

Abstract
Pick disease (PiD) is a frontotemporal lobar degeneration with distinctive neuronal inclusions (Pick bodies) that are enriched in 3-repeat (3R) tau. Although mostly sporadic, mutations in the tau gene (MAPT) have been reported. We screened 24 cases of neuropathologically confirmed PiD for MAPT mutations and found a novel mutation (c.1008G>C, p.Q336H) in one patient. Pathogenicity was confirmed on microtubule assembly and tau filament formation assays. The patient was compared to sporadic PiD and PiD associated with MAPT mutations from a review of the literature. The patient had behavioral changes at 55 years of age, followed by reduced verbal fluency, parkinsonism and death at 63 years of age. His mother and maternal uncle had similar symptoms. Recombinant tau with p.Q336H mutation formed filaments faster than wild type tau, especially with 3R tau. It also promoted more microtubule assembly than wild type tau. We conclude that mutations in MAPT, including p.Q336H, can be associated with clinical, pathologic, and biochemical features that are similar to those in sporadic PiD. The pathomechanism of p.Q336H, and another previously reported variant at the same codon (p.Q336R), appears to be unique to MAPT mutations in that they not only predispose to abnormal tau filament formation but also facilitate microtubule assembly in a 3R tau-dependent manner.
Keywords: Dementia, Frontotemporal dementia, FTDP-17, FTLD-tau, Pick disease, Tau protein, Tau gene
INTRODUCTION
Pick disease (PiD) is a progressive neurodegenerative disease that accounts for less than 5% of all dementia cases (1) and usually occurs before 65 years of age (2). Because original descriptions by Arnold Pick included only clinical and macroscopic neuropathologic evaluations (3), the concept of “Pick disease” is debated (2). Currently, PiD is most often defined by characteristic pathologic and biochemical features and is classified as one of the frontotemporal lobar degenerations with tau pathology (4). The pathognomonic histologic feature is the Pick body, a round and well-circumscribed cytoplasmic inclusion in neurons of allocortex and neocortex (4, 5), which shows immunoreactivity for tau, especially 3-repeat (3R) tau (5). Pick bodies show a paucity of staining with the Gallyas silver impregnation method, despite being argyrophilic with other silver-staining techniques (6).
The most common clinical presentation is behavioral variant frontotemporal dementia, but language variants, both agrammatic and semantic types of primary progressive aphasia, have been reported (7). A less common presentation is the corticobasal syndrome (5, 8).
Pick disease is usually sporadic but discovery of mutations in microtubule-associated protein tau gene (MAPT) has contributed to a better understanding of the concept of hereditary Pick disease. Over 50 mutations have been reported in MAPT in approximately 150 families under the umbrella term of frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) (9). Some of them have been reported to have Pick-like histology (10, 11), with either Pick bodies or Pick body-like neuronal inclusions (11). A competing school of thought is that none of the reported cases with MAPT mutation resemble exactly sporadic PiD with respect to biochemical and neuropathologic criteria (12).
In this study, we screened pathologically confirmed PiD patients for MAPT mutations. We discovered a novel MAPT missense mutation in exon 12 (p.Q336H) in an individual with familial dementia.
MATERIALS AND METHODS
Case Material
The Mayo Clinic brain bank in Jacksonville, Florida, acquired 24 brains from patients with PiD between 2000 and 2014; all were from autopsies performed after approval by the next-of-kin or an individual with legal power of attorney. Genealogical and clinical evaluations were performed by medical chart review and telephone interviews of relatives using a clinical study protocol approved by Mayo Clinic Institutional Review Board.
Tissue Sampling and Neuropathologic Assessment
Brains were evaluated neuropathologically by an experienced neuropathologist (Dennis W. Dickson). Neuropathologic criteria for PiD required focal cortical atrophy and 3R tau-positive Pick bodies, which were negative or at most weakly positive on Gallyas silver stains (4). The fixed left hemibrain divided in the midsagittal plane was available for the proband. The fixed tissue was sampled with a standardized dissection and sampling method and embedded in paraffin blocks. Hematoxylin and eosin-stained sections were used for histologic evaluations. Alzheimer-type pathology was assessed with thioflavin-S fluorescent microscopy. Tau immunohistochemistry was performed using a DAKO Autostainer (Universal Staining System, Carpinteria, CA), with the following anti-tau antibodies: phospho-tau (CP13 - phospho-serine 202; mouse IgG1, 1:1,000, from Dr. Peter Davies, Feinstein Institute for Medical Research, North Shore LIJ Health Care System, Manhasset, NY); 3R tau (RD3, Millipore, Temecula, CA); 4R tau (RD4, Millipore); and 12E8 (phospho serine 262 and 356; from Dr. Peter Seubert, Elan Pharmaceuticals, South San Francisco, CA). Sections were stained for ubiquitin (Ubi-1, 1:60,000; Millipore, Billerica, MA) and a midbrain section with the substantia nigra was stained with α-synuclein (NACP, 1:3000, rabbit polyclonal, Mayo Clinic Jacksonville). Formalin-fixed hippocampus was processed for electron microscopy according to published methods (13).
DNA Sequencing
Genomic DNA was isolated from frozen brain using the Gentra Puregene kit (Qiagen, Venlo, The Netherlands). Polymerase chain reactions were performed by using primer sets designed to amplify exons 0–5, 7, 9–13 of MAPT as well as at least 30 base pairs of intronic sequence flanking each of these exons, as previously described (13). MAPT H1/H2 haplotype was defined by the single nucleotide polymorphism rs1052553 in MAPT exon 9
Biochemical and Tau Functional Studies
Samples of frontal and temporal cortex (150 mg, each) were obtained from frozen brain tissue of the proband and of 2 patients with sporadic PiD. Sarkosyl-insoluble protein fractions were extracted from the temporal and frontal cortex. Then, the fractions and human recombinant tau isoform ladder (rPeptide, Bogart GA), were subjected to polyacrylamide gel electrophoresis on 10% Tris-glycine gels (Invitrogen Life Technologies, Billerica, MA). Separated proteins were transferred to a polyvinylidene difluoride membrane (EMD Millipore) and immunoblotted with a human-specific tau antibody to exon 1 (E1; rabbit Ig, Mayo Clinic Jacksonville) or 3R tau monoclonal antibody (RD3, Millipore).
