

Abstract
Nuclear factor-kappa B (NF-κB) activation is widely implicated in multiple organ failure (MOF); however, a direct inhibitor of IκB kinase (IKK), which plays a pivotal role in the activation of NF-κB, has not been investigated in shock. Thus, the aim of the present work was to investigate the effects of an IKK inhibitor on the MOF associated with hemorrhagic shock (HS). Therefore, rats were subjected to HS and were resuscitated with the shed blood. Rats were treated with the inhibitor of IKK or vehicle at resuscitation. Four hours later, blood and organs were assessed for organ injury and signaling events involved in the activation of NF-κB. Additionally, survival following serum deprivation was assessed in HK-2 cells treated with the inhibitor of IKK. HS resulted in renal dysfunction, lung, liver and muscular injury, and increases in serum inflammatory cytokines. Kidney and liver tissue from HS rats revealed increases in phosphorylation of IKKαβ and IκBα, nuclear translocation of NF-κB and expression of inducible isoform of nitric oxide synthase (iNOS). IKK16 treatment upon resuscitation attenuated NF-κB activation and activated the Akt survival pathway, leading to a significant attenuation of all of the above parameters. Furthermore, IKK16 exhibited cytoprotective effects in human kidney cells. In conclusion, the inhibitor of IKK complex attenuated the MOF associated with HS. This effect may be due to the inhibition of the NF-κB pathway and activation of the survival kinase Akt. Thus, the inhibition of the IKK complex might be an effective strategy for the prevention of MOF associated with HS.
INTRODUCTION
Multiple organ failure (MOF) is common (30%) in the severely injured population and, if not fatal, results in serious outcomes such as prolonged critical care and hospital stays, and poor long-term quality of life (1). Development of MOF occurs early (within 2 d of admission) and correlates with an increase in nosocomial infections and, if it persists, mortality (2). This figure rises rapidly as the degree of blood loss and shock intensifies. Patients requiring at least four units of red cell transfusions have longer critical care (9 versus 5 d) and hospital stays (25 versus 15 d). They also have a higher mortality rate (35% versus 6%). Even modest improvements in these figures would significantly improve patient experience and quality of life and reduce the costs of healthcare. Currently there are no specific treatments for organ failure. Resuscitation after severe hemorrhage improves tissue perfusion but does not alleviate reperfusion injury nor dampen the systemic inflammatory response, both of which importantly contribute to the development of organ injury/dysfunction and ultimately MOF (3). A therapeutic agent that reduces the incidence and severity of multiple organ failure could have a major global impact on patient outcomes and resource utilization.
Nuclear factor-kappa B (NF-κB) is the main transcription factor that regulates the expression of genes involved in the inflammatory response. Functionally active NF-κB is a heterodimer, which is normally sequestered in an inactive cytoplasmic complex by binding to the inhibitory protein IκBα (4). External stimuli such as inflammatory cytokines, bacterial lipopolysaccharides and reactive oxygen species (ROS) activate the IκB kinase (IKK) complex, which in turn causes the rapid phosphorylation of IκBα at Ser32/36 (5). Phosphorylated IκBα becomes ubiquitinated and is subsequently degraded by proteasomes, releasing NF-κB, which is free to translocate to the nucleus to regulate the transcription of a wide variety of genes that encode inflammatory proteins, including iNOS and the proinflammatory cytokines tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6), to name but a few (6,7).
There is now growing evidence that inhibition of the activation of NF-κB may reduce organ injury and dysfunction in septic and hemorrhagic shock (HS) (8–11). The inhibition of IKK complex by IKK16 and the consequent prevention of the activation of NF-κB reduce the cardiac dysfunction, liver injury and renal dysfunction caused by sepsis (12) and ventilation-induced lung injury (13). There is indirect evidence that agents that prevent the activation of NF-κB (for example, calpain inhibitor I) are also of benefit in HS (14). Other examples include ciglitazone (a peroxisome proliferator-activated receptor [PPAR]-γ ligand) and eritoran (a toll-like receptor 4 [TLR4] antagonist), which attenuate organ damage in HS through the indirect inhibition of NF-κB (8,10). However, the effects of the inhibition of IKK in HS have yet to be investigated. Therefore, the aim of the present study was to investigate the effects of IKK16, a potent inhibitor of the IKK complex, on the multiple organ injury and dysfunction induced by HS in the rat. Having found that IKK16 reduced the organ injury/dysfunction associated with HS in the rat, we then investigated the mechanism of action (signaling) of the observed effects of IKK16 in organs (ex vivo) and in cultured cells in vitro.
MATERIALS AND METHODS
Animal Welfare and Ethics Statements
The animal protocols used in this study were approved by the Animal Welfare Ethics Review Board (AWERB) of Queen Mary University of London (PPL: 70/7348) in accordance with the derivatives of Home Office guidance on Operation of Animals (Scientific Procedures Act 1986) published by Her Majesty’s Stationery Office and the Guide for the Care and Use of Laboratory Animals of the National Research Council.
Hemorrhagic Shock Model and Organ Injury Assessment
A timeline of the HS protocol is provided in Figure 1. This study was carried out on 39 male Wistar rats (Charles River Ltd.) weighing 230 to 280 g and receiving a standard diet and water ad libitum. All data from rats that had died during the experiment were recorded but were excluded from the data analysis. Thus, the n numbers provided in figures and text represent those animals that survived the entire experimental protocol. The animals were housed in a temperature-controlled environment with a 12-h light–dark cycle. Hemorrhagic shock was performed as previously described (15). Rats were anaesthetized with sodium thiopentone (120 mg/kg intraperitoneal [IP] maintained using 10 mg/kg intravenous [i.v.]) and cannulation with polyethylene catheters of the trachea, bladder (to collect urine), jugular vein (to administer compounds and blood), left femoral artery (for recording of mean arterial pressure [MAP]) and right carotid artery (for blood withdrawn) was performed. Body temperature was monitored by a rectal thermometer and maintained at 37°C ± 0.5°C by means of a homoeothermic blanket system (Harvard Apparatus). Briefly, blood was withdrawn to achieve a fall in MAP to 30 ± 2 mmHg within 5 min, which was recorded with a pressure transducer coupled to a PowerLab 8/30 (AD Instruments Pty Ltd.). The average ± standard deviation (SD) volumes of blood withdrawn were 9.4 ± 0.74 and 9.61 ± 0.99 mL for the HS and HS + IKK16 groups, respectively (p > 0.05). MAP was maintained at 30 ± 2 mmHg for a period of 90 min either by further withdrawal of blood during the compensation phase or administration of the shed blood during the decompensation phase. At 90 min after initiation of hemorrhage or when 25% of the shed blood had to be reinjected to sustain MAP at 30 mmHg, resuscitation was performed with the remaining shed blood mixed with 100 IU/mL heparinized saline plus then the same volume of blood spent during decompensation of Ringer lactate over a period of 5 min. As different volumes of blood were withdrawn to reach an MAP of 30 ± 2 mmHg, the average ± SD values for the rate of resuscitation were 1.4 ± 0.07 and 1.58 ± 0.24 mL/min for the HS + vehicle and HS + IKK16 groups, respectively (p > 0.05). One hour after resuscitation an infusion of Ringer lactate (1.5 mL/kg/h; i.v.) was started as fluid replacement and it was maintained throughout the experiment for a total of 4 h. The last 3-h urine was obtained for the estimation of creatinine clearance. Then, blood samples were collected via the carotid artery for measurement of lactate (Accutrend Plus Meter, Roche Diagnostics), blood cell counts (IDEXX ProCyte Dx® Hematology Analyzer; IDEXX Laboratories Ltd.) and organ injury parameters. The heart was removed to terminate the experiment. Blood samples were centrifuged to separate serum from which creatinine, aspartate aminotransferase (AST), alanine aminotransferase (ALT), lipase, amylase and creatine kinase (CK) were measured within 24 h (IDEXX Laboratories Ltd). In addition, lung, kidney and liver samples were taken and stored at −80°C for further analysis. Sham-operated rats were used as control and underwent identical surgical procedures but without hemorrhage or resuscitation.
Figure 1.
