

Abstract
PT70 is a diaryl ether inhibitor of InhA, the enoyl-ACP reductase in the Mycobacterium tuberculosis fatty acid biosynthesis pathway. It has a residence time of 24 min on the target, and also shows antibacterial activity in a mouse model of tuberculosis infection. Due to the interest in studying target tissue pharmacokinetics of PT70, we developed a method to radiolabel PT70 with carbon-11 and have studied its pharmacokinetics in mice and baboons using positron emission tomography.
Keywords: Positron emission tomography, InhA enoyl-ACP reductase, Residence time, Carbon-11, PK-PD
Graphical Abstract
Mycobacterium tuberculosis, the causative agent of human tuberculosis (TB),2 infects one-third of the world’s population and is responsible for 9.0 million new infections and 1.5 million deaths in 2013.3 The situation is complicated by HIV/AIDS infection, multidrug resistant TB (MDR-TB) and extensively drug resistant TB (XDR-TB).4 In 2008, 22% of new TB cases were reported to be MDR-TB.5 The basic regimen recommended for TB treatment is comprised of the four front-line drugs: isoniazid (INH), rifampicin (RIF), pyrazinamide (PZA), and ethambutol (EMB). The regimen starts with two months initial phase, followed by INH and RIF for another four months. MDR-TB is resistant against INH or RIF, contributes to the spread and worsens the situation by lengthening the treatment from 6 months to nearly 2 years. XDR-TB is resistant to both front-line and second-line drugs and is extremely difficult to cure.6 Taken together, novel drugs with activity against drug-resistant TB are therefore urgently needed.
InhA, the enoyl-ACP reductase involved in the M. tuberculosis fatty acid biosynthesis (FAS II) pathway,7 catalyzes the last reaction in the elongation cycle, and is the target for INH, one of the most effective and widely used anti-tubercular drugs.7, 8 INH is a prodrug that is activated by the mycobacterial catalase-peroxidase enzyme KatG.7, 9–11 Activation of INH leads to the formation of the INH-NAD, which is a slow, tight-binding inhibitor of InhA.8 Because a substantial fraction of all clinical isolates that are resistant to INH result from KatG mutations,12–15 compounds that target InhA, but that do not require activation by KatG, are promising candidates for drug-resistant M. tuberculosis.
Based on the above hypothesis, we and others have developed direct inhibitors of InhA that do not require activation.16–20 In our own studies, we have used the diaryl ether scaffold of the enoyl-ACP reductase triclosan as a starting point for the generation of InhA inhibitors. Our initial studies resulted in a series of compound with nanomolar potency for InhA and minimum inhibitory concentrations (MICs) of 1 – 2 μg/mL against both drug-sensitive and drug-resistant strains of M. tuberculosis.21 In separate studies, we examined the in vitro and in vivo antibacterial activity of these compounds against other pathogens, and discovered that the residence time (tR) of the diphenyl ethers on the Francisella tularensis FabI correlated with the efficacy of the compounds in an animal model of tularemia,22 supporting the importance of drug-target kinetics in drug action.23–26 Our initial series of InhA inhibitors were not time dependent,21 but based on the F. tularensis data, and the knowledge that the INH-NAD adduct is a slow onset inhibitor of InhA with a residence time of 1 h on the enzyme,8 we set out to develop time-dependent diaryl ether inhibitors of InhA.
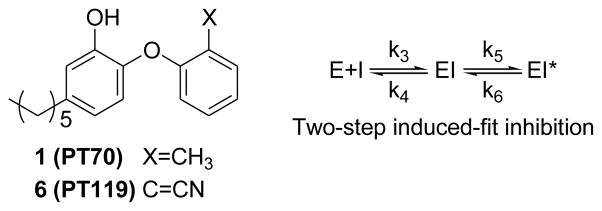
Slow-onset inhibition of the FabI (InhA) enoyl-ACP reductases is coupled to ordering of an active site loop (substrate-binding loop), which closes over the substrate-binding pocket.27–30 Using this information we developed PT70, a slow-onset inhibitor of InhA that binds preferentially to the InhA:NAD+ complex and has a residence time of 24 min on the target.28 PT70 binds to InhA through a two-step induced-fit mechanism, in which the rapid formation of the initial EI complex is followed by the slow formation of the final enzyme-inhibitor complex (EI*) (Scheme 1), and X-ray crystallography confirmed that the substrate-binding loop had closed over the inhibitor in the EI* complex.28, 30
Scheme 1.