Recombinant tau was expressed and purified as previously described (14). Wild type (WT) tau, the novel p.Q336H mutant, and a control p.Q336R mutant each in both 3R0N and 4R0N cDNAs were cloned into pET30a and expressed in competent BL21 (DE3) cells. After induction, the cells were lysed with three freeze and thaw cycles, and the tau proteins were purified by heating lysates for 10 minutes at 80°C and isolating the tau proteins from clarified supernatants using ion exchange chromatography. The purity of the tau preparations was analyzed by SDS-polyacrylamide gel electrophoresis and Coomassie blue staining.
Microtubule assembly with recombinant tau proteins was measured by turbidity assay in 96 well plates in a final volume of 100 µl, as previously described (13). Ice-cold tubulin at 3.0 mg/ml (60 µM) (Cytoskeleton Inc., Denver, CO) was added to an equal volume of 0.24 mg/ml (6 µM) recombinant 4R0N tau or 0.30 mg/ml (8 µM) 3R0N tau in assembly buffer (80 mM PIPES, 2 mM MgCl2, 0.5 mM EGTA, 1 mM GTP, pH 6.8). The extent of microtubule assembly was monitored by turbidity assay, with absorbance measured at 340 nm on SpectraMax M5 Multi-Mode Microplate Readers (Molecular Devices, Sunnyvale, CA). Reactions were run in triplicate, and the rate and extent of microtubule assembly were measured.
Tau filament assembly was carried out as previously described (14). Recombinant tau (8 µM) was incubated with 0.04 mg/ml of low molecular weight dextran sulfate in 10 mM HEPES at pH 7.4, 100 mM NaCl at 37°C and analyzed at 4, 24, 48 and 72 hours. Tau filament polymerization was measured by adsorption of 10 µl of reaction mixture onto a 400-mesh carbon/Formvar grid (EM Sciences Inc., Hatfield PA), and staining with 2% uranyl acetate. Electron micrographs were captured randomly from low magnification fields at 3 predetermined grid coordinates using a Phillips EM208S electron microscope and a Gatan digital camera. Tau filament length was measured and quantified using NIH Image J (Version 1.47n). For each sample, 12 images were taken and analyzed. The number of filaments per EM field was counted and then the individual filament lengths were measured yielding a total polymer mass for each image.
RESULTS
Screening for MAPT Mutations in Pick Disease
Sequence analysis of the MAPT coding region in 24 cases of autopsy-confirmed Pick disease revealed 1 case with a novel missense mutation in exon 12 (c.1008G>C), which was predicted to result in a p.Q336H (glutamine to histidine) substitution in tau protein. In the remaining 23 cases, no MAPT mutations were identified. The proband was homozygous for the H1 MAPT haplotype. No DNA was available from any of his relatives to determine if the mutation segregated with affected family members. His mother, who had similar clinical features and died at 65 years of age, and maternal uncle were diagnosed with Alzheimer disease; however, this diagnosis was not pathologically confirmed. His daughter and his 4 siblings, including his fraternal twin brother, were unaffected at the time of manuscript preparation (pedigree structure in Fig. 1).
Figure 1.

Pedigree structure. Standard symbols were used. Round symbols indicate female patients, squares indicate male patients, and diagonal lines indicate that the individual is deceased. The arrowhead indicates the proband. Black symbols indicate individuals with clinical features of dementia based on family history.
Clinical and Laboratory Findings of Patient with Novel MAPT Mutation
The proband was a right-handed man who died at the age of 63 years following an 8-year disease course. His first symptoms included short-term memory deficits and personality changes with disinhibition. Later, he developed speech hesitancy, word-finding difficulties and incontinence for both bowel and bladder. Two years after symptomatic onset, magnetic resonance imaging showed symmetric dilatation of the lateral ventricles. On neuropsychological evaluation, his speech was fluent and well-articulated, but he demonstrated lack of insight. His appearance suggested lack of concern for personal hygiene. The patient had high-average premorbid intelligence and an average score on a test of global cognition. In contrast, he had borderline-slow phonemic and semantic word generation, reduced fine motor dexterity, and markedly impaired verbal and nonverbal memory. His memory profile suggested impairment in initial acquisition and delayed recall, with some benefit when presented with contextual, narrative information or recognition cues. In the context of his progressive decline in interpersonal conduct, the results were consistent with behavioral variant frontotemporal dementia or Alzheimer disease with prominent frontal involvement. His neurological examination showed fine, bilateral and intermittent resting hand tremors with reduced arm swing, rigor and decreased hand dexterity on the left. Five years after disease onset, his Folstein Mini-Mental State Examination score was 27/30, but he showed marked decline in semantic and phonemic verbal fluency with diminished verbal output. His bradykinesia and rigor became more severe; the gait was unsteady. Trials of antioxidants and vitamins, (vitamin E, folic acid, cyanocobalamin and pyridoxine, coenzyme Q), anti-dementia (donepezil 10 mg/day, galantamine 24 mg/day, memantine 20 mg/day), and anti-parkinsonian drugs (amantadine 100 mg/day, levodopa 300 mg/day/carbidopa 75 mg/day) were unsuccessful. The final clinical diagnosis was atypical Alzheimer disease with parkinsonism.
Neuropathologic Findings of Patient with Novel MAPT Mutation
The brain weighed 1120 grams and had frontal lobar atrophy, especially the medial frontal lobe (Fig. 2A). Medial temporal lobe atrophy was moderate. The corpus callosum, particularly the mid-third, was thinned, and the lateral ventricle, especially the frontal horn, was markedly dilated (Fig. 2B, C). The cortical gray mantle was thinned in the superior frontal gyrus, anteromedial temporal lobe, and insular cortex (Fig. 2B, C). There was atrophy of the hippocampal formation, more severe in anterior than posterior sections (Fig. 2C). The amygdala had mild atrophy. There was mild flattening of the head of the caudate nucleus (Fig. 2B). Horizontal sections of the midbrain, pons and medulla showed visible, but decreased pigmentation in substantia nigra (Fig. 2D). The sections of cerebellum showed no unusual features.
