

Abstract
Osteogenesis imperfecta (OI) is a heritable bone disease with dominant and recessive transmission. It is characterized by a wide spectrum of clinical outcomes ranging from very mild to lethal in the perinatal period. The intra- and inter-familiar OI phenotypic variability in the presence of an identical molecular defect is still puzzling to the research field. We used the OI murine model Brtl+/− to investigate the molecular basis of OI phenotypic variability. Brtl+/− resembles classical dominant OI and shows either a moderately severe or a lethal outcome associated with the same Gly349Cys substitution in the α1 chain of type I collagen. A systems biology approach was used. We took advantage of proteomic pathway analysis to functionally link proteins differentially expressed in bone and skin of Brtl+/− mice with different outcomes to define possible phenotype modulators. The skin/bone and bone/skin hybrid networks highlighted three focal proteins: vimentin, stathmin and cofilin-1, belonging to or involved in cytoskeletal organization. Abnormal cytoskeleton was indeed demonstrated by immunohistochemistry to occur only in tissues from Brtl+/− lethal mice. The aberrant cytoskeleton affected osteoblast proliferation, collagen deposition, integrin and TGF-β signaling with impairment of bone structural properties. Finally, aberrant cytoskeletal assembly was detected in fibroblasts obtained from lethal, but not from non-lethal, OI patients carrying an identical glycine substitution. Our data demonstrated that compromised cytoskeletal assembly impaired both cell signaling and cellular trafficking in mutant lethal mice, altering bone properties. These results point to the cytoskeleton as a phenotypic modulator and potential novel target for OI treatment.
Introduction
Osteogenesis imperfecta (OI) is a hereditary skeletal dysplasia characterized by reduced bone mineral density (BMD), abnormal bone microarchitecture and frequent fractures in the absence of or in response to minor trauma (1). Extraskeletal manifestations are also reported affecting skin, ligament, heart and lung (2,3).
Osteogenesis imperfecta was traditionally considered a dominantly inherited disease affecting the genes encoding the α chains of type I collagen. More recently, recessive forms of OI have also been described, caused by mutations in a variety of genes mainly affecting type I collagen quantity, structure, synthesis, post-translational modification, secretion or extracellular processing (4,5).
The disorder covers a wide spectrum of clinical severity ranging from very mild osteoporosis to perinatal lethality, and the genotype/phenotype relationship is still poorly understood, both in dominant and in recessive forms (6,7). The phenotypic severity in the presence of type I collagen-mutated genes seems to be at least in part correlated to the gene involved. Correlating with type I collagen stoichiometry, COL1A2 mutations generally result in a less severe phenotype than COL1A1 mutations. Also, the position of mutations along the α-chains can modulate the outcome, with mutations at the N-terminus usually being less severe than substitutions in the middle or at the C-terminus of the chains. Specific regions in the triple helix were identified as important for the interaction between collagen and extracellular matrix proteins (Major Ligand Binding Regions), and mutations at these sites are particularly harmful (6). Furthermore, phenotypic variability associated with identical mutations, a common recurrent feature in hereditary diseases, has been described in dominant and recessive OI (8).
The dissection of the molecular basis of OI phenotypic variability is expected to significantly contribute to the understanding of the molecular mechanisms that characterize the disease, leading not only to the identification of novel biomarkers for diagnosis, therapy follow-up and drug design but also toward the delineation of new targetable pathways for novel therapeutic approaches.
The knock-in murine model Brtl+/− represents an important tool for understanding OI phenotypic variability in the presence of an identical defect (9). Brtl+/− mice carry a heterozygous point mutation in Col1a1, responsible for the Gly349Cys (p.Gly527Cys) substitution in one α1(I) chain, which was described to be causative in human dominant OI (10). Interestingly, these mutant mice can have either a moderately severe phenotype, resembling human OI type IV, or perinatal lethal OI, resembling OI type II (9).
In an attempt to understand the molecular basis of OI phenotypic variability, we previously performed deep analysis of collagen and tissue extracts from bone, skin and lung of Brtl+/− mice with lethal (ML) and non-lethal (MA) outcomes. Based on our results, we excluded that the variability of Brtl+/− outcome was attributable to differences in collagen structure, physical properties or interaction between mutant collagen helices (11,12). Instead we demonstrated that a difference in intracellular response to mutant collagen retention could be involved. We found, both in skin and in bone, differential expression of chaperones, proteasomal subunits, metabolic enzymes and proteins related to cellular fate in mice with different outcomes. Our data revealed that the intracellular machinery in lethal mice is less effective at coping with the intracellular retention of mutant collagen, favoring up-regulation of molecules involved in apoptosis and protein degradation, in contrast to the mice with a moderately severe outcome, in which chaperone up-regulation is predominant (13,14).
In the present report, using as a starting point our previously obtained transcriptomic and proteomic data from skin and bone tissues of newborn WT and Brtl+/− mice with moderately severe or lethal outcome, we took advantage of pathway analysis to functionally crosslink differentially expressed proteins to define possible phenotype modulators. Tissue ‘hybrid nets’ were generated by combining all the differentially expressed genes and proteins identified in mutant bone with several proteins detected with aberrant expression in mutant skin, and vice versa. These networks not only highlighted that OI similarly affected common pathways in both tissues but, in particular, they allowed a more comprehensive systems biology approach to accurately visualize functional cross-talk that exists between proteins detected with altered expression in mutants. This enabled a proper investigation of protein differences that were undervalued in our previous analyses and allowed further elucidation of the molecular basis of OI phenotypic variability. In particular, we focused on different expression patterns of three proteins that acquired central roles in hybrid nets and that belong to or are involved in cytoskeletal organization. Abnormal cytoskeleton was then experimentally detected in different tissues from Brtl+/− mice with lethal outcome, but not in surviving mutant mice. Calvarial and long-bone osteoblasts revealed an aberrant cytoskeleton affecting cell proliferation, collagen deposition, and integrin and TGF-β signaling with consequent impairment of bone structural properties. Lastly, abnormal cytoskeletal assembly was detected in fibroblasts obtained from lethal, but not from non-lethal, OI patients carrying a substitution at the same glycine.
Results
Hybrid networks of proteins differentially expressed in bone and skin reveal a role of the cytoskeleton in OI disease
Bone/skin and skin/bone hybrid networks were generated by processing together the differential expression data we previously obtained from transcriptomic and proteomic analyses separately performed on bone and skin samples from 1-day-old wild-type (WT) and Brtl+/− mice, with lethal (ML) or moderately severe (MA) OI phenotype (13,14). We performed MetaCore shortest path algorithm pathway analysis and obtained a first ‘bone/skin hybrid network’ (Fig. 1A) by co-processing genes and proteins differentially expressed in bone (Supplementary Material, Tables S1A and B) (13), and some proteins differentially expressed in skin, which were validated as differentially expressed also in bone by western blot (Supplementary Material, Table S2) (14). Among these latter proteins, only Oct3/4 was excluded from the analysis because of a ‘form defect’ of functional data processing. As known, this transcriptional factor is involved in several cellular mechanisms (15); thus, its inclusion in the list of proteins to be processed leads to the generation of pathways exclusively focused on it, obscuring other factors and processes that may be relevant in the pathogenic definition of the disease. All the proteins identified as differentially regulated in skin proteomic investigations not only entered into the hybrid net, but two of them, maspin and, in particular, vimentin became central hubs, as highlighted in Figure 1A, where proteins from skin analysis are encircled in red.
Figure 1.

Continued
Figure 1.
