

Abstract
Objective:
To evaluate, by skin biopsy, dermal nerve fibers in 31 patients with 3 common Charcot-Marie-Tooth (CMT) genotypes (CMT1A, late-onset CMT1B, and CMTX1), and rarer forms of CMT caused by mutations in RAB7 (CMT2B), TRPV4 (CMT2C), and GDAP1 (AR-CMT2K) genes.
Methods:
We investigated axonal loss by quantifying Meissner corpuscles and intrapapillary myelinated endings and evaluated morphometric changes in myelinated dermal nerve fibers by measuring fiber caliber, internodal, and nodal gap length.
Results:
The density of both Meissner corpuscles and intrapapillary myelinated endings was reduced in skin samples from patients with CMT1A and all the other CMT genotypes. Nodal gaps were larger in all the CMT genotypes though widening was greater in CMT1A. Perhaps an altered communication between axons and glia may be a common feature for multiple forms of CMT. Internodal lengths were shorter in all the CMT genotypes, and patients with CMT1A had the shortest internodes of all our patients. The uniformly shortened internodes in all the CMT genotypes suggest that mutations in both myelin and axon genes may developmentally impede internode formation. The extent of internodal shortening and nodal gap widening are likely both important in determining nerve conduction velocities in CMT.
Conclusions:
This study extends the information gained from skin biopsies on morphologic abnormalities in various forms of CMT and provides insights into potential pathomechanisms of axonal and demyelinating CMT.
Charcot-Marie-Tooth (CMT) disease is the most frequent neurologic hereditary disorder and shows great clinical and genetic heterogeneity.1,2 The advent of genetic testing has made sural nerve biopsy unnecessary for diagnosing most cases of CMT, particularly because the technique is somewhat invasive. This has led to an inability to evaluate morphologic effects by the various mutations on human nerves. Since animal models do not always faithfully reproduce human pathology, the lack of human nerve tissue has limited investigations into pathogenic mechanisms of CMT. Skin biopsies increasingly offer a minimally invasive approach to overcome this problem. Beginning with studies of glabrous3 and hairy skin,4 these biopsies have already provided pathogenic information in sensory nerves from patients with CMT1A, such as reduced Meissner corpuscles (MC) density, shortened internodal length, and abnormal paranodal-juxtaparanodal architecture.5,6
In the present study, we extend these observations in a larger series of patients. We performed skin biopsies on 31 patients with 3 common CMT genotypes (i.e., PMP22 [CMT1A], MPZ [late-onset CMT1B], and GJB1 [CMTX1] genes) and rarer forms of CMT caused by mutations in RAB7 (CMT2B), TRPV4 (CMT2C), and GDAP1 (AR-CMT2K) genes. We investigated axonal loss by quantifying MC and intrapapillary myelinated endings (IME) and evaluated morphometric changes in myelinated dermal nerve fibers by measuring fiber caliber, internodal, and nodal gap length. Results were compared among CMT genotypes and to those of controls who did not have neuropathy.
METHODS
Patients.
Thirty-one patients with CMT and 45 healthy controls were included in the study after screening to rule out abnormalities of glucose metabolism or endocrine function; vitamin E, B12, or folic acid deficiency; hepatic or renal failure; HIV infection; or connective tissue disorders. All patients were older than 18 years at enrollment. Based on molecular genetic analysis, the following CMT genotypes were included: CMT1A (n = 10), CMTX1 (n = 9), late-onset CMT1B (n = 5), CMT2B (n = 3), CMT2C (n = 3), and AR-CMT2K (n = 1).
Twenty-three patients were recruited at the University Federico II of Naples and 8 patients at the University of Verona. All patients underwent neurologic examination and standard nerve conduction study (NCS), which consisted of at least 2 motor nerves and one sensory nerve for upper and lower limbs. For motor NCS, median or ulnar nerves were investigated at upper limbs, peroneal or tibial nerves at lower limbs. For sensory NCS, median or ulnar nerves were investigated at upper limbs, sural or superficial peroneal nerve at lower limbs. Neurologic disability was assessed using the Charcot-Marie-Tooth Neuropathy Score (CMTNS).7
Standard protocol approvals and patient consents.
