

Abstract
The lack of inhibitors that are selective for individual poly-ADP-ribose polymerase (PARP) family members has limited our understanding of their roles in cells. Here, we describe a chemical genetics approach for generating selective inhibitors of an engineered variant of PARP10. We synthesized a series of C-7 substituted 3,4-dihydroisoquinolin-1(2H)-one (dq) analogues designed to selectively inhibit a mutant of PARP10 (LG-PARP10) that contains a unique pocket in its active site. A dq analogue containing a bromo at the C-7 position demonstrated a 10-fold selectivity for LG-PARP10 compared to its WT counterpart. This study provides a platform for the development of selective inhibitors of individual PARP family members that will be useful for decoding their cellular functions.
Graphical abstract
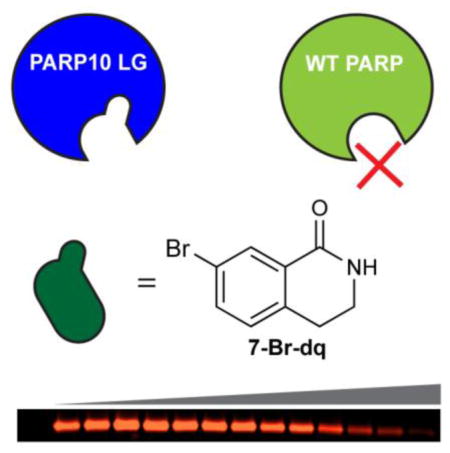
Poly-ADP-ribose polymerases (PARPs) are a family of 17 ubiquitously expressed enzymes in humans that catalyze the transfer of the ADP-ribose moiety from nicotinamide adenine dinucleotide (NAD+) to their target proteins, a process known as ADP-ribosylation (ADPr). Only four PARPs catalyze poly-ADPr (PARP1, 2, 5a, and 5b), whereas the majority of the PARPs catalyze mono-ADPr.1 PARPs are found throughout the cell and play roles in diverse cellular processes, including transcriptional2 and post-transcriptional regulation,3 protein degradation,4 cell signaling5,6 and the unfolded protein response.7 However, the cellular functions and targets of most PARPs–especially those that catalyze mono-ADPr–remain unknown and current strategies to study PARP function are insufficient to provide a comprehensive view of their role in cellular processes. This is due, in large part, to the dearth of inhibitors that selectively inhibit an individual PARP family member.
A major hurdle for developing selective PARP inhibitors is the high structural conservation of the catalytic domain among PARPs. While the primary sequences are semi-divergent (displaying ~50% similarity),8 their structures are highly conserved in the nicotinamide-binding site, which is the major binding site for the majority of PARP inhibitors.9 To address this challenge, we implemented a chemical genetics strategy, commonly referred to as the “bump-hole” method, for the development of selective PARP inhibitors. This strategy has been successfully used for identifying highly selective inhibitors of individual enzyme within a highly conserved family,10 most notably protein kinases.11 In this letter, we describe a bump-hole approach for identifying selective inhibitors of individual PARPs.
We recently described a bump-hole method for identifying the direct protein targets of PARPs that catalyze poly-ADPr.12 This method involved mutating a lysine residue (Lys903 in human PARP1, which we refer to as the “ceiling” position) in the nicotinamide-binding site to an alanine to create a unique pocket for accommodating a C-5 ethyl group on the nicotinamide ring of the NAD+ analogue. We sought to adopt this chemical genetics strategy to generate selective inhibitors of PARP10. We focused our initial efforts on PARP10 since its mono-ADPr activity has been well characterized in vitro.13 While inhibitors of PARP10 have been previously reported, they are not selective among the PARP family.14
We first examined the crystal structure of PARP10 bound to the nicotinamide bioisostere 3-aminobenzamide (3-AB) (PDB ID: 3HKV) to identify potential inhibitor-sensitizing positions within the nicotinamide binding site (Fig. 1a). We focused on Leu926 (human PARP10 numbering) because it occupies the same space in the nicotinamide-binding pocket as the ceiling lysine of PARP1. We reasoned that mutation of Leu926 to either an alanine or glycine would create a unique pocket for orthogonal inhibitors. We first determined if mutation of Leu926 to an alanine or glycine (LA-PARP10 or LG-PARP10, respectively) affects the activity of the catalytic domain of PARP10 (PARP10cat). To test the activity of LA-, LG-, and WT-PARP10cat we monitored ADP-ribosylation of SRSF protein kinase 2 (SRPK2), a previously characterized PARP10 substrate,15,16 using N-6 alkyne-NAD+ (6-a-NAD+)12,17 and click conjugation to a fluorescent azide reporter (Fig. 1b). Using this assay, we found that both engineered PARP10cat mutants exhibited similar activity to the WT-PARP10cat (Fig. 1c). A more detailed analysis using a range of 6-a-NAD+ concentrations revealed that LG-PARP10cat exhibited ~60% activity of WT-PARP10cat (Fig. S1).
