

Abstract
Transgenic overexpression of Galgt2 (official name B4Galnt2) in skeletal muscle stimulates the glycosylation of α dystroglycan (αDG) and the up-regulation of laminin α2 and dystrophin surrogates known to inhibit muscle pathology in mouse models of congenital muscular dystrophy 1A and Duchenne muscular dystrophy. Skeletal muscle Galgt2 gene expression is also normally increased in the mdx mouse model of Duchenne muscular dystrophy compared with the wild-type mice. To assess whether this increased endogenous Galgt2 expression could affect disease, we quantified muscular dystrophy measures in mdx mice deleted for Galgt2 (Galgt2−/−mdx). Galgt2−/− mdx mice had increased heart and skeletal muscle pathology and inflammation, and also worsened cardiac function, relative to age-matched mdx mice. Deletion of Galgt2 in wild-type mice also slowed skeletal muscle growth in response to acute muscle injury. In each instance where Galgt2 expression was elevated (developing muscle, regenerating muscle, and dystrophic muscle), Galgt2-dependent glycosylation of αDG was also increased. Overexpression of Galgt2 failed to inhibit skeletal muscle pathology in dystroglycan-deficient muscles, in contrast to previous studies in dystrophin-deficient mdx muscles. This study demonstrates that Galgt2 gene expression and glycosylation of αDG are dynamically regulated in muscle and that endogenous Galgt2 gene expression can ameliorate the extent of muscle pathology, inflammation, and dysfunction in mdx mice.
Duchenne muscular dystrophy (DMD) is a severe X-linked myopathy of childhood that arises from the loss of dystrophin protein expression in cardiac and skeletal muscles.1–4 Muscle disease in DMD results from increased damage to contracting myofibers as the result of dystrophin protein deficiency.5,6 In response to muscle injury, immune cells, predominantly macrophages, invade the damaged muscle to clear necrotic tissue.7–9 This inflammatory response and a regenerative response, whereby satellite cells, the predominant stem cells in skeletal muscle, divide to generate myoblasts that ultimately fuse to form new myofibers, chronically occur in DMD muscles.10–13 The regenerative response diminishes, however, as the disease progresses, leading to the replacement of muscle tissue with extracellular matrix or fat, termed muscle wasting.14,15 Muscle wasting is the primary cause of the profound skeletal muscle weakness, loss of ambulation, and respiratory impairment in DMD. Similar to skeletal myofibers, dystrophin-deficient cardiomyocytes also become damaged in DMD and produce dilated cardiomyopathy, which, along with respiratory insufficiency, are the major drivers of premature mortality.16
Despite the fact that dystrophin was identified as the defective gene causing DMD >20 years ago,2,3 no therapy that alters the ultimate disease course has yet been shown to be effective in patients. Prednisone and deflazacort therapy have been shown to prolong ambulation by several years, but this benefit is not sustained and the adverse effects of long-term therapy are significant.17,18 Improvements in supportive respiratory and cardiac care have prolonged patient survival, but do not affect the underlying muscle pathology.19 Although several potentially impactful therapeutic approaches, such as gene replacement therapy,20–24 exon skipping therapies,1,4,25 stem or myoblast cell therapies,26,27 proteasome inhibitor,28,29 and muscle building therapies,30–32 show promise, these approaches have significant challenges. Surrogate gene or protein therapies that cause increased expression of proteins not mutated or lost in the disease can impart increased protection to heart and skeletal muscle or can increase muscle mass. Such therapies provide an alternative to gene replacement for DMD and other forms of muscular dystrophy that may bypass T-cell responses that are known to occur to dystrophin protein in DMD patients.33 Several surrogate gene therapies, including utrophin, sarcospan, integrin α7, and follistatin, can inhibit muscle pathology when overexpressed and exacerbate muscle pathology when deleted in mdx mice.34–44 Our laboratory first showed that overexpression of β1,4-N-acetylgalacosaminyl transferase 2 (Galgt2; official name B4Galnt2), a glycosyltransferase normally confined to the neuromuscular and myotendinous junctions in adult skeletal muscle that generates the cytotoxic T-cell (CT) glycan,45,46 can protect both wild-type and dystrophic skeletal myofibers from injury.47 Galgt2 overexpression has been shown to inhibit the development of muscular dystrophy in three mouse models of human disease, dystrophin deficiency (mdx48,49), α sarcoglycan deficiency (Sgca−/−50), and laminin α2 deficiency (dyW49), in part by increasing expression of normally synaptic extracellular matrix proteins, particularly laminin α4, laminin α5, and agrin, and increasing their respective binding to Galgt2-glycosylated α dystroglycan (αDG).51 Galgt2 overexpression also increases the expression of dystrophin surrogates (utrophin and plectin1) and can greatly strengthen the muscle membrane's resistance to injury, both in mdx and wild-type mice.50,51
Although several studies have shown a benefit of transgenic Galgt2 overexpression on disease,47–50,52,53 no data yet exist on the potential impact of endogenous muscle Galgt2 gene expression, which we explored. The most used mouse model for DMD is the mdx mouse, which fails to express dystrophin protein in almost all skeletal and cardiac muscle cells because of a missense mutation in exon 23 of the mouse dystrophin gene,54,55 and endogenous Galgt2 gene expression is normally elevated in mdx skeletal muscles relative to wild-type muscles.56 To determine the impact of elevated endogenous Galgt2 gene expression in DMD, we have generated Galgt2-deficient mdx mice (Galgt2−/−mdx). The experiments presented herein suggest that Galgt2 is similar to other endogenous surrogate gene disease modifiers because it can inhibit disease pathology when overexpressed and increase disease pathology when deleted.
