

Abstract
The P2Y14R is a Gi/o-coupled receptor of the P2Y family of purinergic receptors that is activated by extracellular UDP and UDP-glucose (UDPG). In an earlier report we described a P2Y14R fluorescent probe, MRS4174, based on the potent and selective antagonist PPTN, a naphthoic acid derivative. Here, we report the design, preparation, and activity of an agonist-based fluorescent probe MRS4183 (11) and a shorter P2Y14R agonist congener, which contain a UDP-glucuronic acid pharmacophore and BODIPY fluorophores conjugated through diaminoalkyl linkers. The design relied on both docking in a P2Y14R homology model and established structure activity relationship (SAR) of nucleotide analogues. 11 retained P2Y14R potency with EC50 value of 0.96 nM (inhibition of adenylyl cyclase), compared to parent UDPG (EC50 47 nM) and served as a tracer for microscopy and flow cytometry, displaying minimal nonspecific binding. Binding saturation analysis gave an apparent binding constant for 11 in whole cells of 21.4±1.1 nM, with a t1/2 of association at 50 nM 11 of 23.9 min. Known P2Y14R agonists and PPTN inhibited cell binding of 11 with the expected rank order of potency. The success in the identification of a new P2Y14R fluorescent agonist with low nonspecific binding illustrates the advantages of rational design based on recently determined GPCR X-ray structures. Such conjugates will be useful tools in expanding the SAR of this receptor, which still lacks chemical diversity in its collective ligands.
Keywords: G protein-coupled receptor, nucleotides, pyrimidines, homology modeling, docking, P2Y receptor
Graphical abstract

G protein-coupled receptors (GPCRs) that respond to extracellular uracil and adenine nucleotides (P2Y receptors, P2YRs) constitute important and ever-growing targets for pharmacological exploration. Eight nucleotide GPCRs are divided into two subfamilies of P2YRs, based on sequence and functional similarity: P2Y1R-like, P2Y1,2,4,6,11; and P2Y12R-like, P2Y12–14. The P2YRs are broadly distributed among many tissues and are expressed in immune cells, intestine, kidney, lung, nervous systems, and others.1
The P2Y14R is involved in inflammation, hypoxia2 and mechanical pain hypersensitivity3, and its activation enhances neutrophil chemotaxis4 and promotes the release of mediators from mast cells.5 Studies with P2Y14R−/− mice suggested that P2Y14R antagonism might be a target for diabetes therapy; another study suggested that P2Y14R activation enhances insulin release.6,19 Thus, the P2Y14R is a potential pharmaceutical target for inflammation, hypoxia, and endocrine mis-function.
Given the diversity of expression of the P2Y14R and the ubiquitous nature of its endogenous activators, i.e. UDP (Chart 1, 1, EC50 160 nM20) and UDP-sugars (2, EC50 261 nM8; 4, EC50 370 nM8), the development of ligands selectively targeting this receptor is a considerable challenge and an important goal for pharmacological studies and potential therapeutic applications. A limited number of chemical classes have been identified as ligands and explored for their effects on the P2Y14R: synthetic nucleotide analogues of 1 and 2, such as 3 (EC50 11 nM20),7, 8 derivatives of 2-naphthoic acid, most notably the potent and selective antagonist 4-(4-(piperidin-4-yl)-phenyl)-7-(4-(trifluoromethyl)-phenyl)-2-naphthoic acid (PPTN, 5, KB 0.43 nM),9, 10, 11 and substituted pyrido[4,3-d]pyrimidines including potent analogue 6 (IC50 10 nM).12
Chart 1.

Representative P2Y14R agonists 1–4, antagonists 5, 6 and fluorescent antagonist MRS 4174 7.