Timeline of the experimental protocol.
Experimental Design
Rats were randomly allocated into the following groups: Sham + Vehicle (n = 9), Sham + IKK16 (n = 8); HS + Vehicle (n = 11) and HS + IKK16 (n = 11). Rats were administered i.v. vehicle (10% dimethyl sulfoxide [DMSO]) or IKK16 (1 mg/mL/kg) upon resuscitation. The dose of IKK16 was chosen based on our previous experience (12).
Lung Myeloperoxidase Activity
The activity of myeloperoxidase was determined as an indicator of neutrophil accumulation into the lungs. Samples were homogenized in a 5 mmol/L phosphate buffer and centrifuged for 30 min at 13,000g at 4°C. The supernatant was allowed to react with a solution of o-dianisidine (0.167 mg/mL) and H2O2 (0.1 mmol/L) in 50 mmol/L phosphate buffer. The rate of change in absorbance was measured spectrophotometrically at 460 nm. Myeloperoxidase activity was defined as the quantity of enzyme degrading 1 μmol of peroxide/min at 37°C and was expressed in milliunits/g of wet tissue.
Lung N-Acetyl-β-d-Glucosaminidase Determination
Lung N-acetyl-β-d-glucosaminidase (NAG) was measured as an index of macrophage influx. Lung samples were homogenized in Tris-HCl buffer containing NP-40 (1%), sodium deoxycholate (0,25%), ethylene glycol tetraacetic acid (EGTA; 1 mmoL/L) and protease inhibitor cocktail (1 μL/mL; P-8340; Sigma-Aldrich). The homogenates were centrifuged at 10,000g at 4°C for 10 min and the supernatants were stored at –80°C. The protein content was determined in each sample using a BCA protein assay (Thermo-Fisher Scientific, Inc.). N-acetyl-β-d-glucosaminidase (NAG) was determined on the lung homogenates using a commercial immunoassay kit (Elabscience Biotechnology Co., Ltd.) according to the protocol provided by the manufacturer. The results were expressed as ng/mg of protein.
Immunohistochemistry
Kidney, liver and lung samples were obtained from rats after the end of the experiment and were fixed in formalin for 48 h. Then, tissues were transferred to 70% ethanol solution, embedded in paraffin and processed to obtain 4-μm sections. After deparaffinization, the antigens were retrieved by incubation with 1× citrate buffer (pH 6.0, antigen retriever, Sigma-Aldrich) for 15 min at microwave (700 watts). After cooling, sections were incubated with 0.03% H2O2 for inactivation of endogenous peroxidase. Nonspecific adsorption was minimized by incubating the sections with 10% goat serum for 15 min. The slides were then incubated with rabbit anti-myeloperoxidase antibody (1:25, catalog no. ab9535; Abcam) or anti-CD68 antibody ED1 (1:400; catalog no. MCA341R; AbD Serotec) for 1 h at 37°C. After washing with phosphate-bufferd saline (PBS), slides were incubated for 30 min with labeled polymer-HRP antibody, washed and incubated with DAB (3,3′-diaminobenzidine) chromogen solution (EnVision+ System-HRP (DAB); K4010; Dako). A negative control was performed through the omission of primary antibody (data not shown). The reaction was stopped by immersing slides in water. Counterstaining was performed with Harris hematoxylin. Images were acquired using a NanoZoomer Digital Pathology Scanner (Hamamatsu Photonics K.K.) and analyzed using the NDP Viewer software. The numbers of CD68-and myeloperoxidase (MPO)-positive cells were counted in 10 randomly selected fields (400×) in a double-blinded manner.
Western Blotting
Briefly, kidney and liver samples were homogenized in buffer and centrifuged at 1,300g for 5 min at 4°C. To obtain the cytosolic fraction, supernatants were centrifuged at 16,000g at 4°C for 40 min. The pelleted nuclei were resuspended in extraction buffer and centrifuged at 16,000g for 20 min at 4°C. Protein content was determined on both nuclear and cytosolic extracts using a bicinchoninic acid protein assay (Thermo-Fisher Scientific Inc.). Proteins were separated by 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinyldenediflouoride (PVDF) membrane, which was incubated with a primary antibody (goat anti-ICAM1 [1:200]; rabbit anti-total IKKαβ [1:200]; rabbit anti-pIKKαβ Ser176/180 [1:1000]; mouse anti-total IκBα [1:1000]; mouse anti-IκBα pSer32/36 [1:1000]; rabbit anti-NF-κB p65 [1:1000]; rabbit anti-total Akt [1:1000]; mouse anti-pAkt Ser473 [1:1000]; rabbit anti-total iNOS [1:200]). Membranes were incubated with a secondary antibody conjugated with horseradish peroxidase (1:2000) for 30 min at room temperature and developed with an enhanced chemiluminescence (ECL) detection system. The immunoreactive bands were visualized by autoradiography and the densitometric analysis was performed using Gel Pro Analyzer 4.5, 2000 software (Media Cybernetics). The membranes were stripped and incubated with β-actin monoclonal antibody (1:5000) and subsequently with an anti-mouse antibody (1:10000) to assess gel-loading homogeneity. Densitometric analysis of the related bands is expressed as relative optical density, corrected for the corresponding β-actin contents, and normalized using the related mean value of sham-operated band.
Serum Nitrite and Nitrate
Nitric oxide synthesis was estimated through the measurement of serum nitrite + nitrate (NOx) in plasma. Briefly, nitrate was converted to nitrite using nitrate reductase in the presence of cofactors. The total nitrite in plasma was assayed by adding Griess reagent (0.05% [wt/vol] naphthalethylenediamine dihydrochloride and 0.5% [wt/vol] sulphanil-amide in 2.5% [vol/vol] phosphoric acid) to each sample and read at 550 nm. NOx concentrations were calculated by comparison with a standard solution of sodium nitrate prepared in water. Values are expressed as μmol/L NOx.
Serum Cytokines
Serum cytokines TNF-α, IL-6 and IL-10 were determined using commercial immunoassay kits (R&D Systems) according to the manufacturer protocol.
Cell Culture
Proximal tubular epithelial cell line cells from human kidney, HK-2 cells (European Collection of Cell Cultures, Salisbury, UK), were grown in media comprising Dulbecco’s Modified Eagle’s Medium (DMEM)/Nutrient F-12 Ham (Sigma Aldrich) supplemented with 10% fetal calf serum (FCS) and antibiotics (100 U/mL penicillin G, 100 μg/mL streptomycin and 0.25 μg/mL amphotericin). Cells were seeded in a 96-well tissue culture plate and allowed to adhere for 18 h in an incubator at 37°C (5% CO2 in 95% air). Cells were serum deprived for 24 h and concomitantly incubated with IKK16 (10 nmol/L to 100 nmol/L). Two or 4 h later, IKK16 were removed and new media (with or without serum) were added into the wells. Twenty-four hours later, cell viability was determined by using the CellTiter 96 nonradioactive cell proliferation assay kit (Promega) according to the manufacturer’s instruction. Absorbance was measured at 490 nm and survival was estimated related to control group (percentage).
Reagents
IKK16 was purchased from Tocris Bioscience (R&D Systems Europe). Unless otherwise stated, all compounds used in this study were purchased from Sigma-Aldrich. All stock solutions were prepared using nonpyrogenic saline (0.9% [wt/vol] NaCl (Baxter Healthcare Ltd.). Ringer lactate was also purchased from Baxter Healthcare Ltd. The bicinchoninic acid protein assay kit and SuperBlock blocking buffer were from Thermo-Fisher Scientific Inc. Antibodies were from Cell Signaling Technology Inc.
Data Analysis
All values are expressed as box and whiskers (minimum to maximum) and the central line represents the median. Values on the tables are expressed as mean ± SD. Statistical analysis was carried out using Graph Prism 5.03 (Graph Pad Software). Data were assessed by one-way ANOVA followed by Bonferroni’s post hoc test. A p value of less than 0.05 was considered to be significant.
