

Abstract
HIV infection increases the risk of many types of cancer, including lymphoma. Combination antiretroviral therapy (cART) has reduced, but not eliminated, the risk of HIV-associated lymphoma. There has been a substantial shift in the subtypes of lymphoma observed in HIV-infected patients treated with cART. In this review, we will first outline these changes based on epidemiological studies and describe the impact of cART on lymphoma risk and mortality. Then, we will discuss some immunological factors that may contribute to the increased risk of lymphoma persisting after the administration of cART, including immunological non-response to therapy, chronic B-cell activation and dysfunction, T follicular helper cells, natural killer cells and altered lymphopoiesis. A better understanding of the pathophysiologic mechanisms of HIV-associated lymphoma under effective cART will inform future treatment strategies.
Keywords: HIV-1, virus, lymphomagenesis, antiretroviral drugs, immune activation, chronic infection
This comprehensive and informative review of HIV-related lymphoma explores how HIV infection may promote the development of lymphoma despite effective antiretroviral therapy, highlighting B-cell interaction with persistent HIV proteins, infected T follicular helper cells and altered immunity.
Graphical Abstract Figure.
This comprehensive and informative review of HIV-related lymphoma explores how HIV infection may promote the development of lymphoma despite effective antiretroviral therapy, highlighting B-cell interaction with persistent HIV proteins, infected T follicular helper cells and altered immunity.
SUBTYPES OF HIV-ASSOCIATED LYMPHOMA
Lymphomas in HIV-infected patients are heterogeneous and can be subdivided into different histologic types (Carbone 2002). Non-Hodgkin lymphoma (NHL) is one of three AIDS-defining cancers. Prior to cART, the incidence of NHL in AIDS patients was approximately 100-fold higher than in the general population, occurring in 2–10% of cases of AIDS (Goedert et al. 1998; Carbone 2002). Overall, Epstein–Barr virus (EBV) is found in ∼40% of HIV-associated NHL, though it is more common in some subtypes (Dunleavy and Wilson 2012). The major NHL subtypes include Burkitt's lymphoma (BL, characterized by MYC activation), and diffuse large B-cell lymphoma (DLBCL), which has distinct genetic lesions in HIV+ subjects as compared to seronegatives (Capello et al. 2010). DLBCL can be further divided into two groups based on gene expression profiling: centroblastic/germinal center B-cell type and immunoblastic/activated B-cell type (Dunleavy and Wilson 2012). Primary central nervous system lymphoma (PCNSL), a form of DLBCL found in the brain, is uniformly EBV positive and represented ∼20% of NHL cases pre-cART, but the incidence of this disease has decreased significantly with cART treatment (Carbone 2002). Other rare subtypes, such as primary effusion lymphoma and plasmablastic lymphoma of the oral cavity, are typically EBV and human herpesvirus-8 positive and found in severely immunocompromised individuals including immunosuppressed transplant recipients and thus are not limited to HIV infection. Finally, HIV patients had a 12-fold increased risk for Hodgkin's lymphoma (HL) in the pre-cART era (Patel et al. 2008), prevalently EBV positive, while only 48% of HL is EBV positive overall (Thompson et al. 2004; Lee et al. 2014). The role of EBV in HIV lymphoma has been reviewed elsewhere (Carbone et al. 2009; Dolcetti et al. 2013; White, Pagano and Khalili 2014). Given that the majority of HIV lymphomas are EBV-negative (Chao et al. 2012; Dunleavy and Wilson 2012), this review will explore other immune factors that may contribute to lymphomagenesis.
LYMPHOMA AFTER CART: RISKS AND MORTALITY
The introduction of cART has drastically reduced the risk of NHL and other AIDS-defining cancers and has improved survival. For example, a recent French cohort study found that the incidence of HIV-NHL was 117-fold higher than the general population in the period before cART (1992–96), but was 9-fold higher during the period when a majority of HIV patients were undergoing treatment with cART (2005–09) (Hleyhel et al. 2013). Decreased viral load and improved immunity are often proposed as key factors mediating this decline. Yet, the risk of NHL in people with HIV after cART became available is still elevated 9-fold compared to the general population (Hleyhel et al. 2013). Underlying causes of the elevated risk for NHL in cART-treated HIV patients are unclear, but a recent report suggests that poor response to treatment, either virological or immunological, is responsible. In the same French cohort study, NHL risk was similar to the general population when including only those HIV patients on cART with HIV RNA <500 copies ml−1 and restored CD4 counts >500 cells μl−1 for at least 2 yr (Hleyhel et al. 2013). Furthermore, a Swiss cohort study of approximately 13 000 HIV-positive patients found that risk of NHL in HIV patients was halved in the first 5 months of cART, and declined to a hazard ratio of 0.1 after continuing therapy for 2–10 yr compared to non-users (Polesel et al. 2008). The most striking difference between cART vs non-cART users was the loss of influence of CD4 count at enrollment on NHL risk, indicating that the incidence of NHL in cART-treated patients, albeit low, was the same regardless of immune status at the beginning of the study. Therefore, cART seems to be effective at preventing most cases of NHL by improving immune status, even among the severely immunocompromised. Nonetheless, during the period of 2005–09, when 86% of HIV patients studied were treated with cART, NHL diagnosis occurred at a younger median age (41 yr) than in the general population (73 yr) (Hleyhel et al. 2013). The reasons for the younger age at diagnosis are unclear, but may be related to specific effects of HIV infection despite treatment, or to cART treatment itself (see below).