All subjects signed a written informed consent before enrolling in the study. The study protocol was approved by the local ethical committee.
Skin biopsy.
All subjects underwent 2-mm punch biopsies from the fingertip (at the vortex of III finger). Samples were fixed in cold Zamboni solution, cryoprotected in 20% sucrose phosphate-buffered saline, and cut in 50-μm-thick sections by means of a sliding microtome. Free-floating sections were incubated overnight with a panel of primary antibodies to mark MC and myelinated nerve fibers. Secondary antibodies conjugated with cyanine 2, 3, and 5 were used to visualize the markers. Antibody sources and dilutions are listed in table e-1 on the Neurology® Web site at Neurology.org.
Quantification of MCs and myelinated nerve fibers.
MC and IME were counted in fingertips as previously described.3 Values of density/mm2 were obtained. Receptors whose morphology appeared completely disrupted with loss of their coiled shape in protein gene product (PGP)–stained sections were considered atrophic and therefore were not included in the count.
Morphometric analysis of myelinated nerve fibers.
3D digital images of all myelinated fibers present in 3 PGP/myelin basic protein (MBP) double-stained sections were acquired using confocal microscopy (Apotome2; Zeiss, Oberkochen, Germany) and a 20× objective for the measurements of caliber, internode, and nodal gap length (figure 1A). Internodal and nodal gap lengths were measured on the stack of z-series images using Neurolucida software (MBF Bioscience; Williston, VT). Nodal gap was measured as the length of the fiber not marked by MBP (figure 1, B.a and B.b) and included nodal and part of paranodal regions.3,4 The measurement of internodal length was performed only when 2 consecutive nodes were identified by tracing the internode on the Z-stack of confocal images (figure 1B.c). Four caliber measurements for each internode (figure 1B.d) were obtained and a mean value was reported.
Figure 1. Confocal images show morphometric analysis methods.

(A, B) Protein gene product (PGP)/myelin basic protein (MBP) double-stained images exemplify the procedures used to measure node (B.b), internode (B.c), and fiber diameter (B.d), calculated as the mean value of 4 random measures along the internode. Measurements were performed on MBP channel, as shown by arrowheads, while PGP staining (arrow in B.a) was necessary to check on axonal continuity. (C) The nodal region on an image double-stained with neurofascin (NF) and MBP: branching originates always from nodes (arrowhead). (D.a, D.b) The same image at higher magnification after splitting the 2 channels. Scale bar: 50 μm (A); 100 μm (C).
The Pan-Neurofascin (NF)/MBP double-stained sections (1 section for each biopsy) (figure 1, C and D.a) were used to verify that all the myelin gaps along the fiber course corresponded to nodes and to qualitatively evaluate the nodal/paranodal region.
Statistical analysis.
We compared morphologic and morphometric parameters between CMT and controls and among the CMT genotypes.
Sural nerve biopsy studies show that patients with PMP22 duplication have demyelinating histopathology, patients with late-onset MPZ and GJB1 mutations have exclusively or predominantly axonal pathologic features, and patients with axonal CMT genotypes (namely CMT2) have typically axonal features.8 Therefore, we classified our patients into 2 main groups, the demyelinating (i.e., CMT1A) and the axonal, which included all the other CMT genotypes (late onset-CMT1B, CMTX1, CMT2B, CMT2C, and AR-CMT2K). We compared findings of CMT1A with those of the axonal group as whole.
Moreover, we also compared data from patients with axonal CMT due to mutations in myelin genes (i.e., MPZ and GJB1) with those from patients with CMT due to mutations in axonal genes (i.e., RAB7, TRPV4, and GDAP1).
