

Abstract
Traumatic brain injury affects the whole body in addition to the direct impact on the brain. The systemic response to trauma is associated with the hepatic acute-phase response. To further characterize this response, we performed controlled cortical impact injury on male mice and determined the expression of serum amyloid A1 (SAA1), an apolipoprotein, induced at the early stages of the acute-phase response in liver and plasma. After cortical impact injury, induction of SAA1 was detectable in plasma at 6 hours post-injury and in liver at 1 day post-injury, followed by gradual diminution over time. In the liver, cortical impact injury increased neutrophil and macrophage infiltration, apoptosis, and expression of mRNA encoding the chemokines CXCL1 and CXCL10. An increase in angiotensin II AT1 receptor mRNA at 3 days post-injury was also observed. Administration of the AT1 receptor antagonist telmisartan 1 hour post-injury significantly decreased liver SAA1 levels and CXCL10 mRNA expression, but did not affect CXCL1 expression or the number of apoptotic cells or infiltrating leukocytes. To our knowledge, this is the first study to demonstrate that SAA1 is induced in the liver after traumatic brain injury and that telmisartan prevents this response. Elucidating the molecular pathogenesis of the liver after brain injury will assist in understanding the efficacy of therapeutic approaches to brain injury.
Traumatic brain injury (TBI) triggers tissue damage within the central nervous system and a subsequent strong and rapid inflammatory response that may result in sustained inflammation.1,2 This inflammatory response is not restricted to the brain. Inflammatory mediators are released from the damaged brain tissue into the circulation to produce a systemic inflammatory response in the peripheral organs. The identity of many of these mediators is still unclear, although it is known that TBI has an important impact on several peripheral organs, including the lung, spleen, and liver.3–6 TBI induces an acute-phase response (APR), principally managed through the liver.7 This APR, initiated mainly by the circulating cytokines IL-1β and IL-6, can lead to a coordinated program of hepatic transcriptional responses,8 including a strong activation of NF-κB regulated acute phase proteins and release of many cytokines and chemokines into the bloodstream.9 Serum amyloid A1 (SAA1) is one of the most abundant acute phase proteins, whose expression can be induced >1000-fold during the APR.10 It is therefore a reliable marker of the APR.11 Clinical studies have detected increased SAA1 in the plasma of TBI patients; this increase has been shown to correlate with acute inflammation and the severity of the injury.12 SAA1 has been previously suggested as a biomarker in trauma patients.13,14
The function of the vast increase in SAA1 during the APR is not clear. SAA1 is an apolipoprotein,11,15 incorporated into high-density lipoprotein, and thereby may alter high-density lipoprotein metabolism and modulate cholesterol transport.16 Hepatocytes are the principal source of SAA1. Increase of SAA1 expression, synthesis, and release to the circulation is induced by inflammation, trauma, or stress that activate the APR.17,18 Although the liver is the principal organ that expresses SAA1 protein, particularly in the APR, production in different cell types and tissues has been reported.18 SAA1 has some functions of a cytokine, modulating responses of monocytes and neutrophils. It can also induce expression of cytokines, chemokines, and extracellular matrix degrading enzymes that are necessary for repair of tissue damage.19–22 SAA1 can bind to, and potentially signal through, Toll-like receptor-2, formyl peptide receptor 2, type B scavenger receptor I, and receptor for advanced glycation end product.23–29 Chronic elevation of SAA1 expression can therefore exacerbate inflammatory conditions, and its deposition in different tissues results in secondary amyloidosis.30
Angiotensin II (Ang II), produced by the renin-angiotensin system (RAS), contributes to inflammation in various pathologies throughout the body.31,32 Ang II signaling through the Ang II type 1 receptor (AT1R) is a major generator of reactive oxygen species through activation of NADPH oxidase, and inducer of NF-κB–dependent gene transcription.33,34 AT1Rs, highly expressed in the liver,35 are often up-regulated in response to stress and inflammation.36 Increased Ang II signaling through the AT1R has been implicated in disorders ranging from cardiovascular disease through hepatic fibrosis to septic shock.35,37,38 The molecular intermediates responsible for transmitting many detrimental effects of AT1R signaling are poorly understood. In a rodent model of Ang II–induced muscle loss, Ang II infusion induced hepatic SAA1 expression in an IL-6–dependent manner.39 The induction of IL-6 and SAA1 in the liver was necessary for Ang II–dependent muscle loss.39 Thus, SAA1 may represent one intermediate through which Ang II contributes to inflammatory signaling, with detrimental consequences.
Blockade of AT1R with the Ang II receptor blockers (ARBs) reduces inflammation in many tissues.40 In the central nervous system ARBs have neuroprotective effects.41 We have previously reported that the systemic administration of the ARB, candesartan, reduces cerebral cortical lesion volume and inflammation, and improves motor and cognitive behavior after TBI.42 We found similar neuroprotective results after TBI with another ARB, telmisartan.43 Telmisartan, a highly selective AT1R antagonist, has been reported to exert potent systemic antioxidative and anti-inflammatory effects.44 Both candesartan and telmisartan, in addition to blocking AT1R signaling, also act as partial agonists of the nuclear peroxisome proliferator-activated receptor-γ,45,46 and consequently increase PPARγ-dependent gene expression.
Telmisartan is active in liver because it reduces oxidative stress and fibrogenesis in patients with nonalcoholic fatty liver disease and chronic hepatitis C,47 reduces hepatic glucose output,48 and protects against arsenic hepatotoxicity.49 Systemic administration of ARBs after TBI may reduce peripheral inflammatory responses and dampen the APR. We hypothesized that such action could contribute to the efficacy of ARBs in ameliorating deficits after TBI.
The effects of ARBs on the liver response to TBI have not yet been explored. Here, we investigate the effect of TBI on the hepatic APR through monitoring SAA1 expression and the ability of the ARBs to interfere with the hepatic response to TBI. We demonstrate that TBI does induce a dramatic increase in hepatic and serum expression of SAA1, and that telmisartan reduces this expression, albeit with a delayed time course. Telmisartan also interferes with other selected hepatic responses to TBI. Understanding the molecular pathogenic connections between liver and brain injury will assist in designing efficient therapeutic approaches for modulating the secondary tissue damage that follows acute inflammation.
Materials and Methods
Animals, TBI, and Drug Treatment
Nine-week-old C57BL/6 male mice (National Cancer Institute, Frederick, MD) were housed in regular cages with access to food and water ad libitum and a 12:12 light/dark cycle. Mice were anesthetized with isoflurane (3% for induction, 1.5% for maintenance) and securely positioned in a mouse stereotaxic frame (Stoelting Co, Wood Dale, IL). We performed mild to moderate controlled cortical impact injury (CCI; coordinates, 2-mm bregma; 2 mm lateral to the suture line) at an impact depth of 1 mm, with a 2-mm-diameter round impact tip (speed, 3.6 m/second; dwell time, 100 milliseconds) using an electromagnetically driven CCI device (Impact One stereotaxic impactor CCI; Leica Microsystems GMBH, Wetzlar, Germany). Mice were administered daily with 1 mg/kg telmisartan (Sigma-Aldrich, St. Louis, MO) or saline by oral gavage, starting 1 hour after CCI injury (n = 5 to 12 per group). All animal studies were approved by the Uniformed Services University of the Health Sciences Institutional Animal Care and Use Committee and were conducted in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals.50
Tissue and Serum Collection
Mice were anesthetized with ketamine/xylazine and sacrificed by rapid decapitation with sharp scissors at 6 hours and 1, 3, 7, and 30 days after CCI. Control mice were not injured but underwent identical anesthesia. Liver and brain samples were removed, cut into fractions, quickly frozen on isopentane, and stored at −80°C. Serial liver or brain sections (30 μm thick) were cut using a cryostat, placed on gelatin slides, and stored at −80°C until use. Additional liver fragments were dissected, frozen on isopentane on dry ice, and stored at −80°C until use. Trunk blood was collected and allowed to clot for 10 minutes at room temperature, and serum was obtained by centrifugation (14,000 × g) at 4°C for 10 minutes and stored at −80°C.