Nucleotide analogues of endogenous P2Y14R agonist 1 would be poor drug candidates due to their low bioavailability associated with high charge and their chemical instability due to hydrolysis by endonucleotidases. Derivatives of nonnucleotide P2Y14R antagonist 5 also suffer from poor drug-like characteristics due to high lipophilicity.9, 13 However, the chemical stability, high affinity and selectivity of 5 toward the P2Y14R attracted our attention and inspired its use as a template for the development of fluorescent antagonist affinity probes.10, 14 We applied virtual docking to design analogues of 5 that retain or enhance P2Y14R affinity and tolerate the increase in molecular size and interactions associated with fluorophore conjugation. A human (h) P2Y14R homology model was constructed based on the recently reported structures of the hP2Y12R.15, 16 This model was successfully utilized in predicting a suitable site on 5 for fluorophore conjugation and the minimum linker length required to provide adequate spacing between the pharmacophore and the fluorophore. The resulting PPTN-Alexa Fluor 488 conjugate (MRS4174, 7) displayed sub-nanomolar antagonist affinity at the hP2Y14R and a very low level of nonspecific binding. Its binding to the hP2Y14R stably expressed in mammalian cells was inhibited by other P2Y14R ligands with the appropriate rank order of potency.
In the present study, we designed with the aid of molecular modeling and synthesized a pharmacologically complementary high affinity fluorescent agonist-based probe. The suitability of the site of attachment and the dependence of potency on the length of the spacer chain were predicted by a structure-based analysis using a homology model of the P2Y14R.14,21 Our previous modeling study concluded that the glucose moiety of 4 was the most structurally permissive region of this endogenous agonist since it bound in the second subpocket of the P2Y14R binding site, which is accessible to the extracellular medium. Therefore, two fluorescent derivatives of 4 were designed and prepared (Scheme 1) based on predictions from computational modeling of P2Y14R agonist binding.21 The strategy behind the selection of these target compounds is described below. The conjugation of a fluorophore was accomplished via condensation of commercially available fluorescent amines with the carboxylic group of 4.
Scheme 1.
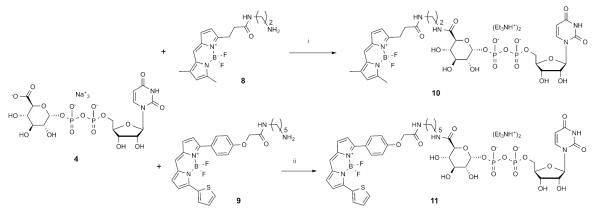
Synthesis of fluorescent conjugates of 4. aReagents and conditions: i: HATU, Et3N, DMF, 23 °C, 2% yield; ii: COMU (1.5 eq.), DIPEA (2 eq.), DMF, 0 °C, 0.3% yield.
Based on previous structure activity relationship (SAR) studies of 2, two designs of fluorescent affinity probes of varying chain length were compared. The structures of 1 and 2, now identified as endogenous P2Y14R agonists, have been extensively probed by chemical modification with regard to their ability to activate the P2Y14R, as well as P2Y2, P2Y4 and P2Y6Rs. 7, 8, 17. The C6 carbon of the hexose moiety of UDPGA 4 was found suitable for chain extension with retention of P2Y14R agonist activity. A number of amide derivatives at the glucose C6 have been constructed, and most of these derivatives retained agonist activity. It was also found that P2Y14R agonist activity was retained with attachment of large groups such as unprotected, as well as acetyl- and Boc-protected aminoethylamides, and polyamidoamine (PAMAM) dendrimers of generation (G) 3 and 6 attached to the C6 carbon.17 Moreover, it was found that dendrimer conjugates possessed enhanced potency when compared to 4.
Thus, we chose the structure of 4 as a starting point for designing and building fluorescent probes. Our objectives were to facilitate the availability of such affinity probes and to validate further the previously constructed computational models. Hence, we chose as our primary fluorophore boron-dipyrromethene (BODIPY), which has been used in other fluorescent probes of GPCRs.18 Unlike the chemical series of hydrophobic P2Y14R antagonist 5, in which a less hydrophobic fluorophore, AlexaFluor 488, in conjugate 7 was optimal, the restriction of choosing a hydrophilic fluorophore was relieved due to the inherent high polarity and hydrophilicity of 4. Additionally, various BODIPY dyes with built-in reactive amine linkers of varying length are readily available commercially. Two amide-bound conjugates, 10 and MRS4183 11 (Scheme 1), were docked into a homology model of the hP2Y14R to predict their fit prior to synthesis. The target molecules 10 and 11 contained linkers consisting of 7 and 14 (including benzene ring) atoms, respectively, between C6 of hexose and the pyrrole ring of BODIPY.