In contrast to NHL, the incidence of HL has not dropped after the introduction of cART (Hleyhel et al. 2014). In a large American cohort, the standardized rate ratio for HL in HIV infected vs the general population actually increased from 12-fold before cART to 18-fold after cART was available (Patel et al. 2008). Furthermore, the risk of HL among HIV patients with controlled viral load and CD4 recovery on cART remains 9-fold higher than the general population (Hleyhel et al. 2014), suggesting that recovery of CD4 cells is initially insufficient to control elevated risk for HL after HIV infection; however, the incidence of HL declines after the first year of cART, suggesting immune reconstitution may be linked to HL development (Kowalkowski et al. 2014).
Examination of an American cohort of HIV patients with lymphoma shows that CD4 count at lymphoma diagnosis is increasing over time, and more patients have controlled viral loads with cART (Gopal et al. 2013). After stratifying lymphoma cases by histological subtype, the authors found that the relative distribution of subtypes is changing. In particular, the proportion of BL has more than doubled, and there was a trend toward more HL and less DLBCL (Gopal et al. 2013). The authors noted that CD4 counts at diagnosis differed between lymphoma types, with the highest CD4 count among BL and HL and the lowest CD4 count among PCNSL. These data highlight the heterogeneity of lymphomas with respect to immune status.
Along with appropriate chemotherapy, cART has improved the prognosis of HIV-associated lymphoma, closing the survival gap between HIV-positive and -negative patients (Diamond et al. 2006; Bohlius et al. 2009; Montoto et al. 2012). However, HIV infection remains an independent risk factor for NHL mortality in the United States, in addition to low CD4 count (Chao et al. 2010). A large European study, subsequently confirmed in an American cohort, found that NHL that develops while on cART is more aggressive, since cART-naïve patients had better survival than those previously treated with cART (Bohlius et al. 2009; Gopal et al. 2013). While the benefits of cART are undisputed, these data underscore the need for new prognostic factors associated with NHL in treated patients and for the study of immunological changes after cART and their consequences (Fig. 1).
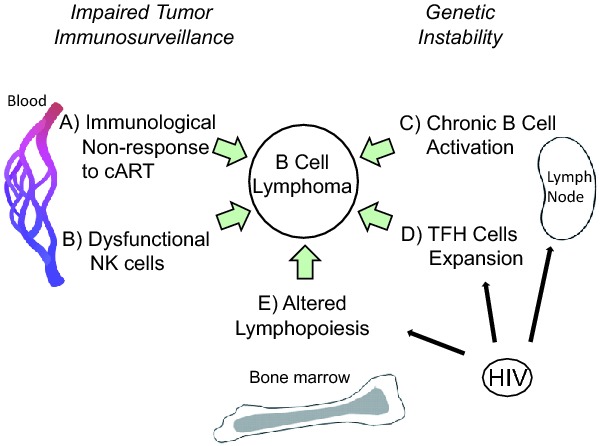
Figure 1.

Development of HIV-associated lymphoma after cART: contribution of immunological changes. After cART, many aspects of immunity remain altered in HIV-infected individuals that may predispose to B-cell lymphoma. (A) Immunological non-response to cART. About 20% of patients experience limited CD4 T-cell recovery or none at all. (B) Dysfunctional NK cells. Despite effective cART, cytotoxic NK cells in aviremic patients have impaired ADCC, an activated phenotype, and are excluded from lymph nodes, an important site of both HIV replication and B-cell transformation. (C) Chronic B cell activation. Residual immune activation, i.e. IL-6, CD40L and/or TLR ligands, leads to polyclonal B-cell activation. Prolonged stimulation may lead to genetic errors via aberrant immunoglobulin class switching or somatic hypermutation in germinal center- or post-germinal center B cells. (D) TFH cells expansion. High numbers of TFH cells in lymph nodes from treated or untreated HIV patients correlate with B-cell abnormalities, including increased germinal center B cells, increased plasma cells and reduced memory B cells. (E) Altered lymphopoiesis. In the bone marrow, increased HPC turnover may lead to accumulation of genetic errors. Also, persistent HIV infection of stroma or HPCs may functionally alter responses to normal cytokine signals for B-cell development and may change the bone marrow microenvironment to encourage lymphoma colonization. Persistent or latent HIV infection of lymph node and bone marrow cells may play a direct role in C, D and E.
IMMUNOLOGICAL CHANGES AFTER CART AND RELATION TO LYMPHOMA
Immunological non-responders
Despite controlling viral replication and improving immune status in a majority of patients, cART does not always result in immune restoration. About 20% of HIV patients experience immunological non-response (INR), defined by no or limited CD4 T-cell rebound to pre-cART levels (Wilson and Sereti 2013). CD4 T-cell recovery is best predicted by nadir counts pre-cART, emphasizing the need for early initiation of therapy prior to severe impairments in T-cell homeostasis, i.e. collagen deposition in lymphoid tissues (Wilson and Sereti 2013). The mechanisms to explain INR are being elucidated, and include exhausted lymphopoiesis, reduced thymic output, homing defects and/or NK-cell killing of CD4 cells (Gaardbo et al. 2012; Sennepin et al. 2013). Considering that immunological non-responders are 2.4-fold more likely to develop serious clinical events or death (Lapadula et al. 2013), further research to understand INR isxbrk warranted.