Immunohistochemistry
Liver and brain sections were postfixed by 1-hour incubation in 4% paraformaldehyde, washed in 0.1% Triton X-100 (Sigma-Aldrich) in 0.1 mol/L phosphate-buffered saline with Tween 20 (PBS-T), blocked in PBS-T/5% normal horse serum for 1 hour, then incubated overnight at 4°C in PBS-T/1% normal horse serum containing goat polyclonal anti-SAA1 antibody (1:200, AF2948; R&D Systems, Minneapolis, MN), rabbit polyclonal anti-CD14 antibody (1:200, sc-9150; Santa Cruz Biotechnology, Dallas, TX), rabbit polyclonal anti-myeloperoxidase (1:50, ab9535; Abcam, Cambridge, MA), or mouse monoclonal anti-CD68 antibody (1:500, MCA1957T; Serotec, Raleigh, NC). Sections were then washed in PBS-T three times for 5 minutes and incubated with the appropriate secondary antibody (1:1000), anti-goat IgG fluorescein isothiocyanate (catalog number 705-095-147; Jackson ImmunoResearch Laboratories, West Grove, PA) (green) or anti-rabbit or anti-mouse Alexa Fluor 568–conjugated IgG (A11036; Invitrogen, Carlsbad, CA) (red) for 2 hours at room temperature. Brain sections were incubated in 0.3% H2O2 and blocked with 3% normal goat serum in PBS with 0.25% Triton X-100 for 1 hour, and incubated overnight in PBS with 0.25% Triton X-100 and 1% normal goat serum with anti-SAA1 antibody. After three 5-minute washes in 1× PBS, sections were incubated in biotinylated anti-goat secondary antibody (1:2000, catalog number BA-100; Vector Laboratories, Burlingame, CA) in PBS-T, followed by the Vectastain avidin/biotinylated enzyme complex kit (1:400, catalog number PK-6100; Vector Laboratories) and 3,3′-diaminobenzidine (Sigma-Aldrich) substrate to visualize brain SAA1 expression. Sections were rinsed once with PBS-T, followed by a distilled water rinse, before coverslipping with ProLong Gold antifade reagent (Invitrogen, Carlsbad, CA).
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick-End Labeling Assay
Liver sections were processed for DNA strand breaks (labeling of fragment DNA) using the Fluorescence in Situ Cell Death Detection Kit (catalog number 11,684,795; Roche, Chicago, IL), as previously described.41
RNA Isolation and Quantitative PCR Analysis
RNA was isolated from liver fractions from CCI-injured and control mice using TRIzol reagent (catalog number 15596-026; Invitrogen, Carlsbad, CA). We added 200 μL TRIzol reagent per sample, sonicated to disrupt the liver tissue, followed by the addition of 0.2 volumes of chloroform and centrifugation at 16,000 × g for 15 minutes. The aqueous layer was removed, and RNA was further purified with the RNeasy Mini Kit (catalog number 74904; Qiagen Sciences, Germantown, MD). Contaminating DNA was digested with RNase-free DNase, according to the manufacturer's instructions, and quantified with a Nanodrop spectrophotometer (Thermo Scientific, Wilmington, DE). Quantitative PCR was performed with RT2 SYBR Green qPCR MasterMix (catalog number 330609; Qiagen Sciences, Germantown, MD) on a BioRad CFX96 with primers designed in our laboratory. Primer sequences are shown in Table 1. Denaturation was performed at 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds, and 60°C for 60 seconds. The relative amount of the target mRNA was normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase.
Table 1.
Sequence of Primers Used for Real-Time PCR
| Target gene | Sequence |
|---|---|
| AT1R | F: 5′-AAGGGCAAGGAACCTTGTTT-3′ R: 5′-CAAAGGGCTCCTGAAACTTG-3′ |
| SAA1 | F: 5′-TCAGACAAATACTTCCATGCT-3′ R: 5′-AAAGGCCTCTCTTCCATCACT-3′ |
| IL6 | F: 5′-GAGGATACCACTCCCAACAGA-3′ R: 5′-CACAACTCTTTTCTCATTTCC-3′ |
| IL1Β | F: 5′-GTGTCTTTCCCGTGGACCTT-3′ R: 5′-TCGTTGCTTGGTTCTCCTTG-3′ |
| IFNG | F: 5′-CTTCTTCAGCAACAGCAAGGCGAAA-3′ R: 5′-CATTGAATGCTTGGCGCTGGA-3′ |
| CCL2 | F: 5′-ACTCATTCACCAGCAAG-3′ R: 5′-TTGAGCTTGGTGACAAAAACT-3′ |
| CXCL10 | F: 5′-TCATCCTGCTGGGTCTGAGTGG-3′ R: 5′-CTCATCATTCTTTTTCATCGT-3′ |
| CXCL1 | F: 5′-ACCCAAACCGAAGTCATAGCC-3′ R: 5′-ACCTTTTAGCATCTTTTGGAC-3′ |
| TNFA | F: 5′-GAACTTCGGGGTGATCGGTCCC-3′ R: 5′-TTGCTACGACGTGGGCTACAGG-3′ |
| GAPDH | F: 5′-ACCATCTTCCAGGAGCGAGA-3′ R: 5′-GGCGGAGATGATGACCCT-3′ |
F, forward; R, reverse.
Protein Extraction and Western Blot Analysis
Liver fragments were homogenized 1:10 wt:vol in a lysis buffer for total protein extraction (T-PER Tissue Protein Extraction Reagent; Thermo Scientific, Rockford, IL) supplemented with protease inhibitor cocktail (Roche, Indianapolis, IN), and centrifuged at 14,000 × g for 10 minutes at 4°C. Serum samples were diluted 1:5 with PBS. Protein concentration in the supernatant was determined by bicinchoninic acid assay assay (Pierce, Rockford, IL). The protein extracts were combined with loading buffer and boiled for 5 minutes, before loading onto a 10% SDS-polyacrylamide gel. Separated proteins were transferred onto nitrocellulose membranes. The membranes were blocked in casein-based blocking buffer (Sigma-Aldrich) for 60 minutes at room temperature and incubated overnight at 4°C with goat anti-SAA1 (1:200). After incubation, the membranes were washed and exposed to secondary horseradish peroxidase–conjugated donkey anti-goat antibody (1:2000; GE Healthcare, Buckinghamshire, UK) for 2 hours at room temperature, developed with SuperSignal enhanced chemiluminescence reagent (Thermo Scientific, Rockford, IL), and detected using a Fuji LAS-3000 image acquisition system (Fuji, Stamford, CT) equipped with a cooled charge-coupled device camera. After SAA1 detection, the membranes were stripped for 15 minutes at room temperature in Restore Western Blot stripping buffer (Thermo Scientific, Rockford, IL), blocked, reincubated with anti-rabbit β-actin antibody (1:10,000; Sigma-Aldrich) for liver samples or anti-rabbit glyceraldehyde-3-phosphate dehydrogenase (1:1000, ab139322; Abcam) for serum samples, washed, incubated with the anti-rabbit secondary as before, and developed as above.
SAA1 Aggregate Analysis
Liver fractions were homogenized using buffer A (tris-buffered saline, pH 7.5, 2 mmol/L EDTA, and 10% sucrose) with 1.5× protease inhibitors (Complete Protease Inhibitor Cocktail Tablets; Roche, Indianapolis, IN), 1 mmol/L phenylmethylsulfonyl fluoride, and sodium orthovanadate. Liver samples were homogenized (three times for 3 seconds), chilled on ice for 5 minutes, and spun at 500 × g for 5 minutes at 4°C before the addition of 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS. Homogenates were mixed by pipetting, sonicated three times for 5 seconds using a Branson sonicator at 50% amplitude, and incubated for 20 minutes on ice. Homogenates were further treated with 0.1 mg/mL RNase A, 5 mmol/L dithiothreitol, and 5 mmol/L MgCl2 for 20 minutes at 4°C. Samples were centrifuged at 10,000 × g for 13 minutes at 4°C, and the supernatants were loaded onto the top of the 30% sucrose + 0.1% Triton X-100 pad in 3-mL ultracentrifuge tubes. Liver homogenates from control mice, as well as from mice at 1 and 3 days post-injury (dpi), underwent high-speed centrifugation. Protein complexes were sedimented in an SW55 rotor (Beckman Coulter, Fullerton, CA) for 1 hour at 45,000 rpm at 4°C. Pellets were resuspended in 1× tris-buffered saline, 2% SDS, containing protease inhibitor cocktail (Roche, Nutley, NJ), 1 mmol/L phenylmethylsulfonyl fluoride at room temperature by intensive pipetting, briefly sonicated, and incubated at 37°C for 20 minutes to solubilize protein complexes. Preparations were loaded onto an acrylamide gel without boiling and subjected to SDS-PAGE under standard conditions, followed by immunostaining with the SAA1 antibody (R&D Systems).