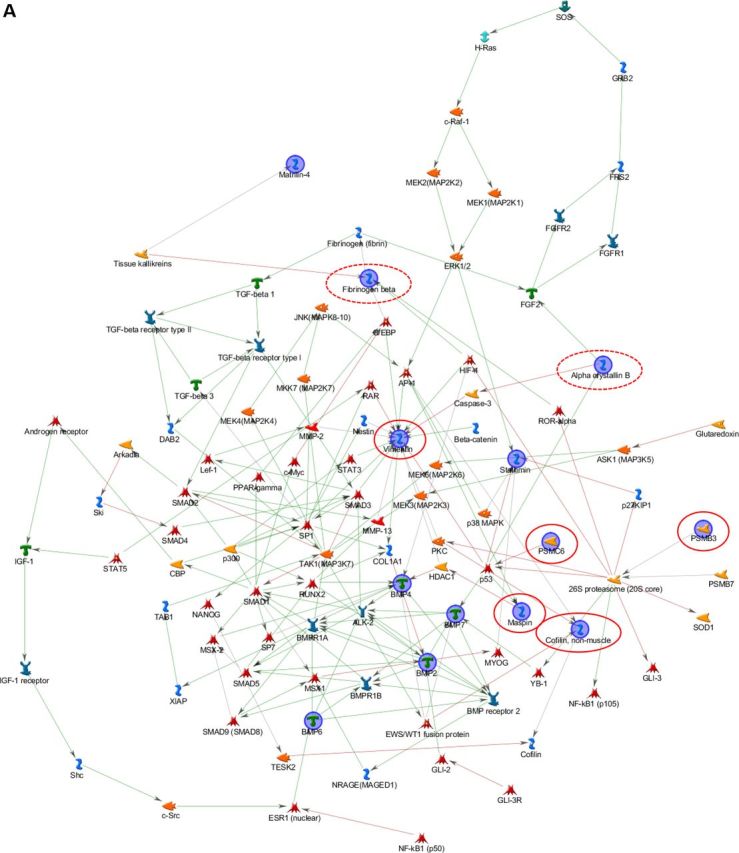
Hybrid networks generated by MetaCore analysis of proteins differentially expressed in bone and skin of lethal and surviving mutant mice and WT littermates. (A) Bone/skin hybrid network. Proteins detected as differentially expressed in skin, and experimentally proven to be deregulated also in bone by western blot assays, are highlighted by continuous red line. Proteins that were found differentially expressed both in skin and bone are circled in dashed red. (B) Skin/bone hybrid network. Proteins that were found differentially expressed in bone are surrounded by continuous green line, whereas proteins observed differentially expressed both in skin and bone are circled in dashed green. Network proteins are visualized by specific symbols, which define the functional nature of the protein (network caption). The edges indicate the relationships existing between individual proteins, and the arrowheads represent the direction of the interaction. The following line colors designate the nature of the interaction: red: negative effect; green: positive effect; gray: unspecified effect. PSMC6: 26S protease regulatory subunit S10B; PSMB3: proteasome subunit beta type-3; BMP: bone morphogenic protein.
Based on the promising results obtained combining bone data with a subset of skin data, we also applied pathway hybridization in a skin functional study. All of the differentially expressed proteins identified in skin from newborn mutant mice (Supplementary Material, Table S3) (14) were processed by the MetaCore shortest path algorithm in combination with all factors differentially expressed in calvarial samples, detected by proteomics and transcriptomics analyses (Supplementary Material, Table S1A and B) (13). Contrary to the bone/skin hybrid net, all of the bone deregulated factors were included among proteins to be processed by the software without experimentally proving their deregulation in skin specimens. With the exception of gadd153, which was excluded from pathway analysis because it is not supported by the MetaCore in-house database, and of matrilin 4 and magp2, all factors with altered expression in mutant bone samples entered into the skin/bone hybrid net (Fig. 1B). Such proteins, shown by green circles in Figure 1B, were perfectly integrated into the net, and only agrin and thrombospondin 3 acquired marginal positions. In particular, stathmin became, with vimentin, one of the main central hubs of the network. Moreover, the integration of bone deregulated proteins did not cause substantial alteration of the original skin network obtained by processing proteins exclusively identified as differentially expressed in skin samples (14).
Clearly, skin and bone share altered cellular processes in which the same proteins are modulated. Hence, hybrid nets consistently support the hypothesis of the systemic nature of OI.
Vimentin and stathmin resulted in two principal central hubs in both hybrid analyses. They are likely to play some key roles in modulating and crosslinking OI-affected pathways. Vimentin is a class-III intermediate filament commonly expressed in various cell types, and its disassembly was described to be correlated to cytoskeleton collapse (16). We previously proved its down-regulation in skin and bone from Brtl+/− mice (14). Stathmin, which we found up-regulated at transcriptional level in bone from mutant lethal mice (13), is instead a microtubule filament-regulating factor that prevents tubulin dimer assembly and promotes disassembly of microtubules (17,18). Based on their functional relevance in both the hybrid nets and on their involvement in correlated cytoskeleton dynamics, we hypothesized the occurrence of an abnormal cytoskeleton organization in Brtl+/− animals.
In the original mutant skin proteomic analysis, we detected the deregulation of another protein with cytoskeleton remodeling activity that was also included into the skin net: cofilin-1 (14). This protein is known to regulate actin dynamics inducing F-actin depolymerization and inhibiting G-actin polymerization (19,20). Interestingly, cofilin-1 not only entered into the skin/bone hybrid net, but it was also included into the bone/skin net, when we added it to the list of skin proteins to be co-processed with all the bone-affected molecules (Fig. 1B).
According to our data, the three main cytoskeletal components, microtubules, microfilaments, and intermediate filaments, are apparently altered in OI. This disease was traditionally considered a disorder of bone extracellular matrix and only recently an intracellular effect on OI phenotype was demonstrated (13,14). Our hybrid pathway analysis further supports this latter concept pointing to cytoskeleton structures involved in OI outcome.
Different expression of stathmin and cofilin-1 in ML, MA and WT mice
In order to experimentally support the in-silico data, we analyzed stathmin and cofilin-1 expression levels in bone and skin samples from WT, ML and MA Brtl+/− mice by western blotting. As shown in Figure 2A for stathmin and in Figure 2B for cofilin-1, the expression profile was consistent in both tissues revealing an overexpression in ML mice with respect to both WT and MA animals. Cofilin-1 was also slightly up-regulated in MA versus WT. As ML Brtl+/− mice die when they were 1 day old owing to respiratory stress, the expression of stathmin and cofilin-1 was also evaluated in lung, where indeed the expression of both proteins was found to be up-regulated. However, while cofilin-1 was strongly over-expressed, a moderate increase of stathmin was detected in this tissue (Fig. 2A and B).
Figure 2.

Expression analysis of some focal hubs from hybrid nets in skin, bone and lung tissues. (A) Representative western blots of stathmin and corresponding densitometric analysis are shown. In all the three analyzed tissues, stathmin is significantly up-regulated in Brtl+/− lethal (ML) mice with respect to surviving Brtl+/− mice (MA) and WT littermates. (B) Representative western blots of cofilin-1 and corresponding densitometric analysis are shown. In all the three analyzed tissues, cofilin-1 is significantly up-regulated in Brtl+/− ML mice. Histograms visualize normalized mean relative-integrated density ± SD values. *, ¥ and § symbols indicate the significance of expression changes occurring between WT and ML, WT and MA, and ML and MA mice, respectively.
Cytoskeletal organization is altered in Brtl+/− lethal mice
The up-regulation of stathmin and cofilin-1 demonstrated in ML mice tissues together with the decrease of vimentin in ML skin and bone samples, as reported in Bianchi et al. (14), suggested a potential alteration of cytoskeletal organization in mice with different phenotypic outcome. Thus, in order to evaluate the cytoskeleton, immunohistochemistry was performed on calvaria and long bones, skin and lung tissues obtained from newborn WT and lethal and non-lethal mutant mice. Being a specific marker for actin filaments, fluorescent phalloidin was applied for this purpose. In each analyzed sample, ML tissues revealed a strongly disorganized cytoskeleton that lost its typical framework when compared with MA and control mice (Fig. 3A). Actin fibers showed a punctuate organization rather than a well-organized reticular net.
Figure 3.
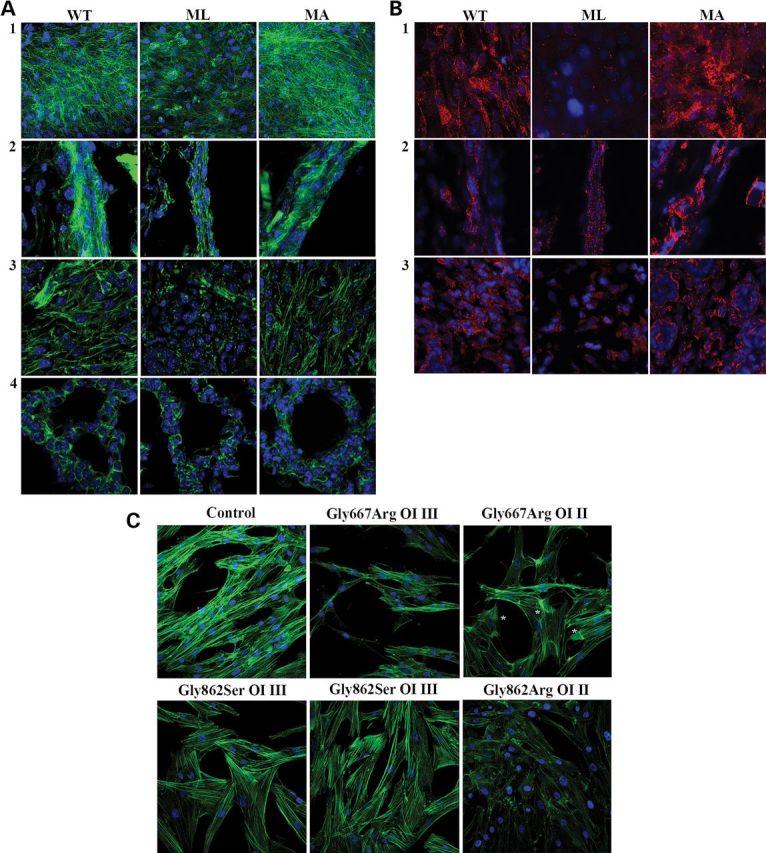
Analysis of the cytoskeletal organization in different tissues from WT, ML and MA mice and in human dermal fibroblasts from OI patients. (A) Fluorescently labeled phalloidin (green), specifically interacting with actin, was used to analyze the cytoskeletal organization of calvarial bone (1), long bone (2), skin (3) and lung (4) in WT, ML and MA animals. A disorganized network is evident in samples from mutant lethal mice. (B) The focal adhesions (FAs) were evaluated in calvarial bone (1), long bone (2) and skin (3) of WT, ML and MA animals using the FAKpY397 antibody (red). Altered distribution of FAs, indicating an abnormal integrin-mediated signal, is evident in lethal mice. (C) Fluorescently labeled phalloidin was used to analyze the cytoskeletal organization of fibroblasts obtained from control, two patients carrying the same α1(I) Gly667Arg substitution, but with different phenotype (severe OI type III and lethal OI type II, respectively), two patients carrying the same Gly862Ser substitution in α1(I) and affected by severe OI type III and in a patient with a Gly862Arg substitution in α1(I) and affected by lethal OI type II. Again a severely compromised cytoskeletal organization is evident only in the cells from the patients with lethal outcome. Asterisks indicate regions of dense fibers in the cytosol of cells from the lethal OI patient carrying Gly667Arg substitution. Counterstaining of the nuclei was performed using DAPI (blue).