B-cell activation and dysfunction
Chronic polyclonal B-cell activation, increased B-cell turnover and hypergammaglobulinemia are hallmarks of untreated HIV infection. Prolonged B-cell activation may increase chances for genetic errors through aberrant immunoglobulin gene class-switch recombination or somatic hypermutation, possibly leading to lymphomagenesis. In HIV-positive individuals, it is thought that B cells are stimulated largely via antigen-independent pathways involving cytokines and TLR ligands (Haas, Zimmermann and Oxenius 2011). Non-specific activation of B cells may result from gut microbial translocation and/or HIV viremia. The hypergammaglobulinemia found in HIV viremic patients is partially reduced after cART-induced viral suppression (Regidor et al. 2011; Buckner et al. 2013). However, less than 1.5% of circulating antibody-secreting cells are specific to viral envelope/gp140 after early or chronic HIV infection, and contribute ∼1% to total IgG (Buckner et al. 2013). Various HIV proteins persist in lymph nodes for at least 1 yr post-cART and represent a potential source of chronic antigenic stimulation (Popovic et al. 2005).
Apart from an antigenic role, extracellular HIV proteins can directly promote B-cell hyperproliferation (Lefevre et al. 1999) and class-switch recombination (He et al. 2006). Matrix protein/p17 is of particular interest in this context, as it has been shown to lead to B-cell growth via signaling through cellular receptors (De Francesco et al. 2002; Fiorentini et al. 2006; Giagulli et al. 2011; Caccuri et al. 2012) and has angiogenic activity, which might facilitate spread of lymphomas (Caccuri et al. 2012, 2014; Basta, Latinovic and Lafferty 2015). Interestingly, expression of HIV proteins in transgenic mouse models correlates with the development of B-cell lymphoma (Kundu et al. 1999; Curreli et al. 2013). In addition, Tat may lead to epigenetic dysregulation of the cell cycle (Luzzi et al. 2014), and induce elements of human endogenous retrovirus K, which are found in plasma of lymphoma patients (Contreras-Galindo et al. 2008; Gonzalez-Hernandez et al. 2014). CD40L stimulation from activated CD4 T cells and virion-associated CD40L could also contribute to hyperactivation of B cells (Martin et al. 2007).
In a study of 290 HIV-infected men, cART reduced, but did not normalize, some biomarkers of B-cell activation including sCD27, sCD30, CXCL13 and serum IgG after two years of therapy (Regidor et al. 2011). However, the same study showed that IL-6 (a biomarker preceding HIV-NHL; Vendrame et al. 2014) and C-reactive protein levels were unaffected by cART, suggesting that B-cell hyperactivation persists. Thus, these results help to explain the remaining elevated risk for NHL in cART-treated individuals. Another study has evaluated the effect of cART on spontaneous apoptosis of naïve and memory B cells from HIV-infected patients (Titanji et al. 2005). After 6 months of therapy, cART reduced spontaneous apoptosis of both B-cell subsets, but survival of memory B cells did not return to normal (Titanji et al. 2005). Therefore, B-cell dysfunctions remain after cART. Recovery of total B-cell counts is generally observed after cART (Moir et al. 2008); however, older individuals treated with long-term cART exhibit expansion of naïve B cells in peripheral blood above control levels (Van Epps et al. 2014). This finding is counterintuitive, since a decline in naïve B-cell numbers is observed in older uninfected individuals. Lymphopoiesis is altered after HIV infection, despite suppression of viral replication, as discussed below (Sauce et al. 2011). The relevance of an expanded naïve B-cell population in older individuals on cART to risk of B-cell lymphoma remains unclear.
T follicular helper cells
T follicular helper cells (TFH) are a specialized T-cell subset within lymph nodes that support the differentiation of B cells into long-lived memory cells and plasma cells via development and maintenance of germinal centers (Crotty 2011). Remarkably, TFH cells are expanded 10-fold in lymph nodes from HIV-infected patients compared to HIV-negative controls (Lindqvist et al. 2012). cART reduced the frequency of TFH cells ∼2-fold, suggesting a link between viremia and their expansion. Regardless of cART treatment, TFH frequency in lymph nodes correlated with several B-cell abnormalities, including loss of memory B cells and increase in germinal center B cells and plasma cells. It is possible that increased TFH cell numbers, together with persistent antigen, lowers competition for B-cell selection. Theoretically, high frequencies of germinal center B cells undergoing somatic hypermutation may increase the risk for genetic errors and lymphoma. Subsequently, it was found that TFH cells represent a major site of HIV replication and persistence, potentially affecting their interactions with B cells (Perreau et al. 2013). Crosstalk between TFH and germinal center B cells is bidirectional, as high expression of programed cell death ligand 1 (PD-L1, which can be upregulated by interferons and TLR ligands (Liu et al. 2007) on lymph node germinal center B cells from HIV-infected individuals is involved in deregulation of TFH cell functions in vitro (Cubas et al. 2013). One study of 22 aviremic cART-treated patients found that PD-L1 levels remain high on peripheral blood lymphocytes compared to uninfected controls (Rosignoli et al. 2007). Nevertheless, lowering the viral burden may help to partially ameliorate TFH–B-cell dysfunctions in germinal centers.
Natural killer cells
NK cells can directly lyse tumor and virally infected cells via recognition through a diverse array of activating and inhibitory receptors to activate natural cytotoxicity, or via CD16/FcγRIIIa to mediate antibody-dependent cellular cytotoxicity (ADCC). Functional ADCC is associated with protection from HIV disease progression and represents a major mechanism in anti-CD20 mAb therapy for lymphoma (Forthal et al. 1999; Glennie et al. 2007). It has long been known that phenotype and function of peripheral blood NK cells is altered after HIV infection (Hu et al. 1995). cART can restore the blood NK-cell compartment in terms of distribution of subsets, expression of inhibitory and activating receptors, and cytotoxic functions (Mavilio et al. 2003). However, certain NK-cell defects remain after cART, such as ADCC and activation status.
