

Abstract
Sialic acids (SAs) are widely expressed on immune cells and their levels and linkages named as sialylation status vary upon cellular environment changes related to both physiological and pathological processes. In this study, we performed a global profiling of the sialylation status of macrophages and their release of SAs in the cell culture medium by using flow cytometry, confocal microscopy and liquid chromatography tandem mass spectrometry (LC-MS/MS). Both flow cytometry and confocal microscopy results showed that cell surface α-2,3-linked SAs were predominant in the normal culture condition and changed slightly upon treatment with atorvastatin for 24 h, whereas α-2,6-linked SAs were negligible in the normal culture condition but significantly increased after treatment. Meanwhile, the amount of total cellular SAs increased about three times (from 369 ± 29 to 1080 ± 50 ng/mL) upon treatment as determined by the LC-MS/MS method. On the other hand, there was no significant change for secreted free SAs and conjugated SAs in the medium. These results indicated that the cell surface α-2,6 sialylation status of macrophages changes distinctly upon atorvastatin stimulation, which may reflect on the biological functions of the cells.
Keywords: confocal microscopy, flow cytometry, LC-MS/MS, macrophage, sialic acid
Introduction
Sialic acids (SAs), a family of 9-carbon containing acidic monosaccharides, often terminate cell surface and secreted glycoconjugates, such as glycoproteins and glycolipids (Varki et al. 2009). So far, more than 50 SAs with acetylation at different hydroxyl groups have been found in nature in a variety of linkages to other sugars including themselves on either N-glycans or O-glycans, thus providing a considerable degree of molecular diversity and specificity (Schauer 2000; Angata and Varki 2002; Varki et al. 2009). Given the location and ubiquitous distribution on the cell surface, SAs are involved in a wide range of physiological and pathological processes, such as immunological process, hormonal response, signal transduction, tumor progression, cell adhesion and infection (Schauer 2000; Varki NM and Varki A 2007). SAs expressed on immune cells are closely related to their activities. An early study found that macrophages form rosettes with normal sheep erythrocytes but not the one after sialidase pretreatment (Crocker and Gordon 1986). Also, guinea pig peritoneal macrophages pretreated with sialidase bound more 125I-IgG than non-treated cells (Gorczyca et al. 1989). Further, sialidase treatment can substantially enhance the capacity of resting B cells to stimulate the proliferation of allogeneic and antigen-specific syngeneic T cells (Cowing and Chapdelaine 1983; Frohman and Cowing 1985; Bagriaçik and Miller 1999). All these studies reveal a fact that cell surface SAs on immune cells are involved in a diverse regulation of immune cell interactions and play important roles related to both physiological and pathological processes.
So far, a number of studies have been performed on the SA-binding proteins in the immune system. The SAs recognizing proteins are categorized to the family of siglecs, which represents SAs recognizing Ig superfamily lectins. The founding members of the siglec family include sialoadhesin (siglec-1), a macrophage adhesion molecule (Crocker et al. 1994; Hartnell et al. 2001; Varki and Angata 2006); CD22 (siglec-2), a B-cell inhibitory receptor (Stamenkovic et al. 1990); CD33 (siglec-3), a marker of myeloid cells (Simmons and Seed 1988) and myelin-associated glycoprotein (or siglec-4), expressed by oligodendrocytes and Schwann cells in the nervous system (Arquint et al. 1987). Despite the extensive studies on the SA-binding proteins in the immune system, the SA profile of immune cells themselves was largely overlooked for decades. In particular, the sialylation status related to the linkages and the amount of SAs on the immune cell surface is less known so far.
It was reported that the cell surface sialylation pattern changes during apoptosis in several cell types, including colon carcinomas and thymocytes (Morris et al. 1984; Rapoport and Pendu 1999). Also, cell surface sialylation plays a role in modulating sensitivity towards APO-1-mediated apoptotic cell death (Peter et al. 1995). A recent study found that decreasing of SA residues on the surface of apoptotic lymphocytes was recognized as an “Eat-Me” signal (Meesmann et al. 2010). In addition, it was reported that β-galactoside α-2,6 sialyltransferase I (ST6Gal-I) regulates macrophage apoptosis via α-2,6 sialylation of the TNFR1 death receptor (Liu et al. 2011).
Statin drugs, as inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A reductase, are widely used in the treatment of hypercholesterolemia for more than a decade. In addition to their lipid-lowering effect, convincing evidence demonstrated that statins have a wide effect on macrophages. For example, statins were found to reduce the cell numbers of macrophages in vivo and in vitro (Aikawa et al. 2001; Liang et al. 2006; Croons et al. 2010), and one mechanism could be attributed to its apoptosis induction effect on macrophages at even low dosages (Liang et al. 2006; Croons et al. 2010). Moreover, the immunomodulatory roles of statin have been studied in macrophages on the cytokine and corresponding protein levels, such as inhibiting inducible nitric oxide synthase (Pahan et al. 1997; Huang et al. 2003), augmenting lipopolysaccharides-induced proinflammatory responses (Matsumoto et al. 2004) and inducing Th2 cytokines secretion while suppressing Th1 cytokines production (Youssef et al. 2002). Although detail mechanisms have been elucidated in each study, most of these pleiotropic effects are mediated by the inhibition of the synthesis of isoprenoid intermediates such as farnesylpyrophosphate and geranylgeranylpyrophosphate, which are essential for many cellular functions like differentiation and proliferation (Takemoto and Liao 2001).
In this study, we systematically investigated the sialylation status of macrophages in the normal culture condition and after atorvastation treatment. In particular, we globally profiled SAs linkages and levels of respective SA forms on the cell surface by confocal microscopy and flow cytometry with SA-specific lectins Sambucus nigra agglutinin (SNA) and Maackia amurensis agglutinin (MAA). Further, free, conjugated and total SAs in the cell culture medium and total SAs in the cells were quantified in both normal and statin-treated conditions by a newly developed liquid chromatography tandem mass spectrometry (LC-MS/MS) methods. The combination of confocal microscopy, flow cytometry and LC-MS/MS provides the first time of global investigation of the cellular sialylation status of macrophages in the entire cell culture system, which will contribute to a better understanding of the physiological and pathological roles of SAs in macrophages and in the immune system as well.