Microscopy and Quantitative Analysis
Cortical brain lesion area was determined from every third brain section, stained with cresyl violet, as previously described.42 Lesion volume was obtained by multiplying the sum of the lesion areas by the distance between sections, with between 9 and 15 brain sections required to bridge the lesion. Percentage lesion volume was calculated by dividing each lesion volume by the total ipsilateral hemisphere volume. To obtain cell counts of specific labeled cells, we quantified five microscopic fields per brain section positioned around the impact site, and 6 to 10 randomly selected fields per liver section. Between three and eight animals per group were analyzed at each time point after injury for either naïve or CCI mice. For each tissue section (at 256-μm intervals), the number of terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)-positive cells, macrophages, or neutrophils in each field was quantified, averaged, and expressed as the number of cells/mm2. Images were acquired at ×20 magnification on an Olympus BX61 with an attached qImaging Retiga EXi Aqua charge-coupled device camera and iVision software version 4.0.9 (BioVision Technologies, Exton, PA) and cropped and adjusted using Adobe Photoshop CS5. Cell counts were performed using ImageJ software version 1.43u (NIH, Bethesda, MD; http://imagej.nih.gov/ij).
Statistical Analysis
All data in this study are expressed as means ± SEM. P < 0.05 was considered statistically significant. Intergroup differences were evaluated by one-way analysis of variance, followed by the Newman-Keuls multiple-comparison test and the Mann-Whitney test. All statistics were performed with Prism 5 software (GraphPad, San Diego, CA).
Results
SAA1 Increases in Serum as a Rapid Response to TBI
To determine whether SAA1 levels are altered in serum after brain injury, we injured mice by CCI, and analyzed the amount of SAA1 at different time points by Western blot analysis. Immunoblot analysis revealed a single band of molecular weight 12 kDa corresponding to SAA1. SAA1 expression was low in control mice and up-regulated approximately 26-fold at 6 hours post-injury (hpi), 11.5-fold at 3 dpi, and 13-fold at 7 dpi (Figure 1). Hence, brain injury leads to significant elevation of SAA1 in the serum, suggesting that increased hepatic SAA1 is rapidly transported in the blood after brain injury.
Figure 1.

Serum amyloid A1 (SAA1) is up-regulated in serum after brain injury. A: Western blot analysis showing SAA1 protein increase in serum at different time points after cortical impact injury (CCI) compared with control naïve mice. One representative experiment of five is shown. B: Quantitation of Western blot analyses showing SAA1 expression in serum was highest 6 hours after brain injury, with a smaller induction at 3 and 7 days after injury. SAA1 expression was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Values are expressed as means ± SEM (B). n = 5 to 8 per group (B). ∗∗P < 0.005, ∗∗∗∗P < 0.0001 for injured mice compared with control mice.
AT1 Receptor Gene Expression Up-Regulates in the Liver after TBI
To investigate whether the hepatic RAS is altered by brain injury, we examined the expression of the mRNA encoding the AT1R. Expression of this receptor is usually induced in response to stress and inflammation.35,51 We found mRNA expression of AT1R to be up-regulated in the liver at 3 dpi but not at 6 hpi, compared with expression in naïve control mice (Figure 2).
Figure 2.

Liver angiotensin II type 1 receptor (AT1R) mRNA expression increases on induction by brain injury. Quantitative PCR analysis of AT1R mRNA expression in liver of control and cortical impact injury (CCI) mice. AT1R mRNA is up-regulated in liver tissue 3 days postinjury (dpi). Values are expressed as means ± SEM. n = 5 to 8 per group. ∗∗P < 0.005, ∗∗∗∗P < 0.0001. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Expression Levels of SAA1 mRNA in Liver Increase by TBI
We next determined whether SAA1 expression was elevated in the liver, as opposed to extrahepatic sites. We found SAA1 mRNA expression in the liver to be considerably increased in injured mice, with minimal expression of SAA1 protein in hepatic cells of control mice. We observed a rapid and dramatic increase in SAA1 mRNA expression of 60-fold by 6 hpi, and peak expression at 1 dpi of 180-fold, which decreased by 3 dpi to 15-fold greater than expression in control mice (Figure 3A). Treatment of mice with telmisartan, initiated 1 hour after injury, had little effect at early time points (Figure 3, B and C). However, by 3 dpi, telmisartan treatment led to a nonsignificant reduction in SAA1 mRNA expression (Figure 3D).
Figure 3.
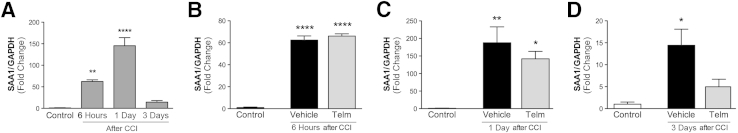
Brain injury induces serum amyloid A1 (SAA1) mRNA expression in the liver. A: Quantitative PCR analysis indicates that liver SAA1 mRNA expression is highest 1 day postinjury (dpi). B–D: Vehicle or telmisartan (Telm) was administered to mice daily until sacrifice. No changes at 6 hours after injury (B). At 1 dpi (C) and 3 dpi (D), telmisartan treatment trends toward a decrease in SAA1 expression compared with mice treated with vehicle (not significant, P = 0.06). SAA1 mRNA expression was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA. Values are expressed as means ± SEM. n = 5 to 8 per group. ∗P < 0.05, ∗∗P < 0.005, and ∗∗∗∗P < 0.0001 for SAA1 in injured mice compared with control mice. CCI, cortical impact injury.
Brain SAA1 Expression, Lesion Volume, and Inflammatory Cell Infiltration into the Injured Brain
To determine whether SAA1 expression was increased in the brain after injury, we examined injured brain sections by immunohistochemistry for SAA1 at various time points after injury. We only observed SAA1 expression at 1 dpi, localized in cells with vacuolated morphology (Supplemental Figure S1A). We did not detect SAA1 expression at any other time after CCI, or in naïve brains. As expected, the size of the brain lesion increased at 1 and 3 dpi (Supplemental Figure S1B), with increased density of CD14-positive monocytes and polymorphonuclear leukocytes detected from 6 hpi until 3 dpi (Supplemental Figure S1C). CD68-positive macrophages also increased at 3 dpi (Supplemental Figure S1D). These parameters correlate with increased liver SAA1 expression after brain injury during the first 24 hours. However, liver SAA1 expression was considerably lower by 3 dpi, whereas the secondary inflammatory response associated with the injured brain was still strong at 3 dpi and beyond (Supplemental Figure S1E).
Specific Accumulation of SAA1 Amyloid Aggregates Is Absent after TBI
We have shown that TBI sharply increases the amount of SAA1 protein in serum (Figure 1) and SAA1 mRNA in liver (Figure 3). SAA1 protein is up-regulated, with a peak of expression at 1 dpi, and decreases at 3 dpi (Figure 4, A and B) in liver. This rapid and pronounced elevation of SAA1 monomers could lead to formation of amyloidogenic oligomers and further high molecular weight aggregates, as were shown for SAA amyloidosis.52 To detect the SAA1 amyloid species, we used a recently developed approach53 where aggregates are detected after enrichment on the basis of their detergent resistance and high molecular weight. This procedure was previously used to detect polyglutamine amyloid aggregates from cells, as well as amyloid aggregates from yeast cells.53 The resistance to strong anionic detergents, such as SDS at room temperature, was reported for many amyloids and appears to be a universal characteristic that distinguishes amyloid aggregates from other protein complexes.54 We did not detect high-molecular-weight SDS-resistant species of SAA1; instead, we only observed accumulation of 30-kDa SAA1 species that might represent SAA1 dimers or trimers (Figure 4A). These species were equally present in preparations from control and post-CCI mice. Analysis of liver homogenates from mice at 7 dpi also did not show any specific accumulation of amyloid SAA1 (data not shown), indicating that brain injury did not generate amyloid deposition in the liver at the analyzed post-TBI time intervals.
Figure 4.