Figure 2.

Macroscopic findings of the proband. (A) There is marked frontal atrophy, with widening of sulcal spaces (arrow) and narrowing of gyri. The frontal horn of the lateral ventricle (asterisk) is enlarged and the third ventricle is dilated. Brainstem and cerebellum are unremarkable. (B, C) Coronal sections show enlargement of the frontal horn of the lateral ventricle (asterisks) with atrophy of the corpus callosum and mild atrophy of the ventromedial caudate nucleus, as well as thinning of cortical ribbon in superior frontal and cingulate gyri (arrowheads in [B]) and insular cortices (arrowhead in [C]). There is also marked atrophy of the anteromedial temporal lobe, including the hippocampus (arrow in [C]). (D) A transverse section of the midbrain shows decreased pigmentation in the substantia nigra (arrow).
On microscopic examination, the neocortex, particularly in frontal and temporal lobes, had thinning of the cortical ribbon with neuronal loss, spongiosis and fibrillary gliosis. Many ballooned achromatic neurons (“Pick cells”) were present in middle and lower cortical layers of the frontal and temporal lobes. Pick bodies were frequent in upper cortical layers. A few non-neuritic senile plaques and a few neurofibrillary tangles (NFTs) were detected with thioflavin S fluorescent microscopy in mid-frontal, superior temporal and inferior parietal cortical sections (Braak neurofibrillary tangle stage IV and Thal amyloid phase 2). The NFTs showed immunoreactivity for 4R and 3R tau. There was mild focal amyloid angiopathy in leptomeningeal vessels.
The hippocampus had severe neuronal loss and gliosis throughout the Sommer sector and the endplate. The dentate fascia had neuronal loss and dispersion of the granule cells. Pick bodies were frequent in the dentate fascia and hippocampal pyramidal cell layer (Fig. 3A). They were mostly negative on the Gallyas silver stain (Fig. 3B). They were immunopositive for 3R tau (Fig. 3C), but variably positive for ubiquitin, 4R tau (Fig. 3D) and 12E8. In contrast to Pick bodies, NFTs in CA2 sector were positive for 3R and 4R tau, as well as 12E8; they were also argyrophilic on both Bielschowsky and Gallyas silver stains. In addition to Pick bodies, sections immunostained for tau revealed sparse tau-positive oligodendroglia, tau positive neuropil threads and a few tau-positive astrocytic lesions. The severity and neuroanatomical distribution of tau-immunoreactive lesions was similar in the proband compared to 9 cases of sporadic PiD evaluated with the same methods in the same time period (Supplemental Table 1). No pathologic changes were identified with α-synuclein staining of the midbrain section.
Figure 3.

Light microscopy, immunohistochemistry and electron microscopy findings. (A) Bielschowsky silver impregnation of hippocampal dentate fascia shows argyrophilic Pick bodies (arrows). (B) Only a few Pick bodies show weak staining with Gallyas silver stain (arrows); most Pick bodies are negative (arrowheads). (C) Virtually every Pick body is positive for 3R tau (arrows). (D) A subset of Pick bodes is positive with 4R tau (arrows), while others are negative (arrowheads). (E) Electron micrograph of a Pick body (right arrow) in a hippocampal granular layer neuron. The nucleus (N, arrow) and other cytoplasmic organelles are pushed to the periphery by the inclusion. (F) Higher magnification of the Pick body in (E) shows randomly oriented filaments mixed with dense granular material. Arrows indicate rough endoplasmic reticulum along the left edge of the inclusion. (G) Higher magnification of filaments in Pick body shows straight filaments (arrows) and twisted filaments with >100 nm half periodicity (arrowheads). (H) Electron micrograph of a neurofibrillary tangle (arrow) in a hippocampal granular layer neuron. N, nucleus. (I) Enlargement of the neurofibrillary tangle in (H) shows densely packed parallel filaments with sparse granular material. Scale bar: 0.2 µm. (J) Higher magnification of paired helical filaments in (H) shows half periodicity of ~50 nm. Scale bars: A–D, 20 µm; E, 1 µm; F, 0.25 µm; G, 50 nm; H, 1 µm; J, 50 nm.
Ultrastructurally, Pick bodies were relatively well circumscribed, but not membrane-bound, cytoplasmic inclusions composed of randomly oriented straight filaments with a diameter of 10 to 15 nm (Fig. 3E–G). A few twisted filaments with >100 nm half periodicity were interspersed. Alzheimer-type NFTs were clearly different from Pick bodies in the same section of the hippocampus (Fig. 3H–J). The NFTs contained bundles of densely packed, paired helical filaments with 20- to 22-nm diameter and <80-nm half periodicity.
Biochemical and Functional Studies of Novel MAPT Mutation
Sarkosyl-insoluble fractions extracted from frontal and temporal cortices from the patient and 2 sporadic cases of PiD showed tau biochemical profile was consistent with PiD, with major bands at ~64 and ~60 kDa, migrating at about the same molecular weight as 3R2N and 3R1N tau (15) (Fig. 4A). A similar pattern was observed when the blot was reprobed with 3R tau-specific monoclonal antibody (Fig. 4B).
Figure 4.

Western blot analysis of sarkosyl-insoluble fractions from frontal cortex (F. ctx) and temporal cortex (Temp. ctx). Lane 1 is the proband (p.Q336H); lanes 2 and 3 are from cases of sporadic Pick disease. (A) Immunoblotting with E1 polyclonal anti-tau, which recognizes all isoforms of tau. Note the bands at ~64 and ~60 kDa characteristic of 3R tauopathies. (B) Reprobing with monoclonal antibody specific to 3R tau indicates insoluble tau consists mostly of 3R2N and 3R1N. The p.Q336H carrier profile is similar to that of 2 cases of sporadic Pick disease.