Statistical analysis was performed using STATA 12.1 for Windows (StataCorp LP, College Station, TX). The analysis of variance (ANOVA) test with the Bonferroni post hoc test for parametric variables or the Kruskal-Wallis test for nonparametric variables were used. For 2-group comparison, Mann-Whitney U test was performed for nonparametric variables. The Spearman rank-order correlation test was used to investigate the correlations between nonparametric variables. The robust test of equality of variance was used for verifying the homogeneity of data between groups. A p value of <0.05 was considered significant.
RESULTS
Patients.
Demographic, NCV, CMTNS, and skin biopsy features of patients and controls are summarized in table 1.
Table 1.
Clinical and skin biopsy features of patients with CMT and healthy controls
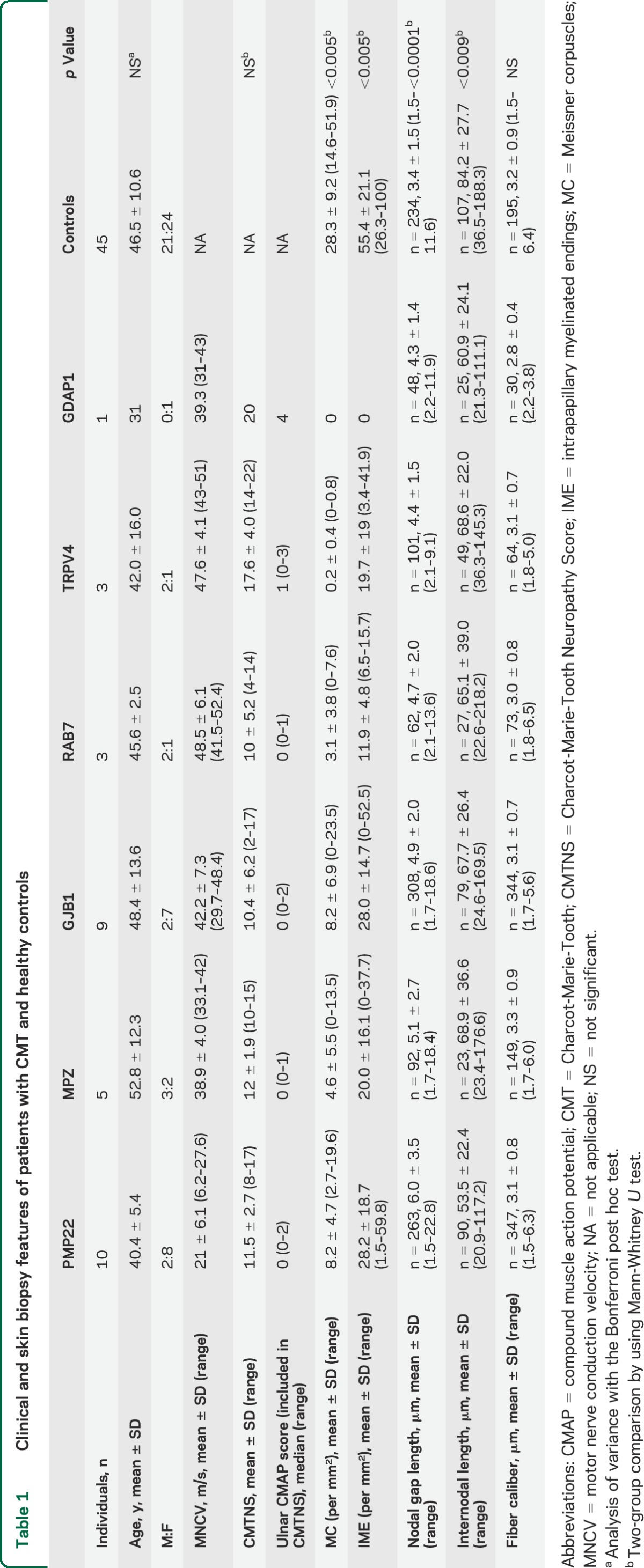
The ANOVA analysis with the Bonferroni post hoc test did not detect statistical difference in mean age among CMT genotypes and between CMT and healthy controls.