A homology model of the hP2Y14R was constructed as we reported, based on the high resolution X-ray crystallographic structure of the P2Y12R complex with 2-MeSADP.15, 16, 21 Various known P2Y14R agonists were docked in the orthosteric binding site. The attempted docking of the fluorescent agonists 10 and 11 did not provide a satisfactory binding pose for conjugate 10, suggesting the inability of the binding pocket to accommodate both pharmacophore and fluorophore connected by a short, 7-atom linker containing ethylene diamine. However, the hypothetical binding mode of 11 revealed that the longer, 14-atom linker containing 1,5-diaminopentane is sufficiently long to connect the pharmacophore inside the deep binding site with the fluorophore presented on the extracellular side of the receptor (Figure 1). According to the obtained binding mode, the nucleobase binds inside the pocket formed by TMs 3–5, including aromatic stacking with Y1023.33 (using standard numbering relative to the TMs26) and with carbonyl oxygen atoms forming hydrogen bond contacts with side chains of H1845.35 and N1885.39. The ribose hydroxyls form contacts with N1564.57 side chain and the backbone carbonyl of C943.25. The diphosphate moiety forms salt bridges with positively charged K772.60, K2777.35, and R2536.55, and makes polar contacts with Y1023.33 and Q2606.60 side chains. The amide functionalities of the linker were found to form hydrogen bond contacts with the side chain of K171 (EL2) on the interior of the receptor and the backbone of K270 (EL3) on the extracellular part. The linker projects toward the extracellular region via a channel along TM3 and through the window formed by EL1 and EL2.
Figure 1.

Hypothetical binding mode of 11 in a hP2Y14R homology model based on the structure of the nucleotide-bound hP2Y12R.14,21 12 The BODIPY fluorophore is predicted to interact with residues of the ELs, including K270. Other residues shown correspond to TMs: 2 (K772.60, D812.64); 3 (Y1023.33); 5 (H1845.35, N1885.39); 6 (R2536.55, Q2606.62); 7 (K2777.35).
The synthesis of derivatives 10 and 11 was accomplished using standard coupling conditions involving the primary amine congeners of the dyes (8 and 9) and the carboxylate group of UDP-glucuronic acid 4. In particular, 10 was synthesized initially using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) as coupling agent and NEt3 as a base, while in the case of 11 different conditions were tried (Table S1, Supporting Information) to increase the reaction yield and simplify the purification process. Indeed, the first attempt (condition A) to synthetize 11 resulted in an impure product even after several successive purification attempts, due to an impurity co-eluting with the desired product. The higher reactivity of a different coupling agent, [[(Z)-(1-cyano-2-ethoxy-2-oxoethylidene)amino]oxy-morpholin-4-ylmethylidene]-dimethylazanium hexafluorophosphate (COMU, conditions B and C), compared to HATU led to the formation of numerous side products during the reaction that complicated the purification process (2 or 3 purifications) without leading to a significant detectable amount of title compound. Nevertheless, the best performing procedure (condition D), using excess 4 (1.5 equivalents), COMU (1.5 equivalents) as coupling agent and DIPEA (2 equivalents) as base, provided the desired, pure product after one purification step using a C18 HPLC column, however, in a very low yield (0.3%). 11 was found to have excitation and emission maxima at 590 and 616 nm, respectively (Figure S2, Supporting information).