We assessed the effects p.Q336H mutation on its ability to promote tubulin polymerization into microtubules and found that the p.Q336H mutation in 3R0N tau promoted MT polymerization at a rate greater than WT 3R0N tau; it also had a greater steady state level, representing a 41% and 72% increase, respectively (Fig. 5A.) Similar effects were observed with 4R0N tau but the effects of the mutation were even larger, with the rate increasing 125% and the steady state extent increasing 164% (Fig. 5B).
Figure 5.
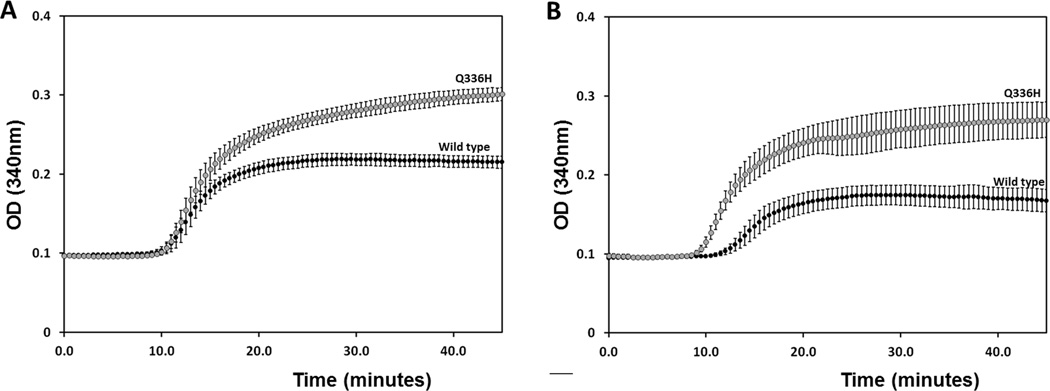
p.Q336H increases microtubule polymerization. (A) Incubation of p.Q336H 3R0N tau with tubulin increases the rate of microtubule assembly compared with wild type tau and increased steady state extent of microtubule polymerization. (B) Qualitatively similar effects were observed for p.Q336H in 4R0N, with even larger increases in the rate and final extent of microtubule polymerization. These were significant at p < 0.05 (A) and p < 0.01 (B) levels.
We next examined the effect of the p.Q336H mutation on tau filament formation; recombinant 3R0N and 4R0N isoforms containing the mutation were compared to WT tau and to another pathogenic mutation at this codon, p.Q336R (16). As shown in Figure 6A, p.Q336H promoted tau filament assembly better than WT tau (3R0N and 4R0N), and it increased polymerization more for 3R than 4R tau. The p.Q336H and p.Q336R increase tau filament assembly by 32% and 35%, respectively, compared to 4R0N WT tau (Fig. 6C, E). The increases in 3R tau polymerization were more robust, with p.Q336R leading to a 5.4-fold increase in tau polymerization compared to 3R0N WT tau, whereas the p.Q336H mutant had a 13.8-fold increase (Fig. 6B, D). These results suggest that mutations at codon 336 have a greater effect on 3R than 4R tau. The increases in total polymer mass for p.Q336H compared to WT (Fig. 6B, C) appeared to be nucleation-dependent in that while the average filament length was only marginally changed (data not shown), there was a significant increase in filament number (Fig. 6D, E), which paralleled increases in total polymer mass.
Figure 6.

Polyglycosaminoglycan-induced filament assembly. (A) Representative electron micrographs of filament formation for 3R tau (top row) or 4R tau (bottom row) for wild type (WT) and mutant (p.Q336H and p.Q336R) tau isoforms after 1 day of in vitro polymerization promoted by polyglycosaminoglycan from which filament number and lengths were assessed with arrows showing typical filaments. (B–E) Graphical display of total filament length per field measurements for 3R0N (B) and 4R0N (C) tau isoforms and of filament number counts for 3R0N (D) and 4R0N (E) for WT (both 3RWT and 4RWT), p.Q336H (both 3RQH and 4RQH) and Q336R (both 3RQR and 4RQR). The p.Q336H mutation has an isoform-specific effect (3R greater than 4R) on both a summed filament length and number. (*p < 0.001 vs. WT; #p < 0.001 vs. p.Q336R).
DISCUSSION
In this study we report clinical and pathologic findings of a patient with autopsy-confirmed PiD with a positive family history of dementia and a novel p.Q336H MAPT (c.1008G>C) mutation. Functional studies provide support for this being a pathogenic mutation. This is the ninth MAPT mutation in exon 12 and second mutation at codon 336.
The novel p.Q336H mutation, and the 6 previously reported MAPT mutations in exons 9–12 (p.K257T [10, 17], p.G272V [11], p.ΔK280 [18], p.S320F [19], p.Q336R [16], and p.K369I [20]) meet stringent criteria for PiD (Table 1). These criteria included cases that clinically had one of the frontotemporal dementia syndromes ideally at onset, but at least at end stage. Pathologically, those cases had to have focal atrophy of frontal or temporal lobes, or both, usually affecting the hippocampus and amygdala, as well. The tau pathology had to include description of well-circumscribed, round neuronal inclusions consistent with Pick bodies. Tau-positive glia could be present, but neuronal lesions had to be the predominant lesion type. A supportive finding was minimal or no immunoreactivity with monoclonal antibody 12E8 (21). Pick bodies had to be described in the hippocampal dentate fascia and pyramidal cell layer. While the presence of argyrophilia was usually reported, it was not essential if not described. Biochemical studies or immunohistochemistry with isoform-specific antibodies had to indicate predominance of 3R tau or at least a ratio of 3R tau equal to 4R tau. Ultrastructural characteristics of the filaments within the inclusions were not a criterion given lack of standardization in methodology, measurement, and nomenclature for filament types.
Table 1.
