

Abstract
Microbial colonization of the human gastrointestinal tract plays an important role in establishing health and homeostasis. However, the time-dependent functional signatures of microbial and human proteins during early colonization of the gut have yet to be determined. To this end, we employed shotgun proteomics to simultaneously monitor microbial and human proteins in fecal samples from a preterm infant during the first month of life. Microbial community complexity increased over time, with compositional changes that were consistent with previous metagenomic and rRNA gene data. More specifically, the function of the microbial community initially involved biomass growth, protein production, and lipid metabolism, and then switched to more complex metabolic functions, such as carbohydrate metabolism, once the community stabilized and matured. Human proteins detected included those responsible for epithelial barrier function and antimicrobial activity. Some neutrophil-derived proteins increased in abundance early in the study period, suggesting activation of the innate immune system. Likewise, abundances of cytoskeletal and mucin proteins increased later in the time course, suggestive of subsequent adjustment to the increased microbial load. This study provides the first snapshot of coordinated human and microbial protein expression in a preterm infant’s gut during early development.
Keywords: microbial colonization, infant gut, microbiome, metaproteomics
1 Introduction
Microbial communities in the gastrointestinal tract play important roles in human health by processing essential nutrients, protecting against pathogenic bacteria, promoting angiogenesis, and regulating host immune responses [1–5]. Initially near sterile, the infant gastrointestinal tract assembles a microbial community of diverse species composition in the first 2.5 years of life [5–10]. This symbiotic relationship requires a careful balance; it has been postulated that disruption of the host-microbe relationship in the gut can lead to diseases such as inflammatory bowel disease and neonatal necrotizing enterocolitis (NEC) [11, 12]. Initial temporal colonization patterns and species distributions vary between individual infants and may be influenced by environmental exposures, delivery mode, diet, and health [6, 7, 9]. In preterm infants, the microbial community compositions in the gastrointestinal tract are low in diversity, highly variable between infants, and change over time at the species and strain level [13–19]. The lack of microbial community complexity in preterm infants provides a powerful opportunity to study the microbiome development at high resolution.
Previously, the gut microbial compositional patterns of a preterm infant during the first month of life were characterized in an rRNA gene and metagenomics-based study [14]. Through 16S rRNA gene-based analysis of fecal samples, the dominant taxa were identified, and community compositional changes revealed three distinct colonization phases. Specifically, days five through nine of the infant’s life were dominated by Leuconostoc, Weisella, and Lactococcus species while days ten through fourteen, consisted primarily of Pseudomonas and Staphylococcus. The third phase (days of life 15 – 21) was primarily composed of members of the Enterobacteriaceae family, including Citrobacter and Serratia species. This pattern coincided with dietary adjustments at days nine and fifteen, and was similar to premature infants from other studies [14, 16, 19]. Sequencing of whole community DNA, followed by reconstruction and intensive curation of population genomic datasets of the dominant microbial members from days 10, 16, 18, and 21, revealed three major species in samples from these days: a Serratia strain (UC1SER), an Enterococcus strain (UC1ENC), and two closely related Citrobacter strains (UC1CITi and UC1CITii). Also present were plasmids UC1CITp, UC1ENCp, and bacteriophage UC1ENCv (as well as many other incompletely resolved phage/plasmids). While critically important, genomic information provides the inventory of possible gene products, but does not reveal the actual metabolic activities. Thus, here we employed shotgun proteomics via nanospray-two dimensional liquid chromatography coupled with tandem mass spectrometry (nano-2D-LC-MS/MS) to elucidate functional signatures of translated gene products (i.e., proteins) from samples of the same preterm infant for which metagenomic data were available.
The use of mass spectrometry-based proteomics allows characterization and quantification of thousands of proteins within a microbial community [20–25]. While early attempts using metaproteomics for the characterization of the infant microbiome demonstrated feasibility, protein identifications were limited due to insufficient genome information [26]. More recently, due to the availability of highly resolved genomes and the advancement in mass spectrometry instrumentation, proteins can be confidently identified at the species and strain level allowing deep proteomic analysis of microbial communities [20, 21, 23, 24, 27–29]. While prior studies have focused on characterizing microbial genes and proteins, most current methodologies prohibit global analyses of microbial proteins in conjunction with human proteins. Here, we report results of the first proteomics-based investigation of the coordinated expression of human and microbial proteins during initial microbial colonization of a preterm infant’s gut microbiome.
2 Materials and Methods
2.1 Description of Preterm Infant
A female infant born at 28 weeks gestation due to premature rupture of membranes was delivered by cesarean section and given antibiotics for the first 7 days of life [14]. Enteral feedings with breast milk were given on days 4–9, and then on days 9–13, feedings were withheld due to abdominal distension. After day 13, enteral feedings were re-administered in the form of artificial infant formula. The baby also received supplemental parenteral nutrition until day 28. The baby had no major anomalies or comorbidities and was discharged to home on day of life 64. Fecal material was collected on days 5–21 as available, with institutional approval, and was immediately stored at −80 °C until analysis. Metagenomic and 16S rRNA data was analyzed in a companion study from matched samples [14]. Based upon sample availability, proteomic measurements were performed on fecal samples from days 7, 13, 15, 16, 17, 18, 20, and 21 after birth.