RESULTS
Effects of IKK16 Treatment on HS
Compared with sham-operated rats, rats subjected to HS treated with vehicle exhibited a significant decrease in creatinine clearance (Figure 2B), as well as a rise in serum creatinine (Figure 2A) indicating the development of renal dysfunction. Compared with sham-operated rats, HS-rats treated with vehicle also had elevated plasma concentrations of AST (Figure 2C) and ALT (Figure 2D) as well as amylase (Figure 2E) and lipase (Figure 2F), indicating the development of liver and pancreatic injury, respectively. Compared with sham-operated rats, HS-rats treated with vehicle also had elevated plasma concentrations of CK (Figure 2G) and lactate (Figure 2H), indicating the development of skeletal muscular injury and global tissue hypoxia, respectively. Treatment of HS-rats with the inhibitor IKK16 significantly attenuated the renal dysfunction, liver injury, pancreatic injury and rise in lactate, but not the skeletal muscular injury associated with HS (Figure 2). The administration of IKK16 to sham-operated rats had no significant effect on any of the biochemical markers evaluated (Figure 2).
Figure 2.

IKK16 protects against multiple organ failure and dysfunction. Rats were subjected to HS and received IKK16 (1 mg/kg; i.v.) or vehicle (Veh) (10% DMSO) at resuscitation. Four hours later, blood was obtained and serum creatinine (A), estimated creatinine clearance (B), serum AST (C), serum ALT (D), amylase (E), lipase (F), CK (G) and lactate (H) were determined. Sham animals were used as control. Data are presented as box and whiskers (minimum to maximum) and the central line represents the median (n = 8–11 animals per group). *p < 0.05 versus sham group and #p < 0.05 versus HS group.
Effects of IKK16 Treatment on Lung Injury
Compared with sham-operated rats, HS-rats treated with vehicle showed an increase in MPO-positive cells, a marker for neutrophils (Figures 3A, C, E), and CD68-positive cells, a marker for macrophages (Figures 3F, H, J). Treatment of HS-rats with IKK16 attenuated the increase in the number of both cell types in the lung (Figures 3D–E, 3I–J). To confirm the data obtain by immunohistochemistry, the activity of MPO (indicator of neutrophil number; Figure 3K) and NAG (indicator of macrophage number; Figure 3L) were also determined in lung tissue. Using this method, we confirmed that HS leads to an increase in neutrophils and macrophages in the lung, and that this neutrophil/macrophage accumulation was significantly reduced in HS-rats treated with IKK16.
Figure 3.

IKK16 attenuates lung injury. Rats were subjected to HS and received IKK16 (1 mg/kg; i.v.) or vehicle (Veh) (10% DMSO) at resuscitation and 4 h later lungs were obtained. Sham animals were used as control. The representative images for the immunostaining for MPO as a neutrophils marker (A–D), CD68 as a macrophage marker (F–I), and the quantitative analysis of the number of cells/mm2 (E and J) are shown. The scale bar represents 50 μm. The activities of MPO (K) and NAG (L) were also determined in lung tissue. The expression of ICAM-1 was determined by Western blotting. Densitometric analysis of the related bands is expressed as relative optical density, corrected for the corresponding β-actin contents, and normalized using the related mean value of sham-operated band (M). Data are presented as box and whiskers (minimum to maximum) and the central line represents the median (n = 5–6 animals per group). *p < 0.05 versus sham group and #p < 0.05 versus HS group.
We have also investigated the expression of the intercellular adhesion molecule (ICAM-1), which may be a key driver for the observed accumulation of neutrophils in the lung. ICAM-1 expression increased in lungs of HS-rats compared with sham-operated rats. This increase was attenuated by the treatment of HS-rats with IKK16 (Figure 3M). No difference was found in sham animals treated with IKK16 compared with sham control animals for any of the parameters measured (Figure 3).
Effects of IKK16 Treatment on Inflammatory Cells in Liver and Kidney
Compared with sham-operated rats, HS-rats treated with vehicle showed an increase in MPO-positive cells (indicator of neutrophil number) in kidney medulla (Figures 4A, C, E) and liver (Figures 4K, M, O). No difference among the groups was observed in the kidney cortex (data not shown). The treatment of HS-rats with IKK16 attenuated the increase of neutrophils in both liver and kidney (Figures 4D, E, N, O). In contrast, no difference was found among the groups in the number of CD68-positive cells (indicator of macrophage number) in these organs (Figures 4F–J, P–T).
Figure 4.

IKK16 effects on inflammatory cells in kidney and liver. Rats were subjected to HS and received IKK16 (1 mg/kg; i.v.) or vehicle (Veh) (10% DMSO) at resuscitation and 4 h later kidney and liver were obtained. Sham animals were used as control. The representative images for the immunostaining for MPO as a neutrophil marker (A–D, K–N), CD68 as a macrophage marker (F–I, P–S), and the quantitative analysis of the number of cells/mm2 (E, J, O, and T) are shown. The scale bar represents 50 μm. Data are presented as box and whiskers (minimum to maximum) and the central line represents the median (n = 5–6 animals per group). *p < 0.05 versus sham group and #p < 0.05 versus HS group.
Effects of IKK16 Treatment on Blood Cells
The following hematological parameters were measured in all animal groups: Red blood cell count (RBC), hemoglobin (HGB), hematocrit (HCT), mean RBC volume (MCV), mean cell hemoglobin (MCH), mean cell hemoglobin concentration (MCHC), red cell distribution width-SD (RDW-SD), red cell distribution width–coefficient of variation (RDW-CV), total reticulocyte count (RET#) and percentage (RET%), platelet count (PLT), platelet distribution width (PDW), mean platelet volume (MPV), platelet large cell ratio (P-LCR), plateletcrit (PCT) and white blood cell counts (WBCs).
Compared with to sham-operated animals, HS-rats treated with vehicle showed a slight increase in lymphocyte cell number, but this rise in lymphocyte count associated with HS was not affected by IKK16 (Table 1). Neither HS nor IKK16 had any other significant effect on any of the other parameters measured (Tables 1, 2).
Table 1.
Effect of hemorrhagic shock and IKK16 treatment on white blood cells.a
| Sham + Vehicle | Sham + IKK16 | HS + Vehicle | HS + IKK16 | |
|---|---|---|---|---|
| WBC (k/μL) | 5.34 ± 0.11 | 5.10 ± 1.38 | 7.34 ± 1.55 | 8.17 ± 1.94 |
| Neutrophils (k/μL) | 2.11 ± 1.02 | 1.86 ± 0.66 | 1.19 ± 1.26 | 1.95 ± 2.32 |
| Lymphocytes (k/μL) | 2.83 ± 1.02 | 3.03 ± 1.20 | 5.58 ± 2.10*b | 5.6 ± 1.45* |
| Monocytes (k/μL) | 0.33 ± 0.19 | 0.28 ± 0.03 | 0.47 ± 0.21 | 0.56 ± 0.17 |
| Eosinophils (k/μL) | 0.06 ± 0.10 | 0.02 ± 0.02 | 0.04 ± 0.06 | 0.03 ± 0.02 |
| Basophils (k/μL) | 0.00 ± 0.00 | 0.01 ± 0.01 | 0.06 ± 0.06 | 0.03 ± 0.04 |
Rats were subjected to HS and received IKK16 (1 mg/kg; i.v.) or vehicle (10% DMSO) at resuscitation and blood was obtained 4 h later. White blood cells, neutrophils, lymphocytes, monocytes, eosinophils and basophils were evaluated. Data are presented as mean ± S.D. (n = 8–11 animals per group).
p < 0.05 versus sham group.
Table 2.
