

Summary
The generation of pancreas, liver and intestine from a common pool of progenitors in the foregut endoderm requires the establishment of organ boundaries. How dorsal foregut progenitors activate pancreatic genes and evade the intestinal lineage choice remains unclear. We here identify Pdx1 and Sox9 as cooperative inducers of a gene regulatory network that distinguishes the pancreatic from the intestinal lineage. Genetic studies demonstrate dual and cooperative functions for Pdx1 and Sox9 in pancreatic lineage induction and repression of the intestinal lineage choice. Pdx1 and Sox9 bind to regulatory sequences near pancreatic and intestinal differentiation genes and jointly regulate their expression, revealing direct cooperative roles for Pdx1 and Sox9 in gene activation and repression. Our study identifies Pdx1 and Sox9 as important regulators of a transcription factor network that initiates pancreatic fate and sheds light on the gene regulatory circuitry that governs the development of distinct organs from multi-lineage-competent foregut progenitors.
Keywords: pancreas, development, progenitor, Sox9, Pdx1, Cdx2
Graphical abstract
Introduction
During mammalian development, naïve endodermal progenitors are directed toward different organ fates, including lung, pancreas, liver, and intestine. At developmental junctures, multipotent progenitors must be allocated to different lineages, exemplified by progenitors in the foregut endoderm, which give rise to pancreas, stomach, duodenum, liver, and the hepatobiliary system. Organ lineage choices are initiated by cross-repressive interactions between transcription factors (TFs) driving alternative lineage programs, followed by feed-forward induction of additional TFs to further execute the differentiation process (Holmberg and Perlmann, 2012). A large body of work has identified numerous TFs that are required for the early development of individual organs, in particular the pancreas and liver (Seymour and Sander, 2011; Zaret, 2008). Despite these significant advances, it is still poorly understood which regulatory networks induce specific organ fates, and how organ boundaries are established in the foregut endoderm. Identifying the mechanisms responsible for specifying individual organ fates is important for devising cell reprogramming strategies, which are still lacking for ex vivo production of pancreatic cells.
The pancreas arises as two buds on opposing sides of the gut tube at the boundary between the stomach and duodenum, the most rostral portion of the intestine (Shih et al., 2013). The anatomical location of the pancreas implies that an organ boundary must be established that distinguishes pancreatic from stomach and intestinal progenitors. The TF Cdx2 is exclusively expressed in intestinal epithelial cells, spanning the length of the alimentary tract from the proximal duodenum to the distal rectum. Cdx2 is essential for intestinal development and induces intestinal epithelial differentiation by activating the transcription of intestine-specific genes, such as MUC2, sucrase, and carbonic anhydrase I (Gao et al., 2009; Verzi et al., 2011). However, the mechanisms preventing expansion of the Cdx2 expression domain beyond the duodenal boundary in the foregut endoderm remain undefined.
The TFs Pdx1, Foxa2, Mnx1 (Hb9), Onecut-1 (Hnf6), Prox1, Tcf2, Gata4/6, Sox9, and Ptf1a, each play an important role in early pancreas development, yet deletion of no single factor alone is sufficient to abrogate pancreatic lineage induction (Carrasco et al., 2012; Harrison et al., 1999; Haumaitre et al., 2005; Jacquemin et al., 2000; Kawaguchi et al., 2002; Lee et al., 2005; Offield et al., 1996; Seymour et al., 2007; Wang et al., 2005; Xuan et al., 2012). These observations imply either that the inducer of the pancreatic fate remains to be identified or that the pancreatic fate is specified through a cooperative mechanism involving multiple TFs.
Combining genetic, cistrome, and transcriptome analysis, we here identify the TFs Pdx1 and Sox9 as cooperative inducers of the pancreatic lineage. The combined inactivation of Pdx1 and Sox9 leads to an intestinal fate conversion of the pre-pancreatic domain, illustrated by expansion of the field of Cdx2 expression. Conversely, ectopic expression of Sox9 in intestinal progenitors is sufficient to induce Pdx1 and repress Cdx2. At a mechanistic level, we show that Pdx1 and Sox9 function as direct and cooperative activators of pancreatic genes and repressors of intestinal lineage regulators. Together, these findings shed light on the transcriptional mechanisms that induce the pancreatic fate and establish the pancreatic-to-intestinal organ boundary.
Results
Pdx1 and Sox9 cooperatively induce the pancreatic lineage program
To identify TFs most closely associated with pancreatic lineage induction, we compared expression levels of TFs represented in the RNA-seq data from pancreatic progenitor cells and closely related endodermal cell populations. These comprised human embryonic stem cell (hESC)-derived definitive endoderm, gut tube progenitors, posterior foregut, pancreatic progenitors, hepatic progenitors, and endocrine cells, as well as primary human fetal pancreatic anlagen and primary cadaver pancreatic islets (Fig. 1A). Principal component analysis of TF expression data clustered the different cell populations by developmental proximity, effectively reconstructing the dynamics of endodermal development and underscoring the importance of TF levels in successfully delineating these cell types (Fig. 1B). Two TFs, PDX1 and SOX9, most strongly distinguished pancreatic progenitors from other cell populations (Fig. 1B), suggesting possible cooperative roles for PDX1 and SOX9 in pancreatic lineage specification.
Figure 1. Principal component analysis for expression of transcription factors in endodermal cell populations.

(A) Experimental strategy for principal component analysis of transcription factors in various endodermal cell populations. (B) Principal component (PC) analysis of the expression values (RPKM) characterizing the variance explained by transcription factors expressed in human embryonic stem cell (hESC)-derived populations and primary human cells. Each vector emanating from the origin represents an individual gene. Each dot represents a sample and each color represents the type of sample.
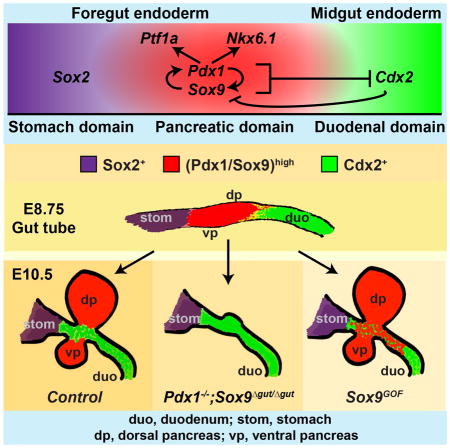
First, to define the domains of Pdx1 and Sox9 expression during pancreatic specification, we performed co-immunofluorescence staining for Pdx1 and Sox9 together with the anterior foregut marker Sox2 or the mid-/hindgut marker Cdx2, respectively, at embryonic day (E) 8.75 (15–17 somites). The Sox2+ domain, from which the stomach develops (McCracken et al., 2014; Sherwood et al., 2009), formed a boundary with both the Pdx1+ and Sox9+ domains (Fig. 2A–A″). Very few cells co-expressing Sox2, Pdx1, and Sox9 were observed at this boundary (Fig. 2A–A″). Cells in the presumptive proximal duodenum expressed high levels of Cdx2 and also Sox9 (Fig. 2B–B″). In contrast to Sox9, which spanned the proximal duodenal and pre-pancreatic domains, Pdx1 was restricted to the pre-pancreatic domain (Fig. 2B–B′). At the boundary between the duodenal and pre-pancreatic domain, we observed a transition from a Cdx2high to a Cdx2low state (Fig. 2B–B″, dashed line; Movie S1). Consistent with previous studies (McCracken et al., 2014), Cdx2 was largely absent from the pancreatic buds (Fig. 2C), showing that Cdx2 is gradually excluded from the pancreatic domain.