Both 10 and 11 were evaluated functionally in Chinese hamster ovary (CHO) cells stably expressing the Gi-coupled human P2Y14R (CHO- P2Y14R) for capacity to inhibit forskolin-stimulated 3′,5′-cyclic adenosine monophosphate (cAMP) accumulation (Figure 2).10 UDPGABODIPY conjugate 11 retained its agonist activity with a nanomolar EC50 value. The EC50 value of 0.96 nM for inhibition of cAMP production in hP2Y14R-expressing CHO cells, compared to 47 nM for 4, constitutes a 50-fold potency enhancement in the fluorescent conjugate. Thus, 11 improved upon the potency of parent UDP-sugars despite the presence of a large fluorophore attached to its hexose C6 position. Accordingly, 11 demonstrated strong specific binding to P2Y14R-CHO cells in flow cytometry (FCM) (Figures 3A, B & C). The level of nonspecific binding determined by coincubating with up to 200 nM 5 was very low. We also quantified saturation of binding of 11 to CHO cells expressing P2Y14R using FCM. Increasing concentrations of 11 from 1 nM to 500 nM were used to calculate the total binding, and nonspecific binding was determined in the presence of 20 μM 5. From the saturation curve, an apparent binding constant (Kd app) was determined to be 21.4±1.1 nM (Figure 4A). The kinetics of 11 binding in CHO cells expressing P2Y14R was studied using FCM, and the t1/2 of binding of 50 nM 11 was 23.9 min (Figure 4B).
Figure 2.

Concentration-response curves for endogenous agonist 2 (blue), short-chain conjugate 10 (red) and long-chain conjugate 11 (black) for inhibition of forskolin-stimulated cAMP formation in P2Y14R-expressing CHO cells, as described.14 Incubations were at 37 °C for 15 min and were terminated by aspiration of medium and addition of 500 μL of ice-cold 5% trichloroacetic acid. All assays with 6, 22, and 30 also contained 30 μM forskolin and 200 μM IBMX, 3-isobutyl-1-methylxanthine, a phosphodiesterase inhibitor. Data points for IBMX alone (indicated by IBMX in the legend) and forskolin + IBMX (indicated by forskolin in the legend) also are shown.
Figure 3.

(A) Inhibition of binding of 11 in P2Y14R-CHO cells by antagonist PPTN 5 and agonists UDP 1 and UDPG 2 (10 μM), analyzed by flow cytometry. Incubation of cells for 30 min with antagonist 5 or agonist 1 or 2 (all at 10 μM) followed by incubation with 11 (50 nM) for 30 min was performed. (B) Dependence of fluorescent labeling of P2Y14R-CHO cells on the concentration of 11, analyzed by flow cytometry. Incubation of cells with a variable concentration of 11 (1 nM – 500 nM) was performed following 60 min of incubation in the absence or presence of PPTN 5 (200 nM). Nearly complete inhibition of binding was observed up to 100 nM 11. (C) Fluorescence ligand binding experiments using FCM in P2Y14R-CHO cells with 11 (50 nM) after 30 min pre-incubation at 37 °C with known P2Y14R agonists 1 and 2 or P2Y14R antagonist 5 (each 10 μM). Each column shows the fluorescent intensity of each compound using 11 normalized to 100% for each time point and after correcting the mean fluorescence intensity values for auto fluorescence. Results are expressed as mean±S.E (n=3).
Figure 4.

(A) Saturation binding assays (B) binding kinetics with tracer 11 using FCM following incubation for 30 min with CHO cells expressing the hP2Y14R. Mean fluorescent Intensity values from FCM were converted to MESF values (Figure S6) using Quickcal program v. 2.3 (Bangs Laboratories Inc., Fishers, IN). Nonspecific binding was measured in the presence of 11 and 20 μM 5. Results are expressed as mean±S.E (n=3). In (A), the Kd value is 26.1±3.6 nM, Bmax = 44,400 MESF (n=3). In (B), 100% corresponds to a MESF value of 26,900.
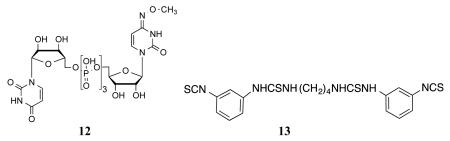
Competition for fluorescent binding with known P2Y14R ligands and P2Y6R ligands was also performed. These two types of ligands were compared because both receptors are activated by UDP, and no other P2YRs respond to UDP. To determine the Ki values of known ligands, we used 11 as a tracer, at a concentration of 50 nM. Competitive binding results with known nucleotide agonists and nonnucleotide antagonist 5 shown in Table 1 roughly followed the expected rank order of P2Y14R potency (5 >> 3 > 1 > 2 > 4). The P2Y6R agonist MRS2957 12, a dinucleoside triphosphate, also inhibited binding of 11, but with a Ki value of 5.18 μM, which is about 500-fold higher than its EC50 value (12 nM) for activation of the P2Y6R.24 The nonnucleotide antagonist of P2Y6R, MRS2578 13,25 did not show any significant binding to P2Y14R at 10 μM, thus confirming its P2Y6R selectivity. As predicted by computational modeling, conjugate 10 was much weaker than 11, with an EC50 of 91 nM for inhibition of cAMP accumulation in P2Y14R-CHO cells. However, over a range of concentrations (0.05 −1 μM) it did not demonstrate significant fluorescence binding in comparison to nonspecific binding in FCM experiments (data not shown).