Effect of hemorrhagic shock and IKK16 treatment on red blood cells and platelet parameters.a
| Sham + Vehicle | Sham + IKK16 | HS + Vehicle | HS + IKK16 | |
|---|---|---|---|---|
| RBC (M/μL) | 6.29 ± 0.38 | 6.11 ± 0.56 | 6.67 ± 0.64 | 6.61 ± 0.59 |
| HGB (g/L) | 13.15 ± 0.59 | 12.73 ± 0.83 | 13.89 ± 1.24 | 13.76 ± 1.38 |
| HCT (%) | 40.45 ± 1.64 | 39.13 ± 3.84 | 42.88 ± 5.20 | 42.30 ± 4.45 |
| MCV (fL) | 64.43 ± 2.29 | 64.07 ± 0.64 | 64.20 ± 3.67 | 63.98 ± 2.48 |
| MCH (pg) | 20.93 ± 0.74 | 20.90 ± 0.61 | 20.85 ± 0.81 | 20.81 ± 0.74 |
| MCHC (g/L) | 32.52 ± 0.87 | 32.60 ± 1.14 | 32.52 ± 1.16 | 32.53 ± 0.52 |
| RDW-SD (fL) | 32.12 ± 1.51 | 33.67 ± 1.65 | 32.68 ± 1.61 | 32.10 ± 1.64 |
| RDW-CV (%) | 16.13 ± 1.09 | 16.80 ± 1.75 | 17.45 ± 1.73 | 16.92 ± 1.29 |
| RET# (k/μL) | 462.68 ± 67.6 | 522.3 ± 147.8 | 501.7 ± 72.4 | 479.8 ± 59.4 |
| RET (%) | 7.40 ± 1.27 | 8.49 ± 1.85 | 7.53 ± 0.90 | 7.29 ± 0.95 |
| PLT (k/μL) | 990.50 ± 177.2 | 1087.3 ± 49.7 | 830.9 ± 300.7 | 954.8 ± 126.6 |
| PDW (fL) | 8.38 ± 0.57 | 8.57 ± 0.47 | 8.85 ± 0.76 | 8.60 ± 0.50 |
| MPV (fL) | 7.42 ± 0.29 | 7.43 ± 0.23 | 7.75 ± 0.52 | 7.60 ± 0.23 |
| P-LCR (fL) | 8.87 ± 1.92 | 9.03 ± 1.23 | 11.05 ± 3.24 | 9.47 ± 1.51 |
| PCT (%) | 0.73 ± 0.10 | 0.80 ± 0.04 | 0.64 ± 0.22 | 0.72 ± 0.09 |
Rats were subjected to HS and received IKK16 (1 mg/kg; i.v.) or vehicle (10% DMSO) at the resuscitation and blood was obtained 4 h later. Data are presented as mean ± S.D. (n = 8–11 animals per group).
Effects of IKK16 Treatment on NF-κB Pathway
Because the largest (beneficial) effect of IKK16 was observed in kidney (for example, reduction in renal dysfunction) and liver (for example, reduction in liver injury) (Figure 2), we subsequently investigated the effects of IKK16 on the signaling pathways involved in the activation of NF-κB and/or cell survival in renal or liver biopsies obtained from all animal groups. When compared with sham-operated rats, we observed a significant increase in the phosphorylation of Ser176/180 on IKKαβ (Figures 5A, D), Ser32/36 on IκBα (Figures 5B, E) and the nuclear translocation of the p65 NF-κB subunit (Figures 5C, F) in the kidney (Figures 5A–C) and liver (Figures 5D–F) biopsies obtained from HS-rats treated with vehicle. Treatment of the HS rats with IKK16 on resuscitation significantly attenuated the phosphorylation of Ser176/180 on IKKαβ, Ser32/36 on IκBα and the subsequent translocation of the p65 NF-κB subunit to the nucleus in both kidney and liver (Figure 5).
Figure 5.

IKK16 attenuates the phosphorylation of IKKαβ, IκBα and the nuclear translocation of the p65 NF-κB subunit. Rats were subjected to HS and received IKK16 (1 mg/kg; i.v.) or vehicle (Veh) (10% DMSO) at resuscitation and 4 h later the phosphorylation of IKKαβ on Ser176/180 (A and D), IκBα on Ser32/36 (B and E) and the nuclear translocation of the p65 NF-κB subunit (C and F) on the kidney (A–C) and liver (D–F) were determined by Western blotting. Sham animals were used as control. Densitometric analysis of the related bands is expressed as relative optical density (O.D.), corrected for the corresponding β-actin contents, and normalized using the related mean value of sham-operated band. Data are presented as box and whiskers (minimum to maximum) and the central line represents the median (n = 3–4 animals per group). *p < 0.05 versus sham group and #p < 0.05 versus HS group.
Effects of IKK16 Treatment on iNOS Expression and NO Production Induced by HS
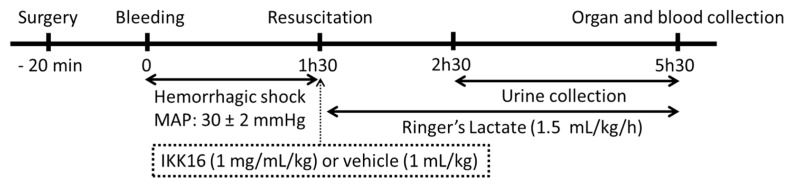
Compared with sham-operated rats, kidneys and livers from rats submitted to HS (treated with vehicle) exhibited a significant increase on iNOS expression (Figure 6A and B, respectively) and consequently an increase in NOx and, hence, NO formation (Figure 6C). Interestingly, treatment of HS-rats with IKK16 at the onset of resuscitation significantly attenuated both iNOS expression in kidney (Figure 6A) and liver (Figure 6B) as well as the associated rise in serum NOx levels (Figure 6C).
Figure 6.
IKK16 attenuates the expression of inducible isoform of nitric oxide synthase (iNOS) and the serum nitrite + nitrate (NOx). Rats were subjected to HS and received IKK16 (1 mg/kg; i.v.) or vehicle (Veh) (Veh) (10% DMSO) at resuscitation and 4 h later the iNOS expression in the kidney (A) and liver (B) was determined by Western blotting and serum NOx (C) was determined through Griess reaction. Sham animals were used as control. Protein expression is measured as relative optical density (O.D.), corrected for the corresponding β-actin contents and normalized using the related mean value of sham-operated band. Data are presented as box and whiskers (minimum to maximum) and the central line represents the median (A, B: n = 3–4 animals per group; C: n = 8–9 per group). *p < 0.05 versus sham group and #p < 0.05 versus HS group.
Effects of IKK16 Treatment on Systemic Cytokines Levels
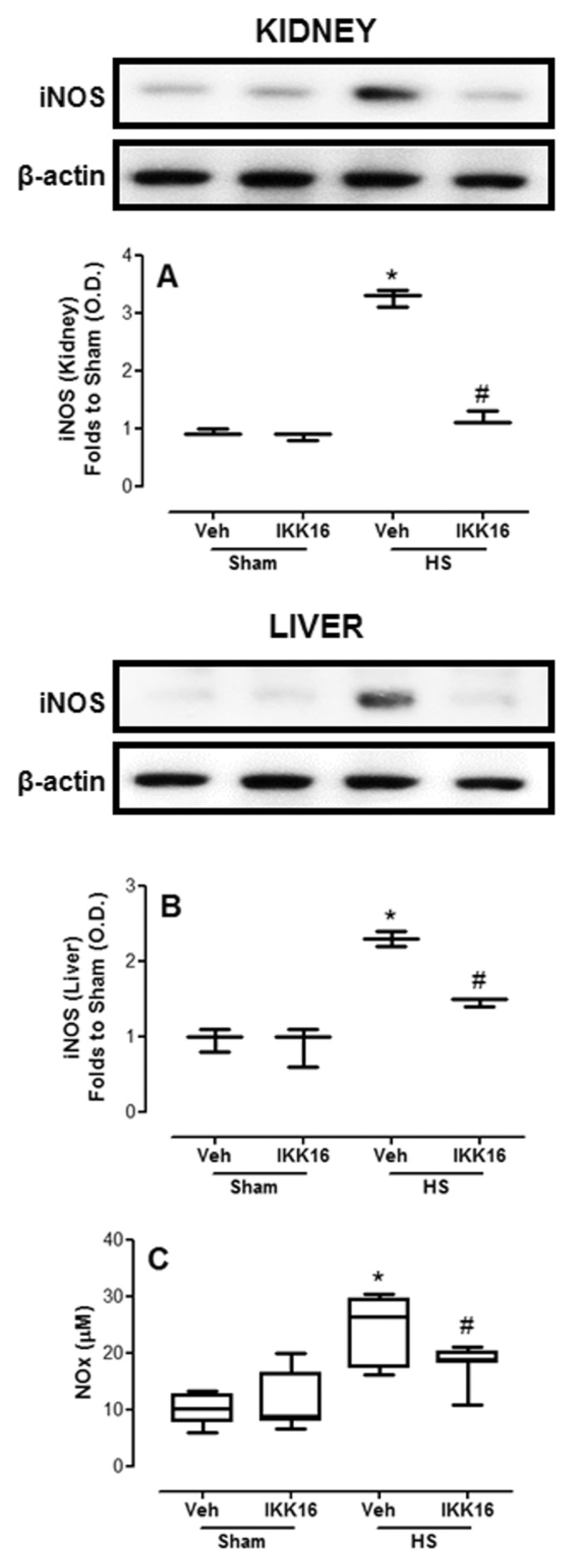
When compared with sham-operated animals, the proinflammatory cytokines TNF-α (Figure 7A) and IL-6 (Figure 7B) and the antiinflammatory cytokine IL-10 (Figure 7C) were significantly increased in the serum of HS-rats treated with vehicle. The administration of IKK16 significantly reduced serum TNF-α, IL-6 and IL-10 (Figure 7) in serum of HS-rats.