Figure 2. Pdx1 and Sox9 are coexpressed in the pancreatic domain in the foregut endoderm.

(A–B″) Immunofluoresence staining for Sox2, Sox9 and Pdx1 (A–A″) and Cdx2, Sox9 and Pdx1 (B–B″) on embryonic sections at embryonic day (E) 8.75. The arrows in A′,A″ and B′,B″ indicate Pdx1+/Sox9+ cells co-expressing Sox2 and Cdx2, respectively. The dashed line in B–B″ demarcates the transition from the presumptive duodenal to the pre-pancreatic region. Fields demarcated by white dashed boxes in A′,A″,B′,B″ are shown at higher magnification in the same panels. Non-specific signal for Cdx2 is evident in the foregut lumen (B,B″, asterisks) due to antibody trapping. (C) Immunofluoresence staining for Cdx2, Sox2, and Pdx1 at E10.5. (D,E) Dams carrying R26mT/mG embryos expressing CreER driven by either the Pdx1 or Sox9 regulatory sequences were injected with tamoxifen at E8.0, embryos sectioned at E10.5, and immunofluorescence staining performed for Cdx2, Pdx1 and GFP. Recombined, membrane-targeted GFP+ (mGFP+) cells trace to the pancreatic epithelium; scattered labeled cells are also detectable in the proximal duodenum in R26mT/mG;Pdx1-CreER (D) and R26mT/mG;Sox9-CreER (E) embryos. dp, dorsal pancreas; vp, ventral pancreas; duo, duodenum; stom, stomach. Scale bars = 50 μm (A–E).
To determine the fate of Sox9- or Pdx1-expressing cells in the foregut endoderm, we performed lineage tracing in embryos carrying the Rosa26mTomato/mGFP (R26mT/mG) reporter allele and an inducible form of Cre-recombinase, CreER, driven by either Sox9 or Pdx1 regulatory sequences. In these mice, tamoxifen administration to pregnant dams turns off constitutive expression of membrane-targeted Tomato (mT) and induces heritable expression of membrane-targeted GFP (mGFP), permitting recombined cells and their progeny to be traced by mGFP labeling. Tamoxifen administration at E8.0 resulted in labeling of the pancreatic epithelium in R26mT/mG;Pdx1-CreER (Fig. 2D) and R26mT/mG;Sox9-CreER (Fig. 2E) embryos at E10.5. Consistent with the incomplete segregation of the Cdx2+ and Pdx1+/Sox9+ domains at E8.75 (Fig. 2B–B″,C), mGFP labeling was also observed in scattered Cdx2+ cells of the proximal duodenum (Fig. 2D,E). mGFP+ cells in the Sox2+ gastric region were extremely rare (data not shown). Together, these findings indicate that the pancreatic-to-stomach boundary is largely established by E8.75, whereas the pancreatic and duodenal domains separate gradually between E8.75 and E10.5.
Previous studies have shown that pancreatic outgrowth and induction of a subset of early pancreatic markers still occur in Pdx1-null mutants (Offield et al., 1996). Similarly, after conditional Sox9 inactivation with a Pdx1-Cre transgene pancreatic buds evaginate (Seymour et al., 2007; Seymour et al., 2012). However, since Pdx1-Cre deletes Sox9 after the pancreatic program has been initiated, it remains unclear whether Sox9 is necessary to initiate the pancreatic program. To determine whether Sox9 is required for pancreatic specification, we generated global Sox9-null mutant embryos (Fig. 3A,C). While hypoplastic, dorsal and ventral pancreatic rudiments arise in Sox9-null embryos (Fig. 3B,B′,D,D′), showing that Sox9 is dispensable for pancreatic fate assignment and outgrowth of the pancreatic buds. Notably, although Pdx1 staining intensity is reduced, Pdx1 is expressed in both dorsal and ventral pancreatic buds of Sox9−/− embryos (Fig. 3B,B′,D,D′), showing that Sox9 is dispensable for Pdx1 induction. Similarly, we have previously found Sox9 to be expressed in Pdx1-deficient dorsal pancreatic progenitors at E10.5 (Seymour et al., 2012). Thus, neither Pdx1 nor Sox9 is required for pancreas specification or induction of the other’s expression.
Figure 3. Sox9 is dispensable for pancreas induction.

(A,C) Confirmation of global Sox9 deletion by WMIF staining of Sox9 in E10.5 tail-tips. (B,D) 2D projections of 3D Imaris-reconstructed z-stacks through trunks of embryos after WMIF for Foxa2 and Pdx1. Although smaller, dorsal and ventral pancreatic buds are present in E10.5 Sox9-null embryos (D,D′). Fields demarcated by white dashed boxes in B,D are shown at higher magnification in B′,D′, respectively. Only single-channel Pdx1 signal is shown in B′ and D′. (E–H″) Immunofluorescence staining of sections through the pancreatic region of Sox9fl/fl;Foxa3-Cre (Sox9Δgut/Δgut) and control Sox9fl/fl embryos at E9.5. Sox9 is efficiently deleted in dorsal (G′,G″) and ventral (H′,H″) pancreatic buds of Sox9Δgut/Δgut embryos. Dashed line in G′ and H′ demarcates the Pdx1+ domain. (I–T) X-Gal staining for β-galactosidase expressed from the Pdx1LacZko allele in embryonic day (E) 10.5 and E12.5 embryos carrying combinations of mutant alleles for Pdx1 and Sox9. With increasing loss of Sox9 dosage on either Pdx1-heterozygous (I–N) or Pdx1-null (O–T) backgrounds, dorsal and ventral pancreatic buds become increasingly hypoplastic. In Pdx1−/−;Sox9Δgut/Δgut embryos (S,T), pancreatic buds are not discernible. Note the reduced ventral pancreas in E12.5 compound heterozygous mutants (L). Asterisks denote absence of ventral pancreas. (U) With decreasing dosage of functional Pdx1 and Sox9 alleles, pancreatic morphogenesis becomes increasingly perturbed. dp, dorsal pancreas; vp, ventral pancreas; duo, duodenum; stom, stomach; li, liver; bd, bile duct. Scale bars = 50 μm (E–H″), 70 μm (B,B′,D, D′), 200 μm (A,C), 250 μm (I–T).
Based on their early expression in pre-pancreatic cells, we postulated that Sox9 and Pdx1 might function together and induce the pancreatic lineage in a cooperative manner. To test this, we generated mice lacking various combinations of either one or two alleles of Pdx1, Sox9, or both. Since early embryonic lethality of Sox9-null embryos precluded the analysis of compound mutants beyond E11.5 (Akiyama et al., 2004), we employed a conditional Sox9 ablation strategy, using the Foxa3-Cre transgenic line (Lee et al., 2005), which ablates Sox9 efficiently in the gut tube by E9.5 (Sox9Δgut) (Fig. 3E–H″).