Table 1.
Inhibition of P2Y14R binding in whole cells by known ligands of P2Y14R and P2Y6R using 11 as a FCM tracer. Results are expressed as mean±S.E (n=3).a
| Compound | hP2Y14R cAMP assay (EC50 or IC50b, μM) |
FCM binding at hP2Y14R (Ki, μM) |
|---|---|---|
| P2Y14 ligands | ||
| 1 UDP | 0.160 | 0.63±0.03 |
| 2 UDP-glucose | 0.261 | 2.23±0.2 |
| 3 MRS2690 | 0.011 | 0.34±0.04 |
| 4 UDP-glucuronic acid | 0.370 | 5.3±0.4 |
| 5 PPTN | 0.00043 | 0.0019±0.0001 |
| P2Y6 ligands | ||
| 12 MRS2957c | ND | 5.18±0.20 |
| 13 MRS2578c | ND | No inhibition at 10 μM |
Confocal fluorescence microscopy indicated specific labeling of P2Y14R-CHO cells by 11 (Figure 5). The fluorescent labeling was inhibited in the presence of competing antagonist 5 and was also absent in control CHO cells lacking P2Y14R expression (not shown). The label appears irregularly distributed on the surface of the cells and partially internalized, consistent with 11 being an agonist of the P2Y14R. Similarly, the internalization of fluorescent agonists of other P2YRs and GPCRs was reported.22,23
Figure 5.

Fluorescence confocal microscopy. Fluorescent labeling of P2Y14R-CHO cells by 11, analyzed by confocal fluorescence microscopy. Incubation of cells with a fixed concentration of 11 (200 nM) was performed following 30 min (A, B) or 120 min (C, D) of incubation in the absence (A, C) or presence (B, D) of antagonist 5 (10 μM). The complete inhibition of binding was observed in the presence of the antagonist. Scale of 60 μm is shown in D. The excitation laser used had a peak at 555 nm and was equipped with a 561 nm filter. Emitted fluorescence was measured without a filter.
In conclusion, we applied computational tools and known P2Y14R agonist SAR toward design of a new agonist fluorescent probe. An insensitive site on the glucose moiety of 4 was covalently derivatized, and two linkers of different length were tested both computationally and experimentally. The demonstrated predictive strength of the utilized approach highlights the validity and usefulness of this particular P2Y14R homology model and the strategy at large when applied toward probe design. Additionally, the lack of fluorescent binding of the analogue 10 with a shorter linker and the strong binding of 11 provide an additional estimate of the shape, size and depth of the orthosteric ligand binding pocket of the P2Y14R. Thus, we have used insights from P2YR structures and modeling to identify 11 as a useful agonist fluorescent probe of the P2Y14R, which has high affinity and low nonspecific binding. Its binding properties and inhibition by known P2YR ligands could readily be determined using FCM. Our previous efforts to characterize this receptor included the design and preparation of a fluorescent probe based on the potent and selective antagonist 5. In order to further the study the role and distribution of this receptor, we introduce here a useful fluorescent probe that is pharmacologically complementary to our reported nonnucleotide fluorescent antagonist 7. The utility of this probe to detect P2Y14R using fluorescence microscopy was also demonstrated. Further studies with regard to fluorescence binding of both agonist and antagonist probes and receptor pharmacology as well as their suitability for ligand affinity screening are currently underway.
