue 3 and 4, which may result from crystallographic packing.28 Most of the interactions of the inhibitor with protease residues in the active site cavity are comparable to those of the protease-darunavir complex.27 The major differences are due to the dimethyl-oxazolylmethyl isophthalamide group in place of the bis-tetrahydrofuranyl (bis-THF) urethane in darunavir. The long P2–P3 ligand is positioned between the side chains of Asp29 and Arg8′ on one side and the carbonyl oxygen of Gly48 on the opposite site, while forming a water-mediated hydrogen bond with the carbonyl oxygen of Pro81 (not shown in Figure 2). A new hydrogen bond is formed between the amide nitrogen of Asp29 and one of the carbonyl oxygen of isophthalamide ligand. Also, C-H…O interactions are evident between the carbonyl oxygen of Gly48 and amide methyl in the P2 ligand, producing a shift in the flap regions 47′–56′. The five-membered oxazole ring at the distal end of the P2 group rotates into two alternative conformations. The major conformation of the oxazole ring forms hydrophobic interactions with the side chains of Leu23 and Val82. The minor conformation of the ring forms C-H…π stacking interactions with the guanidinium moiety of Arg8. These extended interactions of the long P2 group are expected to stabilize the inhibitor in the binding site. The methylbenzene moiety of the P2 ligand forms C-H…π interactions with Ala28 on one side of the binding site and van der Waals contacts with Ile50, Val32′, Ile47′ and Ile84′ on the other side.
Figure 2.
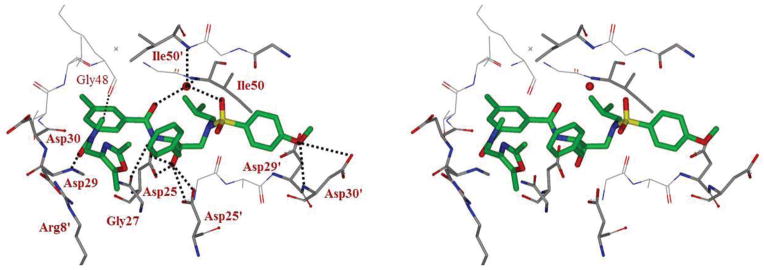
Stereoview of the X-ray structure of inhibitor 5o (green)-bound HIV-1 protease (PDB code: 4ZIP). All strong active site hydrogen bonding interactions of inhibitor 5o with HIV-1 protease are shown as dotted lines.
In summary, we have designed and synthesized a series of novel HIV-1 protease inhibitors and evaluated their enzyme inhibitory and antiviral activity. We designed these inhibitors based upon the X-ray structures of inhibitor-HIV-1 protease complexes. The inhibitors incorporated isophthalamide-derived P2–P3 ligands to interact with the HIV-1 protease active site in the S2 and S3 subsites. We have investigated a variety of acyclic, cyclic, and heterocyclic amide derivatives. In particular, we examined substituted prolines and oxazoles which contain hydrogen bond donor and acceptor groups for specific interactions in the S3 subsite. A number of inhibitors exhibited excellent enzyme inhibitory and antiviral activity. Inhibitor 5n containing isophthalamide dimethyloxazolylmethyl amide displayed an enzyme Ki of 0.14 nM and antiviral IC50 value of 14 nM in MT cells. To obtain molecular insight into the inhibitor’s binding properties in the HIV-1 protease active site, we determined the X-ray structure of 5o-bound HIV-1 at 1.11 Å resolution. Our structural analysis revealed that one of the carboxamide NH formed a strong hydrogen bond with Gly27 carbonyl group of HIV-1 protease. The other carbonyl oxygen was involved in hydrogen bonding with Asp 29 NH in the S2 subsite. Further design and improvement of inhibitor properties utilizing this molecular insight are in progress.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (Grant GM53386, AKG and Grant GM62920, ITW). X-ray data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22BM at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38. This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health, and in part by a Grant-in-Aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan, and the Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Reemerging Infectious Diseases (Renkei Jigyo) of Monbu-Kagakusho. The authors would like to thank the Purdue University Center for Cancer Research, which supports the shared NMR and mass spectrometry facilities.