First, ADCC function is still compromised in treated aviremic patients (Liu et al. 2009). NK ADCC function correlated with CD16/FcγRIIIa cell surface levels, which are reduced in HIV-infected patients regardless of treatment. Impaired ADCC of NK cells from HIV-positive individuals can be restored in vitro by matrix metalloprotease inhibitors (Liu et al. 2009), which may have clinical relevance in therapeutic mAb anti-tumor immunotherapy (Zhou et al. 2013).
Second, blood NK cells in cART-treated patients are qualitatively different from healthy controls. In contrast with T cells, NK cells retain activation markers after cART, including CD38 and HLA-DR (Lichtfuss et al. 2012; Kuri-Cervantes et al. 2014). In addition, despite virologic control, NK cells show high levels of spontaneous degranulation ex vivo and defective CD16-mediated signaling, manifested by downregulation of key signaling molecules (Lichtfuss et al. 2012). Thus, elevated degranulation is not due to heightened ADCC activity, but instead, may be due to recent engagement with target cells that stimulate NK cells through natural cytotoxicity receptors, i.e. NKp30, NKp44, NKp46 or KIRs. Few HIV-infected cells remain after therapy, so the identity of the target cells is unknown. Could NK cells be engaging and killing tumor cells before they are detected clinically? Lichtfuss et al. propose that systemic inflammation from microbial translocation may perpetuate the NK-cell activation indirectly. For example, endotoxin can induce an NKG2D ligand, MIC-A, on monocytes (Kloss et al. 2008). Certain chemokines can also cause spontaneous NK-cell degranulation in the absence of target cells (Taub et al. 1995).
Finally, the anatomical localization of NK cells after cART remains unexplored. Almost all studies utilize peripheral blood samples, but lymphoid tissue and bone marrow are the relevant sites for HIV-infected cells and lymphoma. One study on surgically resected axillary lymph nodes from HIV-infected patients and HIV-negative controls characterized NK-cell distribution after acute infection (Luteijn et al. 2011). Normally, cytotoxic CD56dim NK cells expressing killer cell immunoglobulin-like receptors (KIR) are excluded from lymph nodes, but found in abundance in blood. HIV infection slightly increases the proportion of KIR+ NK cells in blood; however, these effector cells continue to be excluded from lymph nodes, potentially providing HIV an opportunity to escape NK-cell control (Luteijn et al. 2011). One explanation for exclusion may lie in differential chemokine receptor expression. KIR+ blood NK cells show reduced expression of CX3CR1 and CXCR1 in the setting of acute HIV infection, though this did not translate into a significant migratory defect in vitro (Luteijn et al. 2011). Further understanding of how NK cells are reprogramed in treated HIV patients may allow harnessing their potential as critical effector cells to target transformed B cells.
Lymphopoiesis
HIV infection leads to increased lymphocyte apoptosis, cell turnover and eventually widespread lymphopenia. Over time, the bone marrow shows signs of exhaustion, or regenerative failure (Moses, Nelson and Bagby 1998). Lymphopoiesis depends on the interaction of hematopoietic progenitor cells (HPCs) with auxiliary cells, both of which may be infected with HIV (Moses, Nelson and Bagby 1998; Carter et al. 2010). Exactly how HIV perturbs hematopoiesis and why some HIV patients fail to reconstitute immune cells after cART is an area of active research (Corbeau and Reynes 2011; Gaardbo et al. 2012). Compensatory increased cell turnover of precursor cells in bone marrow may increase chances for genetic errors that predispose for HIV-associated lymphoma. In addition, altered cytokine signals during lymphocyte development may result in permanent functional changes and predispose to lymphoma, i.e. altering susceptibility to apoptosis or response to growth factors.
Accumulating evidence suggests that damage to lymphopoiesis after HIV infection may be irreversible, despite effective cART. Bone-marrow-derived CD34+ HPCs from HIV-infected patients express increased proportions of early T- and B-cell markers before cART (Muller et al. 2002). After 6 months of cART, there was a trend toward normalization, but the proportions remained elevated compared to uninfected controls (Muller et al. 2002). There is evidence that a small fraction of HPCs can be latently infected by HIV despite effective cART (Carter et al. 2010), though these cells are not always found (Durand et al. 2012). HIV infection did not alter the potential of HPCs to form colonies in vitro, making it difficult to estimate the impact of HPC infection on lymphopoiesis (Carter et al. 2010).
Due to poor availability of human bone marrow samples, some groups have turned to studying the clonogenic potential of rare circulating CD34+ HPCs directly from blood. The ability of circulating HPCs to form white cells in particular was compromised after HIV infection (Sauce et al. 2011), in agreement with previous studies on bone marrow HPCs (Isgro et al. 2008). The authors surmise that altered lymphopoiesis is a consequence of immune activation, rather than HIV replication, based on (a) a significant inverse correlation between blood CD34+ cell count and the percentage of CD38+CD8+ T cells (a widely used metric of immune activation), and (b) progression toward lymphopenia and defective lymphopoiesis despite control of HIV in rare elite controllers or after successful cART. Interestingly, though cART reduced markers of immune activation to some degree, there was no difference in inflammatory marker levels between patients who progressed and those with restored CD4 counts. Altogether, these data suggest that cART can partially reduce immune activation and restore lymphopoiesis in some patients, but there are additional unidentified factors influencing lymphopoiesis.