Results
Cell surface SAs analyzed by confocal microscopy and flow cytometry
SAs are commonly found α-2,3- and α-2,6-linked to galactose residue on the cell surface (Lehmann et al. 2006). In our study, we first performed confocal microscopy and flow cytometry analysis to distinctively measure SAs with different linkages on the macrophage surfaces by using MAA-FITC (fluorescein isothiocyanate) and SNA-FITC, which specifically bind to α-2,3- and α-2,6-linked SAs, respectively. From the confocal microscopy study, MAA-FITC showed very strong binding compared with SNA-FITC on the cell surface of macrophages cultured under the normal condition (Figure 1A). After cells were treated with atorvastatin for 24 h, MAA-FITC binding remained unchanged (Figure 1B), whereas SNA-FITC binding increased dramatically (Figure 1B).
Fig. 1.
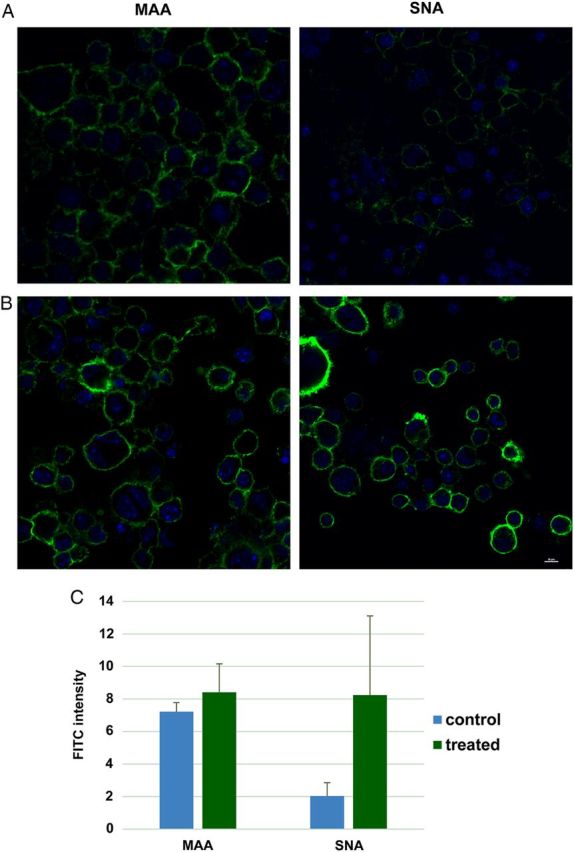
Confocal microscopy analysis of cell surface SA. (A) Raw 264.7 cells at the normal culture condition were stained with MAA-FITC (10 μg/mL) and SNA-FITC (20 μg/mL), respectively. DAPI was used to stain nuclei. (B) Raw 264.7 cells were treated with 20 μM atorvastatin for 24 h followed by staining with lectins and DAPI. The scale bar represents 10 μm. (C) The fluorescent intensity of the confocal images were analyzed by ImageJ obtained from National Institutes of Health. This figure is available in black and white in print and in colour at Glycobiology online.
Next, the lectin-labeled cells were analyzed by flow cytometry. The similar binding pattern was observed from macrophages under the normal and atorvastatin conditions. In particular, SNA-FITC binding for macrophages was significantly increased upon atorvastatin treatment (Figure 2). The dot plots indicated that ∼80% of the cells were MAA positive and 10% were SNA positive in the normal condition (Figure 2A). After 24 h atorvastatin treatment, no apparent change in MAA-FITC labeling was observed, but peak shift was apparently found from SNA-FITC-labeled cells (Figure 2B). The dot plots showed the total ratio of SNA positive cells increased about three times when compared with the untreated cells. Further, the lectin stainings were performed on permeabilized cells, and the peak shift was found in SNA-FITC-labeled cells in both permeabilized and non-permeabilized conditions with atorvastatin treatment, whereas no significant change in MAA-FITC-labeled cells was observed. Interestingly, large peak shift was found in SNA-FITC-labeled cells with permeabilization independent on atorvastatin treatment (Figure 3). These results indicate that 2,6-linked SA increased both on the cell surfaces and inside the cells.
Fig. 2.

Determination of cell surface SAs by flow cytometry. (A) Raw 264.7 cells at normal condition were stained with MAA-FITC (10 μg/mL) and SNA-FITC (20 μg/mL), respectively. PI staining was used to distinguish living cells and dead cells. (B) Raw 264.7 cells were treated with 20 μM atorvastatin for 24 h then stained with lectins and PI. Data are the representative of at least three independent experiments. This figure is available in black and white in print and in colour at Glycobiology online.
Fig. 3.
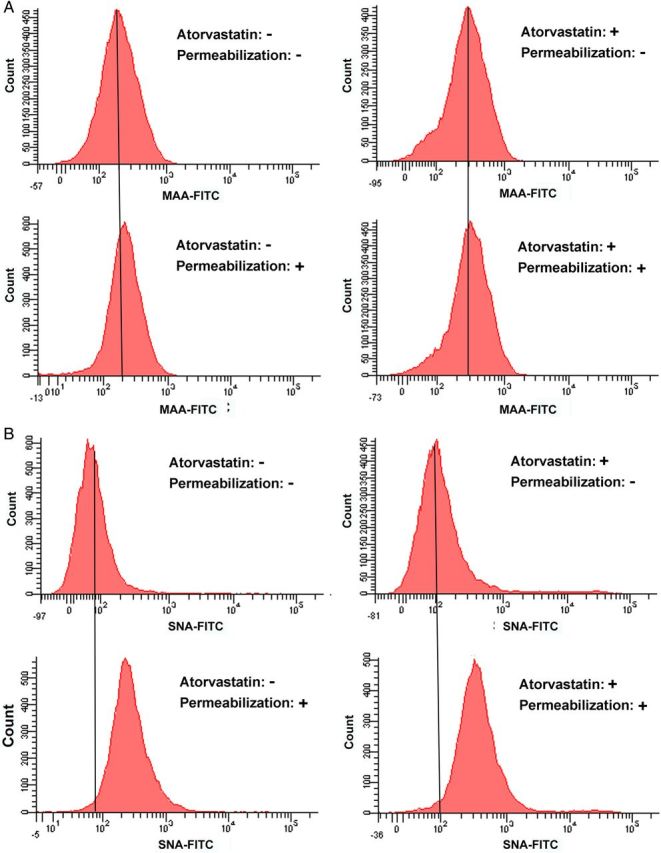
Comparison of the SA level after cell permeabilization. Atorvastatin+: Cells were treated with 20 μM atorvastatin for 24 h. Atorvastatin−: Cells were incubated with vehicle for 24 h. Permeabilization+: Cells were fixed with 4% PFA, followed by permeabilization with 0.2% Triton X-100 and then labeled with lectins. Permeabilization−: Cells were fixed with 4% PFA and then labeled with lectins. (A) Raw 264.7 cells at different conditions were labeled with MAA-FITC (10 μg/mL). (B) Raw 264.7 cells at different conditions were labeled with SNA-FITC (20 μg/mL). This figure is available in black and white in print and in colour at Glycobiology online.