We examined the efficacy of this compound in a mouse model of TB infection. Although PT70 did not affect the bacterial burden in the lungs of the infected animals, the compound reduced the bacterial load in the spleens, whereas a rapid reversible inhibitor of InhA demonstrated no in vivo antibacterial activity.31 Encouraged by these results we set out to examine the metabolism of PT70 as well as the biodistribution of this compound in mice as well as in non-human primates.
Traditionally, due to the limitation of methodologies to access tissue pharmacokinetics (PK), plasma PK has been widely used as a surrogate to evaluate the exposure of candidate drugs and to compare with their corresponding minimum inhibition concentrations in the expectation to predict in vivo efficacies. However, the equilibrium between plasma and target tissue cannot always be taken for granted and drug levels in target tissues are often substantially different from the corresponding plasma levels.32, 33 In the case of antibacterial drugs, failure to reach optimal concentration at target site can not only cause failed therapy but also trigger resistance.34 In order to address these issues, the Food and Drug Administration (FDA) requires target tissue distribution studies at infected and uninfected sites.33
Positron Emission Tomography (PET) has emerged as a powerful tool for studying drug action in humans and laboratory animals.35 This approach quantifies the three-dimensional distribution of drugs in real-time using the non-invasive detection of positron-emitting isotopes.36 It can also be used to validate the mode of drug action by establishing that drugs actually engage their intended targets in vivo.37 Since this is a non-invasive technique, preclinical PK/PD studies can thus be readily translated into humans. Although widely used in fields such as neuroscience and oncology, PET is now beginning to be used to study the biodistribution of antibacterial agents.1, 38–40
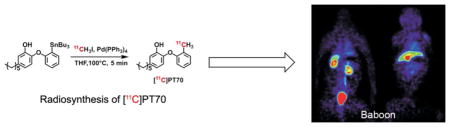
The goal of the present study was to develop a method to incorporate the positron-emitting isotope carbon-11 (half-life: 20.4 min) into PT70 (Scheme 1), and then to study the biodistribution and pharmacokinetics of PT70 in mice and baboons using PET.
The radiosynthesis of [11C]PT70 is shown in Scheme 2. Briefly, 4-ethyl-2-methoxyphenol 3 was prepared from vanillin (2) in three steps as described previously.21, 31 The 2-iodophenoxyphenol 4 was then prepared from 3 in three steps via a diazonium salt intermediate prior to iodide anion attack.41 Subsequently the tributylstannyl intermediate 5 was prepared from 4 using tributylchlorostannane. Introduction of carbon-11 was then achieved from 5 using a modified Stille reaction42 and [11C]CH3I. Initially, the reaction was successfully carried out in toluene at 100°C for 10 min. However this involved a time consuming evaporation before the chromatographic purification due to incompatible solvents, and consequently we explored a range of solvent systems in order to identify conditions that would give the required rate of synthesis, radiochemical yield and solvent compatibility. This screening approach resulted in the use of THF as the solvent with a 5 min reaction time (Scheme 2). This reaction was also tolerant to the phenol group and no major side reactions were observed. The resulting [11C]PT70 was purified by high performance liquid chromatography (HPLC) and concentrated in vacuo. The average decay-corrected yield (DCY), calculated from [11C]CH3I, was 40% – 50% in a total synthesis time of 50 min. Analytical HPLC and TLC demonstrated that the radiolabeled product was over 98% radiochemically pure, with a specific activity of 7 – 13 Ci/μmol at the end of bombardment (EOB).
Scheme 2.