Direct effects of certain antiretroviral drugs on the hematopoietic system are documented, both positive and negative. For example, protease inhibitor ritonavir stimulates bone marrow cell colony formation in vitro, presumably by blocking caspase-dependent apoptosis (Sloand et al. 2000). Zidovudine (AZT) inhibits growth of blood HPCs and erythroid and granulocyte–macrophage colony formation in particular, leading to the common toxicities of anemia and neutropenia (D'Andrea, Brisdelli and Bozzi 2008). To what extent current antiretroviral drugs and their combinations directly affect lymphopoiesis has not been studied in vitro.
Finally, bone marrow provides a stromal niche for lymphoma colonization following lymphomagenesis in secondary lymphoid organs. HIV may induce lymphoma growth and dissemination via the bone marrow stroma. In general, lymphoma cells localize to specific bone marrow niches according to their origin (Sangaletti et al. 2014). Germinal center-derived lymphomas are found in the osteoblastic niche, whereas extrafollicular lymphomas are found in the vascular niche. Evidence for common stromal programs between the lymph node germinal center microenvironment and the bone marrow osteoblast niche includes upregulation of a matricellular protein, secreted protein acidic and rich in cysteine (SPARC), in both tissues (Sangaletti et al. 2014). Using knockout mouse models, the authors demonstrate that SPARC regulates normal B-cell lymphopoiesis, possibly fostering growth of B- cell lymphoma. This finding raises the question of whether expression of SPARC and other key molecules are modified in bone marrow stromal cells after HIV infection, with or without cART.
CONCLUSIONS
cART lowers the risk of most NHL, but not HL, in HIV-infected patients. Yet, an elevated risk for lymphoma persists in cART-treated patients. Beyond its antiviral effects, cART can ameliorate some immune activation and preserve T-, B- and NK-cell counts in the blood in the majority of patients. The evidence suggests that cART is most effective against lymphoma due to improved immunity, as measured by CD4 counts. However, peripheral CD4 count may be a marker for other anti-tumor mechanisms, such as NK cells. In addition, the microenvironment encountered by developing and mature B cells is modulated by both HIV and antiretroviral drugs. The combinatorial effects of HIV and cART on B-cell activation, TFH–B-cell crosstalk in lymph nodes and lymphopoiesis are being elucidated. Finally, individual antiretroviral drugs have direct effects on tumor cells. A direct apoptotic effect of certain protease inhibitors on lymphoma cells has been demonstrated in vitro (Kariya et al. 2014). Further understanding of the successes and pitfalls of cART will help to improve therapeutic options for HIV-associated lymphoma and suggest new avenues for prevention of disease.
Acknowledgments
The authors thank Dr Gregory Carey for his helpful discussion and review of the manuscript, Dr Marvin Reitz for critically reading the manuscript and Dr R. Gallo for supporting this work.
FUNDING
This work was supported by the University of Maryland School of Medicine Deans Challenge Award to Accelerate Innovation and Discovery in Medicine, and by the National Institute of Neurological Disorders and Stroke (NINDS) National Institute of Health (NIH) [grant number NS-066842]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NINDS, or the NIH.
Conflict of interest. None declared.
REFERENCES
- Basta D, Latinovic O, Lafferty MK. Angiogenic, lymphangiogenic and adipogenic effects of HIV-1 Matrix protein p17. 2015 doi: 10.1093/femspd/ftv062. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlius J, Schmidlin K, Costagliola D, et al. Prognosis of HIV-associated non-Hodgkin lymphoma in patients starting combination antiretroviral therapy. AIDS. 2009;23:2029–37. doi: 10.1097/QAD.0b013e32832e531c. [DOI] [PubMed] [Google Scholar]
- Buckner CM, Moir S, Ho J, et al. Characterization of plasmablasts in the blood of HIV-infected viremic individuals: evidence for nonspecific immune activation. J Virol. 2013;87:5800–11. doi: 10.1128/JVI.00094-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccuri F, Giagulli C, Bugatti A, et al. HIV-1 matrix protein p17 promotes angiogenesis via chemokine receptors CXCR1 and CXCR2. P Natl Acad Sci USA. 2012;109:14580–5. doi: 10.1073/pnas.1206605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccuri F, Rueckert C, Giagulli C, et al. HIV-1 matrix protein p17 promotes lymphangiogenesis and activates the endothelin-1/endothelin B receptor axis. Arterioscl Throm Vas. 2014;34:846–56. doi: 10.1161/ATVBAHA.113.302478. [DOI] [PubMed] [Google Scholar]
- Capello D, Scandurra M, Poretti G, et al. Genome wide DNA-profiling of HIV-related B-cell lymphomas. Brit J Haematol. 2010;148:245–55. doi: 10.1111/j.1365-2141.2009.07943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone A. AIDS-related non-Hodgkin's lymphomas: from pathology and molecular pathogenesis to treatment. Human Pathol. 2002;33:392–404. doi: 10.1053/hupa.2002.124723. [DOI] [PubMed] [Google Scholar]
- Carbone A, Cesarman E, Spina M, et al. HIV-associated lymphomas and gamma-herpesviruses. Blood. 2009;113:1213–24. doi: 10.1182/blood-2008-09-180315. [DOI] [PubMed] [Google Scholar]