We next generated compound mutants carrying various combinations of the Pdx1-null (Pdx1LacZko) and Sox9Δgut alleles and visualized the dorsal and ventral pancreatic buds, antral stomach and duodenum by X-Gal staining for β-galactosidase (β-gal) expressed from the Pdx1LacZko allele (Fig. 3I,J). With progressive loss of Sox9 gene dosage (Sox9+/+ > Sox9+/Δgut > Sox9Δgut/Δgut) on the Pdx1-heterozygous mutant background, the pancreatic buds became increasingly hypoplastic (Fig. 3I–N). In E12.5 Pdx1+/−;Sox9Δgut/Δgut embryos, the dorsal pancreas was reduced to a severely hypoplastic remnant and the ventral pancreatic bud was undetectable (Fig. 3N; absent ventral pancreas denoted by asterisk). Notably, the size of the ventral pancreatic bud was significantly reduced in compound-heterozygous mutants (Fig. 3K,L,U), which contrasted with the normal bud size seen in embryos deficient for a single copy of either Pdx1 (Fig. 3I,J) or Sox9 (Seymour et al., 2008). This phenotype in compound-heterozygous mutants demonstrates genetic interaction between Pdx1 and Sox9. The dorsal pancreas remnant (the ventral pancreas is undetectable in Pdx1−/− embryos; Fig. 3P; asterisk) became increasingly smaller with decreasing Sox9 gene dosage on a Pdx1-null background (Fig. 3O–T) and was morphologically almost indiscernible in compound-homozygous Pdx1−/−;Sox9Δgut/Δgut mutants (Fig. 3S–U). Combined, these genetic findings demonstrate cooperative functions of Pdx1 and Sox9 in early pancreas development.
To determine whether deletion of Pdx1 and Sox9 perturbs induction of the pancreatic program, we next analyzed the expression of early pancreatic markers in Pdx1;Sox9 compound mutants. Confirming previous findings (Seymour et al., 2007; Seymour et al., 2012), Sox9 expression was maintained in pancreatic rudiments of Pdx1−/− embryos at E10.5, and conversely, Pdx1 was also expressed in Sox9Δgut/Δgut mutants (Fig. S1A–N; note that the truncated Pdx1 protein expressed from the Pdx1-null allele is detected by the anti-Pdx1 antibody used). Immunofluorescence staining for Foxa2, Mnx1, Onecut-1, Tcf2, Gata4, and Prox1 further revealed maintenance of their expression in embryos lacking either Pdx1, Sox9, or both (Fig. S1O–BB, data not shown).
In contrast, expression of the pancreas-specific TF Ptf1a was drastically reduced in Sox9Δgut/Δgut and Pdx1−/−;Sox9Δgut/Δgut embryos (Fig. S1CC–II), showing that Ptf1a expression is Sox9-dependent. Albeit to a lesser extent, Ptf1a expression was also diminished in Pdx1−/− embryos (Fig. S1HH). Like Ptf1a, the TF Nkx6.1 is pancreas-specific and, together with Ptf1a, governs the endocrine versus acinar cell fate choice (Schaffer et al., 2010). Nkx6.1 was not detected in Pdx1−/− and Pdx1−/−;Sox9Δgut/Δgut embryos and reduced in Sox9Δgut/Δgut embryos (Fig. S1NN–PP). This confirms earlier findings in Pdx1−/− embryos (Pedersen et al., 2005) and suggests that Pdx1 is dominant over Sox9 in regulating Nkx6.1 expression. Together, our findings show that expression of the pancreas-restricted TFs Ptf1a and Nkx6.1 is under the control of Pdx1 and Sox9, whereas the expression of Foxa2, Mnx1, Onecut-1, Tcf2, Gata4, and Prox1 is Pdx1- and Sox9-independent.
PDX1 and SOX9 co-regulate intestinal cell fate determinants
To define the mechanistic basis of the observed cooperativity between Pdx1 and Sox9 in specifying the pancreatic fate, we mapped where PDX1 and SOX9 bind in the genome to explore synergy at the level of gene regulation. As the number of pancreatic progenitors in early mouse embryos is extremely limited, we generated pancreatic progenitors from hESCs (Xie et al., 2013) and performed chromatin immunoprecipitation and sequencing (ChIP-seq) analysis for PDX1 and SOX9. We mapped 55,481 unique binding peaks for PDX1 and 9,767 unique peaks for SOX9 (Fig. 4A). PDX1 and SOX9 peaks exhibited surprisingly limited overlap (Fig. 4B), which was unexpected given that lineage-determining TFs generally bind to cis-regulatory elements, in particular enhancers, as a collective unit (Spitz and Furlong, 2012). To understand the basis for the limited overlap in PDX1 and SOX9 binding sites, we analyzed PDX1 and SOX9 occupancy specifically at promoters and enhancers, using chromatin maps we recently generated based on histone modifications (Wang et al., 2015). This analysis revealed recruitment of both PDX1 and SOX9 to promoters, albeit to not entirely overlapping sites (Fig. S2A). Strikingly, and in stark contrast to PDX1, there was little recruitment of SOX9 to enhancers (Fig. S2B). Other TFs with roles in early pancreatic development, such as FOXA2, ONECUT-1 and TCF2, occupied enhancers together with PDX1 (Fig. S2B), consistent with TFs forming regulatory collectives at transcriptional enhancers (Calo and Wysocka, 2013). Together, these findings show that SOX9 is predominantly recruited to promoter regions, while PDX1 and other early pancreatic TFs co-occupy enhancers.
Figure 4. PDX1 and SOX9 co-occupy pancreatic and intestinal genes.

(A) Genome-wide distribution of PDX1 and SOX9 binding peaks within the human genome from ChIP-seq analysis of human embryonic stem cell (hESC)-derived pancreatic progenitors. (B) Venn diagram of the overlap between PDX1 binding peaks and SOX9 binding peaks (minimum of 1 bp overlap). (C) Venn diagram of the overlap between genes bound by PDX1 and SOX9, showing 2,201 genes to be co-bound by PDX1 and SOX9 (hypergeometric analysis: p-value=4.3×10−9). (D) Gene ontology (GO) analysis of PDX1 and SOX9 co-bound genes (defined as PDX1 and SOX9 binding at enhancers and/or promoters within a 200 kb window). (E) Analysis of co-bound genes revealed that 82% of the co-bound genes are expressed and 18% are not expressed in hESC-derived pancreatic progenitors. (F) ChIP-seq binding profiles (reads per million) for PDX1, SOX9 and histone modifications (H3K4me1, H3K27ac, H3K4me3, H3K27me3) at the PTF1A and CDX2 loci in hESC-derived pancreatic progenitors. Enhancers were identified based on presence of H3K27ac and H3K4me1 and absence of H3K3me3. Black boxes indicate conserved regions in mice. kB, kilobases.
To relate PDX1 and SOX9 binding patterns to gene regulatory functions, we used the Genomic Regions Enrichment of Annotations Tool (GREAT) to predict putative target genes of PDX1-bound enhancers and then catalogued genes with binding peaks for PDX1 and SOX9 around transcriptional start sites and/or at PDX1-bound enhancers. This analysis identified 2,201 PDX1 and SOX9 co-bound genes (Fig. 4C; Supplemental Table 1). Consistent with the cooperative role of Pdx1 and Sox9 in pancreatic fate determination, regulators of pancreatic development are PDX1 and SOX9 co-bound, exemplified by the TFs PTF1A, PAX6 and NEUROG3 (Fig. 4C,F). Interestingly, PDX1 and SOX9 co-bound genes were enriched for Gene Ontology (GO) categories associated with cell developmental processes, including gut and liver development (Fig. 4D). Occupancy of hepatic genes by PDX1 and SOX9 provides a possible explanation for why hepatic genes are ectopically expressed in Pdx1- and Sox9-deficient pancreatic buds (Seymour et al., 2012). PDX1 and SOX9 co-bound genes included several intestinal cell fate-determining TFs, such as CDX2, ONECUT-2 and NKX6-3 (Dusing et al., 2010; Nelson et al., 2005; Pedersen et al., 2005) (Fig. 4C,F), suggesting a possible role for SOX9 and PDX1 in regulating these genes at the lineage bifurcation of pancreas and gut. 18% of all PDX1 and SOX9 co-bound genes were not expressed in pancreatic progenitors (Fig. 4E), indicating that PDX1 and SOX9 could play a role in gene silencing. Combined, these results suggest cooperative roles for SOX9 and PDX1 in the regulation of pancreatic and intestinal genes.
