

Abstract
The human gastrointestinal (GI) tract is a complex, dynamic ecosystem that consists of a carefully tuned balance of human host and microbiota membership. The microbiome is not merely a collection of opportunistic parasites, but rather provides important functions to the host that are absolutely critical to many aspects of health, including nutrient transformation and absorption, drug metabolism, pathogen defense, and immune system development. Microbial metaproteomics provides the ability to characterize the human gut microbiota functions and metabolic activities at a remarkably deep level, revealing information about microbiome development and stability as well as their interactions with their human host. Generally, microbial and human proteins can be extracted and then measured by high performance mass spectrometry (MS)-based proteomics technology. Here we review the field of human gut microbiome metaproteomics, with a focus on the experimental and informatics considerations involved in characterizing systems ranging from low-complexity model gut microbiota in gnotobiotic mice, to the emerging gut microbiome in the GI tract of newborn human infants, and finally to an established gut microbiota in human adults.
Keywords: metaproteomics, human gut microbiome, shotgun proteomics
1. Metaproteomics among the various -omics technologies
The research field of “systems biology” is centered on four main experimental –omics pillars: genomics (DNA), transcriptomics (mRNA), proteomics (proteins) and metabolomics (small molecules/metabolites). Collectively, they comprise the central foundation of the current molecular biology paradigm: genes (DNA) for genetic information storage, transcription (mRNA) for gene expression, proteins for structural and metabolic/enzymatic activities, and metabolites for the substrates/inhibitors/products of metabolism. Recent advances in -omics technologies have facilitated their application to microbial consortia/communities, which have been designated as metagenomics [1], metatranscriptomics [2], metaproteomics [3], and meta-metabolomics [4, 5]. Even a brief inspection of scientific literature over the past 10 years will clearly reveal how these -omics technologies have revolutionized microbial ecology. Presently, researches can uncover in situ metabolic activities of microbial communities in their native environment via a non-targeted, in-depth, high-throughput manner.
Significant research advancements over the past decade have made DNA extraction and sequencing largely routine for many types of environmental samples, including microbial members living in association with the human body. However, a major challenge for metagenomics still exists: the inability to reconstruct long, continuous genomic sequences leading to reasonably complete microbial genomes from the relatively short sequencing reads generated by current sequencing technologies. For example, Illumina sequencing, the most commonly used technology at present, has tremendous throughput and affordable cost, but provides reads only a few hundred base pairs long. As a result, the assembly of microbial genomes from a consortium sample can be confounded by repeat regions within a genome as well as homologous regions between related genomes. To circumvent these shortfalls, the genome assembly process requires iterative assembly, organism binning, and extensive curation to reconstruct relatively continuous genomic sequences [6]. The resulting genomic sequences reveal the taxonomic composition and genetic functional profiles of microbial communities [7]. Although newer sequencing approaches are beginning to appear for producing much longer reads, these technologies have not been fully evaluated yet for complex microbial communities and thus have had very limited impact on metagenomics to date. In total, metagenomics has become a useful tool for microbial community characterizations, and has found significant application as reference information for mapping transcriptomic reads and for searching metaproteome data.
In the context of functional genomics, metatranscriptomics and metaproteomics can be used to examine the level and range of gene expression for microbial communities. In particular, sequencing total mRNA (termed RNAseq) of a microbial community [8] has become very popular, in that one can use the same sequencing technologies for both DNA and RNA. Transcriptomic reads are typically mapped onto assembled genomic sequences and then used for quantification purposes and/or refining genome-derived gene models [2] (for example, identification of operons and transcription start sites). Metaproteomics aims to characterize the complete suite of gene translation products, and provides additional information about post-translational modifications and localization information over transcriptome measurements. In general, the relative abundances of proteins can be determined by label-free, metabolic labeling and isobaric chemical labeling approaches [9, 10]. Unlike DNA and mRNA, there is no “polymerase chain reaction (PCR)-analog” for proteins and therefore it is more challenging to achieve similar biological dynamic ranges typically observed in mRNA sequencing approaches, for example.
Although transcriptomics, proteomics, and metabolomics all generally measure the products of gene expression, one should not necessarily expect exact quantitative correlation among them. Measuring the level of a transcript reflects the production rate of its protein product, but does not accurately predict the concentration pool or stability of the protein product. In fact, the correlation of mRNA abundances with their corresponding protein abundances, while reasonable for some core metabolic processes in some microbial systems, in general is poor or non-existent in most biological systems examined to date [11, 12], suggesting proteomics data is likely more indicative of biological phenotype than mRNA. Although proteomics data provides information about protein production and degradation, it cannot accurately predict a protein’s activity or functional state. To achieve a more complete understanding of metabolic activities, it is usually desirable to integrate -omics data. It is important to realize that while DNA and proteins are relatively stable, transcripts and metabolites often have very short half-lives; thus, there are dramatic temporal information differences in these omics measurements. In total, each –omic technology provides a unique perspective; by integrating these large-scale datasets, researchers can examine cellular metabolism at an unprecedented level. For complex samples such as gut microbiomes, this integrated omics information has potential to provide a detailed molecular view at a resolution and range not previously possible.
The focus of this article is not to provide an extensive discussion or review of metaproteomics in general, as these have been presented elsewhere [13–25], but rather to narrow the consideration to only the human gut microbiomes, with particular attention to key aspects of the experimental and computational approaches used to metaproteome measurements. This article also details the complete list of human gut metaproteome studies published to date, as well as an examination of emerging needs/techniques that will help propel this new area forward.
2. Experimental considerations for metaproteome measurements of human gut microbiota
Metaproteome measurements of gut microbiota are typically conducted with fecal samples, due in large part to the significant amount of microbial biomass in fecal material, and the ease of collecting temporal samples that reflect intestinal conditions under either healthy or disease-related conditions. The most common experimental challenges for this sample type include highly abundant host cells and proteins, endogenous compounds that can interfere with protein measurements, and limited sample sizes (e.g., human infants).
Depending on the focus of the study, protein extraction in fecal samples can be accomplished by either a direct or indirect enrichment protocol. Although feces are a complicated environmental matrix consisting of bacteria, host cells, food particles, and fibrous material, it is possible to extract proteins via a direct cellular lysis of raw fecal material (typically a few grams of material), followed by protein precipitation and cleanup procedures [26]. A unique advantage of a direct extraction is the ability to simultaneously extract and thus monitor both host and microbial proteins, facilitating the characterization of bacterial signatures as well as their interplay/communication with the host. However, the depth of microbial proteome measurement can be limited by the presence of highly abundant host proteins, especially in infant gut samples where microbial colonization is significantly reduced. To circumvent this challenge, indirect enrichment methods, in which bacterial cells are separated/enriched by differential centrifugation [27–30] (low speed centrifugation to remove large fecal debris followed by high speed centrifugation to pellet bacterial cells) or high-speed centrifugation on a Nycodenz density gradient [31–33], facilitate deeper bacterial proteome measurements. A recent study with double filtering strategy, which removes large fibrous material and human cells in the first filter and then collects microbial cells in the second filter, has also shown to effectively enrich microbial populations in the infant fecal samples with dominant human proteins [34]. Overall, enrichment steps successfully enlarge the dynamic range of microbial protein identifications, though at the expense of increased sample losses and possible sample bias.
In contrast to fecal samples, other studies have used an endoscopic saline-lavage technique to study the mucosal luminal interface (MLI) [35, 36]. Whereas fecal samples represent a mixed population of microbiota collected from all intestinal regions, mucosal lavage sampling profiles the microbiota at specific biogeographic regions. These samples have been shown to yield robust recovery of surface microbiota and often do not require any additional preprocessing besides centrifugation to separate the cell pellet from supernatant. One potential complication of this approach is that the collection may yield low microbial biomass, so sample handling is somewhat more difficult and constrained.