Serum amyloid A1 (SAA1) protein expression in liver is induced after brain injury and reduced after telmisartan (Telm) treatment. Representative Western blot analyses showing SAA1 monomers and SDS-resistant SAA1 species in liver homogenates after ultracentrifugation (in dissolved pellet fractions). A: Cortical impact injury (CCI) increases the amount of SAA1 monomers, but not SDS-resistant SAA1 species, in livers of injured mice. Quantification of SAA1 protein expression normalized to that of β-actin. Averages were calculated from three independent replicates. B: SAA1 protein is up-regulated, with a peak of expression at 1 day post-injury (dpi), decreasing at 3 dpi, and returned to control (C) levels by 30 dpi. C: Representative Western blot analyses showing SAA1 monomers (12 kDa) at 3 dpi (duplicate samples). D: Telmisartan (1 mg/kg) significantly reduces SAA1 expression compared with mice treated with vehicle at 3 dpi. E: Immunohistochemical staining of SAA1 (green) and DAPI (blue) in mice liver from control group showing low expression of SAA1; liver tissue from brain-injured, vehicle-treated mice display a significant increase in SAA1 immunofluorescence in the cytoplasm of hepatocytes. Treating brain-injured mice with telmisartan significantly reduces SAA1 immunofluorescence. F: Quantification of immunofluorescence of SAA1 expression shows low SAA1 fluorescence intensity in naïve mice that is increased after 3 dpi. Mice treated with telmisartan after brain injury exhibit a decrease in SAA1 fluorescence intensity. Values are expressed as means ± SEM (B, D, and F). n = 5 to 8 per group (B, D, and F). ∗P < 0.05, ∗∗P < 0.005, and ∗∗∗P < 0.001.
TBI Induction of Hepatic SAA1 Expression Is Reduced with Telmisartan Treatment
To determine whether signaling through the AT1R alters SAA1 expression after TBI, we treated animals with the ARB, telmisartan, starting at 1 hpi, and daily thereafter. Telmisartan significantly reduced hepatic SAA1 expression at 3 dpi (Figure 4, C and D), but not at earlier time points (data not shown). By using immunohistochemical analysis, we observed SAA1 expression in hepatic cells. The intensity of hepatic SAA1 staining increased in mice 3 dpi compared with that in naïve mice. Telmisartan treatment significantly reduced SAA1 immunofluorescence in the cytoplasm of hepatic cells (Figure 4, E and F).
Early Increase in Cytokine and Chemokine Expression of Injured Mice
To examine the effects of brain injury on the hepatic inflammatory response, we measured cytokine and chemokine gene expression by quantitative PCR at different time points after CCI. Specifically, we measured mRNA expression of CXCL1, CXCL10, interferon-γ, IL-6, IL-1β, tumor necrosis factor (TNF)-α, and chemokine ligand 2 in livers taken from naïve mice or from mice at different times after TBI to determine whether cytokines were induced in the liver in response to brain trauma. CXCL1, CXCL10, and IL-1β mRNA levels were significantly increased, and TNF-α mRNA was decreased in the liver at 6 hours after TBI, compared with naïve controls (Table 2). No significant differences in expression between naïve and CCI-injured mice were detected for interferon-γ, IL-6, or chemokine ligand 2 mRNA (Table 2). Telmisartan treatment significantly reduced CXCL10 mRNA when assessed 6 hours after injury but did not alter the expression of CXCL1, IL-1β, or TNF-α (Table 2).
Table 2.
Chemokine and Cytokine Expression Increases in the Liver after Brain Injury
| Time | Control | Vehicle | Telmisartan |
|---|---|---|---|
| IL-1β | |||
| 6 hours | 1.0 ± 0.1 | 2.0 ± 0.2∗ | 1.83 ± 0.4 |
| 1 day | 1.0 ± 0.3 | 0.92 ± 0.1 | 0.89 ± 0.1 |
| 3 days | 1.0 ± 0.2 | 0.82 ± 0.3 | 1.8 ± 0.4 |
| IFN-γ | |||
| 6 hours | 1.0 ± 0.2 | 1.2 ± 0.2 | 0.8 ± 0.1 |
| 1 day | ND | ND | ND |
| 3 days | ND | ND | ND |
| TNF-α | |||
| 6 hours | 1.0 ± 0.2 | 0.5 ± 0.1∗∗ | 0.4 ± 0.1∗∗ |
| 1 day | 1.0 ± 0.2 | 0.66 ± 0.1 | 0.86 ± 0.1 |
| 3 days | 1.0 ± 0.1 | 3.0 ± 0.8 | 3.4 ± 0.9 |
| CXCL10 | |||
| 6 hours | 1.0 ± 0.1 | 1.39 ± 0.1 | 0.98 ± 0.1† |
| 1 day | 1.0 ± 0.2 | 0.83 ± 0.1 | 1.01 ± 0.3 |
| 3 days | 1.0 ± 0.3 | 1.1 ± 0.2 | 1.9 ± 0.2 |
| IL-6 | |||
| 6 hours | 1.0 ± 0.2 | 1.1 ± 0.1 | 1.1 ± 0.3 |
| 1 day | 1.0 ± 0.2 | 0.8 ± 0.1 | 1.1 ± 0.2 |
| 3 days | 1.0 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.2 |
| CCL2 | |||
| 6 hours | ND | ND | ND |
| 1 day | ND | ND | ND |
| 3 days | 1.0 ± 0.1 | 1.4 ± 0.3 | 1.2 ± 0.2 |
| CXCL1 | |||
| 6 hours | 1.0 ± 0.2 | 18.5 ± 1.8∗∗ | 22.1 ± 3.0∗∗∗ |
| 1 day | 1.0 ± 0.3 | 1.56 ± 0.6 | 2.28 ± 0.4 |
| 3 days | 1.0 ± 0.2 | 0.77 ± 0.1 | 1.92 ± 0.5 |
Expression of specific genes in the liver at different time points after traumatic brain injury was analyzed by quantitative PCR. RNA was isolated from livers of naïve mice and mice at 6 hours postinjury (hpi) and 1 and 3 days post-injury (dpi), treated with telmisartan or vehicle. Expression of CXCL1, CXCL10, IFN-γ, IL-6, IL-1β, TNF-α, and CCL2 was normalized to glyceraldehyde-3-phosphate dehydrogenase. CXCL1, IL-1β, and CXCL10 mRNA expression increased at 6 hpi. Telmisartan treatment (1 mg/kg) reduced CXCL10 expression at 6 hpi. TNF-α mRNA expression was reduced at 6 hpi. No differences were observed for hepatic expression of IFN-γ, IL-6, and CCL2 mRNA between control and brain-injured mice. Values are expressed as means ± SEM. n = 5 to 8 per group.
CCL, chemokine ligand; IFN, interferon; ND, not detected; TNF, tumor necrosis factor.
P < 0.05, ∗∗P < 0.005, and ∗∗∗P < 0.001 versus expression in control mice.
P < 0.05 versus expression in vehicle-treated injured mice.
Hepatic Neutrophil and Macrophage Density and Apoptosis Induced by Brain Injury
To determine whether brain injury was associated with cellular changes in the liver, we performed immunohistochemical analysis on sections from the liver of either naïve or CCI-injured mice. Neutrophil infiltration, assessed by staining for myeloperoxidase, was increased in livers taken from CCI-injured mice at 3 dpi compared with those from naïve mice (Figure 5, A and B). Analysis of apoptotic cells in liver tissue, by TUNEL analysis, also revealed that the number of dying hepatic cells with apoptotic morphology (Figure 5C) was significantly increased in brain-injured mice at 3 dpi. Although there were occasional TUNEL-positive cells in liver from naïve mice (approximately 30 cells/mm2), there were many more apoptotic cells in livers taken from CCI-injured mice (approximately 150 cells/mm2) (Figure 5D). There was also increased infiltration of CD68-positive macrophages in livers of CCI-injured mice at 3 dpi, compared with naïve mice (Figure 5, E and F). Telmisartan treatment did not alter neutrophil or macrophage infiltration into the liver after brain injury, nor the number of apoptotic cells.
Figure 5.

Neutrophil and macrophage density and apoptosis increase in liver after brain injury. A: The number of neutrophils [myeloperoxidase (MPO)-positive cells] was lower in liver from control mice compared with in liver from injured mice treated with vehicle, and injured mice treated with telmisartan (Telm). High-power images show hepatic cells with MPO marker (red) and nuclei with DAPI (blue). B: Telmisartan did not reduce the number of neutrophils infiltrating the liver that were induced by cortical impact injury (CCI). C: Images of terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)–positive cells (green) in a single field for control, injured mice treated with vehicle, and injured mice treated with Telm. High-power images show apoptotic cells with the TUNEL marker (green) and nuclei with DAPI (blue) for the vehicle group. D: Telmisartan did not reduce the number of TUNEL-positive cells in the liver that were induced by CCI. E: Immunofluorescence staining of CD68-positive macrophages (red) in liver with nuclei stained with DAPI (blue). F: Macrophages in the liver increase after CCI, but telmisartan treatment does not reduce the number of CD68-positive cells. Data are expressed as means ± SEM (B, D, and F). n = 5 to 8 per group (B, D, and F). ∗P < 0.05, ∗∗∗P < 0.001, and ∗∗∗∗P < 0.0001 comparing liver from injured mice and control mice. Scale bars: 50 μm (A, C, and E, ); 20 μm (A, C, and E, high-magnification images in the insets).