Summary of Clinical and Demographic Information in Hereditary Pick Disease
| MAPT mutation | Exon (haplotype) |
Family history of dementia |
Gender | Age of onset (years) |
Disease duration (years) |
Age of onset (years) |
First symptom | Extrapyramidal symptoms |
|---|---|---|---|---|---|---|---|---|
| Sporadic Pick disease | no | 30F vs. 32M (5) | 42 – 70, mean 60 ± 6.5 (n=21) (8) |
11 ± 6 (n=39) (5); 2 – 16, mean 9 (8) |
mean 68 ± 10 (n=44) (5) |
Pick complex (bvFTD> PPA> mixed or early memory disturbance) (5) | Rare (30) | |
| p.K257T (c.770A>C), (38) |
9 (H1/H1) (1) & (2) | 2 maternal uncles* | M | 47 | 4 | 51 | bvFTD | N/A |
| p.G272V (c.815G>T), (11) |
9 (N/A) | (1): father, brother, sister; (2): father, brother, son |
F (1) F (2) |
45 (1); 52 (2) |
9 (1); 15 (2) |
54 (1); 67 (2) |
bvFTD (1) & (2) | absent (1) & (2) |
| p.ΔK280 (c.838_840delAAG) (18) |
10 (H1/H1) | father | F | 53 | 10 | 63 | bvFTD | absent |
| p.S320F (c.959C>T), (19) |
11 (N/A) | mother | M | 38 | 15 | 53 | initial mild memory problems, later fluent aphasia (PPA) | absent |
| p.Q336R (c.1007A>G), (16) |
12 (N/A) | father, paternal grandfather and 2 uncles | M | 58 | 10 | 68 | initial memory problems, at 4 years bvFTD and PPA |
N/A |
| p.Q336H (c.1008 G>C) | 12 (H1/H1) | mother and maternal uncle | M | 55 | 8 | 63 | bvFTD | hand tremor, unsteady gait, bradykinesia and rigidity |
| p.K369I (c.1106A>T), (20) |
12 (N/A) | N/A | F | 50 | 11 | 61 | bvFTD | absent |
Possibly incomplete penetrance; N/A, not available; F, females; M, males; pos, positive; neg, negative; (1), patient 1, (2), patient 2; bvFTD, behavioral variant of frontotemporal dementia; PPA, primary progressive aphasia.
The proband with the novel p.Q336H mutation had cytoplasmic neuronal argyrophilic inclusions consistent with Pick bodies. They were almost all negative with Gallyas silver stain (Fig. 3B), and consistently immunopositive for 3R tau. Only a subset had 4R tau immunoreactivity. Most of the Pick bodies were also negative with the tau monoclonal antibody 12E8, which recognizes a phospho-epitope in exon 9 (pSer262) (not shown). This profile is typical of Pick bodies in sporadic PiD. Like sporadic PiD (5), neuronal tau pathology was more severe than glial pathology in our proband, as in cases of PiD (Table 2).
Table 2.
Summary of Neuropathological, Biochemical and Functional Data in Hereditary Pick Disease
|
MAPT mutation |
Lobar atrophy; [brain wt.] |
Tau isoform in TB |
Western blot (kDa) |
EM Filament width (nm); (half period) |
Pick bodies | Cell type specificity |
Microtubule assembly | Tau filament assembly | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 12E8 | Other stains | Distribution | ||||||||
| p.K257T (38) |
Severe temporal; moderate frontal; hippocampus; [1120 g] |
3R>4R (TB) |
60, 64 (minor 68, 72) |
14 SF & twisted ribbons (>130 nm) | neg | Palmgren (pos) | Hp: dentate gyrus & pyramidal; Ctx: temporal | neuronal>glial few A |
reduced (95% for 3R; 85% for 4R) (38), reduced (70% for 4R) (39) | increased for 3R, but not for 4R |
| p.G272V (11) |
Severe frontal & temporal; hippocampus; amygdala; [880 g] |
3R>4R (TB) |
60, 64 (frontal also 68) |
20; twisted ribbons (110 nm) | neg | Gallyas (pos); 3R NFT 4R (IM) |
Hp: dentate gyrus & pyramidal; Ctx: frontal | neuronal>glial few A & O |
reduced (70% for 3R; 60% for 4R) (40) | increased for 4R>3R (41) |
| p.ΔK280 (18) |
Severe frontal & temporal; hippocampus; amygdala; [870 g] |
3R>4R front, 3R temp (TB) |
60, 64 | 7 – 8; PHF (95 nm) | neg | Gallyas (neg); 3R (IM) |
Hp: dentate gyrus & pyramidal; Ctx: frontal & temporal | neuronal>glial many A & few O |
reduced for 3R and 4R (42) | increased for 4R (41) |
| p.S320F (19) |
Severe anterior temporal; [1200 g] |
3R=4R (TB) |
60, 64 | 6–8 SF & twisted ribbons (110–160 nm) | neg | Bodian (neg) | Hp: dentate gyrus & pyramidal; Ctx: frontal, temporal, & parietal | neuronal>glial few O |
reduced (90 – 95% for 3R and 4R) | N/A |
| p.Q336R (16) |
Severe temporal; moderate frontal; hippocampus; amygdala; [1100 g] |
3R & 4R (TB) |
N/A | SF & twisted ribbons | mostly neg | 3R>4R (IM) (43) | Hp: dentate gyrus & pyramidal; Ctx: frontal & temporal | neuronal>glial rare A & O | increased (40% for 3R and 4R) | increased (3–4 fold for 3R>4R) |
| p.Q336H | Severe frontal; hippocampus; amygdala; [1120 g] |
N/A | 60, 64 | 10–15 SF & twisted ribbons (100 nm) | mostly neg | Bielschowsky (pos); Gallyas (neg); 3R>4R (IM) | Hp: dentate gyrus & pyramidal; Ctx: frontal & temporal | neuronal>glial few A & O | increased (4R>3R) | increased (13.8 fold; 3R>4R) |
| p.K369I (20) |
Severe temporal; moderate frontal; hippocampus; amygdala; [880 g] |
3R & 4R (TB) (20) |
60, 64, 68 | 28 twisted ribbons (130 nm) few PHF | neg | Bielschowsky (pos): Gallyas (neg); 3R (IM) (43) | Hp: dentate gyrus & pyramidal; Ctx: frontal, temporal, parietal, insular | neuronal>glial rare A & rare O | reduced (90% for 3R and 70% for 4R) | no effect on 3R or 4R |
Wt, weight; g, gram; 4R, four repeat tau isoform; 3R, three repeat tau isoform; IM, tau immunostaining; TB, Tau biochemistry (mostly Western blotting of dephosphorylated sarkosyl-insoluble tau); NFT, neurofibrillary tangles; PB, Pick bodies; PHF, Paired helical filaments; Hp, hippocampus; Ctx, neocortex; A, astrocytes; O, oligodendrocytes; pos, positive; neg, negative; SF, straight filaments; front, frontal cortex; temp, temporal cortex.