Based on these findings, we predicted that decreased Pdx1 and Sox9 levels would induce ectopic activation of intestinal genes in the pancreatic domain. To test this, we identified co-regulated genes of both factors through transcriptional profiling of pancreatic progenitors from embryos with reduced Pdx1 and Sox9 gene dosage. Given that 1.) both pancreatic buds are virtually absent in Pdx1;Sox9 double-homozygous mutants and 2.) evidence of genetic interaction in compound Pdx1;Sox9 heterozygous mutants, we reasoned that mRNA profiling of pancreata from compound Pdx1;Sox9 heterozygous mutants versus either single-heterozygous mutant could identify co-regulated genes. Hence, we performed cDNA microarray profiling of dorsal pancreatic epithelia from Pdx1+/−, Pdx1+/−;Sox9+/Δgut and Sox9+/Δgut littermates at E12.5 when the epithelium is still predominantly comprised of undifferentiated progenitor cells (Fig. 5A).
Figure 5. Pdx1 and Sox9 cooperatively silence genes encoding intestinal cell fate regulators.

(A) Illustration of the experimental strategy for gene expression microarray analysis. The mRNA profiles of embryonic day (E) 12.5 pancreata (n=12 per genotype) from 1.) Pdx1+/− versus Pdx1+/−;Sox9+/Δgut and 2.) Sox9+/Δgut versus Pdx1+/−;Sox9+/Δgut littermates were compared. (B) 3,337 and 4,486 genes were differentially-expressed between 1.) and 2.), respectively. A total of 1,817 genes were common to both sets of significantly-regulated genes (FDR<0.05) with the same sign of change (i.e. up-regulated or down-regulated). (C) Pdx1- and Sox9-co-regulated genes were identified by cross-comparing mRNA profiles of E12.5 pancreata (n=12 per genotype) from 1.) Pdx1+/− versus Pdx1+/−;Sox9+/Δgut and 2.) Sox9+/Δgut versus Pdx1+/−;Sox9+/Δgut littermates. A total of 1,817 genes (denoted by red pixels) were common to both sets of significantly-regulated genes (FDR<0.05) with the same sign of change. (D) Gene ontology analysis of the 1,817 Pdx1- and Sox9-co-regulated genes. (E) The top twenty Pdx1- and Sox9-co-repressed genes with the highest fold-change.
Comparison of gene expression profiles revealed significant differences in the expression of 3,337 genes (False Discovery Rate [FDR] <0.05) between Pdx1+/−;Sox9+/Δgut and Pdx1+/− pancreatic epithelia and 4,486 genes (FDR <0.05) between Pdx1+/−;Sox9+/Δgut and Sox9+/Δgut epithelia (Fig. 5B; Supplemental Tables 2 and 3). We then performed a cross-comparison of these two data sets in order to identify Pdx1- and Sox9-co-regulated genes. A total of 1,817 genes were common to both sets of significantly-regulated genes with the same sign of change (i.e. up-regulated or down-regulated) (Fig. 5B,C: co-regulated genes are denoted by red pixels in Fig. 5C; Supplemental Table 4) and associated with the GO term foregut morphogenesis (Fig. 5D; Supplemental Table 5). Intriguingly, among the top twenty Pdx1- and Sox9-co-repressed genes with the highest fold-change were several genes encoding intestinal cell fate regulators, including Cdx2, Onecut-2 and Nkx6.3 (Fig. 5E), which also showed co-recruitment of PDX1 and SOX9 to their regulatory regions (Fig. 4C,F; Supplemental Table 1). These intestinal markers were all up-regulated in pancreatic epithelia from compound Pdx1;Sox9 heterozygous mutants, suggesting a synergistic and direct role for Pdx1 and Sox9 in repressing genes encoding intestinal lineage regulators.
Pdx1 and Sox9 jointly control the pancreatic versus intestinal cell fate choice
To determine whether Pdx1 and Sox9 indeed control the fate decision between pancreas and intestine, we analyzed the expression of the intestinal marker Cdx2 in the pancreatic region of embryos carrying various combinations of the Pdx1-null and Sox9Δgut alleles. In control embryos at E10.5, cells of the dorsal pancreatic bud can be identified by high levels of Pdx1 expression, whereas prospective duodenal cells express the intestinal marker Cdx2 (Fig. 6A–A″,P). At the duodenal-pancreatic junction, the Pdx1high domain forms a boundary with the Cdx2+ domain; only a few Pdx1high cells express Cdx2 (Fig. 6A–A″,P; note, duodenal precursors express low levels of Pdx1 (Fukuda et al., 2006)). As in control embryos, the Pdx1high and Cdx2+ domains were distinct in embryos deficient for a single copy of either Pdx1 or Sox9, compound Pdx1;Sox9 heterozygous mutant embryos, and Pdx1 or Sox9 single-homozygous mutants (Fig. 6B–F″). In stark contrast, immunofluorescence staining for the truncated Pdx1 protein and Cdx2 in embryos with a combined homozygous deletion of Pdx1 and Sox9 revealed extensive overlap between the Cdx2+ and Pdx1+ domains (Fig. 6G–G″,P). Thus, the presence of either Pdx1 or Sox9 is sufficient to repress the intestinal marker Cdx2 in the pancreatic domain, whereas loss of both Pdx1 and Sox9 results in ectopic Cdx2 expression. In contrast, combined Pdx1 and Sox9 deletion did not result in ectopic expression of the stomach marker Sox2 in the Pdx1+ domain (Fig. S3A–D″), showing that Pdx1 and Sox9 cooperatively repress intestinal but not anterior foregut markers.
Figure 6. Pdx1 and Sox9 are necessary and sufficient to repress the intestinal lineage choice.

(A–G) Immunofluorescence analysis for Pdx1 and Cdx2 on E10.5 embryos carrying various combinations of Pdx1 and Sox9 mutant alleles. In compound Pdx1;Sox9 heterozygous mutant or Pdx1 or Sox9 single-homozygous mutant embryos, Cdx2 expression is restricted to duodenal precursors and excluded from the Pdx1high dorsal pancreas (A–F). In Pdx1−/−;Sox9Δgut/Δgut embryos, a duodenal-pancreatic junction is not discernable and Pdx1 and Cdx2 are co-expressed in a broad domain (arrows in G″). (H–O) Immunofluorescence staining of sections from Sox9GOF and control littermates shows repression of the intestinal markers Cdx2 (J,K) and Onecut-2 (Oc2; L,M) in mCherry+ duodenal precursors in Sox9GOF mice. Pdx1 is upregulated (H,I) but Ptf1a is not induced (N,O) in duodenal precursors in Sox9GOF embryos. Fields demarcated by dashed boxes in A–O are shown at higher magnification in A′–O″. (P) Summary of the phenotypes observed after combined Pdx1 and Sox9 deletion or Sox9 overexpression. (Q) Graphical model summary. Our data support a model whereby Pdx1 and Sox9 cooperatively specify the pancreatic lineage by inducing the pancreatic transcription factors Nkx6.1 and Ptf1a and repressing the duodenal transcription factor Cdx2. A positive regulatory loop between Pdx1 and Sox9 maintains the pancreatic fate choice. Repression of Sox9 by Cdx2 creates bistability of the fate choice (Gao et al. 2009). dp, dorsal pancreatic bud; vp, ventral pancreatic bud; duo, duodenum; stom, stomach. Scale bar = 50 μm.