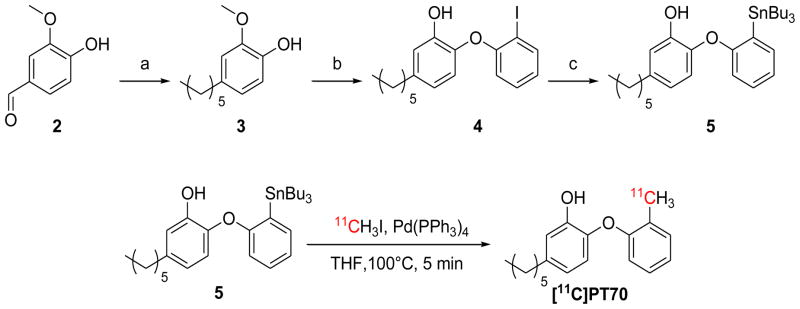
Synthesis of key intermediates and [11C]PT70. Reagents and conditions: (a) i) BnBr, KOH, CH3OH, H2O reflux 4 h, ii) C5H11PPh3BR, n-BuLi, DCM, −78°C to r.t., iii) H2, Pd/C, MeOH, r.t. 16 h (b) i) 1-F-2-NO2-benzene, K2CO3, DMF, 125 °C, 2h. ii) H2, Pd/C, MeOH, r.t., 16 h. iii) HCl, AcOH, NaNO2, 0°C, 40 min then KI, 0 °C 24 h. iv) BBr3, DCM, −78 °C. (c) n-BuLi, Bu3SnCl Et2O, −78°C to r.t. Full details are given in supplementary information.
The lipophilicity (logD) and plasma protein binding (ppb) of PT70 was determined using the radiolabel to report on drug concentration. The logD value determined by measuring the partitioning between octanol and pH 7.4 phosphate buffer was 3.65 which is significantly lower than the clogP value of 6.97 calculated for PT70. This may indicate that the phenolic hydroxyl group of PT70 is partially ionized at pH 7.4, consistent with the knowledge that the pKa of this group in diphenyl ethers is ~ 8.5 depending on ring substitution. Finally, the data also show that PT70 was 99.76% bound to plasma proteins.
Peripheral organ drug distribution was determined in healthy mice (Table 1 and Figure 1) using the methods previously described.39 For all tissues analyzed, the highest concentration of drug occurred 20 min after injection and subsequently decreased. The distribution of [11C]PT70 in blood was also measured. The concentration of [11C]PT70 was much higher in the liver than in other peripheral organs, which might be expected since glucuronidation is the major route of diphenyl ether metabolism.
Table 1.
Biodistribution of [11C]PT70 in healthy mice.
| Time (min) | Spleen | Kidney | Liver | Lung | Blood |
|---|---|---|---|---|---|
| 20 | 1.82 ± 0.28 | 2.21 ± 0.01 | N.D. | 1.36 ± 0.17 | 1.70 ± 0.02 |
| 40 | 0.93 ± 0.14 | 1.45 ± 0.26 | 15.70 ± 1.22 | 1.26 ± 0.16 | 1.46 ± 0.17 |
| 60 | 0.21 ± 0.01 | 0.31 ± 0.03 | 14.09 ± 0.01 | 1.35 ± 0.05 | 0.55 ± 0.14 |
Values given are [%ID/cc].
Figure 1. Biodistribution of [11C]PT70 in mice.

Biodistribution of [11C]PT70 in spleen, kidney, lung and blood in mice at 20, 40 and 60 min after injection. The data are expressed as % injected dose/gram. Each value is the mean ± SD for three animals.
The results for PT70 are broadly similar to data acquired previously for PT119, a cyano derivative of PT70 (Scheme 1).39 PT119 is also an enoyl-ACP reductase inhibitor with a preference for the Staphylococcus aureus FabI (saFabI). PT119 has a residence time of ~12.5 h on saFabI and demonstrates efficacy in a mouse model of MRSA infection. To further compare and contrast the biodistribution of PT70 and PT119, we determined the concentration of [11C]PT70 in a mouse thigh model of MRSA infection. Similar to [11C]PT119, [11C]PT70 showed similar concentrations in the infected compared to the uninfected thighs of mice (data not shown).