A less specific histologic feature of PiD are “Pick cells,” which are also called “swollen achromatic neurons” or “ballooned neurons.” In the proband, ballooned neurons were frequent in middle and lower neocortical layers of the frontal and temporal lobes, a common finding in sporadic PiD (22). Ballooned neurons are also frequent in corticobasal degeneration (23), argyrophilic grain disease (24), and many cases of FTDP-17 (4, 12, 25).
Macroscopic findings in hereditary PiD reflect clinical heterogeneity but all cases had at least frontotemporal atrophy, dilated ventricles and involvement of the allocortex. In a subset of cases (including our proband), there was mild-to-moderate neuronal loss and depigmentation of the substantia nigra. This is the likely correlate of parkinsonism noted later in the proband’s disease course. The density and distribution of Pick bodies usually (but not invariably [26]) correlate with macroscopic atrophy and neuron loss in PiD. Pick disease presenting with frontotemporal behavioral syndromes and severe memory problems have many Pick bodies and neuronal loss in association cortices of the frontal and anterior temporal lobes, as well as allocortex. Pick disease presenting as progressive aphasia, including semantic dementia, has atrophy in temporal lobe and peri-Sylvian areas (7), whereas those patients with corticobasal syndrome have marked pathology in superior frontal and superior parietal areas (27).
Biochemical and ultrastructural findings in our proband were similar to sporadic PiD and to those of hereditary PiD (Table 2). The tau isoforms in detergent insoluble fractions from the brain migrated at about 64 and 60 kDa on Western blots (15) and co-migrated with recombinant 3R tau protein. Electron microscopy of neuronal inclusions showed predominantly straight filaments of a 10- to 15-nm-diameter and fewer twisted filaments with a half period length of 120 to 160 nm (28, 29).
Clinically, most patients with hereditary PiD due to MAPT mutations, including our patient, have behavioral variant frontotemporal dementia, which is also the most common clinical presentation of sporadic PiD (5). Initial neuropsychological evaluation of the proband revealed an amnestic profile instead of a dysexecutive syndrome but profound encoding deficits suggested frontal lobe involvement. In later stages, the proband developed extrapyramidal symptoms that are occasionally seen in PiD (30). He also showed impaired functions attributable to frontal and temporal lobes with features of executive difficulties and semantic impairment.
In all reported cases of hereditary PiD, there is a positive family history of dementia. Although the proband’s maternal uncle and mother were diagnosed with Alzheimer disease, this diagnosis was not autopsy-confirmed. Interestingly, the proband’s fraternal twin brother was unaffected. Inter- and even intra-familial heterogeneity of the same MAPT mutations has been reported (12, 31–33). This suggests coexistence of other modifying loci, stochastic or environmental factors that influence the severity and distribution of pathology, and consequent clinical syndromes (12).
There are many clinical, pathologic and biochemical similarities between p.Q336H and p.Q336R (16). Both had an enhancing effect on tau filament assembly in vitro. The effect was more robust for the 3R p.Q336H mutant than for the 3R p.Q336R mutant. The effect on tau filament assembly and the unique property of increasing microtubule assembly in an isoform-specific manner are very likely to be relevant pathogenetic mechanisms of neurodegeneration that produce the PiD phenotype.
Although exon 12 mutations are missense mutations that affect all 6 tau isoforms, mutations at neighboring codons p.G335S (34) and p.V337M (35) cause distinct microscopic and functional findings. G335-Q336-V337 resides in the microtubule binding domain that is central to regulation of microtubule polymerization, and a hallmark of the structural core tau protein that forms filamentous aggregates isolated from patients with Alzheimer disease (36). Because these mutations have been observed to affect microtubule binding and aggregation properties of tau in an isoform specific manner differentially, this provides a framework to understand how filamentous tau aggregates might differ in structure and isoform composition even when mutations are found on consecutive amino acids. These differences are of significance because there is growing evidence that structurally distinct filamentous aggregates can drive disease-specific processes (37).
Both p.Q336H and p.Q336R (16) are unusual in that they increase microtubule binding for both 3R and 4R isoforms but show dramatic increase tau aggregation selectively for 3R isoforms. This contrasts with p.G335S, which greatly reduces microtubule binding of both 3R and 4R tau isoforms with little effect on tau aggregation (34). In contrast to the 3R tau straight filaments found in p.Q336H and p.Q336R (16), aggregates in p.G335S contain all 6 isoforms and form Alzheimer-type paired helical filaments. Interestingly, the p.V337M (35) mutation also has aggregation of all 6 isoforms as paired helical filaments but it appears that this is because the mutation increases aggregation properties of both 3R and 4R tau with only modest effects on microtubule binding. The results suggest that a small domain of tau protein (G335-Q336-V337) plays an important role in both microtubule binding and tau aggregation and that the mutations in this domain can lead to deposition of tau aggregates that contain either all 6 isoforms or preferentially 3R tau isoforms.