Actin filaments and integrin-based focal adhesions (FAs) form an integrated system mediating cell–cell and cell–matrix interaction and influencing signal transduction. Thus, in order to evaluate whether cytoskeletal disorganization is associated with altered integrin-mediated signaling, immunohistochemistry on skin, long-bone and calvarial bone cryosections from ML, MA and WT mice using FA kinase antibody was performed (Fig. 3B).
In calvarial bone, a reduced number of FAs were present in mutant lethal mice with respect to MA and WT animals, whereas no difference was detected between mutant surviving mice and WT littermates (Fig. 3B, 1). Throughout the cortical bone, a high number of small FAs were instead observed in long-bone osteoblasts from lethal mice (Fig. 3B, 2), whereas in MA and WT animals, FAs were mainly detectable on the endosteal surface. Interestingly, MA samples also showed a peculiar distribution of FAs, which often was concentrated in large spots (Fig. 3B, 2). In skin sections, the mutant lethal samples revealed a reduced number of FAs compared with MA and WT, and no difference was detectable between these last two groups (Fig. 3B, 3).
Cell proliferation is reduced in Brtl+/− lethal mice
Cytoskeleton disorganization is known to potentially affect cell proliferation. Thus, we isolated calvarial murine osteoblasts from WT, ML and MA mice to evaluate their proliferation. Osteoblasts from ML mice showed a proliferation impairment when compared with MA and WT littermates that reached significance at 5 days after plating (P < 0.05) (Fig. 4A).
Figure 4.

Osteoblast proliferation and expression analysis of TGF-β and its effector SMAD2/3 in bone from MA, ML and WT mice. (A) Proliferation in calvarial osteoblasts from ML mice is significantly impaired at d5 from plating (*P < 0.05). Each point was measured in triplicate, and each assay was repeated in three independent experiments. The mean values ± SD are reported. (B) Representative western blots using TGF-β antibody (left panel), SMAD2/3 antibody (center panel) and p-SMAD2/3 antibody (right panel). The densitometric analyses obtained by multiple replicative experiments are reported below western blotting images. TGF-β is up-regulated in ML mice whereas p-SMAD2/3 is up-regulated in MA animals. Histograms visualize normalized mean relative-integrated density ± SD values. *, ¥ and § symbols indicate the significance of expression changes occurring between WT and ML, WT and MA, and ML and MA mice, respectively. In SMAD2/3 histogram, dark gray bars represent SMAD2 and light gray bars represent SMAD3; in p-SMAD2/3 histogram, dark gray bars represent p-SMAD2 and light gray bars represent p-SMAD3.
TGF-β pathway is altered in Brtl+/− lethal mice
Alterations in the cytoskeleton may also compromise the activation of signal transduction pathways necessary to guarantee a proper cellular response to extracellular signals (21). One of the main growth factors involved in bone development is TGF-β. Following binding to its receptor, TGF-β activates a signal transduction pathway mediated by SMAD2/3 phosphorylation and nuclear translocation. Using specific antibodies, the expression levels of TGF-β, SMAD2/3 and activated p-SMAD2/3 were evaluated on calvarial bone lysates from ML, MA and WT mice by western blotting. A significant increase in TGF-β expression was detected in ML mice associated with a normal level of target phosphorylated SMAD2/3. Conversely, MA mice had a significant increase in p-SMAD2/3 signal, although increased expression of TGF-β compared with WT animals was not detected (Fig. 4B).
Expression of the early osteoblast transcription factor Runx2 is decreased in Brtl+/− mice
Based on the TGF-β expression pattern described earlier, and as this factor is a key regulator of osteoblast differentiation, the expression of early (Runx2 and Osterix) and late (Bsp) osteoblast markers was evaluated by qPCR on RNA extracted from calvarial bone of ML, MA and WT mice. Only Runx2 was significantly reduced in both ML and MA Brtl+/− mice compared with WT littermates (ML: 0.46 ± 0.2, MA: 0.55 ± 0.13; WT: 0.90 ± 0.14; for ML, MA versus WT, P < 0.05, Supplementary Material, Fig. S1). Col1a1 was not investigated because we already demonstrated no difference of its expression in a previous study (13).
Long-bone fractures are present and collagen content is decreased in mutant lethal mice
Staining with alizarin red and alcian blue was performed to evaluate skeletal configuration. A severely deformed rib cage with multiple rib fractures was evident in Brtl+/− mutants compared with WT. The rib fracture rate did not significantly differ between ML and MA animals (ML: 8.4 ± 1.1; MA: 7.6 ± 2.3), but both were higher in mutants as compared with WT samples, in which no fractures were present. Fractures were also detected in the ischium of ML (2.4 ± 0.6) and MA (2.6 ± 0.6) mice without significant differences between the two phenotypes, but again absent in WT littermate. Interestingly, fractures of the humerus were present only in ML mice (0.6 ± 0.54), suggesting that ML bones are more fragile (Fig. 5A). Picro-sirius red staining clearly showed a decrease in collagen in ML mice with respect to MA and WT animals in cortical and trabecular long bone (Fig. 5B). Picro-sirius red staining was also performed on lung sections mainly highlighting poorly inflated alveoli in ML mice (Supplementary Material, Fig. S2).
Figure 5.
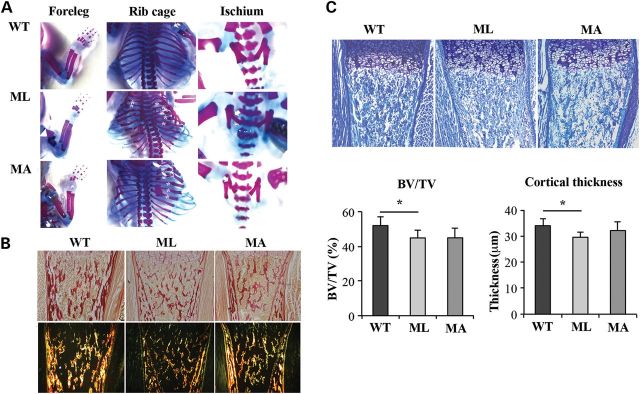
Comparison of skeletal features among ML, MA and WT mice. (A) Skeletal phenotype of Brtl+/− ML, MA and WT mice. Skeletal staining confirmed in mutant mice the characteristic severe OI phenotype including rib fractures (asterisk), flared ribcage and ischium fractures (arrow). Only in ML mice were long-bone fractures present (arrow head). For the skeletal staining, alizarin red, specific for bone, and alcian blue, specific for cartilage, were used. (B) Picro-sirius red staining of type I collagen in tibia sections from ML, MA and WT mice. ML samples revealed reduced collagen content in sections analyzed both under bright field (top panels) and under polarized light (bottom panels) (magnification 10×). (C) Histomorphometric analysis was performed on toluidine blue stained sections of the proximal tibia epiphysis of ML, MA and WT mice. BV/TV and cortical thickness were significantly reduced in ML mice compared with WT (BV/TV, P < 0.05; cortical thickness, P < 0.001) (magnification 10×).
Trabecular bone volume and cortical thickness are reduced in mutant lethal mice
Histomorphometric analysis performed on toluidine-stained sections of femoral distal metaphyses from WT, ML and MA littermates allowed the evaluation of Bone Volume/Total Volume (BV/TV) and of cortical thickness. ML bones showed a significantly reduced BV/TV with respect to WT samples (ML: 44.914 ± 4.621%; WT: 52.040 ± 4.997%, P < 0.05). Similarly, cortical thickness was smaller in ML with respect to WT femurs (ML: 29.536 ± 2.072 µm; WT: 33.989 ± 2.650 µm, P < 0.001). No significant difference was detected for these two parameters between ML and MA (BV/TV, MA: 44.973 ± 5.401%, ML versus MA, P = 0.986; cortical thickness MA: 32.118 ± 3.288 µm, ML versus MA, P = 0.145), or MA and WT (BV/TV: MA versus WT, P = 0.06; cortical thickness MA versus WT, P = 0.462) (Fig. 5C).