2.2 Protein Extraction and Enzymatic Digestion of Fecal Samples
Approximately 250 μg fecal material was boiled for five minutes in 1 ml 100 mM Tris-Cl containing 4% w/v SDS and 10 mM DTT, then underwent continuous bead beating on high setting for 30 minutes, in order to lyse cells and denature/reduce proteins. The supernatant was collected, boiled again, spun down (14,000 g), and precipitated with 20% trichloroacetic acid at −80°C overnight. Protein pellets were washed in ice-cold acetone, re-solubilized in 8 M urea diluted in 100 mM Tris-HCl pH 8, and then sonicated using a Branson sonic disruptor in order to break up the pellet (5 minutes at 20%; 10 seconds on, 10 seconds off). Iodoacetamide (IAA) was added to block disulfide bond reformation. Proteins were quantified using Bicinchronic assay (BCA) and between 1–3 mg protein were diluted to 4 M urea in 100 mM Tris-HCl pH 8, and enzymatically digested into peptides using sequencing grade trypsin (Promega) for four hours at room temperature. Peptides were diluted to 2 M urea, a second dose of trypsin added, and digestion continued overnight. An acidic salt solution (200mM NaCl, 0.1% formic acid), was used to clean up the peptides which were then spun through a 10kDa cutoff spin column filter (VWR). Peptides were quantified by BCA assay and stored at −80°C until further use.
2.3 Nano-2D-LC-MS/MS
A 150 μg peptide mixture was loaded via a pressure cell onto a 150 μm inner diameter split-phase fused silica back column (Polymicro Technologies) hand-packed with reverse phase (C18) and strong cation exchange (SCX) resin (Luna, Phenomenex). The back column was washed off-line with 100% Solvent A (95% H2O, 5% CH3CN and 0.1% formic acid) for 15 minutes at ~140 bar to desalt the column. Peptides were placed in line with a nanospray emitter (New Objective) packed with reverse phase material then separated on-line using high performance two-dimensional liquid chromatography [30–32]. Peptides were eluted from the SCX resin by increasing ammonium acetate salt pulses followed by reverse phase resolution over two hour organic gradients as described previously [20, 21, 28], ionized via nanospray (200 nl/min) (Proxeon, Cambridge MA), and analyzed using an LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific, San Jose, CA). The mass spectrometer was run in data-dependent mode with the top 10 most abundant peptides in full MS selected for MS/MS, and dynamic exclusion enabled (repeat count=1, 60 s exclusion duration). Full MS scans were collected in the Orbitrap at 30K resolution. Two microscans were collected in centroid mode for both full and MS/MS scans. Technical duplicates were run for all samples. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD000114.
2.4 Database Construction and Searching
Metagenomic sequences from matched samples collected on days 10, 16, 18, and 21 were translated, and the predicted protein sequences of the dominant community members were used to generate a search database. These included a Serratia species UC1SER, two closely related Citrobacter strains, UC1CITi and UC1CITii, an Enterococcus species UC1ENC, and associated virus and plasmids UC1ENCp, UC1ENCv, and UC1CITp [14]. Since microbial species from early time points were not represented in the metagenomic sequences, twenty additional isolate sequences were selected, based on 16S rRNA information from matched samples, and added to the search database (acquired from JGI: http://www.hmpdacc-resources.org/cgi-bin/img_hmp/main.cgi in January of 2011). For example, according to the 16S data, Leuconostoc was prevalent on days 5–9 [14]. But since metagenomic sequencing was not performed on days 5–9, Leuconostoc would have been absent from the analysis. 16S sequencing could only identify Leuconostoc at the genus level, so we chose a representative species of this genus, Leuconostoc mesenteroides cremoris, to be included in the database. In addition, human protein sequences (NCBI RefSeq_2011) and common contaminants (i.e. trypsin) were appended to the database. All MS/MS spectra were searched against the concatenated database with the SEQUEST algorithm v.27 (rev.9) [33], and filtered with DTASelect version 1.9 [34] to assemble the identified peptides into their corresponding protein sequences (Supporting Information Table S1). Due to carbamidomethylation effects of IAA, a static cysteine modification (+57) was included in all searches. Only proteins identified with two fully tryptic peptides were considered for further biological study. Reversed protein sequences were appended to the database in order to calculate false discovery rates. The final concatenated search database contained 214,520 protein sequences, including forward and reversed sequences (provided in Supporting Information as search_database.fasta). Conservative cross-correlation filters were used to achieve false discovery rates (FDRs) between 0.5–2.4% at the peptide level [35].
2.5 Database clustering & spectral balancing
Since mass spectrometry based proteomics identifies proteins by their corresponding peptide sequences, data analysis must take into consideration the high levels of protein redundancy within and between species to avoid inflating the total number of proteins identified or misinterpretation of the biological conclusions by over-representing proteins with the same function. Therefore, we applied a bioinformatic clustering algorithm to the database in order improve confidence in protein identification and quantification. Specifically, using the publically-available software, USEARCH v.5.0 [36], microbial proteins were clustered into a protein group if they shared 100% amino acid identity, and human proteins were clustered into a protein group if they contained ≥90% amino acid similarity. These differing similarity thresholds were chosen based on the higher numbers of paralogous proteins present within the human genome, and were supported by plotting similarity thresholds ranging from 0.5–1 against the percent proteome reduction via clustering [37]. Each protein group contained at least one unique peptide. Spectral counts were assigned, balanced, normalized, and adjusted according to methods previously described, yielding adjusted normalized spectral abundance factor (NSAF) values [37–39]. In total, 4,413 microbial and 3,062 human protein groups were detected across the dataset (Supporting Information Table S2). Protein groups range from singletons to groups that contain multiple protein isoforms. All peptides identified throughout the time course are provided in Table S3).