Figure 1.
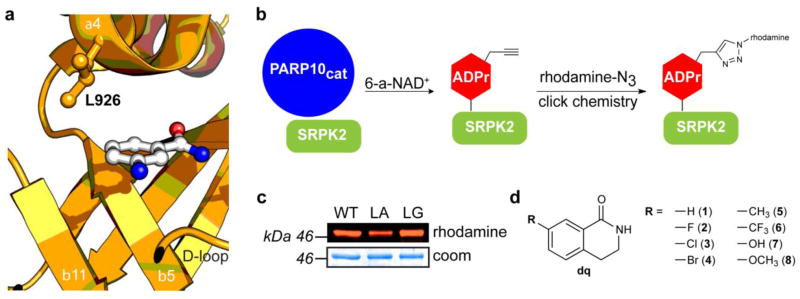
A chemical genetics strategy for generating selective inhibitors of PARP10. (a) Structure of PARP10 with nicotinamide isostere 3-aminobenzamide (3-AB) (PDB ID: 3HKV) with mutated residue (L926) indicated. (b) Schematic showing the PARP10-mediated transfer of alkyne-tagged ADP-ribose onto SRPK2 from 6-a-NAD+, followed by click conjugation with a fluorescent azide reporter (rhodamine-N3). (c) Activity comparison of engineered PARP10cat mutants L926A (LA) and L926G (LG) to WT-PARP10cat. (d) Structure of C-7 substituted dq analogues 1 – 8 designed to selectively inhibit engineered PARP10 mutants.
We next sought to identify compounds that could selectively inhibit our engineered PARP10 mutants. As a starting point for our inhibitor design, we selected 3,4-dihydroisoquinolin-1(2H)-one (dq, 1), a pan-PARP inhibitor scaffold that binds in the same orientation as 3-AB in the nicotinamide binding site.18 We designed a series of dq analogues containing various substituents at the C-7 position to probe the size and shape of the unique pocket in our engineered PARP10 mutants (Fig. 1d). We used two different strategies to synthesize C-7 dq analogues. Analogues 3-8 were synthesized using the Schmidt reaction (Scheme 1). The reaction resulted in a mixture of both N-alkyl and N-aryl regioisomers, which were easily resolved with standard normal phase chromatography. The percentage of the desired N-alkyl isomer obtained from the Schmidt reaction ranged from 26 – 63% (Supporting Information). The yield of the N-alkyl isomer was highest for 1-indanones where the 6-position substituent was highly electron donating (e.g., -OH, -OCH3), whereas N-alkyl regioselectivity decreased with electron withdrawing substituents (e.g., -Cl, -Br, -CF3). The fluoro-substituted dq derivative 2 was synthesized via a previously described route that involves the formation of the methylcarbamate species from p-fluorophenethylamine followed by intramolecular cyclization in triflic acid (Scheme 2 and Supporting Information).19
Scheme 1.
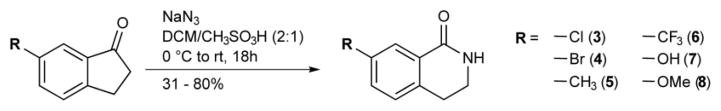
Syntheses of analogues 3 – 8 via Schmidt reaction.