Discussion
In this study we show that TBI induces a rapid increase in SAA1 protein in the blood and in the liver. The complex hepatic response to brain injury includes, in addition to SAA1 elevation, changes in expression of inflammatory genes, neutrophil and macrophage infiltration, and hepatic cell death. The hepatic RAS also responds to brain trauma, and inhibition of AT1R signaling can reduce some of the hepatic responses to injury. Our data demonstrate that liver responds to brain injury and, therefore, suggest that hepatic inflammation and cell death may influence the systemic inflammatory response and, hence, impact back on the damaged brain (Figure 6).
Figure 6.

Interplay of liver inflammation induced by traumatic brain injury. The bidirectional interactions between the injured brain and peripheral tissues, including liver, are controlled by cytokines in the peripheral circulation, neuropeptides, or sympathetic nerve activation. After traumatic brain injury, hepatic inflammation is indicated by higher expression of serum amyloid A1 (SAA1) and proinflammatory cytokines, infiltration of neutrophils and macrophages, and the presence of apoptotic cells. Telmisartan reduces SAA1 and CXCL10 expression. AT1R, angiotensin II type 1 receptor; TNF, tumor necrosis factor.
Hepatic Inflammation in Response to TBI
We have shown that head trauma leads to a hepatic inflammatory response, including highly elevated hepatic SAA1 production. A prior study has shown a close relationship between brain damage and hepatic inflammation.55 It is evident that bidirectional interactions between the brain and peripheral organs, including liver, occur after brain injury, although the messengers through which these interactions happen are not understood. Brain communication to the liver could be mediated by cytokines in the peripheral circulation, neuropeptides, microparticles, or sympathetic nerve activation.55,56 After TBI, serum levels of several proinflammatory cytokines are rapidly elevated, including those of TNF-α and IL-6,57–59 cytokines that can synergistically induce transcription of the SAA1 gene.57 In the liver, we found only a modest increase in IL-1β mRNA, and a decrease in TNF-α mRNA at 6 hpi. Surprisingly, we did not detect induction of IL-6 mRNA expression at any time point examined after injury, suggesting that it is potentially the circulating cytokines that may be responsible for the induction of SAA1 expression. Thus, it is likely that peripheral cytokines contribute to the rapid induction of SAA1 transcription. Cytokine induction of SAA1 gene transcription is mediated by the transcription factors C/EBP and NF-κB.56 Because NF-κB–mediated gene transcription is strongly induced in the liver within 4 hours after a focal inflammatory lesion in the brain,9 it is probable that NF-κB is involved in the rapid induction of SAA1 by CCI in our study. We found that, in addition to induction of SAA1 expression, brain injury induced hepatic neutrophil and macrophage infiltration, enhanced expression of the chemokines CXCL-1 and CXCL-10, and increased the number of apoptotic cells in the liver. Hepatic infiltration of neutrophils is an early response to injury or systemic inflammation60 and may be influenced by CXCL-1, a neutrophil chemoattractant.61 CXCL-1 was shown to amplify cellular hepatic changes after a focal brain inflammatory lesion, and its inhibition reduced the recruitment of neutrophils to the liver, hepatocyte damage, and leukocyte mobilization.61 Thus, inhibition of CXCL-1 after CCI may be an early event to intervene to reduce hepatic amplification of inflammatory signals resulting from TBI.
Increased SAA1 Expression Does Not Induce Amyloid Aggregation
In addition, our results show that rapid hepatic SAA1 production, and its release after brain injury, occurs before the secondary brain inflammatory response that can persist days after brain injury, after SAA1 expression returns to normal levels. These data suggest the possibility that the acute hepatic inflammatory response may contribute to secondary brain injury. SAA1 expression was also detected in the injured brain at 1 dpi, in cells with vacuolated morphology surrounding the lesion border. Further characterization will be needed to identify these cells. Moreover, we have provided evidence that the expression of SAA1 alone in the liver after brain injury does not result in formation of detectable amyloid species. One possibility is that the persistent accumulation of SAA1 over longer time periods is required to initiate amyloid formation. Thus, we hypothesize that TBI-induced inflammation is not robust enough to induce amyloid deposition in the liver; rather, a persistent (chronic) inflammatory response is likely required for amyloid aggregation.
Renin Angiotensin System Involvement in the Hepatic Inflammatory Response to TBI
Little is known of the hepatic RAS in response to TBI. Angiotensinogen, the precursor of Ang II, is synthesized by hepatocytes and is induced during the APR62 in response to circulating proinflammatory cytokines. Expression of the AT1R is also induced by proinflammatory cytokines, mediated by NF-κB activation.63,64 We have shown that AT1R mRNA is induced 3 days after TBI, but not at 6 hours. Because serum cytokine induction after injury is more rapid than AT1R mRNA induction, our data suggest that the mechanism of AT1R induction may be more complex than through cytokine induction alone. Because biological activity of Ang II is mediated by the level of its receptor, this later increase in AT1R mRNA expression may suggest increased importance of Ang II actions in the liver at this later time point. Indeed, activation of the RAS is involved in the pathophysiology of different models of liver tissue injury.65,66
To determine whether AT1R signaling was important in the hepatic response to brain injury, we inhibited Ang II signaling through the AT1R using the ARB, telmisartan. Telmisartan reduced the induction of SAA1 mRNA and protein at 3 dpi, but not at earlier time points. It is, therefore, possible that a different mechanism is responsible for the early stronger induction of SAA1 expression. These data show that telmisartan alters SAA1 mRNA and protein expression at a subacute stage, supporting the involvement of Ang II at this slightly later stage of the APR. Telmisartan did, however, repress TBI-mediated induction of CXCL-10 expression 6 hours after injury, but not that of CXCL-1. These data, therefore, show that telmisartan can specifically alter a subset of hepatic responses to TBI, suggesting involvement of hepatic Ang II signaling in these responses. Telmisartan is effective in reducing the generation of oxidative stress and proinflammatory mediators,40 and specifically has reduced hepatic damage caused by arsenic exposure in mice.49 Likewise, it was demonstrated that another ARB, candesartan, decreases both peripheral inflammation produced by bacterial endotoxin and the corresponding brain inflammation.36
Influence of the Liver on the Brain's Response to Injury
Communication between the brain and liver is bidirectional. The hepatic APR leads to mobilization of leukocytes, and induction of acute-phase proteins, cytokines, and chemokines.10 These cells and signaling molecules enter the bloodstream and will reach the cerebral circulation to affect cells in the central nervous system. Specific inhibition of hepatic NF-κB activity through injection of a repressor into the liver reduced the number of neutrophils recruited to the brain after injury.9 Similarly, neutrophil infiltration into the brain after a focal inflammatory lesion was reduced by selective depletion of hepatic Kupffer cells or by treatment with the peripheral-acting, TNF-α blocker etanercept.67,68 Thus, the hepatic response to brain injury can affect the inflammatory status of the brain, and hence presumably recovery from injury. A critical unanswered question is whether the effect of telmisartan on the hepatic response to injury is involved in its beneficial effect on the brain after TBI.
High levels of circulating SAA1 after TBI may also directly affect the brain response to injury. We found that mRNA and protein expression of SAA1 was increased only 150-fold in injured mice liver compared with controls, considerably smaller than the reported 1000-fold increase generated by lipopolysaccharide.69 SAA1 is considered a cytokine-like molecule, which can induce cytokines that modulate lymphocyte function,70,71 and act as a chemotactic agent for various leukocytes.72 Elevated SAA1 levels are associated with some autoimmune disease phenotypes, such as multiple sclerosis, where SAA1 can influence T-cell phenotype toward type 17 helper T-cell predominance.73 Because SAA1 can signal through several different inflammatory receptors usually found on leukocytes, to alter intracellular signaling, SAA1 can have a broad effect on inflammation, often using NF-κB–regulated pathways.74 Thus, SAA1 in the serum could influence many different organs in the body after brain injury, including the brain itself.
Chronically elevated SAA1 levels are a prerequisite for the development of secondary, or reactive, amyloidosis,75 a disease characterized by the accumulation of insoluble plaques composed principally of proteolytically cleaved SAA1.75,76 Systemic inflammation can increase cytokine expression in the brain and initiate amyloid deposition via activation of microglial cells and amyloid-associated proteins.29,77 We have demonstrated that the expression of SAA1 in the liver after brain injury did not result in the formation of detectable amyloid species. Because SAA1 levels decreased significantly by 3 dpi, presumably persistent accumulation of SAA1 for a longer time period is needed to initiate amyloid formation.75 It would be interesting to determine whether repetitive brain injuries would lead to a larger and more sustained elevation in SAA1 levels that may then lead to amyloid formation.