Supplementary Material
Acknowledgments
We thank Dr. John Lloyd and Dr. Noel Whittaker (NIDDK) for mass spectral determinations. This research was supported by the National Institutes of Health (Intramural Research Program of the NIDDK, Z01 DK031116), NIGMS Postdoctoral Research Associate (PRAT) Program and grant nos. GM38213 to T.K.H. and U54GM094618 to V.K. and R.C.S.
List of abbreviations
- BODIPY
boron-dipyrromethene
- cAMP
3′,5′-cyclic adenosine monophosphate
- CHO
Chinese hamster ovary
- COMU
[[(Z)-(1-cyano-2-ethoxy-2-oxoethylidene)amino]oxy-morpholin-4-ylmethylidene]-dimethylazanium hexafluorophosphate
- DMEM
Dulbecco’s modified Eagle’s medium
- DMF
dimethylformamide
- ECL
extracellular loop
- FCM
flow cytometry
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- IBMX
3-isobutyl-1-methylxanthine
- 2-MeSADP
2-methylthioadenosine-5′-diphosphate
- MESF
molecules of equivalent soluble fluorochrome
- MRS2578
N,N”-1,4-butanediylbis[N’-(3-isothiocyanatophenyl)]thiourea]
- MRS2957
P1-(uridine 5′-)-P3-( N4-methoxycytidine 5′-)triphosphate
- PPTN
4-(4-(piperidin-4-yl)-phenyl)-7-(4-(trifluoromethyl)-phenyl)-2-naphthoic acid
- SAR
structure activity relationship
- UDPG
uridine-5′-diphosphoglucose
- TM
transmembrane helix
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supporting Information Available: Coordinate file of the P2Y14R model-complex with 11, synthetic procedures used for preparation of 10 and 11, NMR and mass spectra of selected compounds and fluorescence spectra of 10 and 11, are included.
References
- 1.Boeynaems J-M, Communi D, Robaye B. Wiley Interdisciplinary Reviews: Membrane Transport and Signaling. 2012;1:581. [Google Scholar]
- 2.Lazarowski ER, Harden TK. Mol. Pharmacol. 2015;88:151. doi: 10.1124/mol.115.098756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kobayashi K, Yamanaka H, Yanamoto F, Okubo M, Noguchi K. Glia. 2012;60:1529. doi: 10.1002/glia.22373. [DOI] [PubMed] [Google Scholar]
- 4.Sesma JI, Kreda SM, Steinckwich-Besancon N, Dang H, Garcia-Mata R, Harden TK, Lazarowski ER. A. J. Physiol. Cell Physiol. 2012;303:C490. doi: 10.1152/ajpcell.00138.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao ZG, Ding Y, Jacobson KA. Biochem. Pharmacol. 2010;79:873. doi: 10.1016/j.bcp.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu J, Morinaga H, Oh D, Li P, Chen A, Talukdar S, Mamane Y, Mancini JA, Nawrocki AR, Lazarowski E, Olefsky JM, Kim JJ. J. Immunol. 2012;189:1992. doi: 10.4049/jimmunol.1103207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ko H, Fricks I, Ivanov AA, Harden TK, Jacobson KA. J. Med. Chem. 2007;50:2030. doi: 10.1021/jm061222w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ko H, Das A, Carter RL, Fricks IP, Zhou Y, Ivanov AA, Melman A, Joshi BV, Kovac P, Hajduch J, Kirk KL, Harden TK, Jacobson KA. Bioorg. Med. Chem. 2009;17:5298. doi: 10.1016/j.bmc.2009.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gauthier JY, Belley M, Deschenes D, Fournier JF, Gagne S, Gareau Y, Hamel M, Henault M, Hyjazie H, Kargman S, Lavallee G, Levesque JF, Li L, Mamane Y, Mancini J, Morin N, Mulrooney E, Robichaud J, Therien M, Tranmer G, Wang Z, Wu J, Black WC. Bioorg. Med. Chem. Lett. 2011;21:2836. doi: 10.1016/j.bmcl.2011.03.081. [DOI] [PubMed] [Google Scholar]