NanoCT reveals less bone in mutant lethal mice
NanoCT analysis was performed on femurs of 1-day-old WT, ML and MA animals to better investigate their morphometric properties. Central diaphyseal BMD, tissue mineral density (TMD), bone volume fraction (BV/TV) and total area encompassed by the cross section were evaluated. BV/TV was significantly reduced in ML mutants when compared with WT (ML: 0.271 ± 0.008; WT: 0.315 ± 0.029, P < 0.05), but no difference was found between ML and MA or MA and WT mice. No difference in BMD, TMD and total area was detected among the three different groups of mice (Fig. 6). ML bones were shorter when measuring mineral-to-mineral surfaces between the proximal and distal secondary ossification centers compared with WT (ML: 2.013 ± 0.339 mm; WT: 2.433 ± 0.094 mm, P < 0.05). In contrast, no difference was detected between ML and MA or between MA and WT. Measurement of mineral-to-mineral length of alizarin red-stained femurs on independent samples confirmed the nanoCT data on length. Also humerus length, calculated by measuring the alizarin stained bone, was significantly shorter in ML than WT (ML: 2.67 ± 0.35 mm; WT: 3.06 ± 0.16 mm, P < 0.05).
Figure 6.

NanoCT was used to assess mineralized bone parameters in 1-day-old mouse femurs. (A) Lethal Brtl+/− mice showed a reduction in mineralized length between proximal and distal secondary ossification centers, and a reduced bone volume fraction of the trabeculated structure (BV/TV). TMD remained unchanged, and no significant differences in total cross sectional area and BMD were observed between genotypes. (E) Representative nanoCT images of femoral cross sections show a highly trabecular architecture.
Cytoskeletal analysis in fibroblast cell lines from OI patients
In order to extend our animal data to human samples, we investigated primary fibroblasts from two OI patients with the same α1(I)Gly667 to Arginine substitution (p.Gly845Arg) and, respectively, lethal (OI type II) or severe (OI type III) phenotype, as well as three OI patients heterozygous for substitutions to Serine (p.Gly1040Ser) or Arginine (p.Gly1040Arg) at α1(I) Gly862, with severe (OI type III) or lethal (OI type II) phenotype, respectively. Immunohistochemistry using labeled phalloidin revealed that cells from the lethal patients had a compromised cytoskeletal organization. Cells from the lethal proband carrying Gly667Arg revealed abnormal condensation of cytoskeletal fibers with irregular distribution in the cytoplasm, whereas Gly862Arg cells showed a completely altered cytoskeletal organization resembling the ML mice. In contrast, no difference from normal control was evident in fibroblasts with Gly667Arg and Gly862Ser substitutions and severe OI phenotype (Fig. 3C).
Discussion
In this study, we identified distinctive cytoskeletal alterations that distinguish different OI phenotypic outcomes. These changes affected cell proliferation and cellular signaling and compromised bone properties.
The molecular basis of lethal versus moderately severe outcomes in Brtl+/− mice has puzzled us since the generation of this murine model for classical OI. Phenotypic variability in the presence of an identical mutation is described in human OI, and the elucidation of its cause is particularly relevant not only to better understand the disease pathophysiology but also to discover potential new targets for treatment. In the present study, we took advantage of powerful in silico tools for functional analysis, combining proteomic and transcriptomic data that were obtained from previous investigations on bone and skin samples from WT and Brtl+/− newborn mice with differing phenotypic outcomes (13,14). Pathway analysis integrates expression differences with known proteins acting in characterized cellular pathways, as properly supported by the scientific literature. The resulting functional analysis suggests the biochemical context in which the proteins of interest act and how their aberrant expression may alter cellular and/or tissue biology in physiological and pathological conditions. To our knowledge, this is the first time that hybrid networks have been generated in OI.
Hybrid networks point to different cytoskeletal organization in Brtl+/− mice with different outcomes
Hybrid networks, generated by MetaCore software, identified two cytoskeletal proteins as consistent central hubs: vimentin and stathmin. Vimentin, as a member of the family of intermediate filaments, is involved in maintaining cell integrity and in supporting and anchoring organelles in the cytosol (Fig. 7A). It has been reported to be involved in cell polarity, motility and signaling (22) and in wound healing (23). Stathmin, an ubiquitous small cytosolic phosphoprotein with cell proliferation regulating activity, is known to affect microtubule dynamics sequestering αβ-tubulin dimers and promoting microtubule disassembly by acting directly on microtubule ends (24,25). Although its exact role is not yet clear, stathmin plays a fundamental function in bone homeostasis by controlling the coupling of osteoblastic and osteoclastic activities. In fact, it promotes osteoblast differentiation and inhibits osteoclast formation (26).
Figure 7.
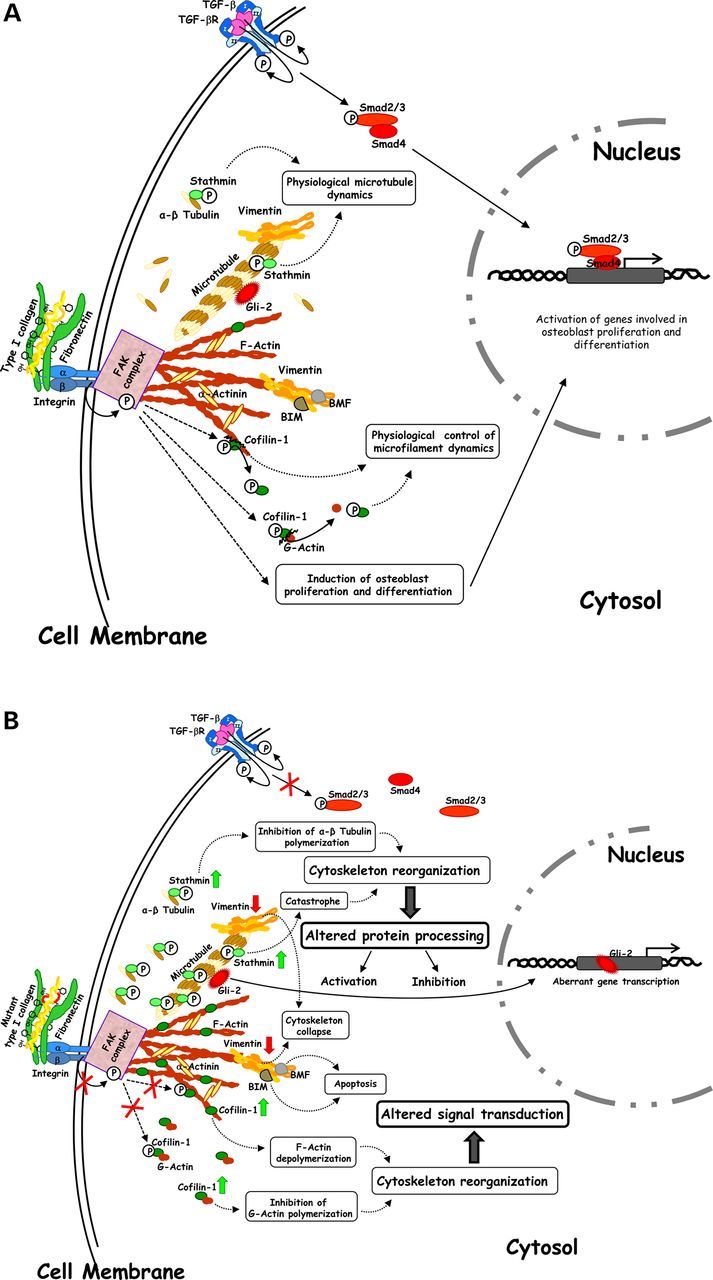
Schematic view of the physiological (A) and aberrant (B) cellular molecular dynamics regulated by cytoskeleton, which is functional in WT and MA Brtl+/− mice (A), and altered in ML Brtl+/− mice (B). (A) Stathmin, vimentin and cofilin-1 and focal adhesion kinase (FAK) complex contribute to control cytoskeleton structural plasticity thus influencing protein processing, signal transduction and gene expression. (B) Increase in the expression of stathmin and cofilin-1, decrease of vimentin and abnormal FAK complex contribute to abnormal cytoskeleton structural plasticity thus influencing protein processing, signal transduction and gene expression.
Vimentin and stathmin are indirectly correlated as microtubule depolymerization leads to the collapse of vimentin and to a disordered distribution of its resulting short agglomerations (16) (Fig. 7A). The down-regulation of vimentin (14) and the up-regulation of stathmin in both skin and bone samples of lethal mice suggested compromised formation and organization of microtubule and intermediate filaments in these mutants Fig. 7B.