2.6 Data Analyses
COG (clusters of orthologous group) assignments for each microbial protein sequence were determined by running rpsblast against the COG database from NCBI, using an E-value threshold of 0.00001 and the top hit used for the assignment [40]. Adjusted NSAFs from all microbial protein groups were summed and grouped into their respective (COG) categories. A linear regression analysis, computed using the basic statistical package in R, was used to model the relationship between the proportions of proteins within each COG category and increasing time points [41]. The major canonical pathways for human proteins detected across the dataset were determined using Ingenuity Pathway Analysis (IPA) software (Ingenuity® Systems, www.ingenuity.com). The significance of the association was measured by calculating the ratio of number of detected proteins that map to the pathway divided by the total number of proteins from that pathway (orange boxes). The Fisher’s exact test was used to calculate a p-value determining the probability that the association between the proteins in the dataset and the canonical pathway is explained by chance alone (y-axis). Hierarchical clustering of individual human proteins was carried out using JMP Genomics using the log transformed NSAFs values above and below the median across all time points for each protein.
3 Results
3.1 Overall proteome characterization
Proteome extracts of fecal samples from a preterm infant on days 7, 13, 15, 16, 17, 18, 20, and 21 after birth were examined via nano-2D-LC-MS/MS. Up to 73,257 mass spectra, 16,605 peptides, and 4,031 proteins were identified per run (Supporting Information Table S1), providing deep proteomic coverage of both microbial and human components. Technical duplicates were run for each sample, with comparable reproducibility between replicates (Supporting Information Figure S1).
By measuring both microbial and human proteins simultaneously in each run, we observed an increased complexity of the microbial composition and a decrease in the ratio of total human/microbial proteins with time (Figure 1). At the earliest time point, when the initial microbial communities were being established, human proteins comprised ~96% of all proteins identified (day 7). The low microbial load may be a consequence of antibiotic administration during the first week of life for this particular infant. Human proteins comprised ~72% of the identified protein dataset on day 13, and by day 15 the percent of human proteins decreased to ~30%, with a concomitant increase in the number of microbial proteins detected. The ratio of human to microbial proteins remained at this level for the remainder of the times measured, with the exception of day 20, when an unexpected rise in human proteins was detected (to be discussed in more detail below). The number of total spectra collected on day 20 was comparable to adjacent days (Supporting Information Figure S2), so the variance was likely not due to a technical issue related to the mass spectrometry measurement.
Figure 1. Distribution of Human and Microbial Proteins.
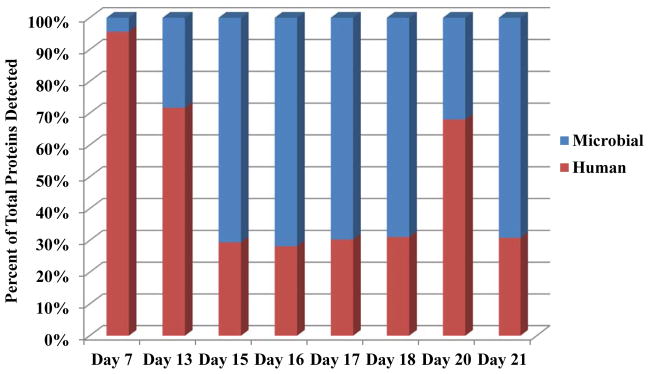
Adjusted NSAFs from microbial (blue) and human (red) protein groups were averaged between two technical replicates, summed for each time point (x-axis), and plotted as a percent of the total proteins detected for each day (y-axis). Technical variability between the two replicates was low as shown by the standard deviations of the microbial and human protein groups: day 7= 0.02%, day 13= 0.82%, day 15= 4.19%, day 16= 7.74%, day17= 7.99%, day 18= 1.71%, day 20= 0.03%, and day 21= 0.57%.
3.2 Microbial Protein Distribution and Functional Categorization
When microbial protein groups from different species were compared across time, the distribution of species was similar to that seen in 16S rRNA and metagenomic data (Supporting Information Figure S3) [14]. At day 7, microbial proteins were very low in abundance, but increased by day 13. This time point was dominated by Pseudomonas and Staphylococcus proteins. However, by day 15 we began to see the emergence of Serratia (UC1SER) and Citrobacter (UC1CIT) proteins, which persisted in days 16–21. In total, this corresponds closely with previous metagenomic data from matched samples, which showed distinct community memberships in colonization phase I (days 5–9), phase II (days 10–15), and phase III (days 16–21) [14]. Proteomic data also suggest UC1SER and UC1CIT were the functionally dominant members of the community during the third colonization phase, as demonstrated by the highest contribution of microbial proteins from these species during these time points.