To directly test whether Pdx1 and Sox9 are sufficient to repress the intestinal fate in vivo, we forcibly expressed Sox9 in Pdx1-expressing foregut progenitor cells, using a Pdx1-driven tetracycline transactivator mouse (Pdx1tTA) and a single copy, tetracycline-regulated Sox9 transgene (mCherry-tetO-Sox9) inserted into the disabled Rosa26 locus (Rosa26mCherry-tetO-Sox9) (Fig. S3E). In this system, Sox9 and the mCherry reporter gene are expressed in the Pdx1+ domain in the absence of doxycycline; administration of doxycycline suppresses transgene expression. In Pdx1tTA;Rosa26mCherry-tetO-Sox9 (Sox9GOF) embryos never exposed to doxycycline, Sox9 expression was enforced in Pdx1+ cells of the pancreatic buds, antral stomach and duodenum (Fig. S3F–G″). In control embryos, Sox9 is detectable in the antral stomach and duodenum, but at much lower levels than in the pancreas (Fig. S3F–F″). Formation of the pancreatic buds and gross gut morphology in Sox9GOF embryos were comparable to controls (Fig. S3H–K).
Consistent with previous observations that Sox9 reinforces Pdx1 expression (Dubois et al., 2011; Seymour et al., 2012), ectopic Sox9 expression resulted in increased Pdx1 staining intensity in the duodenal domain (Fig. 6H–I″), thus creating an extra-pancreatic Sox9 high/Pdx1high domain. In this domain, we observed reduced expression of the intestinal markers Cdx2 and Onecut-2, showing that the concerted activities of Pdx1 and Sox9 are sufficient to repress intestinal cell fate determinants (Fig. 6J–M″,P). Notably, despite induction of a Pdx1high state and repression of intestinal markers in Sox9GOF embryos, Sox9 overexpression failed to induce Ptf1a in intestinal progenitors (Fig. 6N–O″). Previous work has shown that Ptf1a misexpression in the gut tube induces ectopic pancreas formation (Willet et al., 2014). Consistent with the lack of Ptf1a induction, an ectopic pancreatic bud was not observed in Sox9GOF embryos (Fig. 6N–O″). Combined, these results show that a Sox9high/Pdx1high state prevents foregut endoderm progenitor cells from adopting intestinal lineage identity.
Discussion
In this study, we uncover a cooperative role for Pdx1 and Sox9 in governing the lineage choice between pancreas and intestine. Our data suggest a model whereby Pdx1 and Sox9 establish pancreatic lineage identity by excluding intestinal lineage-restricted TFs, such as Cdx2, from foregut endoderm progenitor cells (Fig. 6Q). Our work further shows that the concerted activities of Pdx1 and Sox9 induce pancreatic differentiation programs through regulation of the pancreas-specific TFs Ptf1a and Nkx6.1. Interestingly, although the TFs Foxa2, Mnx1, Onecut-1, Tcf2, Gata4, and Prox1 are also important in early pancreas development (Seymour and Sander, 2011; Shih et al., 2013), their expression was not affected by combined Pdx1 and Sox9 deletion. These findings suggest that Sox9 and Pdx1 together are essential for driving pancreatic gene expression. The pancreatic program is reinforced by both positive autoregulation of Pdx1 (Marshak et al., 2000) and Sox9 (Lynn et al., 2007; Mead et al., 2013) and a positive cross-regulatory loop between Pdx1 and Sox9 (Dubois et al., 2011; Seymour et al., 2012). The mutual reinforcement of expression between Pdx1 and Sox9 appears to be direct, as PDX1 occupied SOX9 regulatory sequences and vice versa (Fig. S2C). Early pancreatic TFs induce a Notchhigh state that is important for maintaining the pancreatic state (Ahnfelt-Ronne et al., 2012; Jensen et al., 2000). For example, Sox9 and Ptf1a both promote expression of the Notch effector Hes1 in the early pancreas, and Hes1 in turn reinforces Ptf1a expression (Ahnfelt-Ronne et al., 2012).
Previous studies have shown that a subset of normally pancreas-fated cells adopt intestinal identity in Ptf1a-null mutant mice (Kawaguchi et al., 2002). This invokes the question of how Pdx1, Sox9 and Ptf1a contribute to the gene regulatory network that establishes pancreatic identity and prevents foregut progenitors from becoming intestinal cells. Together with published observations, findings reported here identify Sox9 and Pdx1 as lying upstream of Ptf1a in the transcriptional regulatory cascade effecting pancreas induction (Fig. 6Q). Several observations support this conclusion. First, combined deletion of Pdx1 and Ptf1a phenocopies the effects of Pdx1 deletion, arguing that Pdx1 is required prior to Ptf1a in pancreatic specification (Burlison et al., 2008). Second, we show that Ptf1a is not expressed in the absence of Sox9 (Fig. S1GG), whereas Sox9 and Pdx1 induction do not depend on Ptf1a (Seymour et al., 2012). We note that Sox9 regulates Ptf1a only during pancreas specification, but not later in pancreas development, when the Sox9 and Ptf1a expression domains are distinct (Shih et al., 2012).
It is important to consider that after combined inactivation of Pdx1 and Ptf1a in mice or Xenopus, the dorsal pancreatic bud still forms and early pancreatic genes are activated (Afelik et al., 2006; Burlison et al., 2008). Furthermore, we found that despite intestinal fate conversion of some Ptf1a-deficient cells (Kawaguchi et al., 2002), Cdx2 remains excluded from the pancreatic domain in Ptf1a-null mutants (data not shown). These findings suggest that the pancreatic-to-intestinal boundary is still established in the absence of Pdx1 and Ptf1a. In contrast, we show that combined deletion of Sox9 and Pdx1 leads to misspecification of progenitors in the foregut endoderm, converting the pancreatic domain into a Cdx2-expressing intestinal domain (Fig. 6G). Moreover, ectopic expression of Sox9 in duodenal precursors was sufficient to induce Pdx1 and repress Cdx2 (Fig. 6I,K). These findings identify Sox9 as a critical early component of the gene regulatory network that governs both the activation of pancreatic genes and the repression of intestinal genes. Consistent with this notion, we found that SOX9 occupies genomic regions near genes required for early pancreatic development (i.e. PTF1A) as well as intestinal development (i.e. CDX2). Mechanistically, our data imply that Sox9 can function as either a transcriptional activator or repressor. Such a dual role for Sox9 is consistent with its ability to recruit both transcriptional coactivators and corepressors (Lee et al., 2012; Leung et al., 2011).