With collected cell pellets, different methods can be used to lyse cells, including chemical (i.e., detergents, acids, alkalis or organic solvents) lysis, mechanical (i.e., homogenization, bead-beating, sonication) disruption, or a combined approach of both, as has been reported for proteome extraction methods from other complex media [37, 38]. While these approaches are moderately comparable in efficacy, each one has distinct advantages and disadvantages that need to be matched to demands of the instrumentation measurement technique employed for protein/peptide identifications.
Proteolytic digestion of proteins extracted from biological samples generally results in complex peptide mixtures, which must then be fractionated to simplify sample complexity prior to a mass spectrometric measurement [39], as illustrated in Figure 1. Early metaproteomic studies on human gut microbiota used 2-dimensional gel electrophoresis (2DGE) for protein separation [32]. Although 2DGE can effectively differentiate protein isoforms and modification states, it has limited dynamic range, bias against membrane protein, low-throughput, and thus has gradually been supplanted with liquid chromatography (LC), at least for peptide-based separation.
Figure 1. Human gut metaproteomics workflow.

Collected fecal samples can be processed by direct or indirect protein extraction methods. For the direct method, the entire fecal material is prepared via chemical/mechanical cell lysis for protein extraction, while indirect method first enriches microbial cells via differential centrifugation or density gradient prior to cellular lysis. The proteolytic peptide mixtures are analyzed via two-dimensional (2D)–LC-MS/MS or by 2D PAGE (polyacrylamide gel electrophoresis) approaches.
The first large-scale study of the human distal gut microbiota involved LC-MS-based metaproteomic characterization [27]. In this study, multi-dimensional liquid chromatography separations that coupled strong cation exchange (SCX) with reverse phase (RP) were used to separate a complex peptide mixture in an automated, high-throughput manner [40]. Coupling these two orthogonal separation techniques dramatically improved resolution, dynamic range, and throughput, which enabled for the first time the identification of thousands of proteins from human fecal samples. This was enabled in large part by the tremendous technical advancements in high performance mass spectrometry over the past decade, in terms of increased sensitivity, dynamic range, resolution, mass accuracies, and speed, all of which have significantly impacted the quality and depth of metaproteome measurements.
While the aforementioned 2D-LC-MS/MS experimental design has been used in several metaproteomics measurements of human gut microbiota, other studies have used 1D gel electrophoresis as the first dimension for differentiating intact proteins prior to mass spectrometric measurements. For example, 1D gel electrophoresis was used to separate proteins extracted from feces of lean and obese adolescents, followed by cutting the gel into seven bands and subsequent in-gel digestion and RP-MS measurements [29]. This approach has seen somewhat wide-spread application, with the main variation being the number of discrete bands excised for examination [26, 31]. Given the obvious proteome complexity in human gut microbiota, additional dimensions or newer separation methods will likely improve proteome measurement depth and should be evaluated.
3. Computational considerations for extracting information from metaproteome measurements of human gut microbiota
The analysis of fragment ion spectra to decode peptide sequences has been significantly facilitated by the development of various database searching algorithms [41–48]. In general, these algorithms are employed to match the collected experimental fragment ion spectra against theoretical fragment ion spectra that have been predicted for peptide sequences from the genome information, as depicted in Figure 2.
Figure 2. Computational metaproteomics workflow.

The vast array of experimental mass spectra is matched against a predicted protein sequence database with an appropriate search algorithm. This process begins by first identifying a list of candidate peptides which appear to match to the experimental spectra. Then each potential match is scored based on the level of similarity between the experimental and predicted fragmentation spectra. The algorithm selects the candidate with the highest score as the identified peptide. The identified peptides are then filtered to control the false discovery rate (FDR). Those peptides that pass the scoring threshold are computationally linked to appropriate proteins using an inference approach. Due to sequence redundancies in the predicted protein sequence database, peptides often cannot be uniquely linked to specific proteins, so they are clustered into protein groups based on either parsimony or sequence homology rules.
The most commonly employed database searching algorithms are SEQUEST [41], Mascot [42], MyriMatch [48], OMSSA [44], and X!Tandem [43]. Understandably, these algorithms were designed for use with single organism proteome datasets. Since hundreds of thousands of fragment ion spectra can easily be acquired per day from a typical MS metaproteome measurement, processing such huge datasets against massive metagenome databases becomes computationally expensive and makes controlling false discovery rates difficult. Although most of these have been scaled for parallel computing on clusters, the performance of these algorithms for metaproteome research is highly variable. To address this concern, one newer search algorithm, Sipros, originally designed for proteomic stable isotopic probing [49] and searching for amino acid mutations [50], has been customized and optimized for metaproteome interrogations [51]. A unique feature of Sipros 3.0 is its scalability for searching huge metaproteomic databases using a large number of CPU cores. Hybrid Message Passing Interface/OpenMP parallelism in the Sipros 3.0 architecture allows database searching to be scalable from desktops to clusters or even supercomputers. Thus, it is possible to use Sipros on a computer cluster to search many MS raw files in parallel, with each one having over 35,000 MS/MS spectra, against a database with millions of protein sequences.
3.1 Construction of protein sequence databases
Metaproteomic databases range in size, and can contain hundreds of thousands to millions of predicted protein sequences from multiple organisms, and therefore appropriate database construction strategy plays a pivotal role in balancing false positive and false negative identification rates [13]. There are essentially three different metagenome constructions that are employed for metaproteome identifications: a “pseudo-metagenome” that consists of selected complete genomes from already sequenced microbial isolates (often guided by 16S-rRNA information of the community), a related but unmatched metagenome (where a similar but not sample-specific metagenome is used as a proxy for protein identifications), and a sample-specific metagenome (where the exact same sample is analyzed by both genomics and proteomics). There are some obvious advantages of using pseudo-metagenomes: First, since many proteomic studies are often conducted on samples whose microbiota have not been deeply sequenced, a reference database constructed by concatenating a number of related sequenced isolate genomes offers an easy glimpse into functional signatures of the microbial membership. This approach is greatly aided by the National Institutes of Health large-scale Human Microbiome Program, which has generated genome sequences for thousands of microbial isolates from various human body sites. Secondly, the quality of isolate genomes is usually higher than metagenomes because the genomes are more completely and accurately assembled. An example of a pseudo-metagenome approach focused on an iterative workflow for database searching, in which spectra were first searched against a synthetic metagenome comprised of over 200 intestinal species [31]. Next a new database was created by blasting the hits from the first search against MetaHIT repository for homologous sequences. This new database was then used for a second search and permitted species-specific protein identifications. Clearly, the major disadvantage of using pseudo-metagenomes is that they do not accurately reflect the actual genome repertoire, since they lack distinct sequence information inherent to a particular microbial population. As a result, the identified metaproteome will be biased toward those organisms included in the database, leading to a skewed representation of the community being investigated.
In contrast to the pseudo-metagenome, the accuracy of protein inference increases when a closely related metagenome is available. Even without being an exact match to a particular sample, this approach improves the accuracy of protein identification and thus has become a common design for gut microbiota studies. For example, an unmatched metagenome is often used when a gut microbiome is being characterized across different humans [27]. In this scenario, the genome is much more reflective of the sample and thus a wider range of microbial membership can be evaluated. The unmatched metagenome can be also augmented with isolate genomes, which can generate even more protein identifications [27].
Of course, the most accurate means to characterize a microbial community involves employment of high quality matched metagenomes [28, 52]. In the context of the fecal proteome of two healthy human individuals, a study compared several assembly and gene finding strategies to increase microbial peptide spectral matching [52]. Overall, searching a matched metagenome facilitated a significant increase in the total number of assigned spectra, peptide identifications as well as protein identifications, as compared to the search with a concatenated database. However, as mentioned previously, there are some challenges in this approach, particularly the depth and coverage of the sequencing, as well as the accuracy of assembly and annotation. This may explain why the iterative search workflow with synthetic metagenomes showed higher spectral identifications when compared to the search with a matched metagenome.