Cofilin-1 is a protein involved in cytoskeletal assembly that we previously described as deregulated in skin specimens from mutants. Here, cofilin-1 also proved dysregulated in Brtl+/− bone and lung. This factor modulates actin filaments by promoting F-actin depolymerization and by inhibiting G-actin polymerization (19) Fig. 7A. Cofilin-1 results well integrated into the two OI hybrid nets thus suggesting its active involvement in OI-affected pathways and perhaps in the pathophysiology of the disorder. In this work, we demonstrated its overexpression in skin, bone and lung in ML mice when compared with MA and WT animals (Fig. 7B). Interestingly, microfilaments have been suggested to directly interact with vimentin filaments (27). Thus, as suggested for stathmin, a concomitantly altered presence of cofilin-1 and vimentin in ML mice may have combined detrimental effects on cytoskeleton properties (Fig. 7 B) (28).
According to our analysis, all three main components of the cytoskeleton, microtubules, microfilaments and intermediate filaments, are likely to be altered in mutants thus suggesting the occurrence of an aberrant cytoskeletal organization in lethal OI.
Cytoskeleton disorganization in Brtl+/− lethal mice affects cell proliferation, differentiation and bone properties
Immunohistochemistry data highlighted the occurrence of a systemic abnormal cytoskeletal network, characterized by punctuated actin fibrils. This was apparent in various Brtl+/− ML tissues expressing high levels of mutant type I collagen, such as skin, calvarial and long bone, and also lung. This latter finding is of particular interest because Brtl+/− lethal mice die a few hours after birth from respiratory failure and poorly inflated lungs, as showed by picro-sirius red staining (Supplementary Material, Fig. S2). However, we did not find any difference in rib cage structure or number of rib fractures between ML and MA mice. Thus, our observation supports a direct effect of the aberrant cytoskeleton on lung tissue, independent from skeletal abnormalities, as recently reported both in human patients and in a different murine model for OI (2).
The cytoskeleton is a dynamic protein network whose structure and reciprocal interactions are necessary for maintenance of cell mechanical properties, cell shape, cell proliferation and differentiation. The cytoskeleton regulates signaling pathways. Intracellular and extracellular stimuli utilize this network to maintain cell homeostasis (29,30). A distinct protein composition of the cytoskeleton has been reported for different cell types depending on specific cellular functions. In particular, the terminal differentiation of osteoblasts to osteocytes is characterized by a change in the distribution of actin binding proteins that are required to ensure the proper shape and mechanosensory functions of these cells (31,32). Perturbation of actin organization has been demonstrated to compromise osteoblast differentiation in C3H10T1/2 murine mesenchymal stem cells and to modulate the ability of murine osteoblastic cells to respond to fluid flow-induced signaling, which mimics mechanical signals in vitro (33,34).
Recently, mutations in plastin 3 (PLS3), a member of the plastin family that regulates the formation of F-actin bundles, have been reported as responsible for X-linked osteoporosis, hence underlining the relevance of cytoskeletal components in bone development (35).
In Brtl+/− lethal mice, the abnormal cytoskeleton affects osteoblast proliferation and is associated with compromised differentiation. qPCR data demonstrated decreased expression of Runx2, an early osteogenic marker, both in ML and MA mice compared with WT. We recently demonstrated a compromised ability to differentiate toward the osteoblast lineage in mesenchymal stem cells obtained from adult Brtl+/− mice (36). The altered cytoskeletal organization may exacerbate this effect by activating cellular apoptosis. As a consequence, the increase of pro-apoptotic factors was actually associated with the lethal outcome in our previous proteomic investigations on bone and skin tissues from Brtl+/− mice (13,14). A disorganized cytoskeleton can initiate or support cell death in various ways. For example, it may favor the release of the pro-apoptotic proteins BIM and BMF, which under normal conditions bind to cytoskeletal components, such as vimentin (37), and are inactivated by these interactions (Fig. 7A and B). Similarly, the detachment of the FAS death receptor from ezrin-mediated actin interaction can cause apoptosis (38).
Similarly to actin, the microtubule cytoskeleton is also known to control functional dynamics of various protein factors involved in gene transcription and cellular differentiation. Among these factors, microtubules regulate proteolytic degradation and processing of the hedgehog signaling mediators GLI2 and GLI3 (36), whose altered activity results in severe impairment of the skeleton (Fig. 7A and B) (39). While a physiological release of GLI2 by microtubule depolymerization results in the expression of genes involved in osteoblast differentiation and bone formation (Fig. 7A) (36), the occurrence of a constitutively activated expression of GLI2 suppresses bone formation in postnatal mice (40). This may be due to the GLI2-induced overexpression of BMP2 (41) (Fig. 7B), which we previously described in ML animals (13).
Reduced osteoblast proliferation and differentiation in Brtl+/− mice is associated with compromised bone properties: BV/TV and cortical thickness were reduced in developing femurs from lethal mice, and the length of mineralized femurs was shorter in ML compared with WT. Consequentially, long-bone fracture rate was increased in ML mice. Although no significant reduction was previously detected at the transcript level (13), less type I collagen was found to be deposited in the extracellular matrix of long bones from ML mice by picro-sirius red staining.
TGF-β and integrin-mediated signaling are altered in Brtl+/− lethal mice
In bone from Brtl+/− lethal mice, we found an increased presence of TGF-β that did not associate with an up-regulation, by phosphorylation, of the signal transduction pathway effector SMAD2/3. As the cytoskeleton is disorganized in ML mice, the TGF-β signaling might be disturbed. This may cause stimulation of TGF-β synthesis and the accumulation of its latent form in the extracellular matrix. In contrast, we found an up-regulation of phosphorylated SMAD2/3 in Brtl+/− alive mice. Interestingly, similar findings have been recently demonstrated for another dominant and a recessive murine model of OI, making the TGF-β pathway a broad target for development of novel OI therapy (42).
In addition, TGF-β signaling, as well as signal transduction by bone morphogenetic proteins (BMPs), is transduced in target cells only when the ‘integrin system’ is functionally active (43). The correct interaction between integrins and matrix proteins as well as integrins and cytoskeleton components has been widely demonstrated as a prerequisite for ‘integrin system’ activity (44–46). Integrins have also been implicated in control of TGF-β activation (47) according to specific functional interactions between integrins and the ECM and/or the cytoskeleton. Purified integrins are not able to activate the latent form of TGF-β (44,45). Therefore, it seems reasonable to assume that TGF-β signaling is negatively affected in mutant animals also by altered integrin activity, which in turn is caused by ECM and cytoskeleton structural and functional aberrances. Indeed, by immunohistochemistry, cells from mutant lethal mice revealed abnormal focal adhesion kinase distribution. Interestingly, fibronectin/integrin and type I collagen/integrin interactions are fundamental during early stages of osteoblast differentiation (48,49). In addition, the association between the cytoskeletal network and integrins may well serve as a reservoir for molecules involved in signal transduction (43).
Identical glycine substitutions altered fibroblast cytoskeleton only in lethal but not in severe OI patients
To investigate phenotypic variability in human OI is quite complicated owing to the difficulty in obtaining samples from families and patients with an identical mutation, but different outcomes. Based on the last review published by the Consortium for Osteogenesis Imperfecta mutations, 11 Gly substitutions in α1(I) and 2 Gly substitutions in α2(I) are responsible for lethal or non-lethal outcomes when substituted by the same amino acid, and 32 Gly substitutions in α1(I) and 7 Gly substitutions in α2(I) cause lethal or non-lethal outcomes when substituted by a different amino acid (6).
We evaluated the cytoskeletal organization in primary fibroblast cell lines from patients carrying a substitution of the same glycine with either an identical (Gly667Arg) or a different (Gly862Ser and Gly862Arg) substituting amino acid with lethal or severe outcomes. Immunohistochemistry using phalloidin revealed a completely disorganized cytoskeleton in lethal cell lines that resembling the disorganized cytoskeleton identified in lethal Brtl+/− mice.
It is difficult to determine whether cytoskeletal organization is a primary or secondary modulator in OI. The presence of induced ER stress was reported to be responsible for cytoskeletal disorganization and apoptosis activation in vitro (50). A compromised ability to cope with mutant intracellular collagen retention causing intracellular stress was demonstrated in ML mice compared with MA (13,14). Thus, we can speculate that in cells producing higher amounts of collagen type I, the failure of chaperones to solve the cellular stress caused by mutant collagen retention may compromise cytoskeleton assembly, resulting in reduced differentiation and proliferation and finally causing increased apoptosis. On the contrary, the ability of the chaperones to properly address intracellular stress might allow cell survival by preserving normal cellular functions. Conversely, a predisposition to abnormal cytoskeletal organization in active secretory cells producing mutant collagen may result in intracellular stress causing altered cellular function.