Of interest is our finding that SOX9 and PDX1 bind to distinct cis-regulatory elements within the genome. While PDX1, FOXA2, ONECUT-1, and TCF2 collectively occupy enhancers, SOX9 was predominantly detected in promoter regions, suggesting a unique role for SOX9 in the regulation of gene expression. This observation could be relevant to gene regulatory mechanisms in multiple contexts, as Sox9 controls cell lineage decisions in several tissues, including gonad, lung and kidney (Reginensi et al., 2011; Rockich et al., 2013; Sekido and Lovell-Badge, 2008). A future direction will be to test whether promoter-specific recruitment of Sox9 is also seen in other tissues and to determine how Sox9 deposition at promoters evokes cooperative effects with tissue-specific TFs bound to enhancers.
Experimental Procedures
Mouse strains
All animal experiments described herein were approved by the University of California San Diego Institutional Animal Care and Use Committees. The following mouse strains have been previously described: Sox9flox (Kist et al., 2002), Pdx1LacZko (herein designated Pdx1−) (Offield et al., 1996), Foxa3-Cre (Lee et al., 2005), Sox9-CreER (Kopp et al., 2011), Pdx1-CreER (Gu et al., 2002), Prm1-Cre (O’Gorman et al., 1997), Zp3-Cre (de Vries et al., 2000), Pdx1tTA (Holland et al., 2002) and R26mT/mG (Muzumdar et al., 2007). To generate Sox9-null mice, germline recombination of the Sox9-flox allele was employed as previously described (Akiyama et al., 2004). Briefly, Sox9fl/+ mice were bred to carry either the oocyte-specific Zp3-Cre (de Vries et al., 2000) or the spermatid-specific Prm1-Cre (O’Gorman et al., 1997) transgenes. One Sox9 allele was deleted in the oocytes or spermatids of Zp3-Cre; or Prm1-Cre;Sox9fl/+ mice, respectively; these mice were then crossed to obtain Sox9-null embryos. To generate Rosa26mCherry-tetO-Sox9 mice, mouse Sox9 coding sequences with MluI and NheI restriction sites on the 5′ and 3′ ends were generated from E15.5 pancreas by linker-primer PCR. The PCR product was then cloned into MluI and NheI sites of pBR322-hygro-ptight-mcherry, screened for orientation, and confirmed for bi-directionality (primers: Sox9-F MluI, tcacgcgtATGAATCTCCTGGACCCCTT; Sox9-R NheI, ggctagcTCAGGGTCTGGTGAGCTGTGT). The bidirectional mCherry-tetO-Sox9 gene was inserted as a single copy transgene into a functionally-disabled Rosa26 gene locus using recombinase-mediated cassette exchange as previously described (Chen et al., 2011; Long et al., 2004). Mice bearing the Rosa26mCherry-tetO-Sox9 allele were obtained after blastocyst microinjections, chimera matings, and FlpE-mediated removal of an FRT-flanked hygromycin resistance cassette.
A single dose of 2 mg/40 g body-weight Tamoxifen (Sigma, St Louis, MO, USA) dissolved at 10 mg/ml in corn oil, was administered by intraperitoneal injection. For each experiment, a minimum of three embryos per genotype was analyzed. Midday on the day of vaginal plug appearance was considered E0.5.
Analysis of ChIP-seq data
Raw Illumina sequencing reads were mapped to reference human genomic database (version hg18) using Bowtie (ver1.1.0, http://bowtie-bio.sourceforge.net/index.shtml) to generate sam files. Sam files were subsequently converted to tag directories using HOMER (http://homer.salk.edu/homer/ngs/index.html). The ChIP-seq peak, peak distribution and gene annotations were also annotated by HOMER analysis. Input sequencing data were used to normalize background reads for peak calling. Overlapping peaks were determined using table browser function in UCSC genome browser website, with minimum of 1 bp overlap. A 200 kb window was used to identify genes associated with the peaks. Transcription factor binding to a promoter was determined by presence of a ChIP-seq peak within 20 kb upstream and 5 kb downstream of a TSS of an annotated gene.
Transcription factor binding to an enhancer was determined based on a minimum of 1 bp overlap between a transcription factor ChIP-seq peak and a predicted enhancer (defined as ±500 bp from the center of the enhancer using the enhancer prediction tool (Rajagopal et al., 2013)). We assigned PDX1-bound enhancers to nearest genes using GREAT (version 2.0, http://bejerano.stanford.edu/great/public/html/) with a basal plus 200 kb extension rule setting. In Fig. 4C–E, PDX1-bound genes were defined as genes with PDX1 binding at either promoters and/or PDX1 binding at enhancers corresponding to the gene. Since SOX9 did not exhibit significant enrichment at enhancers, SOX9-bound genes were defined as genes with SOX9 binding at promoters. Conserved regions were identified using the vista point tool comparing human to mouse (Frazer et al., 2004).
Gene ontology analysis was performed using the web tool David Bioinformatics Database (http://david.abcc.ncifcrf.gov/home.jsp) (Huang da et al., 2009). The complete set of all RefSeq genes was used as a background.
ChIP-seq data for FOXA2, TCF2 and ONECUT-1 in hESC-derived pancreatic progenitors have been previously described (Weedon et al., 2014).
Principal component analysis
The quality of the RNA sequencing data was analyzed using the FastQC v0.10.1 software. Once the samples passed quality control, they were aligned to the hg19 genome using RNA-Star 2.3.0e, with the parameters set to default. After alignment, Sailfish 0.6.3 and Cufflinks 2.2.0 were used to determine gene expression values. Datasets incorporating multivariate sequencing information (commonly gene expression values or splicing scores) were analyzed via the dimensionality reduction method PCA with the intention of uncovering features of the data that can explain variation within the dataset, and as a visual summary of the sample data. The data was stored in pandas dataframes (pandas python package v0.14.1) and visualized using matplotlib v0.13. A detailed description of all methods is available in Supplemental Material.
Supplementary Material
Acknowledgments
We are grateful to G. Scherer, K. Kaestner, R. MacDonald, D. Melton and C. Wright for mouse strains, and R. Behringer and H. Chang for Sox9-null embryos. We also thank M. Wegner, C. Wright, B. Bréant, J. Kehrl and F. Lemaigre for antibodies, M. Jørgensen for expert assistance with confocal imaging, and B. Armstrong for expert assistance with 3D image rendering and P. Serup for support. We acknowledge the Core Facility for Integrated Microscopy, Faculty of Health and Medical Sciences, University of Copenhagen. We acknowledge the support of the UC San Diego Biogem Core Facility for microarray analyses, the University of Pennsylvania Functional Genomics Core for ChIP-seq analysis, Q. Zhang and the Vanderbilt Transgenic Mouse/ESC Shared Resource for deriving Rosa26mCherry-tetO-Sox9 mice, and the Washington Birth Defects Research Laboratory for human fetal pancreas. We thank members of the Sander laboratory for constructive comments on the manuscript. We apologize to our colleagues whose references were omitted owing to space constraints. This work was supported by NIH/NIDDK awards to M.S. (DK078803, DK68471, and DK089567) and M.A.M. (DK89523), JDRF postdoctoral fellowships to P.A.S. (3-2004-608), H.P.S. (3-2009-161), A.W. (3-2012-177), and the California Institute for Regenerative Medicine training grant TG2-01154 to N.A.P and R.X.