Scheme 2.

Synthesis of analogue 2 via intramolecular aromatic substitution reaction.
We first tested our panel of C-7 substituted dq analogues against our engineered PARP10cat mutants. In a preliminary screen, we found that none of the compounds inhibited the LA-PARP10cat mutant (data not shown); therefore, we focused on LG-PARP10cat. We found that most of the C-7 substituted dq analogues inhibited LG-PARP10cat more potently than the parent dq scaffold 1 (Fig. 2a, b, Table 1, and Fig. S2). A clear structure-activity relationship is observed for the halogen series: increasing the size of the halogen group resulted in increased potency against LG-PARP10cat, with the bromo-substituted dq analogue 4 exhibiting the greatest potency (IC50 = 8.6 μM; Fig. 2a, b, Table 1, and Fig. S2). Unlike the halogen-substituted analogues, the methyl (5), methoxy (8), and hydroxyl (7) dq analogues did not exhibit increased potency compared to 1 (Fig. 2b, Table 1, and Fig. S2). The trifluoromethyl dq analogue 6 exhibited a modest, 2-fold increase in potency compared to 1 (Figure 2b, Table 1, and Fig. S2). Importantly, none of the C-7 substituted dq analogues appreciably inhibited WT-PARP10cat at concentrations up to 100 μM (Table 1 and Fig. S3). Moreover, dq analogues with C-7 substituents larger than chlorine exhibited IC50 values greater than 100 μM against WT-PARP1 (Table 1 and Fig. S4), which is consistent with a pharmacophore analysis of PARP1 inhibitors.9 Taken together, these results demonstrate that PARP10 can be engineered to generate unique inhibitor sensitivity for C-7 substituted dq analogues.
Figure 2.

C-7 substituted dq analogues selectively inhibit LG-PARP10cat. (a) Representative fluorescent gel image showing a dose-dependent inhibition of LG-PARP10cat-mediated ADP-ribosylation of SRPK2 by 7-Br-dq (4). SRPK2 (3 μM) was ADP-ribosylated by LG-PARP10cat (500 nM) with 6-a-NAD+ (100 μM) in the presence of analogue 4 (0 – 125 μM) at 30 °C for 1h. Following click conjugation to rhodamine-azide (100 μM), proteins were resolved by SDS-PAGE and detected via in-gel fluorescence. Coomassie (Coom) Brilliant Blue staining was used to demonstrate even loading. (b) IC50 curves for C7-substituted dq analogues against LG-PARP10cat. Activity was determined as described in (a). Error bars represent S.E.M., n = 2.
Table 1.
C-7 substituted dq analogues selectively inhibit the engineered PARP10 mutant, LG-PARP10cat.
| Compound No. | -R | LG-PARP10cat | WT-PARP10cat IC50 (μM) |
WT-PARP1 IC50 (μM) |
|
|---|---|---|---|---|---|
| IC50 (μM) | pIC50 ± S.E.M.a | ||||
| 1 | -H | 130 | −3.89 ± 0.06 | ~30 | ~100 |
| 2 | -F | 72.7 | −4.14 ± 0.04 | ~100 | ~100 |
| 3 | -Cl | 16.2 | −4.79 ± 0.04 | >100 | ~100 |
| 4 | -Br | 8.6 | −5.07 ± 0.05 | >100 | >100 |
| 5 | -CH3 | 112 | −3.95 ± 0.04 | >100 | >100 |
| 6 | -CF3 | 65.5 | −4.18 ± 0.04 | >100 | >100 |
| 7 | -OH | >30 | n.d. | n.d. | n.d. |
| 8 | -OCH3 | 172 | −3.77 ± 0.03 | >100 | >100 |
S.E.M. from two representative dose-response experiments
Several PARPs exhibit auto-ADP-ribosylation activity, including PARP10.1 Auto-ADP-ribosylation of PARP10 is most robust using the full-length protein expressed in mammalian cells (data not shown). We next sought to determine if 4 could selectively inhibit the auto-ADP-ribosylation full-length GFP-tagged LG-PARP10 (GFP-LG-PARP10) expressed in human embryonic kidney (HEK) 293T cells. For these experiments, we developed a modified GFP-immunoprecipitation (IP)-auto-ADP-ribosylation assay1 using 6-alkyne-NAD+ as a substrate. We found that 4 inhibited the auto-ADP-ribosylation of GFP-LG-PARP10 in a dose-dependent manner (Fig. 3). By contrast, auto-ADP-ribosylation of GFP-WT-PARP10 was unaffected by up to 100 μM 4 (Fig. 3). These results demonstrate that 4 can selectively inhibit the auto-ADP-ribosylation activity of the full-length, engineered PARP10 mutant, GFP-LG-PARP10.