Clinically, disease severity, determined by CMTNS, ranged from mild to moderate impairment and no statistical difference was observed among the CMT groups. However, CMTNS did suggest a trend toward greater impairment in CMT2C (CMTNS = 17.6) and AR-CMT2K (CMTNS = 20).
Quantification of MCs and myelinated nerve fibers.
The 2-group comparison (Mann-Whitney U test) showed that a loss (p < 0.005) of MC and IME was present in the fingertip of all patients from each CMT genotype compared to healthy controls (table 1).
The axonal group as whole tended to have a greater loss of MC and IME than CMT1A, and this was especially evident in the CMT2C group and in the AR-CMT2K patient who presented a complete loss of MC and IME (table 1).
The Spearman rank-order correlation test did not reveal significant correlations between MC or IME loss and CMTNS and compound muscle action potential (CMAP) amplitude (included in CMTNS) for any CMT genotype.
Morphometric analysis of myelinated nerve fibers.
A total of 1,007 fibers were analyzed from all patients. We performed 293 measurements of internodal length, 874 of nodal gap length, and 4,028 of fiber calibers on these fibers. Kruskal-Wallis analysis showed a difference in both internodal and nodal gap length among the 7 groups (p = 0.0001).
The 2-group comparison (Mann-Whitney U test) showed that each CMT genotype had shorter internode (p < 0.009) and longer nodal gap (p < 0.0001) compared to healthy controls (table 1).
CMT1A had shorter internodes (p < 0.001) and longer nodal gaps (p < 0.001) than the axonal CMT group. Moreover, CMT1A patients had longer nodal gaps (p < 0.05) than any other CMT genotype. No difference was evident within the axonal group between patients harboring mutations in myelin genes (i.e., MPZ, GJB1) and those with mutations in axonal genes (i.e., RAB7, TRPV4, and GDAP1).
The robust test of equality of variance showed that the variability around the mean internodal length was comparable between each CMT genotype and controls, suggesting that the reduction in internodal length observed in each CMT genotype affected nerve fibers uniformly (figure 2). Moreover, the variability of internodal length was also comparable between demyelinating and axonal CMT, not allowing us to distinguish the primary pathologic event leading to internodal length shortening.
Figure 2. Boxplots of internodal length in each Charcot-Marie-Tooth genotype and controls.
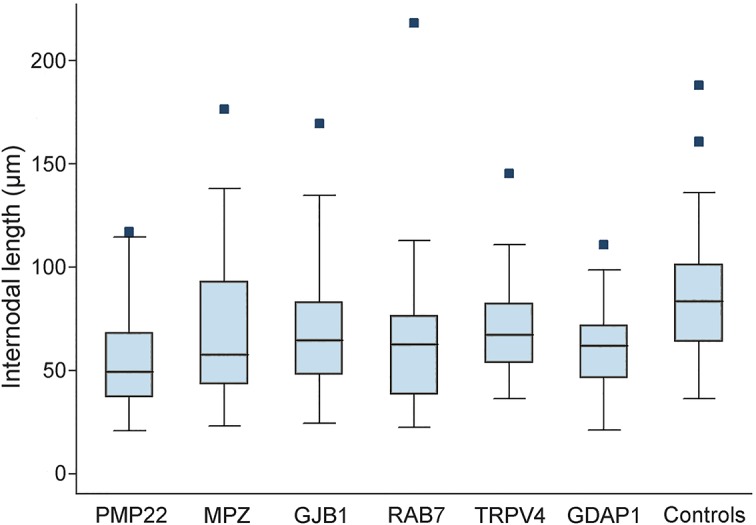
Dark horizontal lines represent the median value, with the box representing the 25th and 75th percentiles, the whiskers the lower and upper adjacent values, and outside values represented by dots.
Fiber caliber was not different among CMT genotypes and between each CMT genotype and controls. One explanation could be that some dermal nerve fibers presented mild and uniform enlargement of caliber such as occurs in predegenerative conditions, and this could have influenced our measurement.