Footnotes
Author Contributions
H.P.S., P.A.S. and M.S. conceived the project. H.P.S., P.A.S., and M.S. designed the experiments and analyzed the data. H.P.S. and P.A.S. performed all analyses of mouse genetic models. R.X. and A.W. performed ChIP-seq experiments. N.A.P. and A.W. analyzed ChIP-seq data. P.P.L. and G.W.Y. performed PCA. M.A.M. designed and generated the Rosa26mCherry-tetO-Sox9 mouse strain. H.P.S., P.A.S. and M.S. wrote the manuscript.
Accession numbers
The NCBI GEO (http://www.ncbi.nlm.nih.gov/geo/) accession numbers for Deep sequencing data reported in this paper are: GSE61945: Human fetal pancreas transcriptome analysis; GSE61946: hESC-derived liver progenitor cell transcriptome analysis; GSE61947: SOX9 cistrome analysis in hESC-derived pancreatic progenitors. The GEO accession number for the MIAME-Compliant Microarray Data set reported in this paper is: GSE62023: Identification of Sox9/Pdx1-coregulated genes during pancreas organogenesis.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afelik S, Chen Y, Pieler T. Combined ectopic expression of Pdx1 and Ptf1a/p48 results in the stable conversion of posterior endoderm into endocrine and exocrine pancreatic tissue. Genes Dev. 2006;20:1441–1446. doi: 10.1101/gad.378706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahnfelt-Ronne J, Jorgensen MC, Klinck R, Jensen JN, Fuchtbauer EM, Deering T, MacDonald RJ, Wright CV, Madsen OD, Serup P. Ptf1a-mediated control of Dll1 reveals an alternative to the lateral inhibition mechanism. Development. 2012;139:33–45. doi: 10.1242/dev.071761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Chaboissier MC, Behringer RR, Rowitch DH, Schedl A, Epstein JA, de Crombrugghe B. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc Natl Acad Sci U S A. 2004;101:6502–6507. doi: 10.1073/pnas.0401711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlison JS, Long Q, Fujitani Y, Wright CV, Magnuson MA. Pdx-1 and Ptf1a concurrently determine fate specification of pancreatic multipotent progenitor cells. Dev Biol. 2008;316:74–86. doi: 10.1016/j.ydbio.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Molecular cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco M, Delgado I, Soria B, Martin F, Rojas A. GATA4 and GATA6 control mouse pancreas organogenesis. J Clin Invest. 2012;122:3504–3515. doi: 10.1172/JCI63240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SX, Osipovich AB, Ustione A, Potter LA, Hipkens S, Gangula R, Yuan W, Piston DW, Magnuson MA. Quantification of factors influencing fluorescent protein expression using RMCE to generate an allelic series in the ROSA26 locus in mice. Dis Model Mech. 2011;4:537–547. doi: 10.1242/dmm.006569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries WN, Binns LT, Fancher KS, Dean J, Moore R, Kemler R, Knowles BB. Expression of Cre recombinase in mouse oocytes: a means to study maternal effect genes. Genesis. 2000;26:110–112. [PubMed] [Google Scholar]
- Dubois CL, Shih HP, Seymour PA, Patel NA, Behrmann JM, Ngo V, Sander M. Sox9-haploinsufficiency causes glucose intolerance in mice. PLoS One. 2011;6:e23131. doi: 10.1371/journal.pone.0023131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dusing MR, Maier EA, Aronow BJ, Wiginton DA. Onecut-2 knockout mice fail to thrive during early postnatal period and have altered patterns of gene expression in small intestine. Physiological genomics. 2010;42:115–125. doi: 10.1152/physiolgenomics.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic acids research. 2004;32:W273–279. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda A, Kawaguchi Y, Furuyama K, Kodama S, Horiguchi M, Kuhara T, Koizumi M, Boyer DF, Fujimoto K, Doi R, et al. Ectopic pancreas formation in Hes1 -knockout mice reveals plasticity of endodermal progenitors of the gut, bile duct, and pancreas. J Clin Invest. 2006;116:1484–1493. doi: 10.1172/JCI27704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, White P, Kaestner KH. Establishment of intestinal identity and epithelial-mesenchymal signaling by Cdx2. Dev Cell. 2009;16:588–599. doi: 10.1016/j.devcel.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- Harrison KA, Thaler J, Pfaff SL, Gu H, Kehrl JH. Pancreas dorsal lobe agenesis and abnormal islets of Langerhans in Hlxb9-deficient mice. Nat Genet. 1999;23:71–75. doi: 10.1038/12674. [DOI] [PubMed] [Google Scholar]
- Haumaitre C, Barbacci E, Jenny M, Ott MO, Gradwohl G, Cereghini S. Lack of TCF2/vHNF1 in mice leads to pancreas agenesis. Proc Natl Acad Sci U S A. 2005;102:1490–1495. doi: 10.1073/pnas.0405776102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland AM, Hale MA, Kagami H, Hammer RE, MacDonald RJ. Experimental control of pancreatic development and maintenance. Proc Natl Acad Sci U S A. 2002;99:12236–12241. doi: 10.1073/pnas.192255099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmberg J, Perlmann T. Maintaining differentiated cellular identity. Nature reviews Genetics. 2012;13:429–439. doi: 10.1038/nrg3209. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jacquemin P, Durviaux SM, Jensen J, Godfraind C, Gradwohl G, Guillemot F, Madsen OD, Carmeliet P, Dewerchin M, Collen D, et al. Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Mol Cell Biol. 2000;20:4445–4454. doi: 10.1128/mcb.20.12.4445-4454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen J, Pedersen EE, Galante P, Hald J, Heller RS, Ishibashi M, Kageyama R, Guillemot F, Serup P, Madsen OD. Control of endodermal endocrine development by Hes-1. Nat Genet. 2000;24:36–44. doi: 10.1038/71657. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32:128–134. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- Kist R, Schrewe H, Balling R, Scherer G. Conditional inactivation of Sox9: a mouse model for campomelic dysplasia. Genesis. 2002;32:121–123. doi: 10.1002/gene.10050. [DOI] [PubMed] [Google Scholar]
- Kopp JL, Dubois CL, Schaffer AE, Hao E, Shih HP, Seymour PA, Ma J, Sander M. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development. 2011;138:653–665. doi: 10.1242/dev.056499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Sund NJ, Behr R, Herrera PL, Kaestner KH. Foxa2 is required for the differentiation of pancreatic alpha-cells. Developmental biology. 2005;278:484–495. doi: 10.1016/j.ydbio.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Lee PC, Taylor-Jaffe KM, Nordin KM, Prasad MS, Lander RM, LaBonne C. SUMOylated SoxE factors recruit Grg4 and function as transcriptional repressors in the neural crest. The Journal of cell biology. 2012;198:799–813. doi: 10.1083/jcb.201204161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung VY, Gao B, Leung KK, Melhado IG, Wynn SL, Au TY, Dung NW, Lau JY, Mak AC, Chan D, et al. SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS genetics. 2011;7:e1002356. doi: 10.1371/journal.pgen.1002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long Q, Shelton KD, Lindner J, Jones JR, Magnuson MA. Efficient DNA cassette exchange in mouse embryonic stem cells by staggered positive-negative selection. Genesis. 2004;39:256–262. doi: 10.1002/gene.20053. [DOI] [PubMed] [Google Scholar]