- 10.Barrett MO, Sesma JI, Ball CB, Jayasekara PS, Jacobson KA, Lazarowski ER, Harden TK. Mol. Pharmacol. 2013;84:41. doi: 10.1124/mol.113.085654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belley M, Deschenes D, Fortin R, Fournier JF, Gagne S, Gareau Y, Gauthier JY, Li L, Robichaud J, Therien M. Patent. 2009
- 12.Guay D, Beaulieu C, Belley M, Crane SN, DeLuca J, Gareau Y, Hamel M, Henault M, Hyjazie H, Kargman S, Chan CC, Xu L, Gordon R, Li L, Mamane Y, Morin N, Mancini J, Therien M, Tranmer G, Truong VL, Wang Z, Black WC. Bioorg. Med. Chem. Lett. 2011;21:2832. doi: 10.1016/j.bmcl.2011.03.084. [DOI] [PubMed] [Google Scholar]
- 13.Robichaud J, Fournier JF, Gagne S, Gauthier JY, Hamel M, Han Y, Henault M, Kargman S, Levesque JF, Mamane Y, Mancini J, Morin N, Mulrooney E, Wu J, Black WC. Bioorg. Med. Chem. Lett. 2011;21:4366. doi: 10.1016/j.bmcl.2010.12.113. [DOI] [PubMed] [Google Scholar]
- 14.Kiselev E, Barrett MO, Katritch V, Paoletta S, Weitzer CD, Brown KA, Hammes E, Yin AL, Zhao Q, Stevens RC, Harden TK, Jacobson KA. ACS Chem. Biol. 2014;9:2833. doi: 10.1021/cb500614p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, Zhang K, Gao ZG, Paoletta S, Zhang D, Han GW, Li T, Ma L, Zhang W, Müller CE, Yang H, Jiang H, Cherezov V, Katritch V, Jacobson KA, Stevens RC, Wu B, Zhao Q. Nature. 2014;509:119. doi: 10.1038/nature13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang K, Zhang J, Gao ZG, Zhang D, Zhu L, Han GW, Moss SM, Paoletta S, Kiselev E, Lu W, Fenalti G, Zhang W, Müller CE, Yang H, Jiang H, Cherezov V, Katritch V, Jacobson KA, Stevens RC, Wu B, Zhao Q. Nature. 2014;509:115. doi: 10.1038/nature13083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das A, Zhou Y, Ivanov AA, Carter RL, Harden TK, Jacobson KA. Bioconjugate Chem. 2009;20:1650. doi: 10.1021/bc900206g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vernall AJ, Hill SJ, Kellam B. Br. J. Pharmacol. 2014;171:1073. doi: 10.1111/bph.12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meister J, Le Duc D, Ricken A, Burkhardt R, Thiery J, Pfannkuche H, Polte T, Grosse J, Schöneberg T, Schulz A. J. Biol. Chem. 2014;289:23353. doi: 10.1074/jbc.M114.580803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das A, Ko H, Burianek LE, Barrett MO, Harden TK, Jacobson KA. J. Med. Chem. 2010;53:471. doi: 10.1021/jm901432g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trujillo K, Paoletta S, Kiselev E, Jacobson KA. Bioorg. Med. Chem. 2015;23:4056. doi: 10.1016/j.bmc.2015.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jayasekara PS, Barrett MO, Ball CB, Brown KA, Kozma E, Costanzi S, Squarcialupi L, Balasubramanian R, Maruoka H, Jacobson KA. Med. Chem. Comm. 2013;4:1156. doi: 10.1039/C3MD00132F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jayasekara PS, Barrett MO, Ball CB, Brown KA, Hammes E, Balasubramanian R, Harden TK, Jacobson KA. J. Med. Chem. 2014;57:3874. doi: 10.1021/jm500367e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maruoka H, Barrett MO, Ko H, Tosh DK, Melman A, Burianek LE, Balasubramanian R, Berk B, Costanzi S, Harden TK, Jacobson KA. J. Med. Chem. 2010;53:4488. doi: 10.1021/jm100287t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mamedova L, Joshi BV, Gao ZG, von Kügelgen I, Jacobson KA. Biochem. Pharmacol. 2004;67:1763. doi: 10.1016/j.bcp.2004.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ballesteros JA, Weinstein H. Methods Neurosci. 1995;25:366. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.