In addition to the nerve fiber loss, several aspects of nerve degeneration were present. Myelinated fibers showed large variability in the caliber, fragmentation (figure 3D), and swellings (figure 3, E and F). NF/MBP double-staining showed asymmetrical distribution (figure 3, H and I) of paranodes compared to control (figure 3G). MC showed abnormalities in their shape (figure 3, B and C, compared to figure 3A) and in their position (located at the base instead of the apex of the dermal papillae).
Figure 3. Confocal images of glabrous skin in patients with Charcot-Marie-Tooth disease and controls.

A moderate loss of Meissner corpuscles is observed in a patient with CMT1A (B) and a severe loss of Meissner corpuscles is present in a patient with RAB7 mutation (C), compared to a control (A). (D–F) Abnormalities of myelinated fibers such as fragmentation (D) and swellings (arrowheads in E and F). (G–I) Disruption of nodal/paranodal architecture with elongation and asymmetry of the paranodes with expansion of neurofascin aggregation along the internode (arrows in H and I) compared to control (G). Scale bar: 200 μm (A, B, C); 30 μm (D, E, F); 50 μm (G, H, I). MBP = myelin basic protein; NF = neurofascin; PGP = protein gene product.
Finally, although segmental demyelination and subsequent remyelination may be a pathologic feature in sural nerve biopsies from CMT1A, we identified no segmental demyelination in any internodes from patients with CMT1A. These data are in keeping with those previously reported in patients with CMT1A.6
DISCUSSION
This study extends the information gained from skin biopsies on morphologic abnormalities in various forms of CMT and provides interesting insights into potential pathomechanisms of axonal and demyelinating CMT.
Axonal degeneration is a major cause of long-term disability in both demyelinating and axonal CMT.9 Skin biopsy studies have previously demonstrated a loss of MC, a consequence of axonal loss, in patients with CMT110 and CMT1A.6 In addition, recent observations using in vivo confocal microscopy demonstrated a correlation between MC loss and CMAP amplitude and clinical disability as assessed by CMTNS.11 We have extended these observations in our studies by observing reductions in the density of both MC and IME in skin samples from multiple patients with axonal (TRPV4, RAB7, GDAP1) and intermediate (late-onset MPZ and GJB1) forms of CMT in addition to patients with CMT1A. We did not find a correlation between MC or IME loss and the CMTNS and CMAP score. The different methodology used in our study might account for the discrepancy with data obtained in a previous study.11 However, our patient with AR-CMT2K who presented with the most severe clinical impairment (CMTNS = 20) had a complete loss of MC and IME at skin biopsy. Taken together, our results demonstrate that axonal loss in CMT can be readily detected by skin biopsy. Since skin biopsies can be performed on multiple occasions, these results also suggest their potential use as an outcome measure to detect axonal loss in longitudinal studies. Nevertheless, further studies on larger CMT samples are needed to better define the sensitivity of MC density as a measure of axonal degeneration as well as to validate skin biopsy as a surrogate biomarker of disease severity.
Nodes of Ranvier, paranodes, and juxtaparanodes contain specialized regions of myelin and the axonal cytoskeleton that play important roles in developing and maintaining the organization of myelin and the ensheathed axon.12 Altered axon–glial communication at this level has been hypothesized to contribute to axonal degeneration in patients with CMT1A and CMTX1.5,13 Abnormalities and reorganization of paranodal regions have been clearly documented in sural and dermal nerve fibers from patients with CMT1A and CMTX1.5,8,14 Retraction of paranodal myelin appears as a widening of the nodal gap, which includes the true nodal space and part of flanking paranodal regions. Thus widening of nodal gaps can be used as a surrogate marker for changes occurring at nodes or paranodes. Evidence of widened gaps was observed in Trembler-J15 and Cx32-deficient mice.16 We found that nodal gaps were larger in patients with CMT1A than in other genotypes, suggesting that widening of the nodal gap may be a feature of CMT subtypes that affect myelin. However, we also found that nodal gaps were wider in all other genotypes compared to controls, suggesting that nodal gaps are abnormal even in patients with axonal forms of CMT. Abnormalities of nodal gaps have not been described previously in patients with CMT2, but experimental models of axonal degeneration have shown that early structural changes include paranodal myelin retraction.17,18 Moreover, retracted paranodal myelin sheaths are frequently associated with swollen axons,19 which are thought to represent predegenerative phenomena.20 These abnormalities were widely observed in dermal nerve fibers from our patients. Perhaps altered communications between axons and glia at paranodal level are common features for multiple forms of CMT, including those that primarily affect axons, though widening may be greater in the demyelinating or dysmyelinating forms such as CMT1A.