- Lynn FC, Smith SB, Wilson ME, Yang KY, Nekrep N, German MS. Sox9 coordinates a transcriptional network in pancreatic progenitor cells. Proc Natl Acad Sci U S A. 2007;104:10500–10505. doi: 10.1073/pnas.0704054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshak S, Benshushan E, Shoshkes M, Havin L, Cerasi E, Melloul D. Functional conservation of regulatory elements in the pdx-1 gene: PDX-1 and hepatocyte nuclear factor 3beta transcription factors mediate beta-cell-specific expression. Mol Cell Biol. 2000;20:7583–7590. doi: 10.1128/mcb.20.20.7583-7590.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCracken KW, Cata EM, Crawford CM, Sinagoga KL, Schumacher M, Rockich BE, Tsai YH, Mayhew CN, Spence JR, Zavros Y, et al. Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature. 2014;516:400–404. doi: 10.1038/nature13863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead TJ, Wang Q, Bhattaram P, Dy P, Afelik S, Jensen J, Lefebvre V. A far-upstream (−70 kb) enhancer mediates Sox9 auto-regulation in somatic tissues during development and adult regeneration. Nucleic acids research. 2013;41:4459–4469. doi: 10.1093/nar/gkt140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Nelson SB, Janiesch C, Sander M. Expression of Nkx6 genes in the hindbrain and gut of the developing mouse. J Histochem Cytochem. 2005;53:787–790. doi: 10.1369/jhc.5B6619.2005. [DOI] [PubMed] [Google Scholar]
- O’Gorman S, Dagenais NA, Qian M, Marchuk Y. Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc Natl Acad Sci U S A. 1997;94:14602–14607. doi: 10.1073/pnas.94.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, Hogan BL, Wright CV. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122:983–995. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- Pedersen JK, Nelson SB, Jorgensen MC, Henseleit KD, Fujitani Y, Wright CV, Sander M, Serup P. Endodermal expression of Nkx6 genes depends differentially on Pdx1. Dev Biol. 2005;288:487–501. doi: 10.1016/j.ydbio.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Rajagopal N, Xie W, Li Y, Wagner U, Wang W, Stamatoyannopoulos J, Ernst J, Kellis M, Ren B. RFECS: a random-forest based algorithm for enhancer identification from chromatin state. PLoS computational biology. 2013;9:e1002968. doi: 10.1371/journal.pcbi.1002968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginensi A, Clarkson M, Neirijnck Y, Lu B, Ohyama T, Groves AK, Sock E, Wegner M, Costantini F, Chaboissier MC, et al. SOX9 controls epithelial branching by activating RET effector genes during kidney development. Hum Mol Genet. 2011;20:1143–1153. doi: 10.1093/hmg/ddq558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockich BE, Hrycaj SM, Shih HP, Nagy MS, Ferguson MA, Kopp JL, Sander M, Wellik DM, Spence JR. Sox9 plays multiple roles in the lung epithelium during branching morphogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2013 doi: 10.1073/pnas.1311847110. [DOI] [PMC free article] [PubMed]
- Schaffer AE, Freude KK, Nelson SB, Sander M. Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev Cell. 2010;18:1022–1029. doi: 10.1016/j.devcel.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekido R, Lovell-Badge R. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature. 2008;453:930–934. doi: 10.1038/nature06944. [DOI] [PubMed] [Google Scholar]
- Seymour PA, Freude KK, Dubois CL, Shih HP, Patel NA, Sander M. A dosage-dependent requirement for Sox9 in pancreatic endocrine cell formation. Dev Biol. 2008;323:19–30. doi: 10.1016/j.ydbio.2008.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour PA, Freude KK, Tran MN, Mayes EE, Jensen J, Kist R, Scherer G, Sander M. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc Natl Acad Sci U S A. 2007;104:1865–1870. doi: 10.1073/pnas.0609217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour PA, Sander M. Historical perspective: beginnings of the beta-cell: current perspectives in beta-cell development. Diabetes. 2011;60:364–376. doi: 10.2337/db10-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour PA, Shih HP, Patel NA, Freude KK, Xie R, Lim CJ, Sander M. A Sox9/Fgf feed-forward loop maintains pancreatic organ identity. Development. 2012;139:3363–3372. doi: 10.1242/dev.078733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood RI, Chen TY, Melton DA. Transcriptional dynamics of endodermal organ formation. Dev Dyn. 2009;238:29–42. doi: 10.1002/dvdy.21810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih HP, Kopp JL, Sandhu M, Dubois CL, Seymour PA, Grapin-Botton A, Sander M. A Notch-dependent molecular circuitry initiates pancreatic endocrine and ductal cell differentiation. Development. 2012;139:2488–2499. doi: 10.1242/dev.078634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih HP, Wang A, Sander M. Pancreas organogenesis: from lineage determination to morphogenesis. Annual review of cell and developmental biology. 2013;29:81–105. doi: 10.1146/annurev-cellbio-101512-122405. [DOI] [PubMed] [Google Scholar]
- Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nature reviews Genetics. 2012;13:613–626. doi: 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- Verzi MP, Shin H, Ho LL, Liu XS, Shivdasani RA. Essential and redundant functions of caudal family proteins in activating adult intestinal genes. Molecular and cellular biology. 2011;31:2026–2039. doi: 10.1128/MCB.01250-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Yue F, Li Y, Xie R, Harper T, Patel NA, Muth K, Palmer J, Qiu Y, Wang J, et al. Epigenetic priming of enhancers predicts developmental competence of hESC-derived endodermal lineage intermediates. Cell Stem Cell. 2015;16:386–399. doi: 10.1016/j.stem.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Kilic G, Aydin M, Burke Z, Oliver G, Sosa-Pineda B. Prox1 activity controls pancreas morphogenesis and participates in the production of “secondary transition” pancreatic endocrine cells. Dev Biol. 2005;286:182–194. doi: 10.1016/j.ydbio.2005.07.021. [DOI] [PubMed] [Google Scholar]
- Weedon MN, Cebola I, Patch AM, Flanagan SE, De Franco E, Caswell R, Rodriguez-Segui SA, Shaw-Smith C, Cho CH, Lango Allen H, et al. Recessive mutations in a distal PTF1A enhancer cause isolated pancreatic agenesis. Nature genetics. 2014;46:61–64. doi: 10.1038/ng.2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willet SG, Hale MA, Grapin-Botton A, Magnuson MA, MacDonald RJ, Wright CV. Dominant and context-specific control of endodermal organ allocation by Ptf1a. Development. 2014;141:4385–4394. doi: 10.1242/dev.114165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie R, Everett LJ, Lim HW, Patel NA, Schug J, Kroon E, Kelly OG, Wang A, D’Amour KA, Robins AJ, et al. Dynamic chromatin remodeling mediated by polycomb proteins orchestrates pancreatic differentiation of human embryonic stem cells. Cell Stem Cell. 2013;12:224–237. doi: 10.1016/j.stem.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan S, Borok MJ, Decker KJ, Battle MA, Duncan SA, Hale MA, Macdonald RJ, Sussel L. Pancreas-specific deletion of mouse Gata4 and Gata6 causes pancreatic agenesis. J Clin Invest. 2012;122:3516–3528. doi: 10.1172/JCI63352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS. Genetic programming of liver and pancreas progenitors: lessons for stem-cell differentiation. Nat Rev Genet. 2008;9:329–340. doi: 10.1038/nrg2318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.