Comparison of SNA and Annexin V staining
It is known that statins stimulate apoptotic cell death in several types of immune cells, such as macrophages (Liang et al. 2006; Croons et al. 2010). Annexin V specifically binds to membrane phosphatidylserine and has been widely used for early apoptosis detection (Koopman et al. 1994; Vermes et al. 1995). In our study above, a significant SNA-FITC staining increase was observed for atorvastatin-treated cells compared with MAA-FITC staining (Figure 1B and 2B). Therefore, we compared the SNA labeling and Annexin V labeling of the cells treated with 20 μM atorvastatin for 14, 24 and 38 h, respectively. As shown in Figure 4A, both SNA and Annexin V positive cells increased dramatically with the incubation time prolonged. Briefly, in the control sample (0 h), the SNA positive cell ratio was only 9.4%, which included both propidium iodide (PI) positive and negative cells, whereas the SNA positive cells increased to 23.5, 33.6 and 41.3% when the cells were incubated with atorvastatin for 14, 24 and 38 h, respectively. Meanwhile, Annexin V positive cells increased in the same time-dependent manner as SNA labeling. However, the change scale of Annexin V labeling was smaller compared with SNA labeling. These results are further supported by the SNA and Annexin V co-stainings of the cells (Figure 4B). All these results indicated that α-2,6-linked SAs were highly expressed on the surface of cells undergoing apoptosis and thus may be used as a marker for apoptosis detection as well. Further study to prove the correlation of macrophage apoptosis and α-2,6-linked SAs status is much deserved.
Fig. 4.
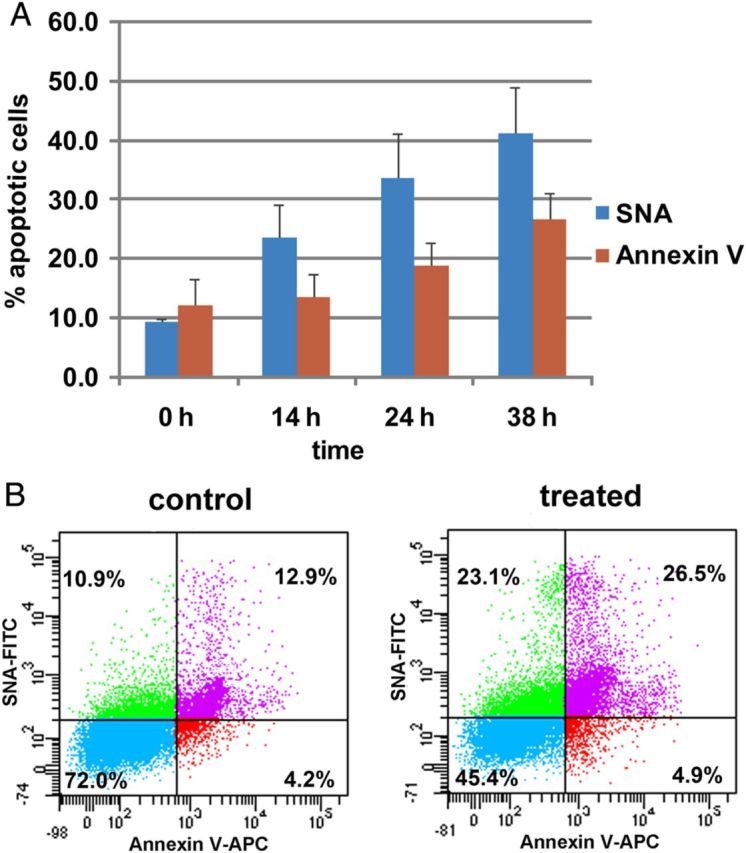
Comparison of SNA and Annexin V labeling of Raw 264.7 cells after cells incubated with atorvastatin. (A) Cells were treated with or without atorvastatin (20 μM) for 14, 24 and 38 h and then labeled with SNA or Annexin V. Both PI positive and negative cells are included here. Values represent the means and standard deviations from three independent experiments. (B) Cells were treated with atorvastatin (or vehicle) for 24 h, and then costained with SNA-FITC and Annexin V-APC. This figure is available in black and white in print and in colour at Glycobiology online.
LC-MS/MS quantification of SAs in the cell culture media and cell lysates
In this study, we developed a new LC-MS/MS method to quantitatively examine the free and conjugated SA levels in the cell culture media, and the total SAs in the cell lysates upon the cell treated with atorvastatin. The mass spectra for both SA and 13C3-SA (internal standard, IS) was obtained in negative electrospray ionization (Supplementary data, Figure S1). The MS detection was carried out in negative electrospray ionization and the multiple reaction monitoring (MRM) mode [Supplementary data, Figure S2—representative chromatograms for the lower limit of quantification (LLOQ) sample and a sample obtained from cells]. The calibration range was obtained from 2.00 to 2.00 × 103 ng/mL and the LLOQ was 2.00 ng/mL. Importantly, the large calibration range and the low LLOQ facilitate the determination of small amount of free SAs in the cell culture medium and total SAs in the cell lysate. This method was validated for matrix effect, precision and accuracy, and stability (Supplementary data, Tables S1–S3). The validated method was successfully used for quantifying the free SAs and conjugated SAs in the cell culture media, and the total SAs in the cell lysates, which would indicate SAs releasing and synthesis fact related to cell function alteration.