Our findings indicate that p.Q336H mutation in exon 12 of MAPT causes hereditary PiD with neuropathologic, biochemical, and clinical phenotype indistinguishable from sporadic PiD. The pathologic effect of p.Q336H is based on at least 2 distinct mechanisms: increased ability of mutant tau to promote microtubule assembly and isoform-specific (3R more than 4R) increase in pathologic filament formation. These are features shared by both mutations at MAPT codon 336.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the patient and his family members who agreed to brain donation. We also acknowledge expert technical assistance of Linda Rousseau and Virginia Phillips for histology, Monica Castanedes-Casey for immunohistochemistry and Patricia Brown for DNA extraction and purification. We appreciate the donation of antibodies for this study from Dr. Peter Seubert, Elan Pharmaceuticals, South San Francisco, CA, and from Dr. Peter Davies, Feinstein Institute for Medical Research, North Shore LIJ Health Care System. We also thank Audrey J. Strongosky for her help with determining the family pedigree. We thank Aleksandra Wojtas, Matthew C. Baker, Dr. Neill Graff-Radford, Dr. Leonard Petrucelli, Dr. Melissa E. Murray, Dr. Naomi Kouri, Dr. Shinsuke Fujioka and Dr. Catherine Labbé for their expertise, comments and assistance with preparing the manuscript.
Sources of support: NIH grants: P50 NS072187 (Dennis W. Dickson, Owen A. Ross, Rosa Rademakers, Zbigniew K. Wszolek), P50 AG016574 (Dennis W. Dickson, Rosa Rademakers), R01 NS078086 (Owen A. Ross). Robert E. Jacoby Professorship in Alzheimer’s Research (Dennis W. Dickson), Max Kade Foundation (Pawel Tacik), Allergan Medical Educational Grant (Pawel Tacik). Gift from Carl Edward Bolch, Jr. and Susan Bass Bolch Foundation (Zbigniew K. Wszolek).
REFERENCES
- 1.Barker WW, Luis CA, Kashuba A, et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord. 2002;16:203–212. doi: 10.1097/00002093-200210000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Uchihara T, Tsuchiya K. Neuropathology of Pick body disease. In: Duyckaerts C, Litvan I, editors. Handbook of Clinical Neurology. Vol. 89. Amsterdam: Elsevier; 2008. pp. 415–430. [DOI] [PubMed] [Google Scholar]
- 3.Pick A. Über die Beziehungen der senilen Hirnatrophie zur Aphasie. Prag Med Wochenschr. 1892;17:165–167. [Google Scholar]
- 4.Dickson DW, Kouri N, Murray ME, et al. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau) J Mol Neurosci. 2011;45:384–389. doi: 10.1007/s12031-011-9589-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kovacs GG, Rozemuller AJM, van Swieten JC, et al. Neuropathology of the hippocampus in FTLD-Tau with Pick bodies: a study of the BrainNet Europe Consortium. Neuropathol Appl Neurobiol. 2013;39:166–178. doi: 10.1111/j.1365-2990.2012.01272.x. [DOI] [PubMed] [Google Scholar]
- 6.Uchihara T. Silver diagnosis in neuropathology: principles, practice and revised interpretation. Acta Neuropathol. 2007;113:483–499. doi: 10.1007/s00401-007-0200-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graff-Radford NR, Damasio AR, Hyman BT, et al. Progressive aphasia in a patient with Pick's disease: a neuropsychological, radiologic, and anatomic study. Neurology. 1990;40:620–626. doi: 10.1212/wnl.40.4.620. [DOI] [PubMed] [Google Scholar]
- 8.Piguet O, Halliday GM, Reid WG, et al. Clinical phenotypes in autopsy-confirmed Pick disease. Neurology. 2011;76:253–259. doi: 10.1212/WNL.0b013e318207b1ce. [DOI] [PubMed] [Google Scholar]
- 9.Ghetti B, Oblak AL, Boeve BF, et al. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 2015;41:24–46. doi: 10.1111/nan.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickering-Brown S, Baker M, Yen SH, et al. Pick's disease is associated with mutations in the tau gene. Ann Neurol. 2000;48:859–867. [PubMed] [Google Scholar]
- 11.Bronner IF, ter Meulen BC, Azmani A, et al. Hereditary Pick's disease with the G272V tau mutation shows predominant three-repeat tau pathology. Brain. 2005;128:2645–2653. doi: 10.1093/brain/awh591. [DOI] [PubMed] [Google Scholar]
- 12.Nasreddine ZS, Loginov M, Clark LN, et al. From genotype to phenotype: A clinical, pathological, and biochemical investigation of frontotemporal dementia and parkinsonism (FTDP-17) caused by the P301L tau mutation. Ann Neurol. 1999;45:704–715. doi: 10.1002/1531-8249(199906)45:6<704::aid-ana4>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 13.Tacik P, DeTure M, Lin WL, et al. A novel tau mutation, p.K317N, causes globular glial tauopathy. Acta Neuropathol. 2015;130:199–214. doi: 10.1007/s00401-015-1425-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adams SJ, DeTure MA, McBride M, et al. Three repeat isoforms of tau inhibit assembly of four repeat tau filaments. Plos One. 2010:5. doi: 10.1371/journal.pone.0010810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sergeant N, Delacourte A, Buee L. Tau protein as a differential biomarker of tauopathies. Biochim Biophys Acta. 2005;1739:179–197. doi: 10.1016/j.bbadis.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 16.Pickering-Brown SM, Baker M, Nonaka T, et al. Frontotemporal dementia with Pick-type histology associated with Q336R mutation in the tau gene. Brain. 2004;127:1415–1426. doi: 10.1093/brain/awh147. [DOI] [PubMed] [Google Scholar]
- 17.Rizzini C, Goedert M, Hodges JR, et al. Tau gene mutation K257T causes a tauopathy similar to Pick's disease. J Neuropathol Exp Neurol. 2000;59:990–1001. doi: 10.1093/jnen/59.11.990. [DOI] [PubMed] [Google Scholar]