The data indicate that PT70 rapidly distributes to the major organs including the spleen, consistent with the ability of this compound to reduce bacterial load in the spleens of mice infected with M. tuberculosis.31 PT70 did not, however, impact bacterial load in the lungs of mice, which is the primary site of infection, despite the rapid distribution of PT70 to the lungs. Since PET imaging reports on the distribution of the parent radiotracer as well as any metabolites that retain the radiolabel, a portion of the signal may derive from radiolabeled metabolites with altered antibacterial activity. In this regard, we previously reported the PK as well as the Phase 1 and 2 metabolism of PT04, a PT70 analog that lacks the B-ring methyl group. 43 These studies demonstrated that O-glucuronidation was a primary route for metabolism, which does not remove the radiolabel from the parent compound. Since the phenolic hydroxyl group is essential for FabI binding, addition of glucuronic acid is expected to reduce the affinity of the compounds for FabI and hence impact the antibacterial activity of the compound.
In addition, PET studies are normally conducted with a single sub-therapeutic microdose of compound, and conclusions about the PK of pharmacological drug doses are made by assuming that drug concentration scales linearly. This is normally true if there are no saturable processes that affect ADME. To provide insight into the effect of a therapeutic drug dose on the biodistribution of [11C]PT70, we studied the tissue concentrations of PT70 following a single 200 mg/kg predose of this compound (Figure 2). We found that pretreatment with PT70 more than doubles the accumulation of radiotracer in the spleen whereas the amount of radiotracer in the lungs is unchanged. Although the efficacy studies are conducted over multiple days of treatment, the observation that predosing raises the level of PT70 in the spleen may provide a potential explanation for the ability of PT70 to reduce bacterial load in this organ in mice infected with M. tuberculosis. A second possibility, raised during review, is that the bacterial population in the spleen, which has disseminated from the primary site of infection in the lung, may be more sensitive to PT70 than the bacterial population in the lung.
Figure 2. Effect of [11C]PT70 biodistribution in mice following a predose of PT70.

Biodistribution of [11C]PT70 following a predose of 200 mg/kg PT70 1 hr prior to imaging. The data were obtained 20 min after injection of [11C]PT70 and are expressed as % injected dose/gram. Each value is the mean ± SD for three animals.
To analyze drug distribution in baboons, dynamic PET scans were performed with [11C]PT70 over 90 min to determine the peripheral organ distribution (Figure 3). The time activity curves (TACs, Figure 4) were generated from the images acquired after i.v. administration of radiotracer by manually drawing the region of interests (ROIs). The injected [11C]PT70 cleared rapidly from the heart, lung and blood, with moderate accumulation in the liver, kidney and spleen, while a large portion of radioactivity accumulated in the gallbladder. The rectilinear scan at around 120 min after administration suggests that another large portion of the injected [11C]PT-70 was eliminated into the bladder. Although we have not conducted efficacy studies with PT70 in non-human primates, the data point to similar drug distribution to that observed in mice, and thus that PT70 might also be expected to reduce bacterial load in the spleen.
Figure 3. Whole body scan of [11C]PT70 in a baboon.
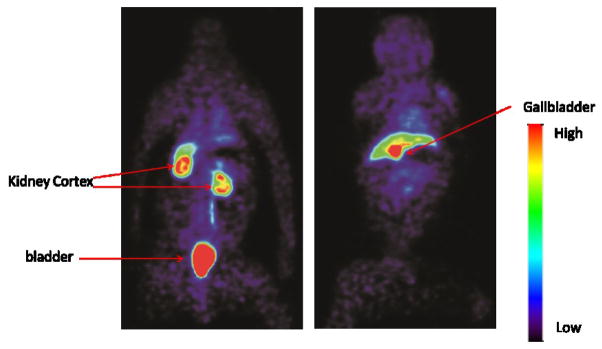
Organ distribution of [11C]PT70 in a healthy baboon using PET imaging. The left and right panels are two different coronal planes that show either the kidneys and bladder, or the liver and gallbladder, respectively. In the right panel, the majority of the radioactivity accumulates in the gallbladder (red) whereas there is less accumulation in the liver (green).
Figure 4. Time-activity curves of [11C]PT70 in the major organs of a baboon.