- Carter CC, Onafuwa-Nuga A, McNamara LA, et al. HIV-1 infects multipotent progenitor cells causing cell death and establishing latent cellular reservoirs. Nat Med. 2010;16:446–51. doi: 10.1038/nm.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao C, Silverberg MJ, Martínez-Maza O, et al. Epstein-Barr virus infection and expression of B-cell oncogenic markers in HIV-related diffuse large B-cell Lymphoma. Clin Cancer Res. 2012;18:4702–12. doi: 10.1158/1078-0432.CCR-11-3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao C, Xu L, Abrams D, et al. Survival of non-Hodgkin lymphoma patients with and without HIV infection in the era of combined antiretroviral therapy. AIDS. 2010;24:1765–70. doi: 10.1097/QAD.0b013e32833a0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras-Galindo R, Kaplan MH, Leissner P, et al. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J Virol. 2008;82:9329–36. doi: 10.1128/JVI.00646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbeau P, Reynes J. Immune reconstitution under antiretroviral therapy: the new challenge in HIV-1 infection. Blood. 2011;117:5582–90. doi: 10.1182/blood-2010-12-322453. [DOI] [PubMed] [Google Scholar]
- Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–63. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- Cubas RA, Mudd JC, Savoye AL, et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med. 2013;19:494–9. doi: 10.1038/nm.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curreli S, Krishnan S, Reitz M, et al. B cell lymphoma in HIV transgenic mice. Retrovirology. 2013;10:92. doi: 10.1186/1742-4690-10-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Andrea G, Brisdelli F, Bozzi A. AZT: an old drug with new perspectives. Curr Clin Pharmacol. 2008;3:20–37. doi: 10.2174/157488408783329913. [DOI] [PubMed] [Google Scholar]
- De, Francesco MA, Baronio M, Fiorentini S, et al. HIV-1 matrix protein p17 increases the production of proinflammatory cytokines and counteracts IL-4 activity by binding to a cellular receptor. P Natl Acad Sci USA. 2002;99:9972–7. doi: 10.1073/pnas.142274699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond C, Taylor TH, Im T, et al. Presentation and outcomes of systemic non-Hodgkin's lymphoma: a comparison between patients with acquired immunodeficiency syndrome (AIDS) treated with highly active antiretroviral therapy and patients without AIDS. Leukemia Lymphoma. 2006;47:1822–9. doi: 10.1080/10428190600658688. [DOI] [PubMed] [Google Scholar]
- Dolcetti R, Dal Col J, Martorelli D, et al. Interplay among viral antigens, cellular pathways and tumor microenvironment in the pathogenesis of EBV-driven lymphomas. Semin Cancer Biol. 2013;23:441–56. doi: 10.1016/j.semcancer.2013.07.005. [DOI] [PubMed] [Google Scholar]
- Dunleavy K, Wilson WH. How I treat HIV-associated lymphoma. Blood. 2012;119:3245–55. doi: 10.1182/blood-2011-08-373738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Ghiaur G, Siliciano JD, et al. HIV-1 DNA is detected in bone marrow populations containing CD4+ T cells but is not found in purified CD34 +hematopoietic progenitor cells in most patients on antiretroviral therapy. J Infect Dis. 2012;205:1014–8. doi: 10.1093/infdis/jir884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentini S, Marini E, Caracciolo S, et al. Functions of the HIV-1 matrix protein p17. New Microbiol. 2006;29:1–10. [PubMed] [Google Scholar]
- Forthal DN, Landucci G, Haubrich R, et al. Antibody-dependent cellular cytotoxicity independently predicts survival in severely immunocompromised human immunodeficiency virus-infected patients. J Infect Dis. 1999;180:1338–41. doi: 10.1086/314988. [DOI] [PubMed] [Google Scholar]
- Gaardbo JC, Hartling HJ, Gerstoft J, et al. Incomplete immune recovery in HIV infection: mechanisms, relevance for clinical care, and possible solutions. Clin Dev Immunol. 2012;2012:670957. doi: 10.1155/2012/670957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giagulli C, Marsico S, Magiera AK, et al. Opposite effects of HIV-1 p17 variants on PTEN activation and cell growth in B cells. PLoS One. 2011;6:e17831. doi: 10.1371/journal.pone.0017831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glennie MJ, French RR, Cragg MS, et al. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol Immunol. 2007;44:3823–37. doi: 10.1016/j.molimm.2007.06.151. [DOI] [PubMed] [Google Scholar]
- Goedert JJ, Coté TR, Virgo P, et al. Spectrum of AIDS-associated malignant disorders. Lancet. 1998;351:1833–9. doi: 10.1016/s0140-6736(97)09028-4. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Hernandez MJ, Cavalcoli JD, Sartor MA, et al. Regulation of the human endogenous retrovirus K (HML-2) transcriptome by the HIV-1 Tat protein. J Virol. 2014;88:8924–35. doi: 10.1128/JVI.00556-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal S, Patel MR, Yanik EL, et al. Temporal trends in presentation and survival for HIV-associated lymphoma in the antiretroviral therapy era. J Natl Cancer I. 2013;105:1221–9. doi: 10.1093/jnci/djt158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas A, Zimmermann K, Oxenius A. Antigen-dependent and -independent mechanisms of T and B cell hyperactivation during chronic HIV-1 infection. J Virol. 2011;85:12102–13. doi: 10.1128/JVI.05607-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Qiao X, Klasse PJ, et al. HIV-1 envelope triggers polyclonal Ig class switch recombination through a CD40-independent mechanism involving BAFF and C-type lectin receptors. J Immunol. 2006;176:3931–41. doi: 10.4049/jimmunol.176.7.3931. [DOI] [PubMed] [Google Scholar]