Microbial community functions were analyzed by grouping proteins into clusters of orthologous groups (COG) categories, and then measuring the relative changes in the proportions of proteins in each category over time (Figure 2 and Figure S4). Because detailed metabolic characterization of microbial membership functions at the strain level was beyond the scope of this paper, and since the main objective of this study was to more broadly compare and link microbial/human host protein signatures over temporal development, we opted to use COG category representation to identify the range of metabolic activities of the microbial community and assess the functional changes over time. At day 7, although there was a very limited level of microbial peptide abundance measured, most of the signal originated from an aspartokinase I-homoserine dehydrogenase protein belonging to Bacteriodes fragilis. This enzyme, from the amino acid metabolism and transport COG category, catalyzes a reaction in the aspartate pathway and may aid in providing essential amino acids from dietary sources to the human host (infant) at this early stage of development [42]. Since there were so few microbial proteins detected from day 7 samples in comparison to other days, we excluded this day from the remainder of the COG analysis.
Figure 2. Analysis of microbial proteins by COG category classifications.

Microbial proteins were assigned to COG (clusters of orthologous groups) categories and adjusted NSAFs for each group summed and plotted as percent of total NSAFs for each time point.[40]. Day seven is removed from the analysis due to low abundance values of microbial proteins from that time point. Categories that significantly increased in proportions of proteins over time included: Carbohydrate transport and metabolism (p=0.015*), Secondary Structure (p=0.002**), and Intracellular trafficking and secretion (p=0.025*). Proportions of proteins in the categories of Cell cycle control and mitosis (p= 0.040*), Lipid metabolism (p= 0.039*), and Translation (p= 0.046*) decreased over time.
Changes in the proportions of proteins belonging to each COG category over time were assessed using a linear regression analysis (Figure S4). The statistical analysis revealed that proportions of proteins belonging to the categories of Lipid transport and metabolism, Cell cycle control and mitosis, and Translation ribosomal structure and biogenesis categories were abundant early, but decreased in relative abundance over time. In contrast, Carbohydrate transport and metabolism, Secondary metabolites biosynthesis, transport, and catabolism, Membrane biogenesis, and Intracellular trafficking and secretion, while lower in proportion initially, increased with time. In general, this information indicates that the microbial community initially focused its resources on biomass growth, protein production, and lipid metabolism (presumably to establish the stable microbiome), and then switched to more complex metabolic functions, such as carbohydrate metabolism, once the community stabilized and matured (around day 15). Interestingly, the functional distribution of the microbiome after about three weeks is very similar to what is observed in the stable adult human gut [20].
3.3 Functional Distributions of Human Proteins
Human proteins detected across all time points were categorized into canonical pathways using Ingenuity Pathway Analysis (IPA) software (Ingenuity® Systems, www.ingenuity.com). The topmost abundant categories were determined based on the number of proteins belonging to that category, and included those related to basic cellular functions such as glycolysis, oxidative phosphorylation, and elongation factor 2 signaling (Supporting Information Figure S5). Other categories, such as inflammatory response, were not displayed since the total number of proteins detected in this category was not among the top 20 overall. However, we detected over 30 inflammatory proteins, and some of these individual proteins were among the most abundantly detected in the samples (Calprotectin [18,385; this is the adjusted NSAFs collected across all time points], ALPI [13,650], SERPINA [37,140] and ANPEP [9,415]) (Table S4).
Some of the most abundant human proteins detected are involved in host-microbe interactions (Table S4). In particular, the most abundant protein detected in our samples, the calcium-activated chloride-channel 1 (CLCA1) protein [17,002], is involved in mucus secretion by goblet cells [43]. Likewise, Fc fragment of the IgG binding protein (FCRPB/Fcgbp) [23,096] is expressed by placental and colonic epithelial cells, and has been reported to bind mucin 2 (MUC2), and play a key role in immune protection and inflammation [44]. In addition, antimicrobial and innate immune proteins including lactoferrin (LTF), intelectin (ITLN1), and olfactomedin (OLFM4) were among the most abundant proteins detected [8,298, 11,214, and 4,875 NSAFs, respectively]. Lactoferrin (aka lactotransferrin), an iron-binding glycoprotein, is a key player in the innate immune system and is abundant and ubiquitous in human secretions such as breast milk. It has been shown to attenuate pathogenic bacteria, interfering with colonization and biofilm formation [45] [46] [47].
3.4 Intestinal Barrier Proteins
Throughout our proteome datasets, we identified numerous human proteins involved in intestinal barrier formation and function (Supporting Information Table S5). The intestinal barrier is composed of enterocytes, absorptive epithelial cells held together by tight junctions, which serve as a physical barrier, and the mucus layer. We detected numerous tight junction proteins including occludin (OCLN), claudins (CLDN18, CLDN23, CLDN3, CLDN7), and tight junction proteins 1, 2, and 3 (TJP1, TJP2, TJP3, or zona occludens 1, 2 and 3). In addition, proteins involved in the tight junction-signaling pathway were identified (Figure 3). Also detected were numerous mucin proteins, including both secretory gel-forming mucins (MUC2, MUC5AC, MUC5B, and MUC6) and membrane-bound mucins (MUC1, MUC3B, and MUC4) (Supporting Information Table S5). Several enzymes in the o-glycan biosynthesis pathway also were detected, including those involved in synthesizing core 3 type glycans, the major type associated with MUC2 [48, 49]. In addition, all three trefoil factor family peptides TFF1, TFF2, and TFF3, a family of proteins which play an important role in maintenance and repair of the intestinal mucosa, were detected [50].
Figure 3. Tight Junction Signaling Pathway.