Overall, the degree of completeness, accuracy, and size of the metagenome will govern the ability to properly assess the quality of a match between observed and predicted peptide mass spectra. Although various approaches are available, an efficient and workable assessment of statistical confidence can be achieved by using a chimeric database search that includes a nonsense reverse entry for every protein. This target-decoy approach consequently allows the determination of a false discover rate (FDR), which is the expected fraction of false positive assignments [53]. Because protein identifications are less definitive in metaproteome measurements, errors are best evaluated at the peptide-spectrum level. Moreover, depending on how protein inference is performed, these errors may propagate to the protein identification level in a non-trivial manner; an issue that is generally not fully appreciated in the literature. In the simplest sense, the FDR estimate assumes that false positive PSMs are equally likely to map to either the target or decoy database. When dealing with metaproteomes, this assumption can be more problematic. The core function of the FDR estimation is to evaluate the ratio of the number of PSMs matching to either target or decoy entries. As database quality diminishes, the level of discrimination between a true PSM and false PSM becomes increasingly blurry, requiring database search algorithms to dynamically increase PSM scoring thresholds; this results in an increase in the number of false negatives. Therefore, careful consideration of database size and completeness is essential. To illustrate this, we compared the false positive and true positive PSM distributions for various database qualities and sample complexities (Figures 3–4). For a well-curated, high-quality, and sample-specific community reference databases, it is possible to accurately identify ~70% of acquired fragment ion spectra (0.3% FDR) [54]. However, a far lower number of acquired spectra can be identified in metaproteomics of microbial community with low-quality metagenome, in spite of the relatively high number of high quality, unassigned spectra. As shown in both figures, better database predictions and lower complexity biological systems facilitate better discrimination between true and false hits, again stressing the importance of quality and curation of metagenome databases with respect to identification sensitivity in metaproteomics.
Figure 3. Impact of database quality on peptide identifications.
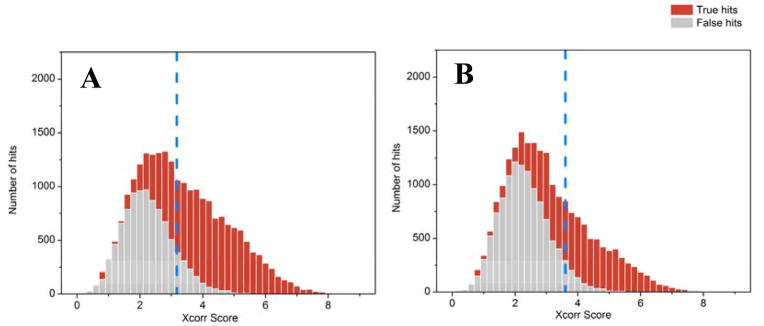
Peptide spectrum matches can be ranked by MyriMatch Xcorr scores to reveal the distribution of true positive (red) vs. false positive (gray) identifications in human adult gut microbiome datasets searched with either a matched metagenome (A) or a pseudo-metagenome assembled from selected microbial isolates (B). An appropriate Xcorr score threshold (indicated by blue dashed line) is chosen to achieve a 1% PSM (peptide spectral match) FDR (false discovery rate; defined by the ratio between false PSMs and total PSMs above the score threshold). The figures reveal that the matched metagenome better differentiates true vs. false distributions, as evidenced by the higher percentage of “red identifications” to the right (i.e. higher Xcorrs) of the dashed line. Even though the pseudo-metagenome likely contains better quality, assembled microbial genomes, the matched metagenome is more closely linked to the actual environmental sample. (Raw data and database details given in reference #70)
Figure 4. Impact of sample complexity on peptide identifications.

Peptide spectrum matches can be ranked by MyriMatch Xcorr scores to reveal the distribution of true positive (red) vs. false positive (gray) identifications for samples of a synthetic mixture of six microbial isolates (all sequenced genomes) (A), a human infant gut microbiome [70], (B), a human adult gut microbiome [54 (C), and an environmental soil (unpublished) (D). An appropriate Xcorr score threshold (indicated by blue dashed line) is chosen to achieve a 1% PSM (peptide spectral match) FDR. The level of true hits is greatest for the synthetic mixture, since the genomes are compete and well annotated. As the complexity of the community increases, the ability to separate true and false hits decreases, as indicated by the superior identification rates in the low complexity infant sample (B) relative to the higher complexity adult gut sample (C). For (B-D), relevant metagenomes were employed, although the metagenome of the soil sample was significantly larger (about 1.3 million genes, which was at least 2X larger than the adult gut microbiome metagenome). This metagenome could not be assembled to a satisfactory level and thus was highly fragmented, which resulted in virtually no distinction between true vs. false hits. This attests to the need for not only matched metagenomes, but well assembled and curated versions, for complex samples.
3.2 Unique vs non-unique proteins / clustering
After a list of peptides has been identified in a metaproteome measurement, the next step is to infer proteins based on the presence of their constituent peptides. In general, a protein isoform can be confidently identified if at least one peptide that exclusively belongs to that protein is observed. However, unlike single microbial isolate databases where most peptides can be uniquely mapped to a single protein, a large amount of inter-protein sequence redundancy makes protein inferences non-trivial in metaproteomics. That is, proteins sharing the same set of peptides cannot be differentiated, and are therefore frequently grouped together. There are essentially two approaches to protein grouping. The first approach uses a parsimony rule with Occam’s razor constraints to identify a minimum set of proteins to explain the identified peptides [55]. While this approach has been widely used in proteomics and is able to minimize over-reporting the number of protein identifications, there is no definitive evidence to determine the presence of any particular protein within a group, and proteins in the same group may not necessarily have a similar biological function, which precludes functional analyses. Furthermore, it is difficult to correctly quantify the abundance of each individual protein in the same group because spectral counting or intensity-based metrics of shared peptides are impossible to assign exclusively to any particular protein in the group.
An alternative approach for protein grouping is based on sequence homology [56]. Proteins with certain level of sequence similarity (e.g., 95-%) are clustered together. Due to such high sequence similarity, all proteins clustered within the same group are likely to have similar biological functions. This grouping scheme allows not only functional analysis using the group as a whole, since all peptides now become unique to the group rather than to individual proteins, but also relative quantification of abundance of the group.
3.3 Taxonomy analysis
Although community composition of gut microbiota can be gleaned from the metagenomics information, whether a community member is active or dormant is not evident in genomics data. By identifying which proteins are observable and under what conditions, metaproteomics can reveal which community members are active, and involved in specific biological processes under a particular ecological context, provided that the identified proteins come from contiguous sequences that have been binned to certain taxon during metagenomic construction. Since taxonomic binning of contiguous sequences is a non-trivial task and the accuracy of binning is highly variable, the use of metaproteomic data for community taxonomic analysis is difficult. Thus, it is much easier to uniquely identify specific proteins in a community sample than to ascribe the species origin of that protein.
4. Metaproteomic studies of human gut microbiota
Trillions of microbes, representing thousands of bacterial species, inhabit the human intestinal tract, making this the most complex human microbial ecosystem [57]. Gut microbiota play an essential role in human health and diseases; for example, the dysfunction of microbiota has been linked with obesity and Crohn’s disease [58–60]. However, to date, relatively little is known about the intricate details and balance of the human gut microbiota. So far, relatively few studies have been conducted on the gut microbial metaproteome, in spite of large numbers of metagenomic interrogations. This is due at least in part by several challenges in gut proteome studies: 1) heterogeneity of bacterial species composition among different individuals; 2) wide dynamic range of protein abundances, especially dominant human proteins that mask the low abundance microbial microbiome; 3) lack of matched metagenomes or low quality metagenome assemblies/annotations that impede comprehensive MS/MS spectrum assignment; 4) informatics hurdles, such as differentiation and quantification of proteins from closely related species and characterization of diverse post-transcriptional modification events. In the following section, we will briefly highlight the range of human gut metaproteomics studies published to date, and how the information they provide is helping to shape our understanding of this unique ecosystem, and its effect on health vs. disease.