Conclusions
We have shown that TBI in mice induces an increase in expression of the acute-phase protein, SAA1, and also AT1R mRNA, together with several other inflammatory changes in liver indicative of a partial APR. Telmisartan can mitigate some, but not all, of these changes, showing that multiple mechanisms are operating to promote these alterations. Thus, we have shown a strong inflammatory response in the liver in response to brain injury that might contribute to the inflammatory response of the brain. It remains to be seen whether therapeutic manipulation of the hepatic response to brain injury will have a beneficial effect on the brain's ability to recover from injury. Peripheral inhibition of inflammation may reduce the brain's own inflammatory cascades and, thus, warrants further investigation.
Footnotes
Supported by Center for Neuroscience and Regenerative Medicine grants (A.J.S.), a postdoctoral fellowship (S.V.) and the Intramural Research Program at the National Institute of Mental Health/NIH (MH 002762-16; J.M.S.).
The opinions and assertions contained herein are the private opinions of the authors and are not to be construed as reflecting the views of the Uniformed Services University of the Health Sciences, the US Department of Defense, or the US government.
Disclosures: None declared.
Supplemental material accompanying this article can be found at http://dx.doi.org/10.1016/j.ajpath.2015.06.016.
Contributor Information
Sonia Villapol, Email: sonia.villapol@georgetown.edu.
Aviva J. Symes, Email: aviva.symes@usuhs.edu.
Supplemental Data
Supplemental Figure S1.

Acute inflammatory response in the brain, cortical lesion, and serum amyloid A1 (SAA1) expression in the liver. A: Immunohistochemical images showing SAA1 expression in the brain in naïve control mice and at 6 hours and 1, 3, 7, and 30 days after injury. Sparse SAA1-positive cells are observed near the border of the lesion, with vacuolated morphology (high-magnification images) at 1 day post-injury (dpi). B: Cortical lesion volume was measured and was greatest at 1 and 3 dpi. C and D: The number of monocytes or polymorphonuclear leukocytes (CD14-positive cells); C) dramatically increases at 6 hours post-injury (hpi) and is maintained up to 3 dpi; however, the number of macrophages (CD68-positive cells) reaches a peak at 3 dpi (D) in the cortical region bordering the impact site. E: Relationship between hepatic SAA1 expression, inflammatory cell density in the brain, and brain lesion volume at 6 hpi and 1 and 3 dpi. Data are expressed as means ± SEM (B–D). n = 5 to 8 per group (B–D). ∗∗P < 0.01, ∗∗∗P < 0.001, comparing injured brains with control brains. CCI, cortical impact injury.
References
- 1.DeLegge M.H., Smoke A. Neurodegeneration and inflammation. Nutr Clin Pract. 2008;23:35–41. doi: 10.1177/011542650802300135. [DOI] [PubMed] [Google Scholar]
- 2.Mayer C.L., Huber B.R., Peskind E. Traumatic brain injury, neuroinflammation, and post-traumatic headaches. Headache. 2013;53:1523–1530. doi: 10.1111/head.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anthony D.C., Couch Y., Losey P., Evans M.C. The systemic response to brain injury and disease. Brain Behav Immun. 2012;26:534–540. doi: 10.1016/j.bbi.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 4.Tobin R.P., Mukherjee S., Kain J.M., Rogers S.K., Henderson S.K., Motal H.L., Rogers M.K., Shapiro L.A. Traumatic brain injury causes selective, CD74-dependent peripheral lymphocyte activation that exacerbates neurodegeneration. Acta Neuropathol Commun. 2014;2:143. doi: 10.1186/s40478-014-0143-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weber D.J., Gracon A.S., Ripsch M.S., Fisher A.J., Cheon B.M., Pandya P.H., Vittal R., Capitano M.L., Kim Y., Allette Y.M., Riley A.A., McCarthy B.P., Territo P.R., Hutchins G.D., Broxmeyer H.E., Sandusky G.E., White F.A., Wilkes D.S. The HMGB1-RAGE axis mediates traumatic brain injury-induced pulmonary dysfunction in lung transplantation. Sci Transl Med. 2014;6:252ra124. doi: 10.1126/scitranslmed.3009443. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Woodcock T., Morganti-Kossmann M.C. The role of markers of inflammation in traumatic brain injury. Front Neurol. 2013;4:18. doi: 10.3389/fneur.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kossmann T., Hans V.H., Imhof H.G., Stocker R., Grob P., Trentz O., Morganti-Kossmann C. Intrathecal and serum interleukin-6 and the acute-phase response in patients with severe traumatic brain injuries. Shock. 1995;4:311–317. doi: 10.1097/00024382-199511000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Ramadori G., Christ B. Cytokines and the hepatic acute-phase response. Semin Liver Dis. 1999;19:141–155. doi: 10.1055/s-2007-1007106. [DOI] [PubMed] [Google Scholar]
- 9.Campbell S.J., Anthony D.C., Oakley F., Carlsen H., Elsharkawy A.M., Blomhoff R., Mann D.A. Hepatic nuclear factor kappa B regulates neutrophil recruitment to the injured brain. J Neuropathol Exp Neurol. 2008;67:223–230. doi: 10.1097/NEN.0b013e3181654957. [DOI] [PubMed] [Google Scholar]
- 10.Kushner I. The phenomenon of the acute phase response. Ann N Y Acad Sci. 1982;389:39–48. doi: 10.1111/j.1749-6632.1982.tb22124.x. [DOI] [PubMed] [Google Scholar]
- 11.Uhlar C.M., Whitehead A.S. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem. 1999;265:501–523. doi: 10.1046/j.1432-1327.1999.00657.x. [DOI] [PubMed] [Google Scholar]
- 12.Gao W., Lu C., Kochanek P.M., Berger R.P. Serum amyloid A is increased in children with abusive head trauma: a gel-based proteomic analysis. Pediatr Res. 2014;76:280–286. doi: 10.1038/pr.2014.86. [DOI] [PubMed] [Google Scholar]
- 13.Hergenroeder G., Redell J.B., Moore A.N., Dubinsky W.P., Funk R.T., Crommett J., Clifton G.L., Levine R., Valadka A., Dash P.K. Identification of serum biomarkers in brain-injured adults: potential for predicting elevated intracranial pressure. J Neurotrauma. 2008;25:79–93. doi: 10.1089/neu.2007.0386. [DOI] [PubMed] [Google Scholar]
- 14.Mozes G., Friedman N., Shainkin-Kestenbaum R. Serum amyloid A: an extremely sensitive marker for intensity of tissue damage in trauma patients and indicator of acute response in various diseases. J Trauma. 1989;29:71–74. [PubMed] [Google Scholar]
- 15.Liao F., Lusis A.J., Berliner J.A., Fogelman A.M., Kindy M., de Beer M.C., de Beer F.C. Serum amyloid A protein family: differential induction by oxidized lipids in mouse strains. Arterioscler Thromb. 1994;14:1475–1479. doi: 10.1161/01.atv.14.9.1475. [DOI] [PubMed] [Google Scholar]
- 16.O'Brien K.D., Chait A. Serum amyloid A: the “other” inflammatory protein. Curr Atheroscler Rep. 2006;8:62–68. doi: 10.1007/s11883-006-0066-0. [DOI] [PubMed] [Google Scholar]
- 17.Villapol S., Bonnin P., Fau S., Baud O., Renolleau S., Charriaut-Marlangue C. Unilateral blood flow decrease induces bilateral and symmetric responses in the immature brain. Am J Pathol. 2009;175:2111–2120. doi: 10.2353/ajpath.2009.090257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jensen L.E., Whitehead A.S. Regulation of serum amyloid A protein expression during the acute-phase response. Biochem J. 1998;334(Pt 3):489–503. doi: 10.1042/bj3340489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Badolato R., Wang J.M., Murphy W.J., Lloyd A.R., Michiel D.F., Bausserman L.L., Kelvin D.J., Oppenheim J.J. Serum amyloid A is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J Exp Med. 1994;180:203–209. doi: 10.1084/jem.180.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu L., Badolato R., Murphy W.J., Longo D.L., Anver M., Hale S., Oppenheim J.J., Wang J.M. A novel biologic function of serum amyloid A: induction of T lymphocyte migration and adhesion. J Immunol. 1995;155:1184–1190. [PubMed] [Google Scholar]