Figure 3.
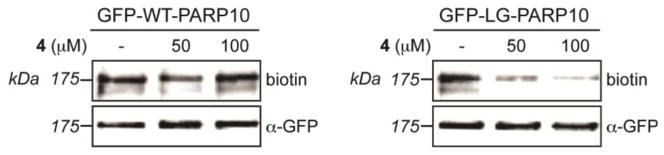
4 selectively inhibits the auto-ADP-ribosylation activity of full-length LG-PARP10. HEK 293T cells overexpressing GFP-WT-PARP10, GFP-LG-PARP10, or GFP-GW-PARP10 were harvested 24 h after transfection, and GFP-tagged PARP10 proteins were immunoprecipitated using an anti-GFP antibody. Auto-ADP-ribosylation assays were performed on immunoprecipitates using 6-a-NAD+. After click conjugation with biotin-azide proteins were resolved by SDS/PAGE and detected by Western blot with either Streptavidin-HRP or an antibody against GFP.
The long-term goal is to use the described chemical genetic strategy to selectively inhibit PARP10 in cells. An important criterion for this strategy is that LG-PARP10 can functionally replace WT-PARP10. We therefore determined if GFP-LG-PARP10 exhibited the same auto-ADP-ribosylation activity as GFP-WT-PARP10 in cells. For this experiment, we used an aminooxy-alkyne (AO-alkyne) clickable probe that can detect ADP-ribosylation in cells.20 AO-alkyne reacts with ADP-ribosylated proteins forming an oxime bond, which can be detected by click chemistry using an azide reporter.20 HEK 293T cells overexpressing either GFP-WT-PARP10, GFP-LG-PARP10, or the catalytically inactive mutant GFP-G888W (GW)-PARP10 were treated with AO-alkyne (100 μM) in the presence of the oxime catalyst p-phenylenediamine (10 mM) followed by click conjugation with biotin-azide. We found that GFP-WT-PARP10 and GFP-LG-PARP10 exhibited similar auto-ADP-ribosylation activity in cells, whereas GFP-GW-PARP10 was inactive (Fig. S5). These results demonstrate that the Leu926 to glycine mutation does not significantly perturb the cellular activity of PARP10.
In conclusion, we used a bump-hole strategy to successfully identify a series of C-7 substituted dq analogues that selectively inhibit the engineered PARP10 mutant, LG-PARP10cat. The most potent C-7 substituted dq analogue is the bromo-substituted analogue 4, which exhibited a greater than 10-fold selectivity for LG- versus WT-PARP10cat. To our knowledge this is the first study that demonstrates the use of halogen-substitution to generate selective inhibitors using the bump-hole approach. In future studies, it will be interesting to explore whether other scaffolds could be adorned with a bromo-substituent to increase potency while maintaining selectivity for LG-PARP10cat. While the bump-hole strategy described here focuses on the development of selective inhibitors of PARP10, our strategy should be generalizable to the other PARPs given the high degree of conservation between family members.