4.1 Human infant gut metaproteome
While the variability of the human gut microbiota is astounding, it is not unexpected, given the influences from genetic variation and diverse cultural environments. Although the human infant gut is thought to be generally sterile at birth, this theory has been recently challenged by new evidence suggesting the presence of microbes in amniotic fluid, placenta, and the infant’s meconium [61–63]. Following birth, rapid microbial colonization occurs and, for the next few years, the microbial composition continues to undergo dramatic changes until a stable microbiota is established [64]. As such, the early microbial composition of the infant gut is relatively simple and of low complexity, and therefore poses fewer analytical challenges (e.g., sampling depth) than the richer, more diverse microbial communities evident in the adult human gut. However, with increasing time, the microbial composition varies tremendously, even from week to week, and therefore a comprehensive profiling of the infant gut requires a greater number of sampling points to effectively capture this inherent variation across time.
Emerging evidence has suggested that not only does the initial colonization of the gastrointestinal tract play a critical role in the development of a stable, healthy ‘adult’ microbiota, but also that deviations from the native early-life bacterial establishment can impact human health and lifestyle across an entire life span [65]. Therefore, it is of great interests to not only capture the genetic diversity of the infant gut microbiome, but to also identify which genetic and external factors alter the molecular composition and activity of the infant gut microbiome.
Although the human infant gut microbiome is a logical place to begin metaproteome studies, to date there have been very little published in this arena. Despite having limited genome information, Klaassens et al. reported the first attempt to use a metaproteomics approach to functionally characterize microbial protein composition changes over time in a human infant fecal sample [32]. Although the level of protein identification was severely limited in this study, this report revealed the need for enhanced experimental (sample preparation as well as measurement methods) and informatics (in particular, more detailed and accurate metagenomes) methodologies. Our understanding of the infant microbiome has since broadened, in part owing to the tremendous improvements in DNA and protein sequencing technologies, as well as significant advancements in the bioinformatic tools used to assemble, annotate, and analyze the data generated. In a more recent study, Young et al. achieved a more comprehensive metaproteome analysis of the infant gut microbiome, providing a rich dataset that has led to a better understanding of the dynamic changes in the functional signature of the infant microbiome [66]. For example, this study demonstrated that the functional signature of the microbial community increased in complexity within 2–3 weeks, stabilized relatively early, and remained remarkably conserved thereafter. Additionally, several microbial-related human proteins were concomitantly observed. In particular, several innate immunity proteins in the same fecal samples revealed a level of human host – microbiome cross-talk.
4.2 Human adult gut metaproteome
Compared to the infant gut microbiota, the human adult gut has been more widely studied. In the following sections, we detail how metaproteomic approaches have been applied to better our understanding of which dominant and key microbial functions are present in a ‘healthy’ human adult gut, how the molecular signature of the microbial community compares between a healthy and diseased state, the longitudinal changes and shifts in microbiota functionality across the gastrointestinal track and the host-microbial interaction located in the mucosal luminal interface.
4.2.1 Insights into the stable microbiome of a healthy human adult gut
The first metaproteomic study of an adult human gut microbiota was performed on a healthy female monozygotic twin pair from a Swedish twin cohort [27]. By employing nano-2D-LC-MS/MS, thousands of identified proteins facilitated the first glimpse of the functional signature of the human gut microbiome, providing insight into the host-bacterial interaction in the gastrointestinal tract. Expectedly, a substantial proportion of the proteins identified in the samples (30%) were human proteins, including but not limited to the functional categories humane innate immunity, cell-to-cell adhesion, and digestion enzymes. Notably, most of the relatively abundant human proteins were similar among the two individuals, yet some differences were found in less abundant proteins, which can be expected due to the stochastic sampling nature of the approach.
A high-level overview of biological processes occurring in gut microbiota was obtained by cataloging identified proteins by Cluster of Orthologous Groups (COGs) [67]. An uneven distribution of relative abundances of each COG in the identified metaproteome relative to metagenome was revealed [27]: the metaproteome was enriched in proteins involved in translation, energy production, and carbohydrate metabolism, whereas the metagenome was dominated by proteins involved in inorganic ion metabolism, cell wall and membrane biogenesis, cell division, and secondary metabolite biosynthesis. Although there are clearly measurement depth differences between these datasets, these observational differences emphasize the important point that in situ functional activities (as measured by the metaproteome) can be significantly distinct from what is predicted from the metagenome information alone.
4.2.2 Microbial functional divergence of healthy versus disease state
The first comparison of the intestinal microbiota between healthy and diseased adults focused on Crohn’s disease (CD) [28]. In brief, CD is an inflammatory bowel disease with evidence converging to suggest that imbalance in the microbiota plays a central role in chronic inflammation associated with CD. In contrast to the healthy twin pair described above, five other twin pairs were selected here, including one concordant colonic CD (CCD) twin pair, two concordant ileum CD (ICD) twins, and two discordant ICD twins were analyzed. Due to advancements in protein sequencing technology as well as sample preparation, this study was able to achieve a more detailed investigation into the presence of microbial and human proteins, identifying 4,120 microbial protein groups and 1,646 human proteins. With a comprehensive cataloging of proteins and their relative abundances across the individuals, this study highlighted key functional signatures of CD, which were associated with alterations in bacterial metabolism (e.g. deficiency in general processes, depleted enzymes for carbohydrates and mucin degradation, and depletion of butyrate and other short-chain fatty acids), bacterial-host interactions (e.g. higher expression of bacterial outer membrane proteins that participate in inflammatory immune responses), and host corresponding response (e.g. impaired epithelial barrier and high abundance of pancreatic enzymes). Consistent with previous 16S rRNA-based phylogenetic analysis and metabolite analysis of the same cohort, the measured metaproteomes clustered according to individuals’ disease status, rather than host genetics. Additionally, reduced protein abundances for butyrate production and degradation of mucin from beneficial bacteria were in agreement with the decreased abundances of these species revealed from previous 16S based phylogenetic profiling. Overall, this study revealed a catalogue of proteins exhibiting the functional signatures of the disease and therefore provided potential targets for future diagnostic and therapeutic research.
In a more targeted investigation, a recent cross-sectional study conducted on six CD patients and six healthy controls also characterized protein signals associated with CD [33]. On the basis of 2D-DIGE followed by LC-MS/MS measurement, a subset of 13 candidate proteins was selected and confirmed by selected reaction monitoring (SRM). 12 bacterial proteins mainly derived from Bacteroides were strongly linked to CD, as well as one depleted human glycoprotein 2 of zymogen granule membranes (GP2), which may promote bacteria binding to host cell receptors and induce inflammatory responses. In total, this study not only discovered but also confirmed and quantified a list of CD-associated microbial proteins, which can serve as candidate targets for IBD treatment.
In effort to identify how the gut microbiota contributes to obesity, Ferrer et al. performed comparative metagenomics and metaproteomics of human fecal samples from one ‘lean’ and one ‘obese’ adolescent [29]. In brief, the proteins identified by shotgun proteomics revealed a drastic change in the total and functionally active microbial community; Bacteroidetes represented the most functional bacteria (81% of total protein) in the lean gut, whereas the obese gut had relatively equal abundances of Firmicutes and Bacteroidetes proteins. This observation is consistent other studies that have shown that the relative abundance of Bacteroidetes increases as obese individuals lose weight [68]. Overall, this study highlighted the importance of comparative metaproteomics approaches to further our understanding of the functional changes that occur in response to obesity.