- 21.Hatanaka E., Pereira Ribeiro F., Campa A. The acute phase protein serum amyloid A primes neutrophils. FEMS Immunol Med Microbiol. 2003;38:81–84. doi: 10.1016/S0928-8244(03)00112-3. [DOI] [PubMed] [Google Scholar]
- 22.Vallon R., Freuler F., Desta-Tsedu N., Robeva A., Dawson J., Wenner P., Engelhardt P., Boes L., Schnyder J., Tschopp C., Urfer R., Baumann G. Serum amyloid A (apoSAA) expression is up-regulated in rheumatoid arthritis and induces transcription of matrix metalloproteinases. J Immunol. 2001;166:2801–2807. doi: 10.4049/jimmunol.166.4.2801. [DOI] [PubMed] [Google Scholar]
- 23.Cheng N., He R., Tian J., Ye P.P., Ye R.D. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J Immunol. 2008;181:22–26. doi: 10.4049/jimmunol.181.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He R., Sang H., Ye R.D. Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood. 2003;101:1572–1581. doi: 10.1182/blood-2002-05-1431. [DOI] [PubMed] [Google Scholar]
- 25.Niemi K., Teirila L., Lappalainen J., Rajamaki K., Baumann M.H., Oorni K., Wolff H., Kovanen P.T., Matikainen S., Eklund K.K. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J Immunol. 2011;186:6119–6128. doi: 10.4049/jimmunol.1002843. [DOI] [PubMed] [Google Scholar]
- 26.Okamoto H., Katagiri Y., Kiire A., Momohara S., Kamatani N. Serum amyloid A activates nuclear factor-kappaB in rheumatoid synovial fibroblasts through binding to receptor of advanced glycation end-products. J Rheumatol. 2008;35:752–756. [PubMed] [Google Scholar]
- 27.Sandri S., Rodriguez D., Gomes E., Monteiro H.P., Russo M., Campa A. Is serum amyloid A an endogenous TLR4 agonist? J Leukoc Biol. 2008;83:1174–1180. doi: 10.1189/jlb.0407203. [DOI] [PubMed] [Google Scholar]
- 28.Su S.B., Gong W., Gao J.L., Shen W., Murphy P.M., Oppenheim J.J., Wang J.M. A seven-transmembrane, G protein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J Exp Med. 1999;189:395–402. doi: 10.1084/jem.189.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu Y., Liu J., Li S.Q., Peng L., Ye R.D. Serum amyloid A differentially activates microglia and astrocytes via the PI3K pathway. J Alzheimers Dis. 2014;38:133–144. doi: 10.3233/JAD-130818. [DOI] [PubMed] [Google Scholar]
- 30.Lindhorst E., Young D., Bagshaw W., Hyland M., Kisilevsky R. Acute inflammation, acute phase serum amyloid A and cholesterol metabolism in the mouse. Biochim Biophys Acta. 1997;1339:143–154. doi: 10.1016/s0167-4838(96)00227-0. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki Y., Ruiz-Ortega M., Lorenzo O., Ruperez M., Esteban V., Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol. 2003;35:881–900. doi: 10.1016/s1357-2725(02)00271-6. [DOI] [PubMed] [Google Scholar]
- 32.Benigni A., Cassis P., Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO Mol Med. 2010;2:247–257. doi: 10.1002/emmm.201000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El Bekay R., Alvarez M., Monteseirin J., Alba G., Chacon P., Vega A., Martin-Nieto J., Jimenez J., Pintado E., Bedoya F.J., Sobrino F. Oxidative stress is a critical mediator of the angiotensin II signal in human neutrophils: involvement of mitogen-activated protein kinase, calcineurin, and the transcription factor NF-kappaB. Blood. 2003;102:662–671. doi: 10.1182/blood-2002-09-2785. [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez-Perez A.I., Borrajo A., Rodriguez-Pallares J., Guerra M.J., Labandeira-Garcia J.L. Interaction between NADPH-oxidase and Rho-kinase in angiotensin II-induced microglial activation. Glia. 2015;63:466–482. doi: 10.1002/glia.22765. [DOI] [PubMed] [Google Scholar]
- 35.Lubel J.S., Herath C.B., Burrell L.M., Angus P.W. Liver disease and the renin-angiotensin system: recent discoveries and clinical implications. J Gastroenterol Hepatol. 2008;23:1327–1338. doi: 10.1111/j.1440-1746.2008.05461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benicky J., Sanchez-Lemus E., Honda M., Pang T., Orecna M., Wang J., Leng Y., Chuang D.M., Saavedra J.M. Angiotensin II AT1 receptor blockade ameliorates brain inflammation. Neuropsychopharmacology. 2011;36:857–870. doi: 10.1038/npp.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huet O., Chin-Dusting J.P. Septic shock: desperately seeking treatment. Clin Sci (Lond) 2014;126:31–39. doi: 10.1042/CS20120668. [DOI] [PubMed] [Google Scholar]
- 38.Steckelings U.M., Rompe F., Kaschina E., Unger T. The evolving story of the RAAS in hypertension, diabetes and CV disease: moving from macrovascular to microvascular targets. Fundam Clin Pharmacol. 2009;23:693–703. doi: 10.1111/j.1472-8206.2009.00780.x. [DOI] [PubMed] [Google Scholar]
- 39.Zhang L., Du J., Hu Z., Han G., Delafontaine P., Garcia G., Mitch W.E. IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J Am Soc Nephrol. 2009;20:604–612. doi: 10.1681/ASN.2008060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang J., Pang T., Hafko R., Benicky J., Sanchez-Lemus E., Saavedra J.M. Telmisartan ameliorates glutamate-induced neurotoxicity: roles of AT(1) receptor blockade and PPARgamma activation. Neuropharmacology. 2014;79:249–261. doi: 10.1016/j.neuropharm.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villapol S., Byrnes K.R., Symes A.J. Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte-vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury. Front Neurol. 2014;5:82. doi: 10.3389/fneur.2014.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Villapol S., Yaszemski A.K., Logan T.T., Sanchez-Lemus E., Saavedra J.M., Symes A.J. Candesartan, an angiotensin II AT(1)-receptor blocker and PPAR-gamma agonist, reduces lesion volume and improves motor and memory function after traumatic brain injury in mice. Neuropsychopharmacology. 2012;37:2817–2829. doi: 10.1038/npp.2012.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Villapol S., Balarezo M.G., Affram K., Saavedra J.M., Symes A.J. Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain. 2015 doi: 10.1093/brain/awv172. [in press] doi: 10.1093/brain/awv172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jung K.H., Chu K., Lee S.T., Kim S.J., Song E.C., Kim E.H., Park D.K., Sinn D.I., Kim J.M., Kim M., Roh J.K. Blockade of AT1 receptor reduces apoptosis, inflammation, and oxidative stress in normotensive rats with intracerebral hemorrhage. J Pharmacol Exp Ther. 2007;322:1051–1058. doi: 10.1124/jpet.107.120097. [DOI] [PubMed] [Google Scholar]
- 45.Villapol S., Saavedra J.M. Neuroprotective effects of angiotensin receptor blockers. Am J Hypertens. 2015;28:289–299. doi: 10.1093/ajh/hpu197. [DOI] [PubMed] [Google Scholar]
- 46.Benson S.C., Pershadsingh H.A., Ho C.I., Chittiboyina A., Desai P., Pravenec M., Qi N., Wang J., Avery M.A., Kurtz T.W. Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating activity. Hypertension. 2004;43:993–1002. doi: 10.1161/01.HYP.0000123072.34629.57. [DOI] [PubMed] [Google Scholar]
- 47.Enjoji M., Kotoh K., Kato M., Higuchi N., Kohjima M., Nakashima M., Nakamuta M. Therapeutic effect of ARBs on insulin resistance and liver injury in patients with NAFLD and chronic hepatitis C: a pilot study. Int J Mol Med. 2008;22:521–527. [PubMed] [Google Scholar]
- 48.Home P.D., Pacini G. Hepatic dysfunction and insulin insensitivity in type 2 diabetes mellitus: a critical target for insulin-sensitizing agents. Diabetes Obes Metab. 2008;10:699–718. doi: 10.1111/j.1463-1326.2007.00761.x. [DOI] [PubMed] [Google Scholar]