Supplementary Material
Acknowledgments
We thank members of the Cohen laboratory for many helpful discussions. We thank P. Chang (MIT) for the GFP-PARP10 plasmid. This work was supported by an Achievement Rewards for College Scientists (ARCS) Scholarship and National Institutes of General Medicine Training Grant T32GM071338 (R.K.M) and National Institutes of Health Grant NS088629 (M.S.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vyas S, Matic I, Uchima L, Rood J, Zaja R, Hay RT, Ahel I, Chang P. Nature Communications. 2014;5 doi: 10.1038/ncomms5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gibson BA, Kraus WL. Nature Reviews Molecular Cell Biology. 2012;13:411. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 3.Leung AKL, Vyas S, Rood JE, Bhutkar A, Sharp PA, Chang P. Molecular Cell. 2011;42:489. doi: 10.1016/j.molcel.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cho-Park PF, Steller H. Cell. 2013;153:614. doi: 10.1016/j.cell.2013.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verheugd P, Forst AH, Milke L, Herzog N, Kremmer E, Kleine H, Lüscher B. Nature Communications. 2013;4:1683. doi: 10.1038/ncomms2672. [DOI] [PubMed] [Google Scholar]
- 6.Huang SA, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, Charlat O, Wiellette E, Zhang Y, Wiessner S, Hild M, Shi X, Wilson CJ, Mickanin C, Myer V, Fazal A, Tomlinson R, Serluca F, Shao W, Cheng H, Shultz M, Rau C, Schirle M, Schlegl J, Ghidelli S, Fawell S, Lu C, Curtis D, Kirschner MW, Lengauer C, Finan PM, Tallarico JA, Bouwmeester T, Porter JA, Bauer A, Cong F. Nature. 2009;461:614. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 7.Jwa M, Chang P. Nature Cell Biology. 2012;15:123. doi: 10.1038/ncb2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amé J, Spenlehauer C, de Murcia G. Bio Essays. 2004;26:882–93. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 9.Ferraris DV. Journal of Medicinal Chemistry. 2010;53:4561. doi: 10.1021/jm100012m. [DOI] [PubMed] [Google Scholar]
- 10.Islam K. ACS Chemical Biology. 2015;10:343. doi: 10.1021/cb500651d. [DOI] [PubMed] [Google Scholar]
- 11.Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- 12.Carter-O’Connell I, Jin H, Morgan RK, David LL, Cohen MS. Journal of the American Chemical Society. 2014;136:5201. doi: 10.1021/ja412897a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kleine H, Poreba E, Lesniewicz K, Hassa PO, Hottiger MO, Litchfield DW, Shilton BH, Lüscher B. Molecular Cell. 2008;32:57. doi: 10.1016/j.molcel.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 14.Ekblad T, Andersson CD, Caraballo R, Thorsell A, Karlberg T, Spjut S, Linusson A, Schüler H, Elofsson M. European Journal of Medicinal Chemistry. 2015;95:546. doi: 10.1016/j.ejmech.2015.03.067. [DOI] [PubMed] [Google Scholar]
- 15.Feijs KL, Kleine H, Braczynski A, Forst AH, Herzog N, Verheugd P, Linzen U, Kremmer E, Lüscher B. Cell Communication and Signaling. 2013;11:5. doi: 10.1186/1478-811X-11-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venkannagari H, Fallarero A, Lüscher B, Lehtiö L. European Journal of Pharmaceutical Sciences. 2013;49:148. doi: 10.1016/j.ejps.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 17.Jiang H, Kim JH, Frizzell KM, Kraus WL, Lin H. Journal of the American Chemical Society. 2010;132:9363. doi: 10.1021/ja101588r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wahlberg E, Karlberg T, Kouznetsova E, Markova N, Macchiarulo A, Thorsell A, Pol E, Frostell Å, Ekblad T, Öncü D, Kull B, Robertson GM, Pellicciari R, Schüler H, Weigelt J. Nature Biotechnology. 2012;30:283. doi: 10.1038/nbt.2121. [DOI] [PubMed] [Google Scholar]
- 19.Kurouchi H, Kawamoto K, Sugimoto H, Nakamura S, Otani Y, Ohwada T. The Journal of Organic Chemistry. 2012;77:9313. doi: 10.1021/jo3020566. [DOI] [PubMed] [Google Scholar]
- 20.Morgan RK, Cohen MS. ACS Chemical Biology. 2015 doi: 10.1021/acschembio.5b00213. article ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.