Proteins in the tight junction signaling pathway as determined by Ingenuity Pathway Analysis (IPA) software. Proteins colored in pink are those detected by proteomics.
Secretory IgA is an important component of the intestinal barrier that specifically binds bacteria, limiting their association with the epithelial cell surface and restricting penetration across the gut epithelia [51–54]. We detected components of secretory Immunoglobulin A, including the two IgA heavy chain constant regions (IgA1 and IgA2), the J chain (15 kDa polypeptide), and the secretory component of the polymeric immunoglobulin receptor (pIgR: 130kDa). The poly Ig-receptor is expressed by epithelial cells, binds to the IgA oligomers and allows transport across the mucosal epithelium.
In addition, we detected several antimicrobial proteins, including alpha defensins (DEFA1, DEFA5), lysozyme (LYZ), and phospholipase A2 (PLA2). These antimicrobial proteins are secreted by a subset of gut epithelial cells, Paneth cells, which directly sense gut commensal bacteria through MyD88- dependent toll-like receptor signaling that triggers expression of certain antimicrobial factors, limits bacterial penetration of host tissues, and maintains microbial-host homeostasis in the intestine [55]. Thus, detection of these proteins might suggest that the premature infant’s gut, even at early stages of development, could have been responding to the introduction of microbial inhabitants and exerted pressure on the community to maintain homeostasis.
3.5 Differential Protein Expression Across Time
Overall, human proteins, when summed across all samples, contributed mostly to generalized maintenance functions (Supporting Information Figure 4). However, when human proteins were clustered based on shared trends in spectral count abundance changes (Figure 4), time shifts were apparent. Several neutrophil derived proteins such as neutrophil elastase (ELANE), calprotectin (S100-A8/S100-A9), and myeloperoxidase (MPO) were most abundant at day 7 (Figure 4, cluster #6) suggesting that activation of the innate immune system occurs early in correspondence with the initial arrival and ensuing establishment of the microbiome. Human cytoskeletal proteins (KRT8, KRT13, KRT18, KRT19, and KRT20) and mucins (MUC2, MUC5B) were more predominant in later time points (days 20–21) (Figure 4, clusters #7 and #10), suggesting structural and epithelial barrier proteins are compensating for the increased microbial load.
Figure 4. Human Proteins Changing Across Time.

Proteins were clustered based on abundance changes across time. The mean of the normalized spectral counts across all time points for each protein was taken. The scale reflects the log transformed value above and below the median.
4. Discussion
In this study we simultaneously monitored both human and microbial proteomes over a time course in early microbiome development of a preterm infant. Microbial proteins detected in this time course are consistent with metagenomic inference of distinct colonization phases with vastly different species composition [14]. The functions of the microbial community shifted over time, with early resources focusing on cell division, protein production, and lipid metabolism. As time progressed, the microbial community increased in size and diversity, stabilized, and switched its focus to breaking down carbohydrates, making secondary metabolites, and secreting and trafficking proteins.
Predominant throughout our measurements were human proteins involved in intestinal barrier function. Breakdown of the intestinal barrier or incomplete formation, as seen in premature infants, can contribute to bacterial translocation and disease states such as NEC [11]. The mucus layer is a major component of the intestinal barrier that helps maintain homeostasis between the gut microbiota and their host by minimizing physical contact between the microbes and intestinal epithelial cells [2]. In the colon, the outer mucus layer harbors commensal bacteria while the thicker, impenetrable inner layer offers protection by providing a physical barrier as well as containing antimicrobial compounds and secretory IgA [48, 56]. The small intestine is composed of only one mucus layer, but still provides a physical barrier with a 50 μm area separating the bacteria from the epithelia [57]. The mucus layer is composed of mucins, glycoconjugates of a polypeptide core covered in O-linked carbohydrate side chains that are secreted by goblet cells. The O-linked glycans provide an energy source for bacteria in the outer mucus layer [4] [58]. In our proteomic analyses, we detected numerous mucin proteins. Most of these were detected at relatively constant abundances throughout all the time points. However, some like the mucin 2 precursor, increased in abundance during the third colonization phase. Mucin 2 (MUC2) is the most abundant mucin in the intestine and has been directly linked to protecting the colonic epithelium from enteric pathogens [48]. It is down regulated in patients with ulcerative colitis and Crohn’s disease [59]. Detection and quantification of the numerous intestinal barrier proteins in this study suggest that comprehensive proteomic analysis of easily obtained fecal samples may represent an effective yet noninvasive means to evaluate gut barrier function in human patients.
As noted, there was a dramatic increase in the numbers of human proteins identified on day 20. Since many of these proteins were keratins, which are important components of both skin cells and gut epithelial cells, there are two possible explanations: 1) human contamination during sample handling, or 2) an increased sloughing event in the GI tract at this time. The 134 human proteins that were solely detected at this time point contribute to a wide range of biological functions (Figure S6A). Thus, we propose that they are more likely to represent sloughed epithelial cells rather than skin cells, and thus conclude that contamination during sample handling was not the most likely explanation. The most highly expressed canonical pathways on this day were those of basic metabolic functions including: EIF2 signaling, pyruvate metabolism, glycolysis (Figure S6A). Additional proteins from the pathways for pyruvate metabolism, glycolysis, and granzyme A signaling were detected on day 20 (Figure S6B & C). Note that the ratio of microbial/human proteins and the range of microbial activities revert back to expected values on day 21, indicating that the microbiome was not highly perturbed by whatever event happened on day 20.