4.2.3 Longitudinal changes and shifts in microbiota functionality
To date, only two studies have examined the change of adult gut metaproteomes as a function of time. In the first study, the metaproteomes of three healthy, omnivorous female subjects were characterized twice within a year [26]. As a novel finding, the fecal metaproteome of each individual was relatively stable during one year period, despite distinct inter-individual differences. In addition, approximately 1,000 proteins were observed in all subjects and likely represent core functional categories, which were also highly representative in other intestinal metaproteome studies [27]. These observations suggested a presumable common functional core in healthy individuals, which is mainly involved in carbohydrate transport and degradation as well as a variety of surface proteins reflecting bacterial adaption to the intestinal environment. A later time-series study examined gut microbial communities over multiple time points from an individual before and after antibiotic (AB) treatment [30]. Based on integrated multi-omics data, the study proposed a presumptive model describing temporal responses of intestinal microbiota to AB therapy, from the perspective of microbial composition dynamics and metabolic activity regulation.
4.2.4 The mucosal luminal interface (MLI)
In general, the intestinal mucosal surface is a barrier layer that prevents the invasion of pathogens and mediates most interactions between the host and luminal intestinal microbiota. Thus far, two studies have profiled MLI metaproteomes in mucosal lavage samples. The first study analyzed 205 lavage samples from six colon regions of 38 healthy subjects [35]. The results were compared with mucosal biopsy transcriptome and showed enrichment in extracellular proteins involved in immune response. Also, metaproteomes from 6 colon regions were further compared, revealing biogeographic features of MLI metaproteome with distinct differences between the proximal colon and the distal colon. The second study investigated the bacteria and metaproteome at the MLI of CD, ulcerative colitis, and healthy subjects, and identified five bacterial phylotypes and a large number of proteins associated with the inflammatory bowel disease (IBD) [36]. Moreover, the relationship between bacteria and metaproteome provided a correlation that could be used to sort most subjects by disease type, supporting the potential role of host-microbe interaction in the etiology of IBD. Investigating the metaproteome of the MLI provides an additional dimension to the characterization of host-microbial interaction, because the approach is capable of analyzing the biogeographic-specific metaproteomes at different locations along the gastrointestinal tract.
The two studies outlined above provide evidence that the bacteria and proteins identified in MLI are clearly involved in host-microbe interactions, which are potentially critical for disease biology. In a somewhat distinct but complementary fashion, fecal microbiota undoubtedly represent a mixture of species from various intestinal regions, thereby presenting an average but broader picture of all microbes and their functional activities along the human gut. Altogether, microbiome studies focused on both fecal and mucosal materials can be complementary to more fully characterize the functions of gut microbiota in human physiology.
4.3 Model gut microbiome systems in gnotobiotic animals
Gnotobiotic mice can be custom-designed with a defined microbial membership and therefore provide a tractable in vivo model to study bacterial and host dynamics. In fact, the microbiome can be ‘humanized’ by inoculating the germ-free gnotobiotic mice with a defined collection of human gut members. For example, to study the adaption of dietary Lactococcus lactis to the digestive tract, Roy et al. colonized gnotobiotic mice with a Lactococcus lactis strain and then analyzed the metaproteomes of fecal and cecal samples by 2-DE [69]. Although increased GroEL expression in fecal samples suggested that the bacteria were adapting to dehydrated environment in the colon, nearly identical protein profiles were identified between bacteria from feces and cecum. As compared to proteins from in vitro culture, the in vivo proteome showed activation of pathways involved in carbon source assimilation, pyruvate catabolism and pentose phosphate, reflecting changes in the fermentative metabolism of L. lactis in the digestive environment. A similar study on the proteome of commensal E. coli in a gnotobiotic mouse was later performed with 2D-GE coupled with ESI-MSMS [70]. In this case, E. coli appeared to express proteins/enzymes that facilitate the utilization of a variety of carbohydrates and amino acids present in the intestinal tract.
Gnotobiotic mice have also been employed to better understand colonization and microbial interactions in the host gut. For example, a model two-member human gut microbiome consisting of E. rectale and B. thetaiotaomicron was created in gnotobiotic mice to study how they interact and respond to host diet [71]. The study mainly focused on the transcriptomic changes after colonization, but proteins present in luminal contents were also analyzed by high-resolution mass spectrometry. In general, the proteome datasets were complementary to the transcriptome information, and revealed proteins abundant in both microbes as ribosomal proteins, elongation factors, chaperones, and proteins involved in energy metabolism.
Moving beyond a two-member community, a higher level of microbial complexity was evaluated by colonizing gnotobiotic mice with a model human gut microbiota comprising 12 human gut bacterial species and feeding them with high-fat vs. low-fat diets [72]. Importantly, as the complexity of the metaproteome increased, the assignment of peptides unique to proteins was affected by homologous proteins and closely related species. Furthermore, the correlation between mRNA and protein data was evaluated for Bacteroides cellulosilyticus WH2 genes revealed a moderate correlation (r =0.53) between overall mRNA and protein levels; yet, the correlations of genes in different functional categories were significantly different. For example, genes involved in translation showed no correlation whereas genes predicted in carbohydrate metabolism had a strong correlation between RNA and protein observations. This further emphasizes the significance of proteome measurement because proteins represent actual functional molecules that may have different temporal and stability characteristics compared with their corresponding transcripts.
The development of “humanized” gut microbiomes in gnotobiotic animals provides a unique ecosystem in which microbial membership can be carefully designed (to control complexity), controlled, and manipulated in a systematic fashion that is not possible in human subject studies. Clearly, the eukaryotic host differences are important here as well, but this system is becoming increasingly important for sorting out and simplifying the complex variables present in human systems.
5. Future directions for human gut metaproteome research
5.1 The need for better assembled metagenomes
When dealing with the tremendous biodiversity inherent to gut microbiota, constructing a high-quality assembled metagenome is a major impediment for deep metaproteomics measurements, as peptide/protein identification rely on the fidelity of the predicted protein sequences. Unlike the remarkable depth and assembly quality achieved in single organism genomes, constructions of metagenomes from the human gut microbiome are often substantially incomplete. Intrinsically, the challenges are due in large part due to the simultaneous sequencing and unambiguous reconstruction of complete microbial genomes from complex, fragmented sequencing data. As such, the effectiveness and impact of metaproteomics will be dictated by the progression of sequencing technologies and computational approaches for more accurately assembling the sequence information should alleviate this limitation [73, 74]. We suspect that newer sequencing technologies that offer much longer read lengths (e.g., PacBio RS II; Pacific Biosciences) should provide a more reliable genetic framework for the assembly of metagenomes. Additionally, as assemblers evolve, so too will the quality of the metagenomes [75].
5.2 The need to characterize protein post-translational modifications (PTMs)
In order to adapt to changing environmental conditions, most organisms, including microbes, have developed complex regulatory systems that are able to adjust the identity and concentration of its molecular machinery for survival. Post-translational modification of proteins is a common and energetically attractive way to satisfy the demand for new cellular functions, because adding to or removing a chemical moiety from a protein for activating, suppressing, or changing its function consumes less energy and other cellular resources than de novo protein synthesis. Furthermore, the functional potential of a genome is greatly enhanced by chemical diversification of its proteome, because one protein can be modified with one or multiple PTMs and each PTM isoform of a protein can assume a different biological function. This route to creation of new cellular functions might be especially important for microorganisms because alternative splicing, a frequently used molecular mechanism for generating protein isoform in eukaryotes, rarely occurs in microbes. Thus, PTM of proteins is quite likely to be a molecular mechanism to compensate for the relative paucity of protein-coding genes in a microbial genome.