Here, we demonstrated that a compromised cytoskeletal assembly impaired both cell signaling and cellular trafficking in mutant lethal mice. This results in altered bone properties thus pointing to the cytoskeleton as a novel target to develop new treatments for OI.
Materials and Methods
Animals
Brtl+/− heterozygous mice and WT littermates were maintained under standard experimental animal care protocol following the Italian Laws in the animal facility of the Department of Molecular Medicine of the University of Pavia (Italy). One-day-old mice were used for the present study. Genomic DNA was extracted from tail clip or skin pieces, and genotyping to distinguish heterozygous mutant animals from WT littermates was performed by PCR (51). Mutant mice were considered lethal when death occurred within 6 h from birth.
Protein network analyses
OI hybrid networks were generated using the MetaCore network building tool (Thomson Reuters, St. Joseph, MI, USA) and applying the shortest path algorithm. MetaCore includes a manually annotated and regularly updated database of protein interactions and metabolic reactions obtained by scientific literature. Gene names of differentially expressed proteins were imported into MetaCore and processed. Hypothetical networks were built combining experimental proteins/genes and additional not experimental proteins, included into the MetaCore protein database, which were judged by the program as essential to crosslink experimental data. According to the shortest path algorithm, only one additional element may enter into the net between two experimental proteins/genes. Hybrid nets thus include only experimental factors that are known to be tightly correlated. The relevant pathway maps were then prioritized according to their statistical significance (P < 0.001), and networks were graphically visualized as nodes (proteins/genes) and edges (the relationship between proteins).
Western blot
Skin, calvarial bone and lung protein extracts from 1-day-old ML and MA Brtl+/− mice, and WT littermates (at least n = 3 per group) were obtained by pestle-pounding minced specimens in the presence of the Laemmli buffer: 100 mm Tris–HCl pH 6.8, 2% (w/v) SDS, 20% (v/v) glycerol, 4% (v/v) β-mercaptoethanol. The sample solutions were then heated at 95°C for 5 min (52). For each sample, proteins (25 µg) were separated on 10% polyacrylamide gel and then transferred onto nitrocellulose (Hybond ECL, GE Healthcare) according to Towbin (53). Before immunodetection, gel sample loading was preliminary proved to be equivalent by Ponceau Red staining (0.2% w/v Ponceau S in 3% w/v trichloroacetic acid) of nitrocellulose. Immunodetection was achieved using the following antibodies: anti-β-Tubulin and anti-Stathmin antibodies from Santa Cruz Biotechnology (San Jose, CA, USA), both 1:1000 diluted; anti-Cofilin-1 from Abcam (Cambridge, UK), 1:1000 diluted; anti-TGF-β pan-specific antibody from R&D Systems (Minneapolis, MN, USA), 1:500 diluted; anti-SMAD2/3 and anti-phospho-SMAD2/3 both from Cell Signaling Technology (Danvers, MA, USA), respectively, 1:1000 and 1:500 diluted; and anti-Rabbit secondary antibody from Sigma–Aldrich (St. Louis, MO, USA), 1:7000 diluted. Hybridization with primary antibodies was performed overnight at room temperature. Incubation with specific HRP-conjugated secondary antibodies was then performed for 2 h at room temperature, and immunostained bands were visualized by chemiluminescence using ECL detection reagents (GE Healthcare, Uppsala, Sweden). Chemiluminescent signals were captured by ImageQuant LAS 4000 (GE Healthcare) digital imaging system. The obtained 1-D western blot images were then analyzed with the ImageQuant 3.0 software (Molecular Dynamics World Headquarters, Sunnyvale, CA, USA). β-tubulin immunoblotting ensured equal loading of samples.
Histology and immunohistochemistry
For histological studies, femurs from WT, ML and MA newborn mice (n = 4 per group) were dissected and fixed in 4% PFA in phosphate-buffered saline (PBS), decalcified in 14% EDTA pH 7.1 for 7 days and processed for light microscopy, according to standard procedures. Longitudinal femur sections of 7 µm were cut using an RM2265 microtome (Leica Microsystems Srl, Milan, Italy), mounted on Superfrost Plus slides (Menzel-Glaser, VWR, Milan, Italy) and stained with toluidine blue or picro-sirius red. Following toluidine blue staining, images were acquired using a DFC480 digital camera (Leica Microsystems Srl) connected to a light microscope (Dialux 20, Leica Microsystems Srl). The trabecular bone was evaluated on an area of 0.07 mm2 manually delineated below the growth plate. The cortical thickness was measured five times for each section on a region of 200 µm, which was manually delineated from the end of the growth plate. All of the analyses were performed with the Leica Application Suite v 3.0 image analysis software (Leica Microsystems Srl) on three sections for each mouse.
For picro-sirius red staining, a 0.1% w/v solution of sirius red (Direct Red 80, Sigma–Aldrich, Milan, Italy) in saturated aqueous solution of picric acid was used. After staining, sections were rinsed in acidified water (87.5 mm acetic acid) and dehydrated in absolute ethanol, cleared and mounted in synthetic resin (DPX Mountant for histology, Sigma–Aldrich). Sirius red-stained sections were analyzed under polarized light, and images were acquired by Leica DM2500 equipped with L ICT/P polarizer and a digital color camera LEICA DFC295 (Leica).
For immunohistochemistry, skin, lung, limb and calvarial bones from ML, MA mutant and WT 1-day-old pups (n = 3) were dissected and, when necessary, cleaned from adherent connective tissue after sacrifice. Calvarial bones were cut into four pieces along the sutures, and parietal bones were used for the assay. Skin and bones were washed in PBS, fixed in 4% PFA for 24 h at 4°C and embedded in OCT. Following dissection and fixation in 4% PFA, lungs were successively incubated in 15 and 30% w/v sucrose in PBS o/n at 4°C, degassed under vacuum and embedded in OCT. Cryosections of 10 µm thickness were obtained using CM1850 UV cryostat (Leica) and mounted on Superfrost Plus slides (Menzel-Glaser, VWR). Sections were permeabilized in 0.2% v/v Triton X-100 in PBS for 90 min at RT, and blocking was performed by incubating the sections in 1% w/v BSA 0.01% v/v Triton X-100 in PBS for other 90 min at RT. Samples were then incubated overnight at 4°C in 0.01% Triton X-100-containing primary antibody Alexa 488-Phalloidin (Molecular Probes, Invitrogen) diluted 1:40 or in PBS, 1% BSA containing primary antibody FAKpY397 (Invitrogen) diluted 1:500. AlexaFluor 633 goat anti-rabbit IgG (Thermo Scientific) was used as secondary antibody diluted 1:500 in PBS, 1% BSA.
For human fibroblasts, 5 × 104 cells were plated on coverslip in 24-well plate in Dulbecco Modified Eagle's Medium (DMEM, Lonza, Milan, Italy) supplemented with 10% heat-inactivated Fetal Calf Serum (hiFCS, Euroclone, Pero, MI, Italy) and antibiotics. The day after the medium was removed, cells were washed in PBS and fixed in 3.7% PFA for 10 min at room temperature. Blocking was done by 30-min incubation in 1% BSA, 0.05% Triton X-100 in PBS. Alexa 488-Phalloidin staining was performed by 1-h incubation at RT in 1% BSA in PBS. The nuclei were counterstained by 4′,6-diamidino-2-phenylindole (DAPI; Sigma–Aldrich). Samples were examined by TCS SP2-Leica confocal microscope (Leica).
Expression analysis
Total RNA was extracted using TriReagent (Sigma–Aldrich) according to the manufacturer's protocol from calvarial bones of WT, ML and MA newborn mice (n = 3). DNase digestion was performed using the Turbo DNA Free Kit (Ambion, Applied Biosystems, Austin, TX, USA), and RNA integrity was verified using the Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). qPCR was performed on the Mx3000P Stratagene thermocycler using commercially available TaqMan primers and probes (Applied Biosystems) and TaqMan Universal PCR Master Mix (Applied Biosystems). All reactions were performed in triplicate. Expression levels for Runx2 (Mm01269515_mH), Osterix (Mm00504574_m1) and Bsp (Mm00492555_m1) were evaluated. Gapdh (Mm99999915_g1) was used as normalizer. Relative expression levels were calculated using the ΔΔCt method.
Calvarial osteoblast culture
Murine osteoblasts were isolated from 1-day-old WT, ML and MA pups. At least five to six animals with the same genotype were pooled together to obtain a sufficient number of cells. After sacrifice, calvariae were removed, cleaned from fibrous tissues and sutures, washed two times with PBS at 37°C for 10 min in a shaker water bath and sequentially digested with 200 U/mL collagenase type II (GIBCO) at 37°C for 15–20 min. Cells obtained from the first two digestions were discharged, whereas cells from digestions 3, 4 and 5 were passed through a 70-μm polypropylene mesh filter and cultured in α-MEM (Lonza) added with 10% hiFCS, 25 μg/ml sodium ascorbate (Fluka) and antibiotics.