Initial microbial colonization of the gastrointestinal tract is a crucial process in a healthy human infant. This process educates the innate immune system and initiates the establishment of a delicate homeostasis between human host and resident microbes. In premature infants, the host-microbe relationship is undoubtedly impacted significantly by underdevelopment of the intestinal barrier, an immature innate immune system, antibiotic administration, and exposure to pathogenic organisms in the intensive care unit [60]. While prior studies have investigated the succession of gut microbiota primarily at the gene level, the functional signatures of microbial and human proteins early in life can provide detailed metabolic activity information [61]. Thus, this study provides detailed information about the microbial and human proteins in fecal samples from a newborn premature infant during the first month of life, and reveals the complex-but-synergistic host adaptation to microbiome establishment.
The infant in this study was born prematurely, treated with antibiotics, and experienced gastrointestinal distention during which feedings were withheld. These and other factors could have influenced microbial colonization. Recent studies have reported high intra-individual diversity in microbial species compositions among preterm infants [15, 62]. As more proteomic data are collected on preterm infants, it will be interesting to see how microbial community functions compare between individuals - between both healthy preterm infants and those who develop NEC. While there may be commonalities or correlations in proteomic responses that predict which preterm infants progress to NEC, there is a strong possibility that due to intra-individual variability, treatment will need to be specialized to each individual. Thus, further research in this area could support personalized medicine for neonatal care.
Supplementary Material
Supporting Information Table S2: All proteins groups detected throughout time-course.
Values represent adjusted NSAFs averaged between two technical replicates
Supporting Information Table S3: All Peptides Identified Throughout Dataset
Acknowledgments
We thank Dr. David Tabb and the Yates Proteomics Laboratory at Scripps Research Institute for DTASelect/Contrast software, Langho Lee for bioinformatics assistance, and the PRIDE team. Oak Ridge National Laboratory is managed by UT-Battelle, LLC, for the U.S. Department of Energy. J.Y. acknowledges stipend support from the Genome Science and Technology program at the University of Tennessee, Knoxville. This work was funded in part by March of Dimes Foundation research grant 5-FY10-103 (M.J.M), NIH grant 1R01-GM-103600, and an NSF Graduate Fellowship to B.B.
Abbreviations
- nano-2D-LC-MS/MS
two dimensional liquid chromatography coupled with tandem mass spectrometry
- COG
clusters of orthologous groups
- NSAFs
normalized spectral abundance factors
Footnotes
DISCLAIMER
This manuscript has been authored by UT-Battelle, LLC under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Conflict of interest: The authors declare no conflicts of interest.
References
- 1.Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci USA. 2002;99:15451. doi: 10.1073/pnas.202604299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annual review of nutrition. 2002;22:283–307. doi: 10.1146/annurev.nutr.22.011602.092259. [DOI] [PubMed] [Google Scholar]
- 3.MacDonald TT, Pettersson S. Bacterial regulation of intestinal immune responses. Inflammatory Bowel Diseases. 2000;6:116–122. doi: 10.1097/00054725-200005000-00008. [DOI] [PubMed] [Google Scholar]
- 4.Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 5.Putignani L, Del Chierico F, Petrucca A, Vernocchi P, Dallapiccola B. The human gut microbiota: a dynamic interplay with the host from birth to senescence settled during childhood. Pediatric research. 2014;76:2–10. doi: 10.1038/pr.2014.49. [DOI] [PubMed] [Google Scholar]
- 6.Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS biology. 2007;5:e177. doi: 10.1371/journal.pbio.0050177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koenig JE, Spor A, Scalfone N, Fricker AD, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci USA. 2011;108:4578. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yatsunenko T, Rey FE, Manary MJ, Trehan I, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergstrom A, Skov TH, Bahl MI, Roager HM, et al. Establishment of intestinal microbiota during early life: a longitudinal, explorative study of a large cohort of Danish infants. Appl Environ Microbiol. 2014;80:2889–2900. doi: 10.1128/AEM.00342-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neu J, Walker WA. Necrotizing enterocolitis. The New England journal of medicine. 2011;364:255–264. doi: 10.1056/NEJMra1005408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morowitz MJ, Poroyko V, Caplan M, Alverdy J, Liu DC. Redefining the role of intestinal microbes in the pathogenesis of necrotizing enterocolitis. Pediatrics. 2010;125:777–785. doi: 10.1542/peds.2009-3149. [DOI] [PubMed] [Google Scholar]