First, the concentrations of free SAs in the cell culture media under the normal culture condition and incubation with atorvastatin for 24 h were measured by LC-MS/MS (Figure 5). As a result, free SAs in the normal culture condition were 6.31 ± 0.45 ng/mL and no apparent change was observed after atorvastatin treatment. Meanwhile, the total and conjugated SAs in the cell culture media were 73.2 ± 3.3 and 66.9 ± 3.6 ng/mL under the normal condition and slightly increased to 80.9 ± 7.1 and 74.3 ± 7.0 ng/mL, respectively, after incubation with 20 μM atorvastatin. Next, the total cellular SAs, including on the cell surfaces and inside the cells were measured by LC-MS/MS after releasing SAs from the cell lysates. As a result, the cellular SAs concentration was 369 ± 29 ng/mL at a density of 1 × 106 cells/mL under the normal culture condition, and it increased to 567 ± 25, 775 ± 32 and 1080 ± 50 ng/mL corresponding to 5, 10 and 20 μM of atorvastatin used, respectively.
Fig. 5.
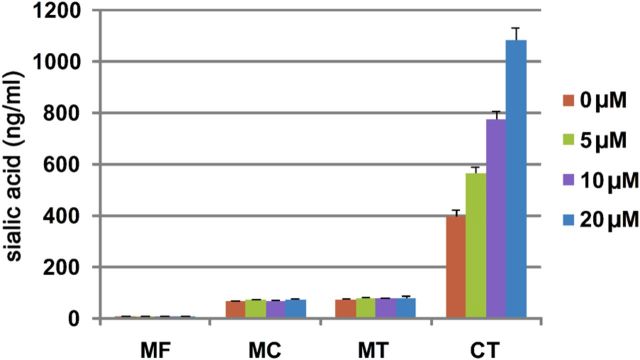
SA concentrations in the cell culture system. Free SAs in the medium were quantified by LC-MS/MS directly. The medium was hydrolyzed with acetic acid followed by LC-MS/MS analysis to determine the total SAs. Conjugated SAs in the medium were calculated by subtracting the free form from total SAs. In order to quantify total SAs of the cell, cells were first ultrasonicated to obtain the homogeneous mixture, followed by acid hydrolysis and LC-MS/MS analysis. MF, medium free SA; MC, medium conjugated SA; MT, medium total SA; CT, cell total SA. Data are the representative of at least three independent experiments. This figure is available in black and white in print and in colour at Glycobiology online.
Western blot analysis of selected α-2,6 sialyltransferase level
The LC-MS/MS results of increased cellular SAs may be in accordance with the increase of α-2,6-linked SAs on the cell surface after atorvastatin treatment as observed by flow cytometry and confocal microscopy above. Therefore, we further performed western blot analysis of the cellular α-2,6 sialyltransferases in normal and statin conditions. These sialyltransferases are grouped into two families according to the carbohydrate linkages they synthesize, including β-galactoside α-2,6 sialyltransferases (ST6Gal-I and -II) and GalNAc α-2,6 sialyltransferases (ST6GalNAc I–VI) (Takashima 2008). ST6Gal-I for N-glycan, ST6GalNAc-II for O-glycan and ST6GalNAc-VI for glycolipids have been investigated on immune cells (Takashima 2008; Videira et al. 2008; Crespo et al. 2013). As shown in Figure 6, ST6GalNAc-II was found highly increased, ST6GalNAc-VI no apparent change, whereas ST6Gal-I decreased upon atorvastatin treatment (with 20 μM for 24 h). Therefore, the elevation of ST6GalNAc-II may contribute to the increased cellular SAs and α-2,6-linked SAs on the cell surface after atorvastatin treatment. There may be other sialyltransferases related to the increase of α-2,6-linked SAs as well, which deserves a further study in future.
Fig. 6.
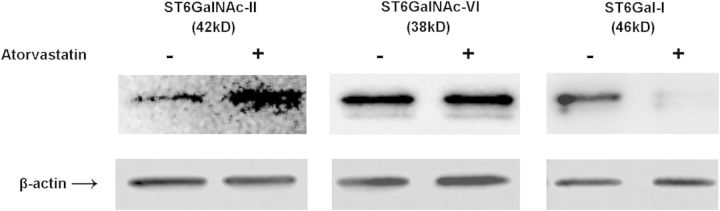
Western blot analysis of Raw 264.7 cells sialyltransferases upon atorvastatin treatment. Raw cells were incubated with 20 μM atorvastatin (or vehicle) for 24 h, and the ST6Gal-I, ST6GalNAc-II and ST6GalNAc-VI were determined by western blot analysis using rabbit anti-ST6Gal-I, rabbit anti-ST6GalNAc-II and rabbit anti-ST6GalNAc-VI antibodies, respectively.
Discussion
Macrophages play very important roles in the immune system related to both physiological and pathological pathways. SAs terminate most glycans on the cell surface and are involved in variety of physiological and pathological activities of immune cells. In this study, we used Raw 264.7 macrophages to comprehensively profile the sialylation status in the cell culture system including cell surface SA levels and their linkages, secreted free, conjugated and total SAs in the culture medium and total SAs in the cells as well. First, cell surface SAs were determined by confocal microscopy and flow cytometry by using SNA and MAA labeling. As a result, α-2,3-linked SAs were found to be predominant on the Raw 264.7 macrophage surfaces, whereas α-2,6-linked SAs were less observed under the normal culture condition (Figure 1A). After cells were treated with atorvastatin for 24 h, α-2,3-linked SAs were unchanged; however, α-2,6-linked SAs increased significantly on cell surface (Figures 1B and 2B). The wide distribution of α-2,3-linked SAs may be associated with the cell–cell interaction and signaling transduction under the normal condition. For example, sialoadhesin (siglec-1), an SA-binding receptor uniquely expressed by macrophage subsets, selectively binds to only α-2,3-linked SAs (Varki and Angata 2006). Although α-2,6-linked SAs may be associated with the cell–cell interaction and signaling transduction in response to external stimuli. The expression level of SAs on the cell surface is affected by its biosynthetic pathway. In particular, the synthetic enzyme, sialyltransferase, functions for adding SAs to the termini of N-linked or O-linked glycans on the cell surfaces (Dall'Olio 2000). In our study, western blot showed a dramatically change of ST6GalNAc-II which may be the major contributor for the increase in cell surface α-2,6-linked SAs.