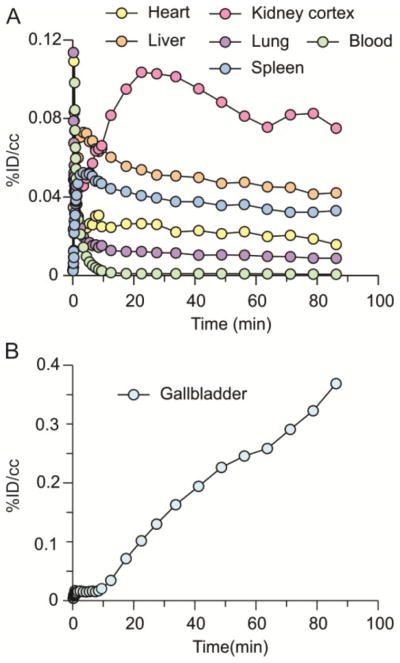
TACs for [11C]PT70 in (A) the heart, lungs, liver, kidneys, blood, spleen and (B) gallbladder.
We next examined the ability of PT70 to penetrate the blood brain barrier (BBB). Images were acquired for 90 min following i.v. administration of radiolabeled drug (Figure 5) and TACs were generated after drawing the ROIs manually (Figure 6). Figures 5 and 6 include data for [11C]PT119, the cyano analog of PT70 (Scheme 1), as well as isoniazid (INH) labeled with carbon-11. The results indicate that both PT119 and PT70 are able to penetrate the brain. Qualitatively, the data also suggest that PT119 has a higher propensity to cross the BBB than PT70, however, further studies are required to fully quantitate the differences in brain levels between the compounds.
Figure 5. PET images for brain uptake of [11C]PT70, [11C]PT119 and [11C]INH.
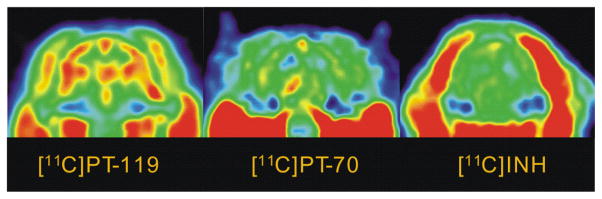
Dose corrected coronal images summed over 15–90 min. The relative concentration of radiotracer is shown using the NIH color scale. Data for INH are taken from Liu et al.1
Figure 6. Time-activity curves of [ C]PT70, [ C]PT119 and [11C]INH in the baboon brain.

Data for INH are taken from Liu et al.1
In summary, PT70, a time-dependent InhA inhibitor, has been labeled with the positron emitting isotope carbon-11. [11C]PT70 was synthesized using a one-step modified Stille reaction, and then purified and formulated within 1 h, with over 98% radiochemical purity. The biodistribution and PK of PT70 was then determined in mice and baboons. The data indicate that PT70 rapidly distributes to the major organs in both mice and baboons shedding light on the correlation between drug concentration and efficacy, and providing a platform for the rational design of novel antibacterial agents.
Supplementary Material
Acknowledgments
This study was supported by the National Institutes of Health grant GM102864 to PJT
Footnotes
Supplementary data associated with this article can be found, in the online version.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu L, Xu Y, Shea C, Fowler JS, Hooker JM, Tonge PJ. J Med Chem. 2010;53:2882. doi: 10.1021/jm901858n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scheindlin S. Mol Interv. 2006;6:124. doi: 10.1124/mi.6.3.1. [DOI] [PubMed] [Google Scholar]
- 3.Zumla A, George A, Sharma V, Herbert RH, Oxley A, Oliver M. Lancet Glob Health. 2015;3:e10. doi: 10.1016/S2214-109X(14)70361-4. [DOI] [PubMed] [Google Scholar]
- 4.Lienhardt C, Glaziou P, Uplekar M, Lonnroth K, Getahun H, Raviglione M. Nat Rev Microbiol. 2012;10:407. doi: 10.1038/nrmicro2797. [DOI] [PubMed] [Google Scholar]
- 5.Vashishtha VM. Indian Pediatr. 2009;46:401. [PubMed] [Google Scholar]
- 6.Jain A, Mondal R. FEMS Immunol Med Microbiol. 2008;53:145. doi: 10.1111/j.1574-695X.2008.00400.x. [DOI] [PubMed] [Google Scholar]
- 7.Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, de Lisle G, Jacobs WR., Jr Science. 1994;263:227. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 8.Rawat R, Whitty A, Tonge PJ. Proc Natl Acad Sci U S A. 2003;100:13881. doi: 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Heym B, Allen B, Young D, Cole S. Nature. 1992;358:591. doi: 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