- 13.Brown CT, Sharon I, Thomas BC, Castelle CJ, et al. Genome resolved analysis of a premature infant gut microbial community reveals a Varibaculum cambriense genome and a shift towards fermentation-based metabolism during the third week of life. Microbiome. 2013;1:30. doi: 10.1186/2049-2618-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morowitz MJ, Denef VJ, Costello EK, Thomas BC, et al. Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proc Natl Acad Sci U S A. 2011;108:1128–1133. doi: 10.1073/pnas.1010992108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharon I, Morowitz MJ, Thomas BC, Costello EK, et al. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome research. 2013;23:111–120. doi: 10.1101/gr.142315.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Hoenig JD, Malin KJ, Qamar S, et al. 16S rRNA gene-based analysis of fecal microbiota from preterm infants with and without necrotizing enterocolitis. The ISME journal. 2009;3:944–954. doi: 10.1038/ismej.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mai V, Torrazza RM, Ukhanova M, Wang X, et al. Distortions in development of intestinal microbiota associated with late onset sepsis in preterm infants. PLoS One. 2013;8:e52876. doi: 10.1371/journal.pone.0052876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mai V, Young CM, Ukhanova M, Wang X, et al. Fecal microbiota in premature infants prior to necrotizing enterocolitis. PLoS One. 2011;6:e20647. doi: 10.1371/journal.pone.0020647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mshvildadze M, Neu J, Shuster J, Theriaque D, et al. Intestinal microbial ecology in premature infants assessed with non–culture-based techniques. The Journal of pediatrics. 2010;156:20–25. doi: 10.1016/j.jpeds.2009.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verberkmoes NC, Russell AL, Shah M, Godzik A, et al. Shotgun metaproteomics of the human distal gut microbiota. ISME J. 2009;3:179–189. doi: 10.1038/ismej.2008.108. [DOI] [PubMed] [Google Scholar]
- 21.Ram RJ, Verberkmoes NC, Thelen MP, Tyson GW, et al. Community proteomics of a natural microbial biofilm. Science. 2005;308:1915–1920. [PubMed] [Google Scholar]
- 22.Knief C, Delmotte N, Chaffron S, Stark M, et al. Metaproteogenomic analysis of microbial communities in the phyllosphere and rhizosphere of rice. ISME J. 2012;6:1378–1390. doi: 10.1038/ismej.2011.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hettich RL, Sharma R, Chourey K, Giannone RJ. Microbial metaproteomics: identifying the repertoire of proteins that microorganisms use to compete and cooperate in complex environmental communities. Current Opinion in Microbiology. 2012 doi: 10.1016/j.mib.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 24.Kolmeder CA, de Vos WM. Metaproteomics of our microbiome - developing insight in function and activity in man and model systems. Journal of proteomics. 2014;97:3–16. doi: 10.1016/j.jprot.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 25.Hettich RL, Pan C, Chourey K, Giannone RJ. Metaproteomics: harnessing the power of high performance mass spectrometry to identify the suite of proteins that control metabolic activities in microbial communities. Anal Chem. 2013;85:4203–4214. doi: 10.1021/ac303053e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klaassens ES, De Vos WM, Vaughan EE. Metaproteomics approach to study the functionality of the microbiota in the human infant gastrointestinal tract. Applied and environmental microbiology. 2007;73:1388–1392. doi: 10.1128/AEM.01921-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erickson AR, Cantarel BL, Lamendella R, Darzi Y, et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS One. 2012;7:e49138. doi: 10.1371/journal.pone.0049138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lo I, Denef VJ, Verberkmoes NC, Shah MB, et al. Strain-resolved community proteomics reveals recombining genomes of acidophilic bacteria. Nature. 2007;446:537–541. doi: 10.1038/nature05624. [DOI] [PubMed] [Google Scholar]
- 29.Wilmes P, Bond PL. Metaproteomics: studying functional gene expression in microbial ecosystems. Trends in microbiology. 2006;14:92–97. doi: 10.1016/j.tim.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 30.McDonald WHOR, Miyamoto DT, Mitchison TJ, JR, YI Comparison of three directly coupled HPLC MS/MS strategies for identification of proteins from complex mixtures: single-dimension LC-MS/MS, 2-phase MudPIT, and 3-phase MudPIT. International Journal of Mass Spectrometry. 2002;219:245–251. [Google Scholar]
- 31.Washburn MP, Ulaszek R, Deciu C, Schieltz DM, Yates JR. 3rd, Analysis of quantitative proteomic data generated via multidimensional protein identification technology. Anal Chem. 2002;74:1650–1657. doi: 10.1021/ac015704l. [DOI] [PubMed] [Google Scholar]
- 32.Washburn MP, Wolters D, Yates JR. 3rd, Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 33.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 34.Tabb DL, McDonald WH, Yates JR., III DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. Journal of proteome research. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng J, Elias JE, Thoreen CC, Licklider LJ, Gygi SP. Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. Journal of proteome research. 2003;2:43–50. doi: 10.1021/pr025556v. [DOI] [PubMed] [Google Scholar]
- 36.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010 doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 37.Abraham P, Adams R, Giannone RJ, Kalluri U, et al. Defining the boundaries and characterizing the landscape of functional genome expression in vascular tissues of Populus using shotgun proteomics. J Proteome Res. 2011;11:449–460. doi: 10.1021/pr200851y. [DOI] [PubMed] [Google Scholar]