- 18.vanSwieten JC, Bronner IF, Severijnen AALA, et al. The delta K280 mutation in MAP tau favors exon 10 skipping in vivo. J Neuropathol Exp Neurol. 2007;66:17–25. doi: 10.1097/nen.0b013e31802c39a4. [DOI] [PubMed] [Google Scholar]
- 19.Rosso SM, van Herpen E, Deelen W, et al. A novel tau mutation, S320F, causes a tauopathy with inclusions similar to those in Pick's disease. Ann Neurol. 2002;51:373–376. doi: 10.1002/ana.10140. [DOI] [PubMed] [Google Scholar]
- 20.Neumann M, Schulz-Schaeffer W, Crowther RA, et al. Pick's disease associated with the novel Tau gene mutation K369I. Ann Neurol. 2001;50:503–513. doi: 10.1002/ana.1223. [DOI] [PubMed] [Google Scholar]
- 21.Taniguchi S, McDonagh AM, Pickering-Brown SM, et al. The neuropathology of frontotemporal lobar degeneration with respect to the cytological and biochemical characteristics of tau protein. Neuropath Appl Neuro. 2004;30:1–18. doi: 10.1046/j.0305-1846.2003.00481.x. [DOI] [PubMed] [Google Scholar]
- 22.Mann DMA, South PW, Snowden JS, Neary D. Dementia of frontal-lobe type - neuropathology and immunohistochemistry. J Neurol Neurosurg Psychiatry. 1993;56:605–614. doi: 10.1136/jnnp.56.6.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dickson DW, Yen SH, Suzuki KI, et al. Ballooned neurons in select neurodegenerative diseases contain phosphorylated neurofilament epitopes. Acta Neuropathol. 1986;71:216–223. doi: 10.1007/BF00688042. [DOI] [PubMed] [Google Scholar]
- 24.Tolnay M, Probst A. Ballooned neurons expressing alphaB-crystallin as a constant feature of the amygdala in argyrophilic grain disease. Neurosci Lett. 1998;246:165–168. doi: 10.1016/s0304-3940(98)00250-x. [DOI] [PubMed] [Google Scholar]
- 25.Lantos PL, Cairns NJ, Khan MN, et al. Neuropathologic variation in frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology. 2002;58:1169–1175. doi: 10.1212/wnl.58.8.1169. [DOI] [PubMed] [Google Scholar]
- 26.Sparks DL, Danner FW, Davis DG, et al. Neurochemical and histopathologic alterations characteristic of Pick's disease in a non-demented individual. J Neuropathol Exp Neurol. 1994;53:37–42. doi: 10.1097/00005072-199401000-00005. [DOI] [PubMed] [Google Scholar]
- 27.Lang AE, Bergeron C, Pollanen MS, et al. Parietal Pick's disease mimicking cortical-basal ganglionic degeneration. Neurology. 1994;44:1436–1440. doi: 10.1212/wnl.44.8.1436. [DOI] [PubMed] [Google Scholar]
- 28.Arima K. Ultrastructural characteristics of tau filaments in tauopathies: immuno-electron microscopic demonstration of tau filaments in tauopathies. Neuropathology. 2006;26:475–483. doi: 10.1111/j.1440-1789.2006.00669.x. [DOI] [PubMed] [Google Scholar]
- 29.Murayama S, Mori H, Ihara Y, et al. Immunocytochemical and ultrastructural studies of Picks disease. Ann Neurol. 1990;27:394–405. doi: 10.1002/ana.410270407. [DOI] [PubMed] [Google Scholar]
- 30.Dickson DW. Pick's disease: A modern approach. Brain Pathol. 1998;8:339–354. doi: 10.1111/j.1750-3639.1998.tb00158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobayashi K, Hayashi M, Kidani T, et al. Pick's disease pathology of a missense mutation of S305N of frontotemporal dementia and parkinsonism linked to chromosome 17: Another phenotype of S305N. Dement Geriatr Cogn. 2004;17:293–297. doi: 10.1159/000077157. [DOI] [PubMed] [Google Scholar]
- 32.Chaunu MP, Deramecourt V, Buee-Scherrer V, et al. Juvenile frontotemporal dementia with parkinsonism associated with tau mutation G389R. J Alzheimers Dis. 2013;37:769–776. doi: 10.3233/JAD-130413. [DOI] [PubMed] [Google Scholar]
- 33.Deramecourt V, Lebert F, Maurage CA, et al. Clinical, neuropathological, and biochemical characterization of the novel tau mutation P332S. J Alzheimers Dis. 2012;31:741–749. doi: 10.3233/JAD-2012-120160. [DOI] [PubMed] [Google Scholar]
- 34.Spina S, Murrell JR, Yoshida H, et al. The novel Tau mutation G335S: clinical, neuropathological and molecular characterization. Acta Neuropathol. 2007;113:461–470. doi: 10.1007/s00401-006-0182-5. [DOI] [PubMed] [Google Scholar]
- 35.Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 36.Wischik CM, Novak M, Edwards PC, et al. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci USA. 1988;85:4884–4888. doi: 10.1073/pnas.85.13.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanders DW, Kaufman SK, DeVos SL, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–1288. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rizzini C, Goedert M, Hodges JR, et al. Tau gene mutation K257T causes a tauopathy similar to Pick's disease. J Neuropathol Exp Neurol. 2000;59:990–1001. doi: 10.1093/jnen/59.11.990. [DOI] [PubMed] [Google Scholar]
- 39.Pickering-Brown S, Baker M, Yen SH, et al. Pick's disease is associated with mutations in the tau gene. Ann Neurol. 2000;48:859–867. [PubMed] [Google Scholar]
- 40.Hasegawa M, Smith MJ, Goedert M. Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett. 1998;437:207–210. doi: 10.1016/s0014-5793(98)01217-4. [DOI] [PubMed] [Google Scholar]
- 41.Goedert M, Jakes R, Crowther RA. Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 1999;450:306–311. doi: 10.1016/s0014-5793(99)00508-6. [DOI] [PubMed] [Google Scholar]
- 42.Barghorn S, Zheng-Fischhofer Q, Ackmann M, et al. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry. 2000;39:11714–11721. doi: 10.1021/bi000850r. [DOI] [PubMed] [Google Scholar]
- 43.de Silva R, Lashley T, Strand C, et al. An immunohistochemical study of cases of sporadic and inherited frontotemporal lobar degeneration using 3R-and 4R-specific tau monoclonal antibodies. Acta Neuropathol. 2006;111:329–340. doi: 10.1007/s00401-006-0048-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.