- 10.Johnsson K, King DS, Schultz PG. J Am Chem Soc. 1995;117:5009. [Google Scholar]
- 11.Basso LA, Zheng RJ, Blanchard JS. J Am Chem Soc. 1996;118:11301. [Google Scholar]
- 12.Heym B, Honore N, Truffot-Pernot C, Banerjee A, Schurra C, Jacobs WR, Jr, van Embden JD, Grosset JH, Cole ST. Lancet. 1994;344:293. doi: 10.1016/s0140-6736(94)91338-2. [DOI] [PubMed] [Google Scholar]
- 13.Stoeckle MY, Guan L, Riegler N, Weitzman I, Kreiswirth B, Kornblum J, Laraque F, Riley LW. J Infect Dis. 1993;168:1063. doi: 10.1093/infdis/168.4.1063. [DOI] [PubMed] [Google Scholar]
- 14.Musser JM, Kapur V, Williams DL, Kreiswirth BN, van Soolingen D, van Embden JD. J Infect Dis. 1996;173:196. doi: 10.1093/infdis/173.1.196. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan G, Post FA, Moreira AL, Wainwright H, Kreiswirth BN, Tanverdi M, Mathema B, Ramaswamy SV, Walther G, Steyn LM, Barry CE, 3rd, Bekker LG. Infect Immun. 2003;71:7099. doi: 10.1128/IAI.71.12.7099-7108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sink R, Sosic I, Zivec M, Fernandez-Menendez R, Turk S, Pajk S, Alvarez-Gomez D, Lopez-Roman EM, Gonzales-Cortez C, Rullas-Triconado J, Angulo-Barturen I, Barros D, Ballell-Pages L, Young RJ, Encinas L, Gobec S. J Med Chem. 2015;58:613. doi: 10.1021/jm501029r. [DOI] [PubMed] [Google Scholar]
- 17.Perryman AL, Yu W, Wang X, Ekins S, Forli S, Li SG, Freundlich JS, Tonge PJ, Olson AJ. J Chem Inf Model. 2015;55:645. doi: 10.1021/ci500672v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manjunatha UH, SPSR, Kondreddi RR, Noble CG, Camacho LR, Tan BH, Ng SH, Ng PS, Ma NL, Lakshminarayana SB, Herve M, Barnes SW, Yu W, Kuhen K, Blasco F, Beer D, Walker JR, Tonge PJ, Glynne R, Smith PW, Diagana TT. Sci Transl Med. 2015;7:269ra3. doi: 10.1126/scitranslmed.3010597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vilcheze C, Baughn AD, Tufariello J, Leung LW, Kuo M, Basler CF, Alland D, Sacchettini JC, Freundlich JS, Jacobs WR., Jr Antimicrob Agents Chemother. 2011;55:3889. doi: 10.1128/AAC.00266-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freundlich JS, Wang F, Vilcheze C, Gulten G, Langley R, Schiehser GA, Jacobus DP, Jacobs WR, Jr, Sacchettini JC. ChemMedChem. 2009;4:241. doi: 10.1002/cmdc.200800261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sullivan TJ, Truglio JJ, Boyne ME, Novichenok P, Zhang X, Stratton CF, Li HJ, Kaur T, Amin A, Johnson F, Slayden RA, Kisker C, Tonge PJ. ACS Chem Biol. 2006;1:43. doi: 10.1021/cb0500042. [DOI] [PubMed] [Google Scholar]
- 22.Lu H, England K, am Ende C, Truglio JJ, Luckner S, Reddy BG, Marlenee NL, Knudson SE, Knudson DL, Bowen RA, Kisker C, Slayden RA, Tonge PJ. ACS Chem Biol. 2009;4:221. doi: 10.1021/cb800306y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Copeland RA, Pompliano DL, Meek TD. Nat Rev Drug Discov. 2006;5:730. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- 24.Lu H, Tonge PJ. Curr Opin Chem Biol. 2010;14:467. doi: 10.1016/j.cbpa.2010.06.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swinney DC. Curr Opin Drug Discov Devel. 2009;12:31. [PubMed] [Google Scholar]