- 38.Giannone RJ, Huber H, Karpinets T, Heimerl T, et al. Proteomic characterization of cellular and molecular processes that enable the Nanoarchaeum equitans-Ignicoccus hospitalis relationship. PLoS One. 2011;6:e22942. doi: 10.1371/journal.pone.0022942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zybailov B, Mosley AL, Sardiu ME, Coleman MK, et al. Statistical Analysis of Membrane Proteome Expression Changes in Saccharomyces cerevisiae. Journal of proteome research. 2006;5:2339–2347. doi: 10.1021/pr060161n. [DOI] [PubMed] [Google Scholar]
- 40.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, et al. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic acids research. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.R Development Core Team. R Foundation for Statistical Computing; Vienna, Austria: 2011. [Google Scholar]
- 42.Viola RE. The central enzymes of the aspartate family of amino acid biosynthesis. Accounts of chemical research. 2001;34:339–349. doi: 10.1021/ar000057q. [DOI] [PubMed] [Google Scholar]
- 43.Loewen ME, Forsyth GW. Structure and function of CLCA proteins. Physiological reviews. 2005;85:1061–1092. doi: 10.1152/physrev.00016.2004. [DOI] [PubMed] [Google Scholar]
- 44.Johansson MEV, Thomsson KA, Hansson GC. Proteomic analyses of the two mucus layers of the colon barrier reveal that their main component, the Muc2 mucin, is strongly bound to the Fcgbp protein. Journal of proteome research. 2009;8:3549–3557. doi: 10.1021/pr9002504. [DOI] [PubMed] [Google Scholar]
- 45.Qiu J, Hendrixson DR, Baker EN, Murphy TF, et al. Human milk lactoferrin inactivates two putative colonization factors expressed by Haemophilus influenzae. Proc Natl Acad Sci USA. 1998;95:12641. doi: 10.1073/pnas.95.21.12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh PK, Parsek MR, Greenberg EP, Welsh MJ. A component of innate immunity prevents bacterial biofilm development. development. 2005;417:552–555. doi: 10.1038/417552a. [DOI] [PubMed] [Google Scholar]
- 47.Legrand D, Pierce A, Elass E, Carpentier M, et al. Lactoferrin structure and functions. Bioactive Components of Milk. 2008:163–194. doi: 10.1007/978-0-387-74087-4_6. [DOI] [PubMed] [Google Scholar]
- 48.Johansson MEV, Larsson JMH, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host–microbial interactions. Proc Natl Acad Sci USA. 2011;108:4659. doi: 10.1073/pnas.1006451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Larsson JMH, Karlsson H, Sjövall H, Hansson GC. A complex, but uniform O-glycosylation of the human MUC2 mucin from colonic biopsies analyzed by nanoLC/MSn. Glycobiology. 2009;19:756–766. doi: 10.1093/glycob/cwp048. [DOI] [PubMed] [Google Scholar]
- 50.Albert TK, Laubinger W, Müller S, Hanisch FG, et al. Human intestinal TFF3 forms disulfide-linked heteromers with the mucus-associated FCGBP protein and is released by hydrogen sulfide. J Proteome Res. 2010;9:3108–3117. doi: 10.1021/pr100020c. [DOI] [PubMed] [Google Scholar]
- 51.Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nature Reviews Immunology. 2010;10:159–169. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- 52.Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science’s STKE. 2004;303:1662. doi: 10.1126/science.1091334. [DOI] [PubMed] [Google Scholar]
- 53.Macpherson AJ, Gatto D, Sainsbury E, Harriman GR, et al. A primitive T cell-independent mechanism of intestinal mucosal IgA responses to commensal bacteria. Science. 2000;288:2222–2226. doi: 10.1126/science.288.5474.2222. [DOI] [PubMed] [Google Scholar]
- 54.Suzuki K, Meek B, Doi Y, Muramatsu M, et al. Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc Natl Acad Sci USA. 2004;101:1981. doi: 10.1073/pnas.0307317101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci USA. 2008;105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rodriguez-Pineiro AM, Post S, Johansson MEV, Thomsson KA, et al. Proteomic study of the mucin granulae in an intestinal goblet cell model. Journal of proteome research. 2012 doi: 10.1021/pr2010988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, et al. The Antibacterial Lectin RegIII {gamma} Promotes the Spatial Segregation of Microbiota and Host in the Intestine. Science’s STKE. 2011;334:255. doi: 10.1126/science.1209791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fu J, Wei B, Wen T, Johansson MEV, et al. Loss of intestinal core 1–derived O-glycans causes spontaneous colitis in mice. The Journal of Clinical Investigation. 2011;121:1657. doi: 10.1172/JCI45538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moehle C, Ackermann N, Langmann T, Aslanidis C, et al. Aberrant intestinal expression and allelic variants of mucin genes associated with inflammatory bowel disease. Journal of molecular medicine. 2006;84:1055–1066. doi: 10.1007/s00109-006-0100-2. [DOI] [PubMed] [Google Scholar]
- 60.Cilieborg MS, Boye M, Sangild PT. Bacterial colonization and gut development in preterm neonates. Early Human Development. 2012 doi: 10.1016/j.earlhumdev.2011.12.027. [DOI] [PubMed] [Google Scholar]
- 61.Lichtman JS, Marcobal A, Sonnenburg JL, Elias JE. Host-centric proteomics of stool: a novel strategy focused on intestinal responses to the gut microbiota. Molecular & cellular proteomics: MCP. 2013;12:3310–3318. doi: 10.1074/mcp.M113.029967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Costello EK, Carlisle EM, Bik EM, Morowitz MJ, Relman DA. Microbiome assembly across multiple body sites in low-birthweight infants. mBio. 2013;4:e00782–00713. doi: 10.1128/mBio.00782-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Table S2: All proteins groups detected throughout time-course.
Values represent adjusted NSAFs averaged between two technical replicates
Supporting Information Table S3: All Peptides Identified Throughout Dataset