Osteoblast proliferation
Proliferation of osteoblasts from WT, ML and MA mice was evaluated at Passage 1 using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA) according to the manufacturer's protocol. Osteoblasts were plated in triplicate in 96-well plates at 2.5 × 103 cells per well in 100 µl of complete α-MEM medium. After 1, 2 and 5 days from plating, 20 μl of MTS solution was added to each well. After 4 h of incubation at 37°C, absorbance at 490 nm was measured using an ELISA plate reader.
Skeletal staining
Skin and internal organs were removed from WT, ML and MA mice (n = 5 for group) within a few hours after birth following sacrifice. The pups were fixed in 95% ethanol for 7 days and then stained with 0.3% w/v Alcian Blue 8GS and 0.1% w/v Alizarin Red S as described in Forlino et al. (1999) (9). Digitalized images were acquired using the digital camera DFC425C (Leica Microsystems Srl) connected to the stereomicroscope M165FC (Leica Microsystems Srl). Long-bone and rib fractures were considered based on the presence of callus to avoid including fractures owing to manual handling of the samples. The length of the mineralized part of the femur and humerus was calculated on the digitized images by measuring the alizarin stained distance between the width midpoint of the segment connecting both epiphyses.
NanoCT
Mouse hind limbs from ML, MA Brtl+/− and WT littermates (n = 5 per group) were scanned using a nano-computed tomography system (nanotom-s, phoenix|x-ray, GE Measurement & Control, Wunstorf, Germany) at 80 kV, 130 μA, 1000 ms integration time, four frame-averaging and a voxel size of 2.5 μm. The total scan time for each sample was 85 min. Images were calibrated to Hounsfield units for densitometry using a hydroxyapatite phantom scan. For all femora, a volume of interest (VOI) was selected by taking 10% of the mineralized length (region defined by onset of mineralization of primary trabeculae, from distal to proximal). The VOI was centered in the mid-diaphyseal region. A region of interest was then selected by using a spline function to manually contour a region encompassing the entire periosteal circumference of the bone. A highly trabecular structure with a thin cortico-trabecular shell was assessed together for total trabecular morphology and densitometry.
Human dermal fibroblasts
For two patients harboring the c.3118G>A mutation in COL1A1 (Gly862Ser; p.Gly1040Ser, severe OI) and one patient carrying the c.3118G>C mutation in COL1A1 (Gly862Arg; p.Gly1040Arg, lethal OI), a fibroblast cell culture was established from a skin biopsy taken from the probands' inner aspect of the upper arm, after informed consent. The fibroblasts from the two patients harboring the c.2533G>A mutation in COL1A1 (Gly667Arg; p.Gly845Arg) associated with severe or lethal OI, respectively, were obtained from Dr Joan C Marini.
Statistics
Statistic differences between groups were evaluated by one-way ANOVA or by two-tailed Student's T-test using Sigma Plot Statistic 11.0 software. A P < 0.05 was considered significant. All data are expressed as mean ± standard deviation.
Supplementary Material
Funding
The work was supported by Fondazione Cariplo (grant No. 2013-0612), Telethon (grant No. GGP13098) and the European Community, FP7, ‘Sybil’ project (grant No. 602300).
Supplementary Material
Acknowledgements
We thank Mr Angelo Gallanti, Department of Molecular Medicine, Biochemistry Unit, University of Pavia, Italy, for support on cell cultures, Dr Patrizia Vaghi, Centro Grandi Strumenti, University of Pavia, Italy, for technical assistance with confocal microscopy and Mrs Aileen M. Barnes, Bone and Extracellular Matrix Branch, NICHD, National Institute of Health, Bethesda, MD, USA, for technical assistance.
Conflict of Interest statement. None declared.
References
- 1.Forlino A., Cabral W.A., Barnes A.M., Marini J.C. (2011) New perspectives on osteogenesis imperfecta. Nat. Rev. Endocrinol., 7, 540–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thiele F., Cohrs C.M., Flor A., Lisse T.S., Przemeck G.K., Horsch M., Schrewe A., Gailus-Durner V., Ivandic B., Katus H.A., et al. (2012) Cardiopulmonary dysfunction in the Osteogenesis imperfecta mouse model Aga2 and human patients are caused by bone-independent mechanisms. Hum. Mol. Genet., 21, 3535–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baldridge D., Lennington J., Weis M., Homan E.P., Jiang M.M., Munivez E., Keene D.R., Hogue W.R., Pyott S., Byers P.H., et al. (2010) Generalized connective tissue disease in Crtap-/- mouse. PLoS One, 5, e10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garbes L., Kim K., Riess A., Hoyer-Kuhn H., Beleggia F., Bevot A., Kim M.J., Huh Y.H., Kweon H.S., Savarirayan R., et al. (2015) Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am J Hum Genet, 96, 432–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Dijk F.S., Sillence D.O. (2014) Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am. J. Med. Genet. A, 164A, 1470–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marini J.C., Forlino A., Cabral W.A., Barnes A.M., San Antonio J.D., Milgrom S., Hyland J.C., Korkko J., Prockop D.J., De Paepe A., et al. (2007) Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum. Mutat., 28, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rauch F., Moffatt P., Cheung M., Roughley P., Lalic L., Lund A.M., Ramirez N., Fahiminiya S., Majewski J., Glorieux F.H. (2013) Osteogenesis imperfecta type V: marked phenotypic variability despite the presence of the IFITM5 c.-14C>T mutation in all patients. J Med. Genet., 50, 21–24. [DOI] [PubMed] [Google Scholar]
- 8.Valadares E.R., Carneiro T.B., Santos P.M., Oliveira A.C., Zabel B. (2014) What is new in genetics and osteogenesis imperfecta classification? J. Pediatr. (Rio J), 90, 536–541. [DOI] [PubMed] [Google Scholar]
- 9.Forlino A., Porter F.D., Lee E.J., Westphal H., Marini J.C. (1999) Use of the Cre/lox recombination system to develop a non-lethal knock-in murine model for osteogenesis imperfecta with an alpha1(I) G349C substitution. Variability in phenotype in BrtlIV mice. J. Biol. Chem., 274, 37923–37931. [DOI] [PubMed] [Google Scholar]
- 10.Sarafova A.P., Choi H., Forlino A., Gajko A., Cabral W.A., Tosi L., Reing C.M., Marini J.C. (1998) Three novel type I collagen mutations in osteogenesis imperfecta type IV probands are associated with discrepancies between electrophoretic migration of osteoblast and fibroblast collagen. Hum. Mutat., 11, 395–403. [DOI] [PubMed] [Google Scholar]
- 11.Kuznetsova N.V., Forlino A., Cabral W.A., Marini J.C., Leikin S. (2004) Structure, stability and interactions of type I collagen with GLY349-CYS substitution in alpha 1(I) chain in a murine osteogenesis imperfecta model. Matrix Biol., 23, 101–112. [DOI] [PubMed] [Google Scholar]
- 12.Forlino A., Kuznetsova N.V., Marini J.C., Leikin S. (2007) Selective retention and degradation of molecules with a single mutant alpha1(I) chain in the Brtl IV mouse model of OI. Matrix Biol., 26, 604–614. [DOI] [PubMed] [Google Scholar]
- 13.Forlino A., Tani C., Rossi A., Lupi A., Campari E., Gualeni B., Bianchi L., Armini A., Cetta G., Bini L., et al. (2007) Differential expression of both extracellular and intracellular proteins is involved in the lethal or nonlethal phenotypic variation of BrtlIV, a murine model for osteogenesis imperfecta. Proteomics, 7, 1877–1891. [DOI] [PubMed] [Google Scholar]
- 14.Bianchi L., Gagliardi A., Gioia R., Besio R., Tani C., Landi C., Cipriano M., Gimigliano A., Rossi A., Marini J.C., et al. (2012) Differential response to intracellular stress in the skin from osteogenesis imperfecta Brtl mice with lethal and non lethal phenotype: a proteomic approach. J. Proteomics, 75, 4717–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao F.Q. (2013) Octamer-binding transcription factors: genomics and functions. Front. Biosci. (Landmark Ed), 18, 1051–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldman R.D., Khuon S., Chou Y.H., Opal P., Steinert P.M. (1996) The function of intermediate filaments in cell shape and cytoskeletal integrity. J. Cell. Biol., 134, 971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Curmi P.A., Gavet O., Charbaut E., Ozon S., Lachkar-Colmerauer S., Manceau V., Siavoshian S., Maucuer A., Sobel A. (1999) Stathmin and its phosphoprotein family: general properties, biochemical and functional interaction with tubulin. Cell. Struct. Funct., 24, 345–357. [DOI] [PubMed] [Google Scholar]