With the current high-performance mass spectrometry-based proteomics, it is now possible to identify tens of thousands of PTM events from a single study [76]. For example, a recent study demonstrated an approach to combine multiple protease digestions, optimized high-resolution mass spectrometry, and high-performance computing for direct identification and quantification of a broad range of PTMs in microbial systems [54], including hydroxylation, methylation, citrullination, acetylation, phosphorylation, methylthiolation, S-nitrosylation, and nitration. In this particular study, 5,000 diverse PTM events from an E. coli proteome and a large number of modified proteins that carried multiple types of PTMs were identified. This approach then was applied to profile PTMs in a natural microbial community, and provided the first experimental evidence that multi-type, multi-site protein modifications are highly prevalent in free-living microorganisms, and that a large number of proteins involved in various biological processes, such as chemotaxis pathway, CRISPR-Cas (Clustered Regularly Interspaced Short Palindromic Repeats and associated proteins) system, and reductive TCA cycle, were dynamically modified during the community succession. Furthermore, the data showed that closely related, but ecologically differentiated bacteria harbored remarkably divergent PTM patterns between their orthologous proteins. The findings of this study revealed the prevalence of PTMs in microorganisms and demonstrated the role of PTM in microbial adaptation and ecology. Although there are challenges to characterizing multiple PTMs simultaneously in complex gut microbiota, enrichment-based approaches that target a specific type of PTM (such as phosphorylation) should be readily applicable to PTM analysis in this ecosystem. As such, this approach would provide an additional level of protein characterization that might be important in unraveling the metabolic activities and functional control of human gut microbiota.
5.3 The need to assess protein stability/turn-over: Stable Isotope Probing (SIP)
Quantitative proteomics measures protein abundance changes as a function of temporal or other altered conditions [9]. However, protein abundance is the outcome of two counteracting processes: protein synthesis and degradation. By maintaining constant mRNA abundance, an increase in protein abundance could be achieved by accelerating protein synthesis, and similarly, a decrease in protein abundance may be caused by an enhanced protein degradation rate. This not only questions the approach of using mRNA abundance alone as a proxy for estimating protein abundance, but also emphasizes the need to measure the protein synthesis and degradation rates to understand the fundamental principles that control the protein abundance changes in the context of gut microbiota. Stable isotope probing has been used for some time to monitor protein turnover [77]. For this approach, cells are first cultivated in non-labeled media. When the cells are then are transferred to media containing a heavy isotope, such as 15N, newly synthesized proteins will incorporate the heavy isotope, resulting in increased abundance of heavy isotope-labeled proteins. Pre-existing proteins remain in unlabeled (light) form and will be constantly degraded, leading to decreased abundance of light isotope-labeled proteins. By quantifying abundance changes of heavy isotope-labeled proteins and light isotope-labeled proteins, protein synthesis rates and degradation rates can be determined.
Furthermore, by feeding microbial communities with heavy isotope-labeled nutrients, the metabolism of those nutrients results in incorporation of those heavy isotopes into proteomes of different organisms [49, 78, 79]. The identification of organisms that are involved in metabolism of a specific labeled nutrient and quantification of the extent of incorporation has been automated by the development of advanced new algorithms [49]. Linking nutrient flows to specific organisms in microbial consortia answers important ecological questions with respect to the major players during nutrient transformations. While it is impractical to label human gut microbiota, this approach has been used to label whole animals, such as mice [80], by feeding them with labeled diets. Proteomic stable isotope probing should be applicable to track isotope flows between host and its gut microbiota in gnotobiotic systems, providing insight into how microorganisms help nutrient digestion.
5.4 The need for high-throughput measurement campaigns
With the increasing availability of individual human genomes as well as human gut metagenomes, various aspects of genetic variation can be deduced for the human population as a whole. At present, large-scale comparative analyses of these metagenomes are beginning to uncover the complicated landscape of human genetic diversity at a population level [81–83], identifying genetic and structural variants across the genome. Although continuing the characterization of genetic variations across a range of populations will undoubtedly predict variants of functional importance, proteome-wide data will be necessary to provide insight into the functional importance of such genetic variants in a given genetic background. Not only can large-scale comparative metaproteomics analyses refine our understanding of how functional variants contribute to phenotypic diversity, such comparative analysis can also better define how the functional signature of the human gut microbiome is shaped by human genetics, diets, cultures, etc. In order to capture such biological variation, future large-scale comparative metaproteomic investigations would be required to analyze hundreds, if not thousands, of samples. Therefore, to keep pace with the exponentially growing number of individual human and metagenomes, it is critical that mass spectrometry-based metaproteomic approaches experience significant improvements in throughput in the coming years.
6. Concluding comments
Recent research advances in high performance mass spectrometry and computational informatics have enabled microbial metaproteomics to become a significant technology platform for characterizing the human gut microbiome. In particular, the integration of advanced chromatographic separations with high performance MS platforms has afforded an unprecedented depth of peptide measurement level. When combined with sophisticated computational tools for searching large peptide datasets and assembling the resulting information into definitive protein information, this approach now opens the door to measuring many thousands of proteins from individual gut microbiome samples, revealing information about both human host and microbial membership metabolic activities. Although this field is in its infancy, this approach has been used to characterize a variety of systems ranging from low complexity, custom-designed microbiomes (such as gnotobiotic mice systems) to moderate complexity infant gut microbiomes, and more recently to complex adult gut systems. Interesting information is beginning to emerge about how the functional information in the metaproteome is distinct and complementary to the genomic potential information in the metagenomes. Clearly, there is significant interest in advancements of the construction of metagenomes, throughput of metaproteomic approaches, and protein stabilities/turn-over for the metaproteomic approach.
Acknowledgments
Stipend support for W.X. was provided by the University of Tennessee-Knoxville Genome Science and Technology Program. Research support for the technical project was provided by NIH grant 1R01-GM-103600. Oak Ridge National Laboratory is managed by UT-Battelle, LLC, for the U.S. Department of Energy.