Figure 7.
IKK16 attenuates serum inflammatory cytokines. Rats were subjected to HS and received IKK16 (1 mg/kg; i.v.) or vehicle (Veh) (10% DMSO) at resuscitation and 4 h later serum concentrations of TNF-α (A), IL-6 (B) and IL-10 (C) were determined. Sham animals were used as control. Data are presented as box and whiskers (minimum to maximum) and the central line represents the median (n = 7–9 animals per group). *p < 0.05 versus sham group and #p < 0.05 versus HS group; n.d.: not determined.
Effects of IKK16 Treatment on Akt Survival Pathway
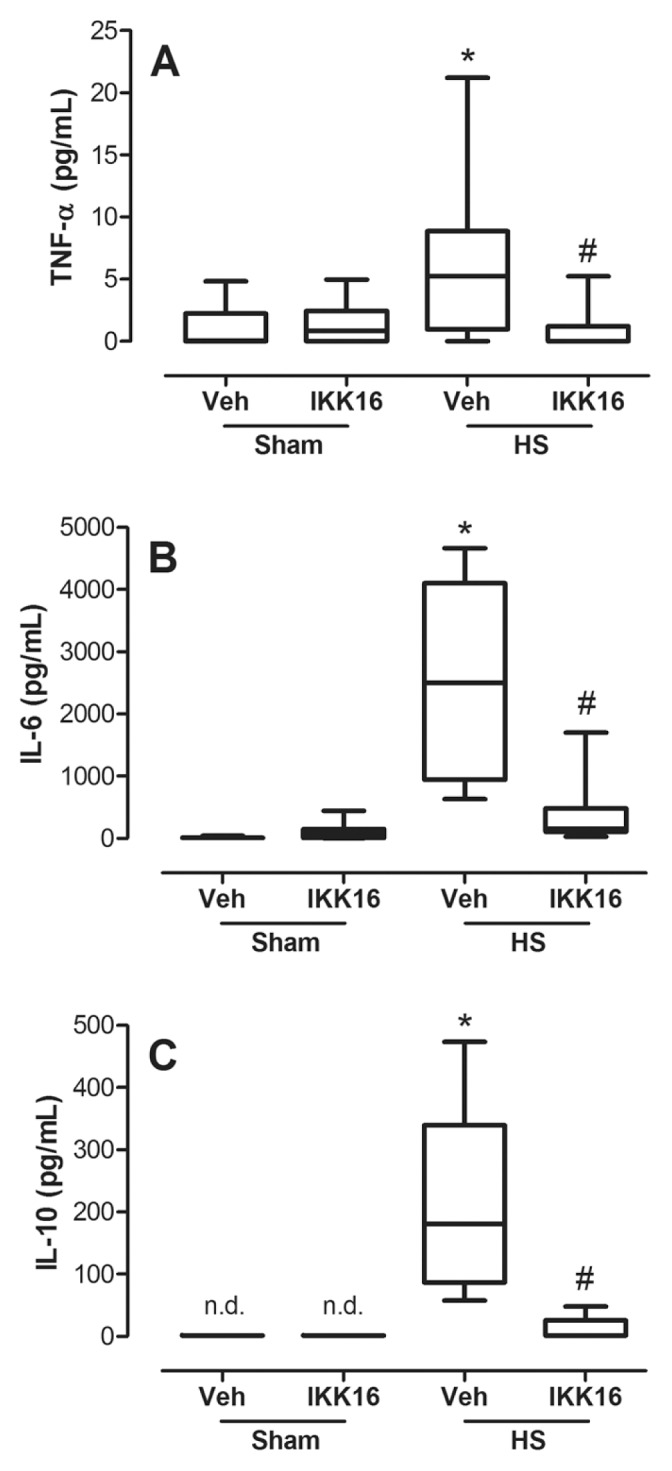
To gain a better insight into the potential mechanisms involved in the beneficial effects of IKK16, we investigated the effects of IKK16 on the Akt-survival pathway, which is known to confer tissue protection (12,13). In HS-rats, HS did not significantly affect the degree of phosphorylation of Akt in either kidney or liver compared with sham-operated rats (Figures 8A, B). Surprisingly, we discovered, however, that the degree of Akt phosphorylation in tissue biopsies of kidney (Figure 8A) and liver (Figure 8B) obtained from HS-rats that had been treated with IKK16 was significantly increased. Interestingly, no such effect was observed in sham-operated rats treated with IKK16.
Figure 8.
IKK16 increases survival. Rats were subjected to HS and received IKK16 (1 mg/kg; i.v.) or vehicle (Veh) (10% DMSO) at resuscitation and 4 h later the phosphorylation of Ser473 and the total Akt on the kidney (A) and liver (B) were determined by Western blotting. Sham animals were used as control. Densitometric analysis of the related bands is expressed as relative optical density, corrected for the corresponding β-actin contents, and normalized using the related mean value of sham-operated band. Data are presented as box and whiskers (minimum to maximum) and the central line represents the median (n = 3–4 animals per group). *p < 0.05 versus sham group and #p < 0.05 versus HS group. (C) HK-2 cells were serum deprived for 24 h and concomitantly incubated with indicates concentrations of IKK16 for the first 4 h. Cell viability was determined by methylthiazoletetrazolium assay and the absorbance was measured at 490 nm. Data are presented as box and whiskers (minimum to maximum) and the central line represents the median of the percentage of survival cells related to complete growth medium (DMEN) control group. *p < 0.05 versus DMEN group and #p < 0.05 versus serum-deprived group.
To gain a better understanding of the role of the potential effects of IKK16 (and its mechanism of action) on cell survival, we investigated the effects of IKK16 on the cell death (apoptosis) caused by serum deprivation in a human kidney (proximal tubule) cell line (HK-2 cells). In HK-2 cells, serum starvation for 24 h resulted in a significant apoptotic cell death (Figure 8C). Interestingly, treatment of these cells with IKK16 (10 nmol/L to 100 nmol/L) during the first 4 h (of the 24 h observation period) significantly reduced the cell apoptosis caused by serum deprivation (Figure 8C).
DISCUSSION
The main findings of this study are that a potent inhibitor of IKK (IKK16) protects rats against the multiple organ dysfunction/failure and inflammation (lung, kidney, liver) associated with HS; inhibits the activation of NF-κB and the expression of NF-κB-dependent proteins (including iNOS and the cytokines TNFα, IL-6 and IL-10, and the adhesion molecule ICAM-1) caused by HS; and activates the Akt survival pathway in animals subjected to HS. We propose that both the inhibition of NF-κB and the activation of Akt contribute to the observed beneficial effects of IKK16 in HS.