Footnotes
Supplementary data associated with this article can be found in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Conway B. Future Virol. 2009;4:39–41. [Google Scholar]
- 2.Hue S, Gifford RJ, Dunn D, Fernhill E, Pillay D. J Virol. 2009;83:2645–2654. doi: 10.1128/JVI.01556-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diffenbach CW, Fauci AS. Ann Intern Med. 2011;154:766–771. doi: 10.7326/0003-4819-154-11-201106070-00345. [DOI] [PubMed] [Google Scholar]
- 4.Cohen MS, Chen YQ, McCauley M. N Engl J Med. 2011;365:493–505. doi: 10.1056/NEJMoa1105243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyd MA. Curr Opin HIV AIDS. 2009;4:194–199. doi: 10.1097/COH.0b013e328329fc8d. [DOI] [PubMed] [Google Scholar]
- 6.Thompson MA, Aberg JA, Hoy JF, Telenti A, Benson C, et al. JAMA. 2012;308:387–402l. doi: 10.1001/jama.2012.7961. [DOI] [PubMed] [Google Scholar]
- 7.Patel K, Hernán MA, Williams PL, Seeger JD, McIntosh K, Van Dyke RB, Seage GR., III Clin Infect Dis. 2008;46:507–515. doi: 10.1086/526524. [DOI] [PubMed] [Google Scholar]
- 8.Gupta R, Hill A, Sawyer AW, Pillay D. Clin Infect Dis. 2008;47:712–722. doi: 10.1086/590943. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh AK, Anderson DD, Weber IT, Mitsuya H. Angew Chem Int Ed. 2012;51:1778–1802. doi: 10.1002/anie.201102762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh AK, Xu CX, Rao KV, Baldridge A, Agniswamy J, Wang YF, Weber IT, Aoki M, Miguel SGP, Amano M, Mitsuya H. Chem Med Chem. 2010;5:1850–1854. doi: 10.1002/cmdc.201000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghosh AK, Dawson ZL, Mitsuya H. Bioorg Med Chem. 2007;15:756–7580. doi: 10.1016/j.bmc.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh AK, Sridhar PR, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H. Chem Med Chem. 2006;1:939–950. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh AK, Chapsal B, Mitsuya H. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2010. pp. 205–243. [Google Scholar]
- 14.Ghosh AK. J Med Chem. 2009;52:2163–2176. doi: 10.1021/jm900064c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh AK, Schiltz GE, Rusere LN, Osswald HL, Walters DE, Amano M, Mitsuya H. Org Biomol Chem. 2014;12:6842–6854. doi: 10.1039/c4ob00738g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh AK, Swanson LM, Cho H, Hussain KA, Leschenko S, Kay S, Walters DE, Mitsuya H. J Med Chem. 2005;48:3576. doi: 10.1021/jm050019i. [DOI] [PubMed] [Google Scholar]
- 17.Kozisek M, Bray J, Rezacova P, Saskova K, Brynda J, Pokorna J, Mammano F, Rulisek L, Konvalinka J. J Mol Biol. 2007;374:1005–1016. doi: 10.1016/j.jmb.2007.09.083. [DOI] [PubMed] [Google Scholar]
- 18.Ghosh AK, Kumaragurubaran N, Hong L, Kulkarni S, Xu X, Miller HB, Reddy DS, Weerasena V, Turner R, Chang W, Koelsch G, Tang J. Bioorg Med Chem Lett. 2008;18:1031–1036. doi: 10.1016/j.bmcl.2007.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh AK, Kumaragurubaran N, Hong L, Kulkarni SS, Xu X, Chang W, Weerasena V, Turner R, Koelsch G, Bilcer G, Tang J. J Med Chem. 2007;50:2399–2407. doi: 10.1021/jm061338s. [DOI] [PubMed] [Google Scholar]
- 20.Horn RK, Gailunas M, Fang LY, Tung JS, Walker DD, Thorsett ED, Jewette NE, Moon JB, Varghese J. J Med Chem. 2004;47:158–164. doi: 10.1021/jm0304008. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh AK, Chapsal BD, Baldridge A, Steffey MP, Walters DE, Koh Y, Amano M, Mitsuya H. J Med Chem. 2011;54:622–634. doi: 10.1021/jm1012787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind JE, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. J Med Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 23.Baker GL, Fritschel SJ, Stille JR, Stille JK. J Org Chem. 1981;46:2954–2960. [Google Scholar]
- 24.Narasimhan S, Prasad KG, Madhavan S. Syn Commun. 1995;25:1689–1697. [Google Scholar]
- 25.Toth MV, Marshall GR. Protease Int J Pept Protein Res. 1990;36:544–550. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 26.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Antimicrob Agent Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. J Mol Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 28.The optimized HIV-1 protease was expressed and purified as described.29 The protease-inhibitor complex was crystallized by the hanging drop vapor diffusion method with well solutions of 0.95 M NaCl, 0.1 M Sodium Acetate buffer (pH 5.0). X-ray diffraction data were collected on a single crystal cooling to 90 K at SER-CAT (22-ID beamline), Advanced Photon Source, Argonne National Lab (Chicago, USA) with X-ray wavelength 0.8 Å, and processed by HKL-200030 with Rmerge of 6.7%. Using one of previous isomorphous structures as the initial model,31 the crystal structure was solved by Molrep in CCP4i Suite and refined by SHELX-97 to 1.11 Å resolution.32,33 PRODRG-2 was used to construct the inhibitor and the restraints for refinement.34 COOT was used for manual modification of the structure.35 Alternative conformations were modeled, anisotropic atomic displacement parameters (B factors) were applied for all atoms including solvent molecules. The final refined solvent structure comprised three Na+ ions, three Cl- ions, one glycerol and 256 water molecules. The coordinates and structure factors for the structure of HIV-1 protease with inhibitor 5o have been deposited in Protein Data Bank with code: 4ZIP.
- 29.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Proteins. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Methods in Enzymology, 276: Macromolecular Crystallography. 1997;(Part A):307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Ghosh AK, Gemma S, Baldridge A, Wang YF, Kovalevsky AY, Koh Y, Weber IT, Mitsuya H. J Med Chem. 2008;51:6021–6033. doi: 10.1021/jm8004543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vagin A, Teplyakov J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- 33.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNichols SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KA. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sheldrick GM. Acta Crystallogr, Sect A: Found Crystallogr. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 35.Schuettelkopf AW, van Aalten DMF. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.