A more detailed investigation of paranodal regions in CMT is warranted in further studies by using antibodies against the paranode-specific protein Caspr. Paranodal alterations such as paranodal asymmetry, which is thought to be an index of demyelination,6 might also differ in CMT due to myelin gene mutations with respect to axonal CMT.
Uniformly short internodes occur in dysmyelinating models such as periaxin null mice and have been hypothesized to cause slow nerve conductions such as are seen in these mice and in patients with periaxin mutations causing CMT4F.21 Uniformly shortened internodes in dermal nerve fibers were previously observed in skin biopsies from patients with CMT1A, suggesting that developmental abnormalities in internode formation contributed to the slow nerve conductions in patients.6 However, shortened internodes have been reported in a sural nerve biopsy from a single late-onset axonal MPZ-mutated patient22 and in peripheral nerves of mutant mice deficient for Cx32.16 Patients with late-onset CMT1B or with CMTX1 often have normal or only mildly slowed nerve conductions and minimal evidence of myelin abnormalities morphologically.23,24 In the current study, we observed shortened internodes in all the CMT genotypes, including those with mutations in axonal genes (i.e., RAB7, TRPV4, and GDAP1). Molecular definition in our series of patients with axonal CMT might account for the difference with respect to a previous report, which did not show shortened internodes in 5 patients with nongenetically defined CMT2.6
Therefore, while uniformly shortened internodes may suggest a developmental abnormality in internode formation, it is unclear whether they are always associated with abnormal myelin and whether they alone always cause slow conductions. We would not find it surprising that developmental abnormalities in axonal neuropathies could result in shortened internodes if we consider that reciprocal interactions between Schwann cells and axons are critical for the development of myelinated axons.25,26 Therefore, mutations in both myelin and axon genes may developmentally impede internode formation. It will be interesting to investigate internodal length in skin biopsies with severe, early-onset dysmyelinating CMT to determine whether it is the extent of the shortening of internodes rather than the presence or absence of shortening that best correlates with abnormalities of myelination.
The question remains of why conduction velocities would be normal or near normal in axonal neuropathies if they, like demyelinating neuropathies, also have shortened internodes. A potential answer to this question is provided in the recent study demonstrating that nerve conduction velocity in myelinated nerves relies on internodal length until a threshold of internodal distance is reached, beyond which conduction speed does not increase further.27 We hypothesize that shortened internodes will therefore not cause slow conduction velocities until the shortening crosses this threshold. Consistent with this hypothesis, our patients with CMT1A had the shortest internodal length of all our patients. Moreover, patients with CMT1A also had the widest nodal gap of our patients. The nodal/paranodal region represents an essential region for saltatory conduction in myelinated nerve fibers. Paranodal demyelination and abnormal exposure of voltage-gated potassium channels at the paranodal regions can delay the rise time of action potentials, and may further contribute to NCV slowing. It may be that the extent of internodal shortening and nodal gap widening are both important in causing uniformly slow nerve conductions in CMT1A.