HS is associated with a high mortality rate and MOF is the leading cause of late mortality and extended intensive care length of stay (16). MOF involves several mediators and effector cells, and neutrophils play an important role in the pathogenesis of MOF after trauma/HS (17). We have shown increased numbers of neutrophils in the main organs affected by HS (lung, kidney and liver). The presence of neutrophils is usually associated with endothelial and epithelial injury. Neutrophils adhere to vascular endothelium by binding to ICAM-1 on endothelial cells and then transmigrate into end organs, causing direct local cytotoxic cellular effects via degranulation and release of substances such as MPO, NO, ROS and cytokines (18). Treatment of HS-rats with IKK16 decreased the number of neutrophils in lung, kidney and liver, and this antiinflammatory effect may have contributed to the beneficial effects of IKK16 in these organs. In the lung, the increase in the neutrophil number in HS-rats was associated with (and probably secondary to) an increase ICAM-1 expression, which was also attenuated by treatment of HS-rats with IKK16. ICAM-1 is critical in transendothelial migration of neutrophils into multiple organs, including the lungs (19,20). The expression of ICAM-1 in lungs is upregulated in inflammation, and antibodies against ICAM-1 are also able to reduce the migration of neutrophils (21,22).
We have investigated the number of CD68-positive cells, and hence, macrophages in lung, kidney and liver of all animals. HS resulted in a significant increase in the number of macrophages in the lung (but not in kidney or liver), which was attenuated by treatment of HS-rats with IKK16. Indeed, alveolar macrophages play a role in the pathophysiology of acute lung injury (and other lung diseases), because these cells secrete proinflammatory mediators and growth factors (23). To confirm that HS indeed resulted in an increased macrophage number in the lung, we also measured the amount of NAG in the lungs of all animals. HS resulted in an increase in NAG in the lung, which was reduced by treatment of HS-rats with IKK16.
The activation of NF-κB results in the transcription of a number of proinflammatory cytokines, chemokines and proteins that are widely implicated in the pathophysiology of MOF. Thus, it is not surprising that much attention has focused on the development of drugs and tools that are able to target this transcription factor (8–11,24). However, the effects of the inhibition of IKK in the MOF induced by HS have not been investigated. Thus, the present finding is the first experimental evidence that the therapeutic administration of an IKK inhibitor at resuscitation attenuates multiple organ dysfunction/injury associated with HS. The activation of the IKK complex is defined as the phosphorylation of Ser176 and Ser180 on IKKα and Ser177 and Ser181 on IKKβ, which leads to a conformation change in the activation loop of the complex, catalytically activating the kinase domain (25). Activation of the IKK complex also requires IKKγ (NF-κB essential modulator [NEMO]), which functions as a chaperone protein that brings the IKK complex to upstream kinases, leading to IKK phosphorylation and activation (26). HS resulted in the phosphorylation of Ser176/180 on IKKαβ, indicating IKK activation, which in turn caused the phosphorylation of Ser32/36 on IκBα and consequently promoted p65 NF-κB subunit translocation to the nucleus. Treatment of HS-animals on resuscitation with IKK16 decreased the phosphorylation of IKKαβ and, hence, the activation of IKK and ultimately NF-κB.
It is well documented that NF-κB regulates iNOS expression, and iNOS-derived NO is involved on the pathogenesis of organ injury in HS and septic shock (27–30). In addition to contributing to organ injury and vascular decompensation in HS (27), NO can react with super-oxide and produce peroxynitrite, which causes cellular damage and subsequent tissue injury (31). We showed that HS caused an upregulation of iNOS expression in kidney and liver, as well as an increase in systemic NOx levels. Most notably, the inhibition of IKK on resuscitation attenuated iNOS expression and NOx production. The reduction of the iNOS/NO pathway may contribute to the organ protection afforded by IKK16, because several strategies which reduce an excessive formation of NO also reduce organ injury (32–35).
The proinflammatory cytokines TNF-α and IL-6 are important mediators of alterations associated with organ dysfunction and even lethality following hemorrhage and resuscitation (3,15,36,37). The use of monoclonal antibodies against TNF-α attenuated the cardiovascular consequences and improved the survival rate in HS (36), and a monoclonal antibody against IL-6 reduced organ dysfunction and inflammation in HS (37). Our results are in line with these studies, because we also report an association between high levels of TNF-α and IL-6 and the development of organ injury and dysfunction. We thus believe that the beneficial effects of IKK16 on organ failure and dysfunction followed by HS may, in part, be due to the reduction of the formation of these proinflammatory cytokines secondary to the inhibition of NF-κB.
Inhibitors of the NF-κB pathway may also prolong the inflammatory response by preventing (or slowing down) the resolution of inflammation (38). Indeed, the fact that inhibitors of NF-κB not only reduce proinflammatory mediators, but also prevent the formation of proteins which play a key role in the resolution of inflammation may well be a concern, as most of these mediators are produced and released at the same time in inflammation, sepsis and septic shock (39–42). Thus, we have also evaluated the effects of IKK16 on the formation of IL-10, a well-known antiinflammatory cytokine. We have observed an increase on IL-10 concentration in HS-rats and a reduction of this cytokine in rats treated with IKK16. Although enhanced levels of IL-10 are associated with a better outcome in some diseases associated with inflammation (43–45), the upregulation of IL-10 has also been associated with immunosuppression after sepsis (46) and trauma-hemorrhage (47,48), resulting in delayed clinical recovery in trauma patients (39) and aggravation of lung injury secondary to posttraumatic pneumonia (49). Thus, we believe that the reduction in IL-10 production may, on balance, contribute to the beneficial effects of IKK16 in trauma/hemorrhage.
To our surprise, we discovered that the treatment of HS-rats with IKK16 increased the phosphorylation and, hence, the activation of the kinase Akt. The kinase Akt is a member of the phosphoinositide 3-kinase signal transduction enzyme family, activation of which plays a pivotal role on the protection of several organs against ischemia-reperfusion injury (50) and sepsis (12). It is known that NF-κB is upstream of the activation of Akt (51), although the functional link between Akt and the NF-κB pathway is still controversial. For instance, the activation of NF-κB by bacterial proteins has been reported to involve Akt activation (52). However, other studies have shown that Akt negatively regulates p65 transactivation induced by activation of receptors that recognize either extracellular or intracellular bacterial products (53,54). In addition, the activation of Akt by IKK16 has been recently reported in sepsis (12) and lung injury (13). Overall, these data suggest a complex cross-talk between the Akt and NF-κB signaling pathways, which still warrants further investigation.
Moreover, we report here that IKK16 attenuates the severe renal dysfunction caused by HS, which is frequently associated with apoptosis or necrosis of proximal tubule cells (55–57). As the activation of Akt is also implicated in cell survival (58–60), we hypothesized that activation of Akt induced by IKK16 may enhance cell survival. Thus, we investigated the effect of IKK16 on the survival of human proximal tubular cells subjected to serum deprivation. Most notably, concentrations of IKK16 as low as 10 nmol/L attenuated the death of proximal tubule cells caused by serum deprivation. Indeed, this is the first study that demonstrates that IKK16 protects proximal tubule cells (and indeed any other cell line) against cell death in vitro. Clearly, the death of proximal tubule cells in response to serum deprivation is not due to excessive inflammation and, hence, the observed beneficial effects of IKK16 are not related to its antiinflammatory effects. Thus, we speculate that activation of Akt by IKK16 improves cell survival (at least of proximal tubule cells) and may contribute to its ability to reduce organ injury and dysfunction induced by HS.
Our experimental study of HS has several limitations that need to be considered. The model used here is a very acute rodent model of severe HS, which leads to MOF and systemic inflammation within a few hours of the onset of resuscitation. Although effective in this acute setting, we cannot conclude that IKK inhibition may be of benefit in other models of HS (possibly conducted in larger animals), in which the animals are resuscitated and followed up for longer periods after the initial insult. Due to the short follow-up time, we were unable to evaluate the effects of IKK16 on long-term mortality. In our experimental model, we use heparin to prevent the formation of clots in our lines. Heparin is not only an anticoagulant, it also has many potentially beneficial effects (61), although these were clearly not sufficient to prevent the development of organ injury/dysfunction in the model of HS used here.