It is known that the glycosylation pattern, including sialylation, changes when cells undergo apoptosis (Rapoport and Pendu 1999; Heyder et al. 2003; Bilyy et al. 2004; Franz et al. 2006; Meesmann et al. 2010; Liu et al. 2011). The extensive studies related to the SA expression level and cell apoptosis have been done by the Dr Susan L. Bellis research group focusing on the ST6Gal-I sialyltransferase (Zhuo et al. 2008; Liu et al. 2011; Swindall and Bellis 2011; Zhuo and Bellis 2011). The decrease in the SA level regulated by ST6Gal-I on the TNFR1 Death Receptor has been reported to induce macrophages apoptosis (Liu et al. 2011). ST6Gal-I regulated sialylation of β1 integrins can block cell adhesion to galectin-3 and protect cells against galectin-3-induced apoptosis (Zhuo et al. 2008). Moreover, Fas-mediated apoptosis in colon carcinoma cells can be diminished by the sialylation of the Fas death receptor by ST6Gal-I (Swindall and Bellis 2011). One explanation of ST6Gal-I protecting cell against apoptosis can be explained by its inhibition of galectins blinding to underling galactose, which is reported to limit the life span of variety of cells (Zhuo and Bellis 2011). In our study, the ST6Gal-I was found to decrease significantly after cells treated with atorvastatin, and meantime the cell apoptosis level elevated dramatically. Based on the Dr Bellis report, the reduction in the ST6Gal-I level can partially contribute to atorvastatin-induced macrophage apoptosis. Conversely, an increase in α-2,6-linked SAs was observed in the late apoptotic lymphocytes (Meesmann et al. 2010). One explanation for this phenomenon was the redistribution of immature (intracellular) membranes onto the cell surface during cell apoptosis (Meesmann et al. 2010). This scenario is supported by an earlier study, which demonstrated the exposure of endoplasmic reticulum (ER) resident proteins and sugars predominantly on the incompletely processed proteins in ER and Golgi (Franz et al. 2007). In our study, we found that α-2,6-linked SAs increased apparently on macrophage cell surface upon atorvastatin activation. The late apoptotic redistribution of the intracellular membrane may, at least in part, explain the increase in α-2,6-linked SAs on the cell surface upon atorvastatin stimulation. A further study to reveal the particular molecular mechanism is highly warranted.
Most studies on the sialylation status have been limited to SAs on the cell surface with the methods such as lectins labeling, metabolic labeling and chemical modification (Chen and Varki 2010). Free and conjugated SA levels in the cell culture medium may change upon cell activation, such as releasing SAs from cells (surface). In addition, SA biosynthesis may increase inside of the cells as well. Therefore, we quantitatively examined the free and conjugated SA levels in the cell culture medium after atorvastatin treatment, which indicated SAs releasing fact related to cell function alteration. In addition, we quantified the total SAs in the cells as well.
So far, few studies tried to quantify the SAs trimmed off from cell surfaces (Gorczyca et al. 1989; Eguchi et al. 2005). However, the quantification methods used were often with complicated sample preparation procedures, such as enzymatic or acid digestion, sample concentration and SA derivatization. 1,2-Diamino-4,5methylenedioxybenzene derivatization was mainly used for the quantification of SAs (Hara et al. 1989; Morimoto et al. 2001; Eguchi et al. 2005; Galuska et al. 2012; Bayer et al. 2013). However, the derivatization involves both high temperature and acidic conditions, which can hydrolyze part of the conjugated SAs, and severely interrupt the quantitative accuracy of a small amount of free SAs in the cell culture medium. In our study, we developed a new LC-MS/MS method for direct quantification of SAs in the cell culture system. In this method, the free SAs in the medium can be directly analyzed after simple centrifugation, and no any sample purification or concentration step is needed. Conjugated SAs in the medium or cell lysate were efficiently hydrolyzed with 2 M acetic acid at 80°C for 90 min (data is not shown) followed by direct LC-MS/MS analysis. As a result, there were no significant changes for free SAs and conjugated SAs in the medium after atorvastatin treatment, indicating that no apparent sialidases activity changed during this process. However, the amount of total cellular SAs increased from 369 ± 29 to 1080 ± 50 ng/mL upon cell activation. Both confocal microscope and flow cytometry studies above revealed increasing α-2,6-linked SAs on the cells, indicating that the α-2,6-linked SAs was the major contributor for the total cellular SAs increase. Our western blot analysis of sialyltrasferases showed the high level of ST6GalNAc-II, indicating that it may be the major contributor for the increase in α-2,6-linked SAs. Overall, the newly developed LC-MS/MS method was successfully applied to sensitively quantify both free and total SAs in the culture medium and cell lysate.
In conclusion, we profiled SAs dynamics in the whole culture system in the normal and atorvastatin-treated conditions. α-2,3-Linked SAs were found predominant on the cell surface of Raw 264.7 cells under the normal condition and unchanged upon atorvastatin-treated. In comparison, α-2,6-linked SAs were obscure but increased dramatically on the cell surface after atorvastatin treatment. In addition, the total SAs of the cells were significantly increased, whereas the SAs in the cell culture medium were unchanged after cells treated with atorvastatin. This is the first time of comprehensively analysis of cell surface SAs of their levels and linkages, free, conjugated and total SAs in the culture medium, and total SAs in the cells by combination of confocal microscopy, flow cytometry and LC-MS/MS, which provide a sensitive, specific and systematic way to globally study SAs in the cell culture system. This work will contribute to a better understanding of the physiological and pathological roles of SAs in macrophages and the immune system as well. In general, the reported methods can be expanded to study the status of SAs of any kind of cells of interests.
Materials and methods
Materials
FITC-labeled maackia amurensis (MAA) lectin was purchased from bioWORLD (Dublin, OH, USA). FITC-labeled sambucus nigra (SNA) lectin was provided by Vector Laboratories (Burlingame, CA, USA). Alexa Fluor 488-labeled Annexin V, APC-labeled Annexin V and ProLong Gold Antifade Mountant were obtained from Life Technologies (Grand Island, NY, USA). Atorvastatin was purchased from A Chem Tek Inc. (Worcester, MA, USA). N-Acetyl-d-neuraminic acid (SA, 98%) was supplied by Rose Scientific Ltd (Edmonton AB, Canada). N-Acetyl-d-neuraminic acid-1,2,3-13C3 (13C3-SA, 99%), 4′,6-diamidino-2-phenylindole (DAPI), PI, radio-immunoprecipitation assay (RIPA) buffer, protease inhibitor cocktail, rabbit anti-ST6Gal-I antibody, rabbit anti-ST6GalNAc-II antibody, rabbit anti-ST6GalNAc-VI antibody and goat anti-rabbit IgG-peroxidase antibody were purchased from Sigma-Aldrich Inc. (St Louis, MO, USA). Acetonitrile (HPLC grade), paraformaldehyde (PFA, 16% w/v) and acetic acid (ACS grade) were obtained from Fisher Scientific (Hanover Park, IL, USA).