This study extends the information gained from skin biopsies on morphologic abnormalities in various forms of CMT. We have demonstrated that myelinated dermal nerves can be used to detect axonal degeneration and changes in nodal and paranodal regions as well as in internodal length in axonal and demyelinating forms of CMT. It will be important to extend these investigations into patients with severe dysmyelinating forms of CMT such as early-onset CMT1B or CMT4 to determine whether widening of nodal gaps or shortening of internodes is even more pronounced in these disorders. Nonetheless, it will also be of interest to evaluate a larger number of patients with axonal or intermediate forms of CMT. Finally, since this minimally invasive procedure can easily be repeated in the same subject, it could provide a morphologic marker of disease progression in both natural history and clinical trials.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Dr. Peter J. Brophy and Diane Sherman (Centre for Neuroregeneration, University of Edinburgh, UK) for providing antibodies against Pan-Neurofascin.
GLOSSARY
- ANOVA
analysis of variance
- CMAP
compound muscle action potential
- CMT
Charcot-Marie-Tooth
- CMTNS
Charcot-Marie-Tooth Neuropathy Score
- IME
intrapapillary myelinated endings
- MBP
myelin basic protein
- MC
Meissner corpuscles
- NCS
nerve conduction study
- NF
neurofascin
- PGP
protein gene product
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
F. Manganelli: study concept and design, drafting and revising the manuscript, statistical analysis, patient recruitment. M. Nolano: study concept and design, drafting and revising the manuscript, data analysis. C. Pisciotta: revising the manuscript, patient recruitment, data acquisition, data analysis. V. Provitera: revising the manuscript, data acquisition, data analysis. G.M. Fabrizi: patient recruitment, data acquisition. T. Cavallaro: patient recruitment, data acquisition. A. Stancanelli: data acquisition, data analysis. G. Caporaso: data acquisition, data analysis. M.E. Shy: interpretation of the data, revising the manuscript. L. Santoro: study concept and design, interpretation of the data, drafting and revising the manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Shy M, Lupski JR, Chance PF, Klein CJ, Dyck PJ. Hereditary motor and sensory neuropathies: an overview of clinical, genetic, electrophysiologic and pathologic features. In: Peripheral Neuropathy, 4th ed Philadelphia: Elsevier Saunders; 2005. [Google Scholar]
- 2.Manganelli F, Tozza S, Pisciotta C, et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes in a Southern Italy population. J Peripher Nerv Syst 2014;19:292–298. [DOI] [PubMed] [Google Scholar]
- 3.Nolano M, Provitera V, Crisci C, et al. Quantification of myelinated endings and mechanoreceptors in human digital skin. Ann Neurol 2003:54:197–205. [DOI] [PubMed] [Google Scholar]
- 4.Provitera V, Nolano M, Pagano A, Caporaso G, Stancanelli A, Santoro L. Myelinated nerve endings in human skin. Muscle Nerve 2007;35:767–775. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Bai Y, Ghandour K, et al. Skin biopsies in myelin-related neuropathies: bringing molecular pathology to the bedside. Brain 2005;128:1168–1177. [DOI] [PubMed] [Google Scholar]
- 6.Saporta MA, Katona I, Lewis RA, Masse S, Shy ME, Li J. Shortened internodal length of dermal myelinated nerve fibres in Charcot-Marie-Tooth disease type 1A. Brain 2009;132:3263–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shy ME, Blake J, Krajewski K, et al. Reliability and validity of the CMT neuropathy score as a measure of disability. Neurology 2005;64:1209–1214. [DOI] [PubMed] [Google Scholar]
- 8.Hattori N, Yamamoto M, Yoshihara T, et al. Demyelinating and axonal features of Charcot-Marie-Tooth disease with mutations of myelin-related proteins (PMP22, MPZ and Cx32): a clinicopathological study of 205 Japanese patients. Brain 2003;126:134–151. [DOI] [PubMed] [Google Scholar]