- Hleyhel M, Belot A, Bouvier AM, et al. Risk of AIDS-defining cancers among HIV-1-infected patients in France between 1992 and 2009: results from the FHDH-ANRS CO4 cohort. Clin Infect Dis. 2013;57:1638–47. doi: 10.1093/cid/cit497. [DOI] [PubMed] [Google Scholar]
- Hleyhel M, Bouvier AM, Belot A, et al. Risk of non-AIDS-defining cancers among HIV-1-infected individuals in France between 1997 and 2009: results from a French cohort. AIDS. 2014;28:2109–18. doi: 10.1097/QAD.0000000000000382. [DOI] [PubMed] [Google Scholar]
- Hu PF, Hultin LE, Hultin P, et al. Natural killer cell immunodeficiency in HIV disease is manifest by profoundly decreased numbers of CD16+CD56+ cells and expansion of a population of CD16dimCD56- cells with low lytic activity. J Acq Immun Def Synd. 1995;10:331–40. [PubMed] [Google Scholar]
- Isgro A, Leti W, De Santis W, et al. Altered clonogenic capability and stromal cell function characterize bone marrow of HIV-infected subjects with low CD4+ T cell counts despite viral suppression during HAART. Clin Infect Dis. 2008;46:1902–10. doi: 10.1086/588480. [DOI] [PubMed] [Google Scholar]
- Kariya R, Taura M, Suzu S, et al. HIV protease inhibitor Lopinavir induces apoptosis of primary effusion lymphoma cells via suppression of NF-kappaB pathway. Cancer Lett. 2014;342:52–9. doi: 10.1016/j.canlet.2013.08.045. [DOI] [PubMed] [Google Scholar]
- Kloss M, Decker P, Baltz KM, et al. Interaction of monocytes with NK cells upon toll-like receptor-induced expression of the NKG2D ligand MICA. J Immunol. 2008;181:6711–9. doi: 10.4049/jimmunol.181.10.6711. [DOI] [PubMed] [Google Scholar]
- Kowalkowski MA, Mims MA, Day RS, et al. Longer duration of combination antiretroviral therapy reduces the risk of Hodgkin lymphoma: a cohort study of HIV-infected male veterans. Cancer Epidemiol. 2014;38:386–92. doi: 10.1016/j.canep.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu RK, Sangiorgi F, Wu LY, et al. Expression of the human immunodeficiency virus-Tat gene in lymphoid tissues of transgenic mice is associated with B-cell lymphoma. Blood. 1999;94:275–82. [PubMed] [Google Scholar]
- Kuri-Cervantes L, de Oca GS, Avila-Ríos S, et al. Activation of NK cells is associated with HIV-1 disease progression. J Leukocyte Biol. 2014;96:7–16. doi: 10.1189/jlb.0913514. [DOI] [PubMed] [Google Scholar]
- Lapadula G, Cozzi-Lepri A, Marchetti G, et al. Risk of clinical progression among patients with immunological nonresponse despite virological suppression after combination antiretroviral treatment. AIDS. 2013;27:769–79. doi: 10.1097/QAD.0b013e32835cb747. [DOI] [PubMed] [Google Scholar]
- Lee JH, Kim Y, Choi JW, et al. Prevalence and prognostic significance of Epstein-Barr virus infection in classical Hodgkin's lymphoma: a meta-analysis. Arch Med Res. 2014;45:417–31. doi: 10.1016/j.arcmed.2014.06.001. [DOI] [PubMed] [Google Scholar]
- Lefevre EA, Krzysiek R, Loret EP, et al. Cutting edge: HIV-1 Tat protein differentially modulates the B cell response of naive, memory, and germinal center B cells. J Immunol. 1999;163:1119–22. [PubMed] [Google Scholar]
- Lichtfuss GF, Cheng WJ, Farsakoglu Y, et al. Virologically suppressed HIV patients show activation of NK cells and persistent innate immune activation. J Immunol. 2012;189:1491–9. doi: 10.4049/jimmunol.1200458. [DOI] [PubMed] [Google Scholar]
- Lindqvist M, van Lunzen J, Soghoian DZ, et al. Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J Clin Invest. 2012;122:3271–80. doi: 10.1172/JCI64314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Hamrouni A, Wolowiec D, et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-gamma and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. 2007;110:296–304. doi: 10.1182/blood-2006-10-051482. [DOI] [PubMed] [Google Scholar]
- Liu Q, Sun Y, Rihn S, et al. Matrix metalloprotease inhibitors restore impaired NK cell-mediated antibody-dependent cellular cytotoxicity in human immunodeficiency virus type 1 infection. J Virol. 2009;83:8705–12. doi: 10.1128/JVI.02666-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luteijn R, Sciaranghella G, van Lunzen J, et al. Early viral replication in lymph nodes provides HIV with a means by which to escape NK-cell-mediated control. Eur J Immunol. 2011;41:2729–40. doi: 10.1002/eji.201040886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzi A, Morettini F, Gazaneo S, et al. HIV-1 Tat induces DNMT over-expression through microRNA dysregulation in HIV-related non Hodgkin lymphomas. Infect Agent Cancer. 2014;9:41. doi: 10.1186/1750-9378-9-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Roy J, Barat C, et al. Human immunodeficiency virus type 1-associated CD40 ligand transactivates B lymphocytes and promotes infection of CD4+ T cells. J Virol. 2007;81:5872–81. doi: 10.1128/JVI.02542-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavilio D, Benjamin J, Daucher M, et al. Natural killer cells in HIV-1 infection: dichotomous effects of viremia on inhibitory and activating receptors and their functional correlates. P Natl Acad Sci USA. 2003;100:15011–6. doi: 10.1073/pnas.2336091100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir S, Malaspina A, Ho J, et al. Normalization of B cell counts and subpopulations after antiretroviral therapy in chronic HIV disease. J Infect Dis. 2008;197:572–9. doi: 10.1086/526789. [DOI] [PubMed] [Google Scholar]