Apparatus
Flow cytometry was performed on a FACSCanto II system consisting of a blue and a red laser, operated through BD FACSDiva software (BD Bioscience, Mountain View, CA, USA). Images were acquired at a 60× magnification using a Nikon A1Rsi confocal microscope, and data analysis was performed using NIS-Elements software (Nikon Instruments Inc., Melville, NY, USA). LC-MS/MS quantification was carried out by a Nexera liquid chromatography system (Shimadzu, Columbia, MD, USA) interfaced to a Qtrap 5500 mass spectrometer equipped with eletrospray ionization source (AB SCIEX, Framingham, MA, USA), and the data acquisition and processing were conducted using Analysis 1.6.1 software.
Cell culture and stimulation
Raw 264.7 cells, obtained from American Type Culture Collection, were cultured in the Dulbecco's modified Eagle's medium (DMEM; Cleveland Clinic Core Facility, Cleveland, OH, USA) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals, Flowery branch, GA, USA) and 1% penicillin/streptomycin (Cleveland Clinic Core Facility) in a humidified atmosphere containing 5% CO2 at 37°C. In order to quantify free SAs, FBS was removed from the culture medium before adding the drug. For atorvastatin simulation, cells were seeded in 6-well plates for overnight, followed by atorvastatin treatment at different concentrations and incubation times. The media without FBS were collected and centrifuged at 15,000 × g for 5 min. The supernatants were stored at −20°C until use. Cells were analyzed by flow cytometry, confocal microscopy or LC-MS/MS.
Flow cytometric analysis
For the determination of cell surface SA, cells were treated with atorvastatin (or vehicle) for 24 h. The cells were harvested and washed three times with cold phosphate-buffered saline (PBS), and then suspended in 50 μL MAA (10 μg/mL) or SNA (20 μg/mL) and 5 μL PI (1000 μg/mL). After incubation for 30 min at room temperature, cells were washed with cold PBS for three times and resuspended in the same buffer for flow analysis. A minimum of 20,000 cells were measured each time.
The cell apoptosis study was performed by staining cells with Annexin V and PI using the dead cell apoptosis kit (Life Technologies). Briefly, cells treated with or without 20 μM atorvastatin for 14, 24 and 38 h were collected and washed three times with cold PBS and then resuspended in 100 μL of 1× binding buffer provided by the manufacturer. Annexin V (5 μL) and PI (1 μL) were added to the cell suspension and incubated at room temperature for 15 min. After incubation, 400 μL of 1× binding buffer was added to each vial and cells were analyzed immediately with flow cytometry.
Confocal microscopy
Cells were seeded on a coverslip in a 6-well plate for overnight and then treated with 20 μM atorvastatin (or vehicle) for 24 h. After incubation, cells on the coverslip were washed for three times with cold PBS and fixed with 4% PFA for 10 min. Excess PFA was removed by another three times wash with cold PBS. Then, cells were stained with MAA-FITC (10 μg/mL) or SNA-FITC (20 μg/mL) for 30 min, followed by 1 min DAPI stain. Unbound lectin was washed off with cold PBS and cover glasses were mounted on the ProLong Gold Antifade reagent.
LC-MS/MS analysis
SA and IS were separated by a Primesep D column (2.1 × 100 mm, 5 μm; SIELC Technologies, Prospect Heights, IL, USA) with a binary linear gradient elution, in which phase A was deionized water containing 10 mM ammonium formate and 0.1% formic acid and phase B was a mixture of 90% acetonitrile, 10% water, 3 mM ammonium formate and 0.04% formic acid. The gradient program started at 10% B and maintained for 1 min and then changed linearly to 100% B in 3 min and maintained for 2 min. In order to equilibrate the column, the gradient sharply dropped to 10% B in 0.1 min and kept for 2.9 min. The total run time was 9 min for each sample. The MS detection was carried out in negative electrospray ionization and the MRM mode. The quantitative MRM transitions were set at m/z 308 → 87 for SA and 311 → 90 for IS. The quality assurance MRM channel for SA was set at m/z 308 → 170.
Method validation sample preparation
SA calibration standards were prepared in the DMEM at concentrations of 2.00, 6.00, 20.0, 60.0, 200, 600 and 2.00 × 103 ng/mL. The quality control (QC) samples were prepared in the same manner at low, mid and high concentrations of 4.00, 100 and 1.60 × 103 ng/mL. IS was prepared in the DMEM at the concentration of 2.00 × 103 ng/mL. For the sample preparation, 5 μL of IS was mixed together with 95 μL of calibration standards or QC samples, and 5 μL of the mixture was subjected to the LC-MS/MS analysis.
Cell culture sample preparation
Free SA in the medium was quantified by spiking 95 μL of culture medium with 5 μL of IS, and then 5 μL of the mixture was injection to the LC-MS/MS system. In order to determine total SA in the culture medium, the sample was first hydrolyzed with 2 M acetic acid (1:1) at 80°C for 90 min, and then 5 μL of the IS was added to 95 μL of the lysate before analysis. For the quantification of total SA of the cell, the sample at a density of 1 × 106 cells/mL was untrosonicated to obtain the homogeneous mixture, then followed the procedure of total SA quantification in the culture medium.
Western blot
Cells were washed twice with ice-cold PBS and lysed with RIPA buffer on ice for 5 min. The cell lysates were collected and clarified by centrifugation at 15,000 × g at 4°C for 10 min. Protein concentrations were measured by the Bradford method using a protein assay kit (Bio-Rad, Hercules, CA, USA). Proteins (30 μg/sample) were fractionated on sodium dodecyl sulfate-15% polyacrylamide gels and transferred to the polyvinylidene difluoride membrane. The membranes were blocked with 5% nonfat milk in Tri-buffered saline containing 0.05% Tween 20 (TBS-T) for 1 h at room temperature and then probed with primary antibodies in the TBS-T buffer with 5% non-fat milk at 4°C overnight. The membranes were washed three times with the TBS-T buffer, followed by incubation with the secondary antibody for 1 h at room temperature. Antibody detection was accomplished by using the ECL Prime kit (Pierce) with the LI-COR Odyssey Fc imaging system.