Footnotes
8. Conflict of Interest: The authors declare no conflict of interest
References
- 1.Tyson GW, Chapman J, Hugenholtz P, Allen EE, et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- 2.Goltsman DSA, Comolli LR, Thomas BC, Banfield JF. Community transcriptomics reveals unexpected high microbial diversity in acidophilic biofilm communities. The ISME journal. 2014 doi: 10.1038/ismej.2014.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ram RJ, VerBerkmoes NC, Thelen MP, Tyson GW, et al. Community proteomics of a natural microbial biofilm. Science. 2005;308:1915–1920. [PubMed] [Google Scholar]
- 4.Dettmer K, Aronov PA, Hammock BD. Mass spectrometry-based metabolomics. Mass spectrometry reviews. 2007;26:51–78. doi: 10.1002/mas.20108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steffen MM, Dearth SP, Dill BD, Li Z, et al. Nutrients drive transcriptional changes that maintain metabolic homeostasis but alter genome architecture in Microcystis. The ISME journal. 2014 doi: 10.1038/ismej.2014.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wrighton KC, Thomas BC, Sharon I, Miller CS, et al. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science. 2012;337:1661–1665. doi: 10.1126/science.1224041. [DOI] [PubMed] [Google Scholar]
- 7.Chai J, Kora G, Ahn TH, Hyatt D, Pan C. Functional phylogenomics analysis of bacteria and archaea using consistent genome annotation with UniFam. BMC evolutionary biology. 2014;14:207. doi: 10.1186/s12862-014-0207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ottesen EA, Young CR, Gifford SM, Eppley JM, et al. Multispecies diel transcriptional oscillations in open ocean heterotrophic bacterial assemblages. Science. 2014;345:207–212. doi: 10.1126/science.1252476. [DOI] [PubMed] [Google Scholar]
- 9.Mosier AC, Li Z, Thomas BC, Hettich RL, et al. Elevated temperature alters proteomic responses of individual organisms within a biofilm community. ISME J. 2014 doi: 10.1038/ismej.2014.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Z, Adams RM, Chourey K, Hurst GB, et al. Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos. Journal of proteome research. 2012;11:1582–1590. doi: 10.1021/pr200748h. [DOI] [PubMed] [Google Scholar]
- 11.Maier T, Guell M, Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS letters. 2009;583:3966–3973. doi: 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 12.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nature Reviews Genetics. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abraham PE, Giannone RJ, Xiong W, Hettich RL. Metaproteomics: Extracting and Mining Proteome Information to Characterize Metabolic Activities in Microbial Communities. Current Protocols in Bioinformatics. 2014:13.26.11–13.26.14. doi: 10.1002/0471250953.bi1326s46. [DOI] [PubMed] [Google Scholar]
- 14.Hettich RL, Sharma R, Chourey K, Giannone RJ. Microbial metaproteomics: identifying the repertoire of proteins that microorganisms use to compete and cooperate in complex environmental communities. Current opinion in microbiology. 2012;15:373–380. doi: 10.1016/j.mib.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 15.Hettich RL, Pan C, Chourey K, Giannone RJ. Metaproteomics: harnessing the power of high performance mass spectrometry to identify the suite of proteins that control metabolic activities in microbial communities. Analytical chemistry. 2013;85:4203–4214. doi: 10.1021/ac303053e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan C, Banfield JF. Environmental microbiology. Springer; 2014. pp. 231–240. [Google Scholar]
- 17.Kolmeder CA, de Vos WM. Metaproteomics of our microbiome—developing insight in function and activity in man and model systems. Journal of proteomics. 2014;97:3–16. doi: 10.1016/j.jprot.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 18.Keller M, Hettich R. Environmental proteomics: a paradigm shift in characterizing microbial activities at the molecular level. Microbiology and Molecular Biology Reviews. 2009;73:62–70. doi: 10.1128/MMBR.00028-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otto A, Becher D, Schmidt F. Quantitative proteomics in the field of microbiology. Proteomics. 2014;14:547–565. doi: 10.1002/pmic.201300403. [DOI] [PubMed] [Google Scholar]
- 20.Seifert J, Herbst FA, Halkjær Nielsen P, Planes FJ, et al. Bioinformatic progress and applications in metaproteogenomics for bridging the gap between genomic sequences and metabolic functions in microbial communities. Proteomics. 2013;13:2786–2804. doi: 10.1002/pmic.201200566. [DOI] [PubMed] [Google Scholar]
- 21.Becher D, Bernhardt J, Fuchs S, Riedel K. Metaproteomics to unravel major microbial players in leaf litter and soil environments: challenges and perspectives. Proteomics. 2013;13:2895–2909. doi: 10.1002/pmic.201300095. [DOI] [PubMed] [Google Scholar]
- 22.von Bergen M, Jehmlich N, Taubert M, Vogt C, et al. Insights from quantitative metaproteomics and protein-stable isotope probing into microbial ecology. The ISME journal. 2013;7:1877–1885. doi: 10.1038/ismej.2013.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muth T, Benndorf D, Reichl U, Rapp E, Martens L. Searching for a needle in a stack of needles: challenges in metaproteomics data analysis. Molecular BioSystems. 2013;9:578–585. doi: 10.1039/c2mb25415h. [DOI] [PubMed] [Google Scholar]
- 24.Seifert J, Taubert M, Jehmlich N, Schmidt F, et al. Protein-based stable isotope probing (protein-SIP) in functional metaproteomics. Mass spectrometry reviews. 2012;31:683–697. doi: 10.1002/mas.21346. [DOI] [PubMed] [Google Scholar]
- 25.Siggins A, Gunnigle E, Abram F. Exploring mixed microbial community functioning: recent advances in metaproteomics. FEMS microbiology ecology. 2012;80:265–280. doi: 10.1111/j.1574-6941.2011.01284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolmeder CA, de Been M, Nikkila J, Ritamo I, et al. Comparative metaproteomics and diversity analysis of human intestinal microbiota testifies for its temporal stability and expression of core functions. PloS one. 2012;7:e29913. doi: 10.1371/journal.pone.0029913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verberkmoes NC, Russell AL, Shah M, Godzik A, et al. Shotgun metaproteomics of the human distal gut microbiota. ISME J. 2009;3:179–189. doi: 10.1038/ismej.2008.108. [DOI] [PubMed] [Google Scholar]
- 28.Erickson AR, Cantarel BL, Lamendella R, Darzi Y, et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PloS one. 2012;7:e49138. doi: 10.1371/journal.pone.0049138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferrer M, Ruiz A, Lanza F, Haange SB, et al. Microbiota from the distal guts of lean and obese adolescents exhibit partial functional redundancy besides clear differences in community structure. Environmental microbiology. 2013;15:211–226. doi: 10.1111/j.1462-2920.2012.02845.x. [DOI] [PubMed] [Google Scholar]
- 30.Ferrer M, Martins dos Santos VA, Ott SJ, Moya A. Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut microbes. 2014;5:64–70. doi: 10.4161/gmic.27128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rooijers K, Kolmeder C, Juste C, Dore J, et al. An iterative workflow for mining the human intestinal metaproteome. BMC genomics. 2011;12:6. doi: 10.1186/1471-2164-12-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klaassens ES, De Vos WM, Vaughan EE. Metaproteomics approach to study the functionality of the microbiota in the human infant gastrointestinal tract. Applied and environmental microbiology. 2007;73:1388–1392. doi: 10.1128/AEM.01921-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Juste C, Kreil DP, Beauvallet C, Guillot A, et al. Bacterial protein signals are associated with Crohn’s disease. Gut. 2014 doi: 10.1136/gutjnl-2012-303786. gutjnl-2012–303786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiong W, Giannone RJ, Morowitz MJ, Banfield JF, Hettich RL. Development of an Enhanced Metaproteomic Approach for Deepening the Microbiome Characterization of the Human Infant Gut. J Proteome Res. 2014 doi: 10.1021/pr500936p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, LeBlanc J, Truong A, Vuthoori R, et al. A metaproteomic approach to study human-microbial ecosystems at the mucosal luminal interface. PloS one. 2011;6:e26542. doi: 10.1371/journal.pone.0026542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Presley LL, Ye J, Li X, Leblanc J, et al. Host-microbe relationships in inflammatory bowel disease detected by bacterial and metaproteomic analysis of the mucosal-luminal interface. Inflammatory bowel diseases. 2012;18:409–417. doi: 10.1002/ibd.21793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma R, Dill BD, Chourey K, Shah M, et al. Coupling a detergent lysis/cleanup methodology with intact protein fractionation for enhanced proteome characterization. Journal of proteome research. 2012;11:6008–6018. doi: 10.1021/pr300709k. [DOI] [PubMed] [Google Scholar]
- 38.Chourey K, Jansson J, VerBerkmoes N, Shah M, et al. Direct cellular lysis/protein extraction protocol for soil metaproteomics. Journal of proteome research. 2010;9:6615–6622. doi: 10.1021/pr100787q. [DOI] [PubMed] [Google Scholar]