- Montoto S, Shaw K, Okosun J, et al. HIV status does not influence outcome in patients with classical Hodgkin lymphoma treated with chemotherapy using doxorubicin, bleomycin, vinblastine, and dacarbazine in the highly active antiretroviral therapy era. J Clin Oncol. 2012;30:4111–6. doi: 10.1200/JCO.2011.41.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses A, Nelson J, Bagby GC., Jr The influence of human immunodeficiency virus-1 on hematopoiesis. Blood. 1998;91:1479–95. [PubMed] [Google Scholar]
- Muller F, Tjønnfjord GE, Nordøy I, et al. Immunophenotypic analyses of CD34(+) cell subsets in bone marrow from HIV-infected patients during highly- active antiretroviral therapy. Eur J Clin Invest. 2002;32:535–40. doi: 10.1046/j.1365-2362.2002.00995.x. [DOI] [PubMed] [Google Scholar]
- Patel P, Hanson DL, Sullivan PS, et al. Incidence of types of cancer among HIV-infected persons compared with the general population in the United States, 1992–2003. Ann Intern Med. 2008;148:728–36. doi: 10.7326/0003-4819-148-10-200805200-00005. [DOI] [PubMed] [Google Scholar]
- Perreau M, Savoye AL, De Crignis E, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med. 2013;210:143–56. doi: 10.1084/jem.20121932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polesel J, Clifford GM, Rickenbach M, et al. Non-Hodgkin lymphoma incidence in the Swiss HIV Cohort Study before and after highly active antiretroviral therapy. AIDS. 2008;22:301–6. doi: 10.1097/QAD.0b013e3282f2705d. [DOI] [PubMed] [Google Scholar]
- Popovic M, Tenner-Racz K, Pelser C, et al. Persistence of HIV-1 structural proteins and glycoproteins in lymph nodes of patients under highly active antiretroviral therapy. P Natl Acad Sci USA. 2005;102:14807–12. doi: 10.1073/pnas.0506857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regidor DL, Detels R, Breen EC, et al. Effect of highly active antiretroviral therapy on biomarkers of B-lymphocyte activation and inflammation. AIDS. 2011;25:303–14. doi: 10.1097/QAD.0b013e32834273ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosignoli G, Cranage A, Burton C, et al. Expression of PD-L1, a marker of disease status, is not reduced by HAART in aviraemic patients. AIDS. 2007;21:1379–81. doi: 10.1097/QAD.0b013e3281de7296. [DOI] [PubMed] [Google Scholar]
- Sangaletti S, Tripodo C, Portararo P, et al. Stromal niche communalities underscore the contribution of the matricellular protein SPARC to B-cell development and lymphoid malignancies. Oncoimmunology. 2014;3:e28989. doi: 10.4161/onci.28989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauce D, Larsen M, Fastenackels S, et al. HIV disease progression despite suppression of viral replication is associated with exhaustion of lymphopoiesis. Blood. 2011;117:5142–51. doi: 10.1182/blood-2011-01-331306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sennepin A, Baychelier F, Guihot A, et al. NKp44L expression on CD4+ T cells is associated with impaired immunological recovery in HIV-infected patients under highly active antiretroviral therapy. Aids. 2013;27:1857–66. doi: 10.1097/qad.0b013e328361a3fe. [DOI] [PubMed] [Google Scholar]
- Sloand EM, Maciejewski J, Kumar P, et al. Protease inhibitors stimulate hematopoiesis and decrease apoptosis and ICE expression in CD34(+) cells. Blood. 2000;96:2735–9. [PubMed] [Google Scholar]
- Taub DD, Sayers TJ, Carter CR, et al. Alpha and beta chemokines induce NK cell migration and enhance NK-mediated cytolysis. J Immunol. 1995;155:3877–88. [PubMed] [Google Scholar]
- Thompson LD, Fisher SI, Chu WS, et al. HIV-associated Hodgkin lymphoma: a clinicopathologic and immunophenotypic study of 45 cases. Am J Clin Pathol. 2004;121:727–38. doi: 10.1309/PNVQ-0PQG-XHVY-6L7G. [DOI] [PubMed] [Google Scholar]
- Titanji K, Chiodi F, Bellocco R, et al. Primary HIV-1 infection sets the stage for important B lymphocyte dysfunctions. AIDS. 2005;19:1947–55. doi: 10.1097/01.aids.0000191231.54170.89. [DOI] [PubMed] [Google Scholar]
- Van Epps P, Matining RM, Tassiopoulos K, et al. Older age is associated with peripheral blood expansion of naive B cells in HIV-infected subjects on antiretroviral therapy. PLoS One. 2014;9:e107064. doi: 10.1371/journal.pone.0107064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendrame E, Hussain SK, Breen EC, et al. Serum levels of cytokines and biomarkers for inflammation and immune activation, and HIV-associated non-Hodgkin B-cell lymphoma risk. Cancer Epidem Biomar. 2014;23:343–9. doi: 10.1158/1055-9965.EPI-13-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MK, Pagano JS, Khalili K. Viruses and human cancers: a long road of discovery of molecular paradigms. Clin Microbiol Rev. 2014;27:463–81. doi: 10.1128/CMR.00124-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson EM, Sereti I. Immune restoration after antiretroviral therapy: the pitfalls of hasty or incomplete repairs. Immunol Rev. 2013;254:343–54. doi: 10.1111/imr.12064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Gil-Krzewska A, Peruzzi G, et al. Matrix metalloproteinases inhibition promotes the polyfunctionality of human natural killer cells in therapeutic antibody-based anti-tumour immunotherapy. Clin Exp Immunol. 2013;173:131–9. doi: 10.1111/cei.12095. [DOI] [PMC free article] [PubMed] [Google Scholar]