- 49.Fouad A.A., Al-Mulhim A.S., Jresat I. Telmisartan treatment attenuates arsenic-induced hepatotoxicity in mice. Toxicology. 2012;300:149–157. doi: 10.1016/j.tox.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 50.Committee for the Update of the Guide for the Care and Use of Laboratory Animals; National Research Council . National Academies Press; Washington, DC: 2011. Guide for the Care and Use of Laboratory Animals: Eighth Edition. [Google Scholar]
- 51.Saavedra J.M. Angiotensin II AT(1) receptor blockers as treatments for inflammatory brain disorders. Clin Sci (Lond) 2012;123:567–590. doi: 10.1042/CS20120078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simons J.P., Al-Shawi R., Ellmerich S., Speck I., Aslam S., Hutchinson W.L., Mangione P.P., Disterer P., Gilbertson J.A., Hunt T., Millar D.J., Minogue S., Bodin K., Pepys M.B., Hawkins P.N. Pathogenetic mechanisms of amyloid A amyloidosis. Proc Natl Acad Sci U S A. 2013;110:16115–16120. doi: 10.1073/pnas.1306621110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kryndushkin D., Pripuzova N., Burnett B.G., Shewmaker F. Non-targeted identification of prions and amyloid-forming proteins from yeast and mammalian cells. J Biol Chem. 2013;288:27100–27111. doi: 10.1074/jbc.M113.485359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kryndushkin D., Wear M.P., Shewmaker F. Amyloid cannot resist identification. Prion. 2013;7:464–468. doi: 10.4161/pri.27503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bundzikova J., Pirnik Z., Lackovicova L., Mravec B., Kiss A. Brain-liver interactions during liver ischemia reperfusion injury: a minireview. Endocr Regul. 2011;45:163–172. doi: 10.4149/endo_2011_03_163. [DOI] [PubMed] [Google Scholar]
- 56.Yoneda M., Watanobe H., Terano A. Central regulation of hepatic function by neuropeptides. J Gastroenterol. 2001;36:361–367. doi: 10.1007/s005350170079. [DOI] [PubMed] [Google Scholar]
- 57.Shimizu H., Yamamoto K. NF-kappa B and C/EBP transcription factor families synergistically function in mouse serum amyloid A gene expression induced by inflammatory cytokines. Gene. 1994;149:305–310. doi: 10.1016/0378-1119(94)90166-x. [DOI] [PubMed] [Google Scholar]
- 58.Ferreira L.C., Regner A., Miotto K.D., Moura S., Ikuta N., Vargas A.E., Chies J.A., Simon D. Increased levels of interleukin-6, -8 and -10 are associated with fatal outcome following severe traumatic brain injury. Brain Inj. 2014;28:1311–1316. doi: 10.3109/02699052.2014.916818. [DOI] [PubMed] [Google Scholar]
- 59.Lumpkins K., Bochicchio G.V., Zagol B., Ulloa K., Simard J.M., Schaub S., Meyer W., Scalea T. Plasma levels of the beta chemokine regulated upon activation, normal T cell expressed, and secreted (RANTES) correlate with severe brain injury. J Trauma. 2008;64:358–361. doi: 10.1097/TA.0b013e318160df9b. [DOI] [PubMed] [Google Scholar]
- 60.Jaeschke H., Hasegawa T. Role of neutrophils in acute inflammatory liver injury. Liver Int. 2006;26:912–919. doi: 10.1111/j.1478-3231.2006.01327.x. [DOI] [PubMed] [Google Scholar]
- 61.Campbell S.J., Hughes P.M., Iredale J.P., Wilcockson D.C., Waters S., Docagne F., Perry V.H., Anthony D.C. CINC-1 is an acute-phase protein induced by focal brain injury causing leukocyte mobilization and liver injury. FASEB J. 2003;17:1168–1170. doi: 10.1096/fj.02-0757fje. [DOI] [PubMed] [Google Scholar]
- 62.Ron D., Brasier A.R., Habener J.F. Transcriptional regulation of hepatic angiotensinogen gene expression by the acute-phase response. Mol Cell Endocrinol. 1990;74:C97–C104. doi: 10.1016/0303-7207(90)90221-s. [DOI] [PubMed] [Google Scholar]
- 63.Cowling R.T., Gurantz D., Peng J., Dillmann W.H., Greenberg B.H. Transcription factor NF-kappa B is necessary for up-regulation of type 1 angiotensin II receptor mRNA in rat cardiac fibroblasts treated with tumor necrosis factor-alpha or interleukin-1 beta. J Biol Chem. 2002;277:5719–5724. doi: 10.1074/jbc.M107515200. [DOI] [PubMed] [Google Scholar]
- 64.Mitra A.K., Gao L., Zucker I.H. Angiotensin II-induced upregulation of AT(1) receptor expression: sequential activation of NF-kappaB and Elk-1 in neurons. Am J Physiol Cell Physiol. 2010;299:C561–C569. doi: 10.1152/ajpcell.00127.2010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 65.Wynn T.A. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bataller R., Sancho-Bru P., Gines P., Brenner D.A. Liver fibrogenesis: a new role for the renin-angiotensin system. Antioxid Redox Signal. 2005;7:1346–1355. doi: 10.1089/ars.2005.7.1346. [DOI] [PubMed] [Google Scholar]
- 67.Campbell S.J., Jiang Y., Davis A.E., Farrands R., Holbrook J., Leppert D., Anthony D.C. Immunomodulatory effects of etanercept in a model of brain injury act through attenuation of the acute-phase response. J Neurochem. 2007;103:2245–2255. doi: 10.1111/j.1471-4159.2007.04928.x. [DOI] [PubMed] [Google Scholar]
- 68.Campbell S.J., Zahid I., Losey P., Law S., Jiang Y., Bilgen M., van Rooijen N., Morsali D., Davis A.E., Anthony D.C. Liver Kupffer cells control the magnitude of the inflammatory response in the injured brain and spinal cord. Neuropharmacology. 2008;55:780–787. doi: 10.1016/j.neuropharm.2008.06.074. [DOI] [PubMed] [Google Scholar]
- 69.Chiba T., Chang M.Y., Wang S., Wight T.N., McMillen T.S., Oram J.F., Vaisar T., Heinecke J.W., De Beer F.C., De Beer M.C., Chait A. Serum amyloid A facilitates the binding of high-density lipoprotein from mice injected with lipopolysaccharide to vascular proteoglycans. Arterioscler Thromb Vasc Biol. 2011;31:1326–1332. doi: 10.1161/ATVBAHA.111.226159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gorevic P.D. Amyloid and inflammation. Proc Natl Acad Sci U S A. 2013;110:16291–16292. doi: 10.1073/pnas.1315112110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Christenson K., Bjorkman L., Tangemo C., Bylund J. Serum amyloid A inhibits apoptosis of human neutrophils via a P2X7-sensitive pathway independent of formyl peptide receptor-like 1. J Leukoc Biol. 2008;83:139–148. doi: 10.1189/jlb.0507276. [DOI] [PubMed] [Google Scholar]
- 72.Selinger M.J., McAdam K.P., Kaplan M.M., Sipe J.D., Vogel S.N., Rosenstreich D.L. Monokine-induced synthesis of serum amyloid A protein by hepatocytes. Nature. 1980;285:498–500. doi: 10.1038/285498a0. [DOI] [PubMed] [Google Scholar]
- 73.Yokote H., Yagi Y., Watanabe Y., Amino T., Kamata T., Mizusawa H. Serum amyloid A level is increased in neuromyelitis optica and atypical multiple sclerosis with smaller T2 lesion volume in brain MRI. J Neuroimmunol. 2013;259:92–95. doi: 10.1016/j.jneuroim.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 74.Li H., Ooi S.Q., Heng C.K. The role of NF-κB in SAA-induced peroxisome proliferator-activated receptor gamma activation. Atherosclerosis. 2013;227:72–78. doi: 10.1016/j.atherosclerosis.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 75.Obici L., Merlini G. AA amyloidosis: basic knowledge, unmet needs and future treatments. Swiss Med Wkly. 2012;142:w13580. doi: 10.4414/smw.2012.13580. [DOI] [PubMed] [Google Scholar]
- 76.Lowell C.A., Stearman R.S., Morrow J.F. Transcriptional regulation of serum amyloid A gene expression. J Biol Chem. 1986;261:8453–8461. [PubMed] [Google Scholar]
- 77.Eikelenboom P., Hoozemans J.J., Veerhuis R., van Exel E., Rozemuller A.J., van Gool W.A. Whether, when and how chronic inflammation increases the risk of developing late-onset Alzheimer's disease. Alzheimers Res Ther. 2012;4:15. doi: 10.1186/alzrt118. [DOI] [PMC free article] [PubMed] [Google Scholar]