Supplementary data
Supplementary data for this article is available online at http://glycob.oxfordjournals.org/.
Funding
This work was supported by Faculty Research Development Fund and the research fund from the Center for Gene Regulation in Health and Disease (GRHD) at Cleveland State University supported by Ohio Department of Development (ODOD). The authors acknowledge the National Science Foundation Major Research Instrumentation Grant (CHE-0923398) for supporting Q-Trap 5500 mass spectrometer instrument, the National Institution of Health for supporting Nikon A1Rsi confocal microscope (1S10OD010381). This work was partially supported by grants from The National Natural Science Foundation of China (31328006). H Nie appreciates the China Oversea Scholar Award from China Scholarship Council.
Conflict of interest
Authors claim no conflict of interest.
Abbreviations
DAPI, 4′,6-diamidino-2-phenylindole; DMEM, Dulbecco's modified Eagle's medium; ER, endoplasmic reticulum; FBS, fetal bovine serum; FITC, fluorescein isothiocyanate; IS, internal standard; LC-MS/MS, liquid chromatography tandem mass spectrometry; LLOQ, lower limit of quantification; MAA, Maackia amurensis agglutinin; MRM, multiple reaction monitoring; PBS, phosphate-buffered saline; PFA, paraformaldehyde; PI, propidium iodide; QC, quality control; SA, sialic acid; SNA, Sambucus nigra agglutinin; ST6Gal-I, β-galactoside α-2,6 sialyltransferase I; TBS-T, Tri-buffered saline containing 0.05% Tween 20.
Supplementary Material
References
- Aikawa M, Rabkin E, Sugiyama S, Voglic SJ, Fukumoto Y, Furukawa Y, Shiomi M, Schoen FJ, Libby P. 2001. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation. 103:276–283. [DOI] [PubMed] [Google Scholar]
- Angata T, Varki A. 2002. Chemical diversity in the SAs and related alpha-keto acids: An evolutionary perspective. Chem Rev. 102:439–469. [DOI] [PubMed] [Google Scholar]
- Arquint M, Roder J, Chia LS, Down J, Wilkinson D, Bayley H, Braun P, Dunn R. 1987. Molecular cloning and primary structure of myelin-associated glycoprotein. Proc Natl Acad Sci USA. 84:600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagriaçik EU, Miller KS. 1999. Cell surface SA and the regulation of immune cell interactions: The neuraminidase effect reconsidered. Glycobiology. 9:267–275. [DOI] [PubMed] [Google Scholar]
- Bayer NB, Schubert U, Sentürk Z, Rudloff S, Frank S, Hausmann H, Geyer H, Geyer R, Preissner KT, Galuska SP. 2013. Artificial and natural SA precursors influence the angiogenic capacity of human umbilical vein endothelial cells. Molecules. 18:2571–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilyy RO, Antonyuk VO, Stoika RS. 2004. Cytochemical study of role of alpha-d-mannose- and beta-d-galactose-containing glycoproteins in apoptosis. J Mol Histol. 35:829–838. [DOI] [PubMed] [Google Scholar]
- Chen X, Varki A. 2010. Advances in the biology and chemistry of SAs. ACS Chem Biol. 5:163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowing C, Chapdelaine JM. 1983. T cells discriminate between Ia antigens expressed on allogeneic accessory cells and B cells: A potential function for carbohydrate side chains on Ia molecules. Proc Natl Acad Sci USA. 80:6000–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo HJ, Lau JT, Videira PA. 2013. Dendritic cells: A spot on sialic Acid. Front Immunol. 4:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker PR, Gordon S. 1986. Properties and distribution of a lectin-like hemagglutinin differentially expressed by murine stromal tissue macrophages. J Exp Med. 164:1862–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crocker PR, Mucklow S, Bouckson V, McWilliam A, Willis AC, Gordon S, Milon G, Kelm S, Bradfield P. 1994. Sialoadhesin, a macrophage SA binding receptor for haemopoietic cells with 17 immunoglobulin-like domains. EMBO J. 13:4490–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croons V, De Meyer I, Houten SM, Martinet W, Bult H, Herman AG, De Meyer GR. 2010. Effect of statins on the viability of macrophages and smooth muscle cells. J Cardiovasc Pharmacol. 55:269–275. [DOI] [PubMed] [Google Scholar]
- Dall'Olio F. 2000. The sialyl-alpha2,6-lactosaminyl-structure: biosynthesis and functional role. Glycoconj J. 17:669–676. [DOI] [PubMed] [Google Scholar]
- Eguchi H, Ikeda Y, Ookawara T, Koyota S, Fujiwara N, Honke K, Wang PG, Taniguchi N, Suzuki K. 2005. Modification of oligosaccharides by reactive oxygen species decreases sialyl lewis x-mediated cell adhesion. Glycobiology. 15:1094–1101. [DOI] [PubMed] [Google Scholar]
- Franz S, Frey B, Sheriff A, Gaipl US, Beer A, Voll RE, Kalden JR, Herrmann M. 2006. Lectins detect changes of the glycosylation status of plasma membrane constituents during late apoptosis. Cytometry A. 69:230–239. [DOI] [PubMed] [Google Scholar]
- Franz S, Herrmann K, Fürnrohr BG, Sheriff A, Frey B, Gaipl US, Voll RE, Kalden JR, Jäck HM, Herrmann M. 2007. After shrinkage apoptotic cells expose internal membrane-derived epitopes on their plasma membranes. Cell Death Differ. 14:733–742. [DOI] [PubMed] [Google Scholar]
- Frohman M, Cowing C. 1985. Presentation of antigen by B cells: Functional dependence on radiation dose, interleukins, cellular activation, and differential glycosylation. J Immunol. 134:2269–2275. [PubMed] [Google Scholar]
- Galuska SP, Geyer H, Mink W, Kaese P, Kühnhardt S, Schäfer B, Mühlenhoff M, Freiberger F, Gerardy-Schahn R, Geyer R. 2012. Glycomic strategy for efficient linkage analysis of di-, oligo- and polySAs. J Proteomics. 75:5266–5278. [DOI] [PubMed] [Google Scholar]
- Gorczyca W, Wieczorek Z, Lisowski J. 1989. Cell surface SA affects immunoglobulin binding to macrophages. FEBS Lett. 259:99–102. [DOI] [PubMed] [Google Scholar]