- 18.Cassimeris L. (2002) The oncoprotein 18/stathmin family of microtubule destabilizers. Curr. Opin. Cell. Biol., 14, 18–24. [DOI] [PubMed] [Google Scholar]
- 19.Bamburg J.R. (1999) Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu. Rev. Cell. Dev. Biol., 15, 185–230. [DOI] [PubMed] [Google Scholar]
- 20.Bamburg J.R., McGough A., Ono S. (1999) Putting a new twist on actin: ADF/cofilins modulate actin dynamics. Trends Cell. Biol., 9, 364–370. [DOI] [PubMed] [Google Scholar]
- 21.Pessoa-Pureur R., Heimfarth L., Rocha J.B. (2014) Signaling mechanisms and disrupted cytoskeleton in the diphenyl ditelluride neurotoxicity. Oxid. Med. Cell Longev., 2014, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivaska J., Pallari H.M., Nevo J., Eriksson J.E. (2007) Novel functions of vimentin in cell adhesion, migration, and signaling. Exp. Cell. Res., 313, 2050–2062. [DOI] [PubMed] [Google Scholar]
- 23.Eckes B., Colucci-Guyon E., Smola H., Nodder S., Babinet C., Krieg T., Martin P. (2000) Impaired wound healing in embryonic and adult mice lacking vimentin. J. Cell. Sci., 113 (Pt 13), 2455–2462. [DOI] [PubMed] [Google Scholar]
- 24.Sobel A., Boutterin M.C., Beretta L., Chneiweiss H., Doye V., Peyro-Saint-Paul H. (1989) Intracellular substrates for extracellular signaling. Characterization of a ubiquitous, neuron-enriched phosphoprotein (stathmin). J. Biol. Chem., 264, 3765–3772. [PubMed] [Google Scholar]
- 25.Charbaut E., Curmi P.A., Ozon S., Lachkar S., Redeker V., Sobel A. (2001) Stathmin family proteins display specific molecular and tubulin binding properties. J. Biol. Chem., 276, 16146–16154. [DOI] [PubMed] [Google Scholar]
- 26.Liu H., Zhang R., Ko S.Y., Oyajobi B.O., Papasian C.J., Deng H.W., Zhang S., Zhao M. (2011) Microtubule assembly affects bone mass by regulating both osteoblast and osteoclast functions: stathmin deficiency produces an osteopenic phenotype in mice. J. Bone Miner. Res., 26, 2052–2067. [DOI] [PubMed] [Google Scholar]
- 27.Esue O., Carson A.A., Tseng Y., Wirtz D. (2006) A direct interaction between actin and vimentin filaments mediated by the tail domain of vimentin. J. Biol. Chem., 281, 30393–30399. [DOI] [PubMed] [Google Scholar]
- 28.Chang L., Goldman R.D. (2004) Intermediate filaments mediate cytoskeletal crosstalk. Nat. Rev. Mol. Cell. Biol., 5, 601–613. [DOI] [PubMed] [Google Scholar]
- 29.Pollard T.D., Cooper J.A. (2009) Actin, a central player in cell shape and movement. Science, 326, 1208–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klymkowsky M.W. (1999) Weaving a tangled web: the interconnected cytoskeleton. Nat. Cell. Biol., 1, E121–E123. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka-Kamioka K., Kamioka H., Ris H., Lim S.S. (1998) Osteocyte shape is dependent on actin filaments and osteocyte processes are unique actin-rich projections. J Bone Miner. Res., 13, 1555–1568. [DOI] [PubMed] [Google Scholar]
- 32.Kamioka H., Sugawara Y., Honjo T., Yamashiro T., Takano-Yamamoto T. (2004) Terminal differentiation of osteoblasts to osteocytes is accompanied by dramatic changes in the distribution of actin-binding proteins. J. Bone Miner. Res., 19, 471–478. [DOI] [PubMed] [Google Scholar]
- 33.Arnsdorf E.J., Tummala P., Kwon R.Y., Jacobs C.R. (2009) Mechanically induced osteogenic differentiation—the role of RhoA, ROCKII and cytoskeletal dynamics. J. Cell. Sci., 122, 546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malone A.M., Batra N.N., Shivaram G., Kwon R.Y., You L., Kim C.H., Rodriguez J., Jair K., Jacobs C.R. (2007) The role of actin cytoskeleton in oscillatory fluid flow-induced signaling in MC3T3-E1 osteoblasts. Am. J. Physiol. Cell. Physiol., 292, C1830–C1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Dijk F.S., Zillikens M.C., Micha D., Riessland M., Marcelis C.L., de Die-Smulders C.E., Milbradt J., Franken A.A., Harsevoort A.J., Lichtenbelt K.D., et al. (2013) PLS3 mutations in X-linked osteoporosis with fractures. N. Engl. J. Med., 369, 1529–1536. [DOI] [PubMed] [Google Scholar]
- 36.Gioia R., Panaroni C., Besio R., Palladini G., Merlini G., Giansanti V., Scovassi I.A., Villani S., Villa I., Villa A., et al. (2012) Impaired osteoblastogenesis in a murine model of dominant osteogenesis imperfecta: a new target for osteogenesis imperfecta pharmacological therapy. Stem Cells, 30, 1465–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galluzzi L., Bravo-San Pedro J.M., Kroemer G. (2014) Organelle-specific initiation of cell death. Nat. Cell Biol., 16, 728–736. [DOI] [PubMed] [Google Scholar]
- 38.Fais S., De Milito A., Lozupone F. (2005) The role of FAS to ezrin association in FAS-mediated apoptosis. Apoptosis, 10, 941–947. [DOI] [PubMed] [Google Scholar]
- 39.Mo R., Freer A.M., Zinyk D.L., Crackower M.A., Michaud J., Heng H.H., Chik K.W., Shi X.M., Tsui L.C., Cheng S.H., et al. (1997) Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development, 124, 113–123. [DOI] [PubMed] [Google Scholar]
- 40.Joeng K.S., Long F. (2013) Constitutive activation of Gli2 impairs bone formation in postnatal growing mice. PLoS One, 8, e55134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hyzy S.L., Olivares-Navarrete R., Schwartz Z., Boyan B.D. (2012) BMP2 induces osteoblast apoptosis in a maturation state and noggin-dependent manner. J. Cell. Biochem., 113, 3236–3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grafe I., Yang T., Alexander S., Homan E.P., Lietman C., Jiang M.M., Bertin T., Munivez E., Chen Y., Dawson B., et al. (2014) Excessive transforming growth factor-beta signaling is a common mechanism in osteogenesis imperfecta. Nat. Med., 20, 670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lai C.F., Cheng S.L. (2005) Alphavbeta integrins play an essential role in BMP-2 induction of osteoblast differentiation. J. Bone Miner. Res., 20, 330–340. [DOI] [PubMed] [Google Scholar]
- 44.Sheppard D. (2005) Integrin-mediated activation of latent transforming growth factor beta. Cancer Metastasis Rev., 24, 395–402. [DOI] [PubMed] [Google Scholar]
- 45.Munger J.S., Huang X., Kawakatsu H., Griffiths M.J., Dalton S.L., Wu J., Pittet J.F., Kaminski N., Garat C., Matthay M.A., et al. (1999) The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell, 96, 319–328. [DOI] [PubMed] [Google Scholar]
- 46.Meredith J.E., Jr., Winitz S., Lewis J.M., Hess S., Ren X.D., Renshaw M.W., Schwartz M.A. (1996) The regulation of growth and intracellular signaling by integrins. Endocr. Rev., 17, 207–220. [DOI] [PubMed] [Google Scholar]
- 47.Wipff P.J., Hinz B. (2008) Integrins and the activation of latent transforming growth factor beta1 - an intimate relationship. Eur. J. Cell Biol., 87, 601–615. [DOI] [PubMed] [Google Scholar]
- 48.Jikko A., Harris S.E., Chen D., Mendrick D.L., Damsky C.H. (1999) Collagen integrin receptors regulate early osteoblast differentiation induced by BMP-2. J. Bone Miner. Res., 14, 1075–1083. [DOI] [PubMed] [Google Scholar]
- 49.Moursi A.M., Globus R.K., Damsky C.H. (1997) Interactions between integrin receptors and fibronectin are required for calvarial osteoblast differentiation in vitro. J. Cell Sci., 110 (Pt 18), 2187–2196. [DOI] [PubMed] [Google Scholar]
- 50.Seyb K.I., Ansar S., Bean J., Michaelis M.L. (2006) beta-Amyloid and endoplasmic reticulum stress responses in primary neurons: effects of drugs that interact with the cytoskeleton. J. Mol. Neurosci., 28, 111–123. [DOI] [PubMed] [Google Scholar]
- 51.Uveges T.E., Collin-Osdoby P., Cabral W.A., Ledgard F., Goldberg L., Bergwitz C., Forlino A., Osdoby P., Gronowicz G.A., Marini J.C. (2008) Cellular mechanism of decreased bone in Brtl mouse model of OI: imbalance of decreased osteoblast function and increased osteoclasts and their precursors. J. Bone Miner. Res., 23, 1983–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 53.Towbin H., Staehelin T., Gordon J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl Acad. Sci. USA, 76, 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.