- 39.Motoyama A, Yates JR., III Multidimensional LC separations in shotgun proteomics. Analytical chemistry. 2008;80:7187–7193. doi: 10.1021/ac8013669. [DOI] [PubMed] [Google Scholar]
- 40.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 41.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 42.Cottrell JS, London U. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 43.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 44.Geer LY, Markey SP, Kowalak JA, Wagner L, et al. Open mass spectrometry search algorithm. Journal of proteome research. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 45.Razumovskaya J, Olman V, Xu D, Uberbacher EC, et al. A computational method for assessing peptide-identification reliability in tandem mass spectrometry analysis with SEQUEST. Proteomics. 2004;4:961–969. doi: 10.1002/pmic.200300656. [DOI] [PubMed] [Google Scholar]
- 46.Sadygov RG, Cociorva D, Yates JR., 3rd Large-scale database searching using tandem mass spectra: looking up the answer in the back of the book. Nature methods. 2004;1:195–202. doi: 10.1038/nmeth725. [DOI] [PubMed] [Google Scholar]
- 47.Tabb DL, Narasimhan C, Strader MB, Hettich RL. DBDigger: reorganized proteomic database identification that improves flexibility and speed. Analytical chemistry. 2005;77:2464–2474. doi: 10.1021/ac0487000. [DOI] [PubMed] [Google Scholar]
- 48.Tabb DL, Fernando CG, Chambers MC. MyriMatch: highly accurate tandem mass spectral peptide identification by multivariate hypergeometric analysis. Journal of proteome research. 2007;6:654–661. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pan C, Fischer CR, Hyatt D, Bowen BP, et al. Quantitative tracking of isotope flows in proteomes of microbial communities. Molecular & Cellular Proteomics. 2011;10:M110. 006049. doi: 10.1074/mcp.M110.006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hyatt D, Pan C. Exhaustive database searching for amino acid mutations in proteomes. Bioinformatics. 2012;28:1895–1901. doi: 10.1093/bioinformatics/bts274. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Ahn TH, Li Z, Pan C. Sipros/ProRata: a versatile informatics system for quantitative community proteomics. Bioinformatics. 2013;29:2064–2065. doi: 10.1093/bioinformatics/btt329. [DOI] [PubMed] [Google Scholar]
- 52.Cantarel BL, Erickson AR, VerBerkmoes NC, Erickson BK, et al. Strategies for metagenomic-guided whole-community proteomics of complex microbial environments. PloS one. 2011;6:e27173. doi: 10.1371/journal.pone.0027173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 54.Li Z, Wang Y, Yao Q, Justice NB, et al. Diverse and divergent protein post-translational modifications in two growth stages of a natural microbial community. Nature communications. 2014;5:4405. doi: 10.1038/ncomms5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nesvizhskii AI, Aebersold R. Interpretation of shotgun proteomic data the protein inference problem. Molecular & Cellular Proteomics. 2005;4:1419–1440. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- 56.Abraham P, Adams R, Giannone RJ, Kalluri U, et al. Defining the boundaries and characterizing the landscape of functional genome expression in vascular tissues of Populus using shotgun proteomics. Journal of proteome research. 2011;11:449–460. doi: 10.1021/pr200851y. [DOI] [PubMed] [Google Scholar]
- 57.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148:1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nell S, Suerbaum S, Josenhans C. The impact of the microbiota on the pathogenesis of IBD: lessons from mouse infection models. Nat Rev Microbiol. 2010;8:564–577. doi: 10.1038/nrmicro2403. [DOI] [PubMed] [Google Scholar]
- 60.Berer K, Mues M, Koutrolos M, Rasbi ZA, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479:538–541. doi: 10.1038/nature10554. [DOI] [PubMed] [Google Scholar]
- 61.Wang X, Buhimschi CS, Temoin S, Bhandari V, et al. Comparative microbial analysis of paired amniotic fluid and cord blood from pregnancies complicated by preterm birth and early-onset neonatal sepsis. PloS one. 2013;8:e56131. doi: 10.1371/journal.pone.0056131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aagaard K, Ma J, Antony KM, Ganu R, et al. The placenta harbors a unique microbiome. Science translational medicine. 2014;6:237ra265. doi: 10.1126/scitranslmed.3008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ardissone AN, de la Cruz DM, Davis-Richardson AG, Rechcigl KT, et al. Meconium microbiome analysis identifies bacteria correlated with premature birth. PloS one. 2014;9:e90784. doi: 10.1371/journal.pone.0090784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matamoros S, Gras-Leguen C, Le Vacon F, Potel G, de La Cochetiere MF. Development of intestinal microbiota in infants and its impact on health. Trends Microbiol. 2013;21:167–173. doi: 10.1016/j.tim.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 65.Groer MW, Luciano AA, Dishaw LJ, Ashmeade TL, et al. Development of the preterm infant gut microbiome: a research priority. Microbiome. 2014;2:38. doi: 10.1186/2049-2618-2-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Young JC, Pan C, Adams R, Brooks B, et al. Metaproteomics Reveals Functional Shifts in Microbial and Human Proteins During Infant Gut Colonization. 2015 doi: 10.1002/pmic.201400563. paper under review at Proteomics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic acids research. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 69.Roy K, Meyrand M, Corthier G, Monnet V, Mistou MY. Proteomic investigation of the adaptation of Lactococcus lactis to the mouse digestive tract. Proteomics. 2008;8:1661–1676. doi: 10.1002/pmic.200700698. [DOI] [PubMed] [Google Scholar]
- 70.Alpert C, Scheel J, Engst W, Loh G, Blaut M. Adaptation of protein expression by Escherichia coli in the gastrointestinal tract of gnotobiotic mice. Environmental microbiology. 2009;11:751–761. doi: 10.1111/j.1462-2920.2008.01798.x. [DOI] [PubMed] [Google Scholar]
- 71.Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5859–5864. doi: 10.1073/pnas.0901529106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McNulty NP, Wu M, Erickson AR, Pan C, et al. Effects of diet on resource utilization by a model human gut microbiota containing Bacteroides cellulosilyticus WH2: a symbiont with an extensive glycobiome. PLoS biology. 2013;11:e1001637. doi: 10.1371/journal.pbio.1001637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haider B, Ahn TH, Bushnell B, Chai J, et al. Omega: an overlap-graph de novo assembler for metagenomics. Bioinformatics. 2014;30:2717–2722. doi: 10.1093/bioinformatics/btu395. [DOI] [PubMed] [Google Scholar]
- 74.Ahn T-H, Chai J, Pan C. Sigma: Strain-level inference of genomes from metagenomic analysis for biosurveillance. Bioinformatics. 2014:btu641. doi: 10.1093/bioinformatics/btu641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yok NG, Rosen GL. Combining gene prediction methods to improve metagenomic gene annotation. BMC bioinformatics. 2011;12:20. doi: 10.1186/1471-2105-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Olsen JV, Mann M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Molecular & Cellular Proteomics. 2013;12:3444–3452. doi: 10.1074/mcp.O113.034181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schoenheimer R, Rittenberg D, Foster G, Keston AS, Ratner S. The application of the nitrogen isotope N15 for the study of protein metabolism. Science. 1938;88:599–600. doi: 10.1126/science.88.2295.599. [DOI] [PubMed] [Google Scholar]
- 78.Justice NB, Li Z, Wang Y, Spaudling SE, et al. 15N-and 2H proteomic stable isotope probing links nitrogen flow to archaeal heterotrophic activity. Environmental microbiology. 2014 doi: 10.1111/1462-2920.12488. [DOI] [PubMed] [Google Scholar]
- 79.Fischer CR, Bowen BP, Pan C, Northen TR, Banfield JF. Stable-Isotope Probing Reveals That Hydrogen Isotope Fractionation in Proteins and Lipids in a Microbial Community Are Different and Species-Specific. ACS chemical biology. 2013;8:1755–1763. doi: 10.1021/cb400210q. [DOI] [PubMed] [Google Scholar]
- 80.Krüger M, Moser M, Ussar S, Thievessen I, et al. SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell. 2008;134:353–364. doi: 10.1016/j.cell.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 81.International HapMap C. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 82.Buchanan CC, Torstenson ES, Bush WS, Ritchie MD. A comparison of cataloged variation between International HapMap Consortium and 1000 Genomes Project data. Journal of the American Medical Informatics Association : JAMIA. 2012;19:289–294. doi: 10.1136/amiajnl-2011-000652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gurdasani D, Carstensen T, Tekola-Ayele F, Pagani L, et al. The African Genome Variation Project shapes medical genetics in Africa. Nature. 2015;517:327–332. doi: 10.1038/nature13997. [DOI] [PMC free article] [PubMed] [Google Scholar]