- 9.Suter U, Scherer SS. Disease mechanisms in inherited neuropathies. Nat Rev Neurosci 2003;4:714–726. [DOI] [PubMed] [Google Scholar]
- 10.Dyck PJ, Winkelmann RK, Bolton CF. Quantitation of Meissner's corpuscles in hereditary neurologic disorders: Charcot-Marie-Tooth disease, Roussy-Levy syndrome, Dejerine-Sottas disease, hereditary sensory neuropathy, spinocerebellar degenerations, and hereditary spastic paraplegia. Neurology 1966;16:10–17. [DOI] [PubMed] [Google Scholar]
- 11.Almodovar JL, Ferguson M, McDermott MP, Lewis RA, Shy ME, Herrmann DN. In vivo confocal microscopy of Meissner corpuscles as a novel sensory measure in CMT1A. J Peripher Nerv Syst 2011;16:169–174. [DOI] [PubMed] [Google Scholar]
- 12.Scherer SS, Arroyo EJ. Recent progress on the molecular organization of myelinated axons. J Peripher Nerv Syst 2002;7:1–12. [DOI] [PubMed] [Google Scholar]
- 13.Sousa AD, Bhat MA. Cytoskeletal transition at the paranodes: the Achilles' heel of myelinated axons. Neuron Glia Biol 2007;3:169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hahn AF, Ainsworth PJ, Bolton CF, Bilbao JM, Vallat JM. Pathological findings in the X-linked form of Charcot-Marie-Tooth disease: a morphometric and ultrastructural analysis. Acta Neuropathol 2001;101:129–139. [DOI] [PubMed] [Google Scholar]
- 15.Devaux JJ, Scherer SS. Altered ion channels in an animal model of Charcot-Marie-Tooth disease type IA. J Neurosci 2005;25:1470–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neuberg DH, Sancho S, Suter U. Altered molecular architecture of peripheral nerves in mice lacking the peripheral myelin protein 22 or connexin32. J Neurosci Res 1999;58:612–623. [DOI] [PubMed] [Google Scholar]
- 17.Spencer PS, Schaumburg HH. Ultrastructural studies of the dying-back process: III: the evolution of experimental peripheral giant axonal degeneration. J Neuropathol Exp Neurol 1977;36:276–299. [DOI] [PubMed] [Google Scholar]
- 18.Veronesi B, Peterson ER, Bornstein MB, Spencer PS. Ultrastructural studies of the dying-back process: VI: examination of nerve fibers undergoing giant axonal degeneration in organotypic culture. J Neuropathol Exp Neurol 1983;42:153–165. [PubMed] [Google Scholar]
- 19.Griffin JW, Drucker N, Gold BG, et al. Schwann cell proliferation and migration during paranodal demyelination. J Neurosci 1987;7:682–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ebenezer GJ, McArthur JC, Thomas D, et al. Denervation of skin in neuropathies: the sequence of axonal and Schwann cell changes in skin biopsies. Brain 2007;130:2703–2714. [DOI] [PubMed] [Google Scholar]
- 21.Court FA, Sherman DL, Pratt T, et al. Restricted growth of Schwann cells lacking Cajal bands slows conduction in myelinated nerves. Nature 2004;431:191–195. [DOI] [PubMed] [Google Scholar]
- 22.Laurà M, Milani M, Morbin M, et al. Rapid progression of late onset axonal Charcot-Marie-Tooth disease associated with a novel MPZ mutation in the extracellular domain. J Neurol Neurosurg Psychiatry 2007;78:1263–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Bai Y, Ianakova E, et al. Major myelin protein gene (P0) mutation causes a novel form of axonal degeneration. J Comp Neurol 2006;498:252–265. [DOI] [PubMed] [Google Scholar]
- 24.Kleopa KA, Abrams CK, Scherer SS. How do mutations in GJB1 cause X-linked Charcot-Marie-Tooth disease? Brain Res 2012;1487:198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woodhoo A, Sommer L. Development of the Schwann cell lineage: from the neural crest to the myelinated nerve. Glia 2008;56:1481–1490. [DOI] [PubMed] [Google Scholar]
- 26.Taveggia C, Feltri ML, Wrabetz L. Signals to promote myelin formation and repair. Nat Rev Neurol 2010;6:276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu LM, Williams A, Delaney A, Sherman DL, Brophy PJ. Increasing internodal distance in myelinated nerves accelerates nerve conduction to a flat maximum. Curr Biol 2012;22:1957–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.