- 26.Walkup GK, You Z, Ross PL, Allen EK, Daryaee F, Hale MR, O’Donnell J, Ehmann DE, Schuck VJ, Buurman ET, Choy AL, Hajec L, Murphy-Benenato K, Marone V, Patey SA, Grosser LA, Johnstone M, Walker SG, Tonge PJ, Fisher SL. Nat Chem Biol. 2015;11:416. doi: 10.1038/nchembio.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stewart MJ, Parikh S, Xiao GP, Tonge PJ, Kisker C. J Mol Biol. 1999;290:859. doi: 10.1006/jmbi.1999.2907. [DOI] [PubMed] [Google Scholar]
- 28.Luckner SR, Liu N, Am Ende CW, Tonge PJ, Kisker C. J Biol Chem. 2010;285:14330. doi: 10.1074/jbc.M109.090373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ward WH, Holdgate GA, Rowsell S, McLean EG, Pauptit RA, Clayton E, Nichols WW, Colls JG, Minshull CA, Jude DA, Mistry A, Timms D, Camble R, Hales NJ, Britton CJ, Taylor IW. Biochemistry. 1999;38:12514. doi: 10.1021/bi9907779. [DOI] [PubMed] [Google Scholar]
- 30.Li HJ, Lai CT, Pan P, Yu W, Liu N, Bommineni GR, Garcia-Diaz M, Simmerling C, Tonge PJ. ACS Chem Biol. 2014;9:986. doi: 10.1021/cb400896g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pan P, Knudson SE, Bommineni GR, Li HJ, Lai CT, Liu N, Garcia-Diaz M, Simmerling C, Patil SS, Slayden RA, Tonge PJ. ChemMedChem. 2014;9:776. doi: 10.1002/cmdc.201300429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langer O, Muller M. Curr Drug Metab. 2004;5:463. doi: 10.2174/1389200043335379. [DOI] [PubMed] [Google Scholar]
- 33.Muller M, Pena AD, Derendorf H. Antimicrob Agents Chemother. 2004;48:1441. doi: 10.1128/AAC.48.5.1441-1453.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasipanodya JG, Srivastava S, Gumbo T. Clin Infect Dis. 2012;55:169. doi: 10.1093/cid/cis353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fowler JS, Volkow ND, Wang GJ, Ding YS, Dewey SL. J Nucl Med. 1999;40:1154. [PubMed] [Google Scholar]
- 36.Fischman AJ, Alpert NM, Babich JW, Rubin RH. Drug Metab Rev. 1997;29:923. doi: 10.3109/03602539709002238. [DOI] [PubMed] [Google Scholar]
- 37.Simon GM, Niphakis MJ, Cravatt BF. Nat Chem Biol. 2013;9:200. doi: 10.1038/nchembio.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weinstein EA, Ordonez AA, DeMarco VP, Murawski AM, Pokkali S, MacDonald EM, Klunk M, Mease RC, Pomper MG, Jain SK. Sci Transl Med. 2014;6:259ra146. doi: 10.1126/scitranslmed.3009815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H, Lu Y, Liu L, Kim SW, Hooker JM, Fowler JS, Tonge PJ. Eur J Med Chem. 2014;88:66. doi: 10.1016/j.ejmech.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weinstein EA, Liu L, Ordonez AA, Wang H, Hooker JM, Tonge PJ, Jain SK. Antimicrob Agents Chemother. 2012;56:6284. doi: 10.1128/AAC.01644-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hodgson HH. Chem Rev. 1947;40:251. doi: 10.1021/cr60126a003. [DOI] [PubMed] [Google Scholar]
- 42.Miller PW, Long NJ, Vilar R, Gee AD. Angew Chem Int Ed Engl. 2008;47:8998. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]
- 43.England K, am Ende C, Lu H, Sullivan TJ, Marlenee NL, Bowen RA, Knudson SE, Knudson DL, Tonge PJ, Slayden RA. J Antimicrob Chemother. 2009;64:1052. doi: 10.1093/jac/dkp307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.