CONCLUSION
In conclusion, our data show for the first time that the potent IKK inhibitor IKK16 attenuates the organ injury/dysfunction associated with HS. This effect is likely to be due to the prevention of excessive inflammation (measured as increase of neutrophil number [lung/kidney/liver], increase in macrophage number [lung], increased expression of ICAM-1 [lung] and iNOS [lung/kidney/liver], and enhanced serum concentration of TNF-α, IL-6, IL-10 and NOx) and also due to increased activation of the Akt-survival pathway. Thus, the inhibition of IKK complex prior to or during resuscitation represents a novel strategy for the prevention of organ injury and dysfunction induced by HS.
ACKNOWLEDGMENTS
R Sordi is supported by the program Science without Borders, CAPES Foundation, Ministry of Education of Brazil, Brasilia/DF, Brazil; NSA Patel is, in part, supported by the Bart’s and The London Charity (753/1722). The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme (FP7/2007-2013) under REA grant agreement n° 608765, from the William Harvey Research Foundation and University of Turin (Ricerca Locale ex-60%). This work contributes to the Organ Protection research theme of the Barts Centre for Trauma Sciences, supported by the Barts and The London Charity (Award 753/1722) and forms part of the research themes contributing to the translational research portfolio of Barts and the London Cardiovascular Biomedical Research Unit, which is supported and funded by the National Institute of Health Research. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Online address: http://www.molmed.org
DISCLOSURE
The authors declare they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Cite this article as: Sordi R, et al. (2015) Inhibition of IκB kinase attenuates the organ injury and dysfunction associated with hemorrhagic shock. Mol. Med. 21:563–75.
REFERENCES
- 1.Krug EG, Sharma GK, Lozano R. The global burden of injuries. Am J Public Health. 2000;90:523–6. doi: 10.2105/ajph.90.4.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Minei JP, et al. The changing pattern and implications of multiple organ failure after blunt injury with hemorrhagic shock. Crit Care Med. 2012;40:1129–35. doi: 10.1097/CCM.0b013e3182376e9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jarrar D, Chaudry IH, Wang P. Organ dysfunction following hemorrhage and sepsis: mechanisms and therapeutic approaches (review) Int J Mol Med. 1999;4:575–83. doi: 10.3892/ijmm.4.6.575. [DOI] [PubMed] [Google Scholar]
- 4.Beg AA, Baldwin AS., Jr The I kappa B proteins: multifunctional regulators of Rel/NF-kappa B transcription factors. Genes Dev. 1993;7:2064–70. doi: 10.1101/gad.7.11.2064. [DOI] [PubMed] [Google Scholar]
- 5.DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature. 1997;388:548–54. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- 6.Hinz M, Arslan SÇ, Scheidereit C. It takes two to tango: IκBs, the multifunctional partners of NF-κB. Immunol Rev. 2012;246:59–76. doi: 10.1111/j.1600-065X.2012.01102.x. [DOI] [PubMed] [Google Scholar]
- 7.Hinz M, Scheidereit C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 2014;15:46–61. doi: 10.1002/embr.201337983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chima RS, et al. Ciglitazone ameliorates lung inflammation by modulating the inhibitor kappaB protein kinase/nuclear factor-kappaB pathway after hemorrhagic shock. Crit Care Med. 2008;36:2849–57. doi: 10.1097/ccm.0b013e318187810e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao C, et al. An exogenous hydrogen sulphide donor, NaHS, inhibits the nuclear factor κB inhibitor kinase/nuclear factor κb inhibitor/nuclear factor-κB signaling pathway and exerts cardioprotective effects in a rat hemorrhagic shock model. Biol Pharm Bull. 2012;35:1029–34. doi: 10.1248/bpb.b110679. [DOI] [PubMed] [Google Scholar]
- 10.Korff S, et al. Eritoran attenuates tissue damage and inflammation in hemorrhagic shock/trauma. J Surg Res. 2013;184:e17–25. doi: 10.1016/j.jss.2013.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patel NS, et al. A nonerythropoietic peptide that mimics the 3D structure of erythropoietin reduces organ injury/dysfunction and inflammation in experimental hemorrhagic shock. Mol Med. 2011;17:883–92. doi: 10.2119/molmed.2011.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coldewey SM, Rogazzo M, Collino M, Patel NS, Thiemermann C. Inhibition of IκB kinase reduces the multiple organ dysfunction caused by sepsis in the mouse. Dis Model Mech. 2013;6:1031–42. doi: 10.1242/dmm.012435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shu YS, Tao W, Miao QB, Zhu YB, Yang YF. Improvement of ventilation-induced lung injury in a rodent model by inhibition of inhibitory κB kinase. J Trauma Acute Care Surg. 2014;76:1417–24. doi: 10.1097/TA.0000000000000229. [DOI] [PubMed] [Google Scholar]
- 14.McDonald MC, et al. Calpain inhibitor I reduces the activation of nuclear factor-kappaB and organ injury/dysfunction in hemorrhagic shock. FASEB J. 2001;15:171–86. doi: 10.1096/fj.99-0645com. [DOI] [PubMed] [Google Scholar]
- 15.Sordi R, et al. ‘Preconditioning’ with low dose lipopolysaccharide aggravates the organ injury/dysfunction caused by hemorrhagic shock in rats. PLoS One. 2015;10:e0122096. doi: 10.1371/journal.pone.0122096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciesla DJ, et al. A 12-year prospective study of postinjury multiple organ failure: has anything changed? Arch Surg. 2005;140:432–8. doi: 10.1001/archsurg.140.5.432. [DOI] [PubMed] [Google Scholar]
- 17.Tsukamoto T, Chanthaphavong RS, Pape HC. Current theories on the pathophysiology of multiple organ failure after trauma. Injury. 2010;41:21–6. doi: 10.1016/j.injury.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 18.Dewar D, Moore FA, Moore EE, Balogh Z. Postinjury multiple organ failure. Injury. 2009;40:912–8. doi: 10.1016/j.injury.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 19.Bochner BS, et al. Adhesion of human basophils, eosinophils, and neutrophils to inter-leukin 1-activated human vascular endothelial cells: contributions of endothelial cell adhesion molecules. J Exp Med. 1991;173:1553–7. doi: 10.1084/jem.173.6.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly M, Hwang JM, Kubes P. Modulating leukocyte recruitment in inflammation. J Allergy Clin Immunol. 2007;120:3–10. doi: 10.1016/j.jaci.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 21.Sorkness RL, et al. Effect of ICAM-1 blockade on lung inflammation and physiology during acute viral bronchiolitis in rats. Pediatr Res. 2000;47:819–24. doi: 10.1203/00006450-200006000-00023. [DOI] [PubMed] [Google Scholar]
- 22.Burns AR, Takei F, Doerschuk CM. Quantitation of ICAM-1 expression in mouse lung during pneumonia. J Immunol. 1994;153:3189–98. [PubMed] [Google Scholar]
- 23.Niesler U, Palmer A, Radermacher P, Huber-Lang MS. Role of alveolar macrophages in the inflammatory response after trauma. Shock. 2014;42:3–10. doi: 10.1097/SHK.0000000000000167. [DOI] [PubMed] [Google Scholar]
- 24.Patel NS, et al. Delayed administration of pyroglutamate helix B surface peptide (pHBSP), a novel nonerythropoietic analog of erythropoietin, attenuates acute kidney injury. Mol Med. 2012;18:719–27. doi: 10.2119/molmed.2012.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amaya M, Keck F, Bailey C, Narayanan A. The role of the IKK complex in viral infections. Pathog Dis. 2014;72:32–44. doi: 10.1111/2049-632X.12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu F, Xia Y, Parker AS, Verma IM. IKK biology. Immunol Rev. 2012;246:239–53. doi: 10.1111/j.1600-065X.2012.01107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thiemermann C, Szabó C, Mitchell JA, Vane JR. Vascular hyporeactivity to vasoconstrictor agents and hemodynamic decompensation in hemorrhagic shock is mediated by nitric oxide. Proc Natl Acad Sci U S A. 1993;90:267–71. doi: 10.1073/pnas.90.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szabó C, Southan GJ, Thiemermann C. Beneficial effects and improved survival in rodent models of septic shock with S-methylisothiourea sulfate, a potent and selective inhibitor of inducible nitric oxide synthase. Proc Natl Acad Sci U S A. 1994;91:12472–6. doi: 10.1073/pnas.91.26.12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandes D, et al. Late, but not early, inhibition of soluble guanylate cyclase decreases mortality in a rat sepsis model. J Pharmacol Exp Ther. 2009;328:991–9. doi: 10.1124/jpet.108.142034. [DOI] [PubMed] [Google Scholar]