- Hara S, Yamaguchi M, Takemori Y, Furuhata K, Ogura H, Nakamura M. 1989. Determination of mono-O-acetylated N-acetylneuraminic acids in human and rat sera by fluorometric high-performance liquid chromatography. Anal Biochem. 179:162–166. [DOI] [PubMed] [Google Scholar]
- Hartnell A, Steel J, Turley H, Jones M, Jackson DG, Crocker PR. 2001. Characterization of human sialoadhesin, a SA binding receptor expressed by resident and inflammatory macrophage populations. Blood. 97:288–296. [DOI] [PubMed] [Google Scholar]
- Heyder P, Gaipl US, Beyer TD, Voll RE, Kern PM, Stach C, Kalden JR, Herrmann M. 2003. Early detection of apoptosis by staining of acid-treated apoptotic cells with FITC-labeled lectin from Narcissus pseudonarcissus. Cytometry A. 55:86–93. [DOI] [PubMed] [Google Scholar]
- Huang KC, Chen CW, Chen JC, Lin WW. 2003. HMG-CoA reductase inhibitors inhibit inducible nitric oxide synthase gene expression in macrophages. J Biomed Sci. 10:396–405. [DOI] [PubMed] [Google Scholar]
- Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. 1994. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 84:1415–1420. [PubMed] [Google Scholar]
- Lehmann F, Tiralongo E, Tiralongo J. 2006. SA-specific lectins: Occurrence, specificity and function. Cell Mol Life Sci. 63:1331–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SL, Liu H, Zhou A. 2006. Lovastatin-induced apoptosis in macrophages through the Rac1/Cdc42/JNK pathway. J Immunol. 177:651–656. [DOI] [PubMed] [Google Scholar]
- Liu Z, Swindall AF, Kesterson RA, Schoeb TR, Bullard DC, Bellis SL. 2011. ST6Gal-I regulates macrophage apoptosis via α2-6 sialylation of the TNFR1 death receptor. J Biol Chem. 286:39654–39662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Einhaus D, Gold ES, Aderem A. 2004. Simvastatin augments lipopolysaccharide-induced proinflammatory responses in macrophages by differential regulation of the c-Fos and c-Jun transcription factors. J Immunol. 172:7377–7384. [DOI] [PubMed] [Google Scholar]
- Meesmann HM, Fehr EM, Kierschke S, Herrmann M, Bilyy R, Heyder P, Blank N, Krienke S, Lorenz HM, Schiller M. 2010. Decrease of SA residues as an eat-me signal on the surface of apoptotic lymphocytes. J Cell Sci. 123:3347–3356. [DOI] [PubMed] [Google Scholar]
- Morimoto N, Nakano M, Kinoshita M, Kawabata A, Morita M, Oda Y, Kuroda R, Kakehi K. 2001. Specific distribution of SAs in animal tissues as examined by LC-ESI-MS after derivatization with 1,2-diamino-4,5-methylenedioxybenzene. Anal Chem. 73:5422–5428. [DOI] [PubMed] [Google Scholar]
- Morris RG, Hargreaves AD, Duvall E, Wyllie AH. 1984. Hormone-induced cell death. 2. Surface changes in thymocytes undergoing apoptosis. Am J Pathol. 115:426–436. [PMC free article] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Namboodiri AM, Singh I. 1997. Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. J Clin Invest. 100:2671–2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter ME, Hellbardt S, Schwartz-Albiez R, Westendorp MO, Walczak H, Moldenhauer G, Grell M, Krammer PH. 1995. Cell surface sialylation plays a role in modulating sensitivity towards APO-1-mediated apoptotic cell death. Cell Death Differ. 2:163–171. [PubMed] [Google Scholar]
- Rapoport E, Pendu JL. 1999. Glycosylation alterations of cells in late phase apoptosis from colon carcinomas. Glycobiology. 9:1337–1345. [DOI] [PubMed] [Google Scholar]
- Schauer R. 2000. Achievements and challenges of SA research. Glycoconj J. 17:485–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons D, Seed B. 1988. Isolation of a cDNA encoding CD33, a differentiation antigen of myeloid progenitor cells. J Immunol. 141:2797–2800. [PubMed] [Google Scholar]
- Stamenkovic I, Seed B. 1990. The B-cell antigen CD22 mediates monocyte and erythrocyte adhesion. Nature. 345:74–77. [DOI] [PubMed] [Google Scholar]
- Swindall AF, Bellis SL. 2011. Sialylation of the Fas death receptor by ST6Gal-I provides protection against Fas-mediated apoptosis in colon carcinoma cells. J Biol Chem. 286:22982–22990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima S. 2008. Characterization of mouse sialyltransferase genes: Their evolution and diversity. Biosci Biotechnol Biochem. 72:1155–1167. [DOI] [PubMed] [Google Scholar]
- Takemoto M, Liao JK. 2001. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler Thromb Vasc Biol. 21:1712–1719. [DOI] [PubMed] [Google Scholar]
- Varki A, Angata T. 2006. Siglecs-the major subfamily of I-type lectins. Glycobiology. 16:1R–27R. [DOI] [PubMed] [Google Scholar]
- Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. 2009. Essentials of Glycobiology. 2nd ed. Cold Spring Harbor: (NY: ): Cold Spring Harbor Laboratory Press. [PubMed] [Google Scholar]
- Varki NM, Varki A. 2007. Diversity in cell surface SA presentations: Implications for biology and disease. Lab Invest. 87:851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. 1995. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 184:39–51. [DOI] [PubMed] [Google Scholar]
- Videira PA, Amado IF, Crespo HJ, Algueró MC, Dall'Olio F, Cabral MG, Trindade H. 2008. Surface alpha 2-3- and alpha 2-6-sialylation of human monocytes and derived dendritic cells and its influence on endocytosis. Glycoconj J. 25:259–268. [DOI] [PubMed] [Google Scholar]
- Youssef S, Stüve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, Bravo M, Mitchell DJ, Sobel RA, Steinman L, et al. 2002. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 420:78–84. [DOI] [PubMed] [Google Scholar]
- Zhuo Y, Bellis SL. 2011. Emerging role of alpha2,6-sialic acid as a negative regulator of galectin binding and function. J Biol Chem. 286:5935–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo Y, Chammas R, Bellis SL. 2008. Sialylation of beta1 integrins blocks cell adhesion to galectin-3 and protects cells against galectin-3-induced apoptosis. J Biol Chem. 283:22177–22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.