- 30.Sordi R, Menezes-de-Lima O, Della-Justina AM, Rezende E, Assreuy J. Pneumonia-induced sepsis in mice: temporal study of inflammatory and cardiovascular parameters. Int J Exp Pathol. 2013;94:144–55. doi: 10.1111/iep.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDonald MC, Izumi M, Cuzzocrea S, Thiemermann C. A novel, potent and selective inhibitor of the activity of inducible nitric oxide synthase (GW274150) reduces the organ injury in hemorrhagic shock. J Physiol Pharmacol. 2002;53(4 Pt 1):555–69. [PubMed] [Google Scholar]
- 32.Shirhan M, Moochhala SM, Kerwin SY, Ng KC, Lu J. Influence of selective nitric oxide synthetase inhibitor for treatment of refractory haemorrhagic shock. Resuscitation. 2004;61:221–9. doi: 10.1016/j.resuscitation.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Ganster F, et al. Effects of hydrogen sulfide on hemodynamics, inflammatory response and oxidative stress during resuscitated hemorrhagic shock in rats. Crit Care. 2010;14:R165. doi: 10.1186/cc9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang H, Huang Y, Xu H, Hu R, Li QF. Inhibition of hypoxia inducible factor-1α ameliorates lung injury induced by trauma and hemorrhagic shock in rats. Acta Pharmacol Sin. 2012;33:635–43. doi: 10.1038/aps.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sordi R, Fernandes D, Heckert BT, Assreuy J. Early potassium channel blockade improves sepsis-induced organ damage and cardiovascular dysfunction. Br J Pharmacol. 2011;163:1289–301. doi: 10.1111/j.1476-5381.2011.01324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bahrami S, et al. Significance of TNF in hemorrhage-related hemodynamic alterations, organ injury, and mortality in rats. Am J Physiol. 1997;272(5 Pt 2):H2219–26. doi: 10.1152/ajpheart.1997.272.5.H2219. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, et al. Delayed neutralization of interleukin 6 reduces organ injury, selectively suppresses inflammatory mediator, and partially normalizes immune dysfunction following trauma and hemorrhagic shock. Shock. 2014;42:218–27. doi: 10.1097/SHK.0000000000000211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. Possible new role for NF-kappaB in the resolution of inflammation. Nat Med. 2001;7:1291–7. doi: 10.1038/nm1201-1291. [DOI] [PubMed] [Google Scholar]
- 39.Xiao W, et al. A genomic storm in critically injured humans. J Exp Med. 2011;208:2581–90. doi: 10.1084/jem.20111354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Osuchowski MF, Craciun F, Weixelbaumer KM, Duffy ER, Remick DG. Sepsis chronically in MARS: systemic cytokine responses are always mixed regardless of the outcome, magnitude, or phase of sepsis. J Immunol. 2012;189:4648–56. doi: 10.4049/jimmunol.1201806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Novotny AR, et al. Mixed antagonist response and sepsis severity-dependent dysbalance of pro- and anti-inflammatory responses at the onset of postoperative sepsis. Immunobiology. 2012;217:616–21. doi: 10.1016/j.imbio.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 42.Sordi R, et al. Dual role of lipoxin A4 in pneumosepsis pathogenesis. Int Immunopharmacol. 2013;17:283–92. doi: 10.1016/j.intimp.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 43.Howard M, Muchamuel T, Andrade S, Menon S. Interleukin 10 protects mice from lethal endotoxemia. J Exp Med. 1993;177:1205–8. doi: 10.1084/jem.177.4.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Laethem JL, et al. Interleukin 10 prevents necrosis in murine experimental acute pancreatitis. Gastroenterology. 1995;108:1917–22. doi: 10.1016/0016-5085(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 45.Aggarwal NR, et al. Immunological priming requires regulatory T cells and IL-10-producing macrophages to accelerate resolution from severe lung inflammation. J Immunol. 2014;192:4453–64. doi: 10.4049/jimmunol.1400146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fumeaux T, Pugin J. Role of interleukin-10 in the intracellular sequestration of human leukocyte antigen-DR in monocytes during septic shock. Am J Respir Crit Care Med. 2002;166:1475–82. doi: 10.1164/rccm.200203-217OC. [DOI] [PubMed] [Google Scholar]
- 47.Adib-Conquy M, et al. Toll-like receptor-mediated tumor necrosis factor and interleukin-10 production differ during systemic inflammation. Am J Respir Crit Care Med. 2003;168:158–64. doi: 10.1164/rccm.200209-1077OC. [DOI] [PubMed] [Google Scholar]
- 48.Roquilly A, et al. Hydrocortisone prevents immunosuppression by interleukin-10+ natural killer cells after trauma-hemorrhage. Crit Care Med. 2014;42:e752–61. doi: 10.1097/CCM.0000000000000658. [DOI] [PubMed] [Google Scholar]
- 49.Carles M, et al. Heat-shock response increases lung injury caused by Pseudomonas aeruginosa via an interleukin-10-dependent mechanism in mice. Anesthesiology. 2014;120:1450–62. doi: 10.1097/ALN.0000000000000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai Z, Semenza GL. Phosphatidylinositol-3-kinase signaling is required for erythropoietin-mediated acute protection against myocardial ischemia/reperfusion injury. Circulation. 2004;109:2050–3. doi: 10.1161/01.CIR.0000127954.98131.23. [DOI] [PubMed] [Google Scholar]
- 51.Meng F, Liu L, Chin PC, D’Mello SR. Akt is a downstream target of NF-kappa B. J Biol Chem. 2002;277:29674–80. doi: 10.1074/jbc.M112464200. [DOI] [PubMed] [Google Scholar]
- 52.Mansell A, Khelef N, Cossart P, O’Neill LA. Internalin B activates nuclear factor-kappa B via Ras, phosphoinositide 3-kinase, and Akt. J Biol Chem. 2001;276(47):43597–603. doi: 10.1074/jbc.M105202200. [DOI] [PubMed] [Google Scholar]
- 53.Guha M, Mackman N. The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124–32. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- 54.Zhao L, Lee JY, Hwang DH. The phosphatidylinositol 3-kinase/Akt pathway negatively regulates Nod2-mediated NF-kappaB pathway. Biochem Pharmacol. 2008;75:1515–25. doi: 10.1016/j.bcp.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 55.Abdelrahman M, et al. Erythropoietin attenuates the tissue injury associated with hemorrhagic shock and myocardial ischemia. Shock. 2004;22:63–9. doi: 10.1097/01.shk.00001276869.21260.9d. [DOI] [PubMed] [Google Scholar]
- 56.Chai W, et al. Exogenous hydrogen sulfide protects against traumatic hemorrhagic shock via attenuation of oxidative stress. J Surg Res. 2012;176:210–9. doi: 10.1016/j.jss.2011.07.016. [DOI] [PubMed] [Google Scholar]
- 57.Faguer S, et al. Hnf-1β transcription factor is an early hif-1α-independent marker of epithelial hypoxia and controls renal repair. PLoS One. 2013;8:e63585. doi: 10.1371/journal.pone.0063585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000;101:660–7. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rajesh KG, et al. Hydrophilic bile salt ursodeoxycholic acid protects myocardium against reperfusion injury in a PI3K/Akt dependent pathway. J Mol Cell Cardiol. 2005;39:766–76. doi: 10.1016/j.yjmcc.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 60.Yu HP, et al. The PI3K/Akt pathway mediates the nongenomic cardioprotective effects of estrogen following trauma-hemorrhage. Ann Surg. 2007;245:971–7. doi: 10.1097/01.sla.0000254417.15591.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tyrrell DJ, Horne AP, Holme KR, Preuss JM, Page CP. Heparin in inflammation: potential therapeutic applications beyond anticoagulation. Adv Pharmacol. 1999;46:151–208. doi: 10.1016/s1054-3589(08)60471-8. [DOI] [PubMed] [Google Scholar]