Materials and Methods
Mice
All mouse experiments were performed using protocols approved by the Institutional Animal Care and Use Committee at The Research Institute at Nationwide Children's Hospital (Columbus, OH). Galgt2−/− mice were obtained by the Consortium for Functional Glycomics and were originally generated by John Lowe (Genentech, South San Francisco, CA). The mdx mice55 (C57Bl/10) were obtained from Jackson Laboratories (Bar Harbor, ME). Galgt2−/− mice were bred to mdx mice to generate Galgt2−/−mdx/mdx female and Galgt2−/−mdx/Y male mice. Age-matched littermates from such crosses were always used as controls. Mice bearing a floxed allele of Dag1 (Dag1lox) and Pax3-Cre transgenic mice (P3Pro-Cre), where Cre is expressed in caudal skeletal muscles but not in rostral skeletal muscles because of the integration site of the transgene,57 were kindly provided by Jeffery Miner (Washington University Medical School, St. Louis, MO). Dag1lox/lox mice were generated by Cohn et al58 (The University of Iowa, Iowa City), and P3Pro-Cre mice were generated by Li et al59 (University of Pennsylvania, Philadelphia). Female Dag1lox/lox mice were bred with male P3Pro-Cre transgenic mice to generate P3Pro-CreDag1lox/+ mice. Male P3Pro-CreTgDag1loxP/+ mice were then bred with female Dag1lox/lox mice to generate P3Pro-CreDag1lox/lox mice, where Dag1 was deleted in caudal skeletal muscles (Dag1−/−). Dag1lox/lox littermates were used as Dag1+/+ controls. P3Pro-Cre mice were also tested as controls and did not differ significantly from Dag1lox/lox mice for the experiments presented herein. Skeletal muscle-specific Galgt2 transgenic mice (mCT), where transgene expression is driven by the skeletal α actin promoter/intron,60,61 and Galgt2 transgenic mdx mice were generated as described previously.48,62 mCT mice were bred to P3ProCreDag1lox/lox mice, and offspring were intercrossed to generate mCTP3ProCreDag1lox/lox mice. For all experimental comparisons, equivalent numbers of male and female mice were used in each grouping.
Ctx-Induced Muscle Regeneration
Gastrocnemius muscles were injected with 10 μmol/L cardiotoxin (Ctx; from Naga mossambica mossambica; C9759; Sigma, St. Louis, MO) in 50 μL sterile phosphate-buffered saline (PBS). Control gastrocnemius muscles were injected with an equivalent volume of sterile PBS alone. Mice were sacrificed by CO2 inhalation at 1, 4, 7, 14, or 28 days after injection, and the gastrocnemius muscle was harvested and snap frozen in liquid nitrogen–cooled isopentane for subsequent histological and biochemical analysis. Young adult (2-month-old) mice were used for all Ctx experiments. For Ctx experiments comparing wild-type with Galgt2−/− muscles, and for non-Ctx experiments comparing mdx with Galgt2−/−mdx muscles, six muscles were averaged per condition, with 1600 to 3600 total myofibers counted per condition.
Semiquantitative Real-Time PCR
Relative transcription levels were assessed by semiquantitative real-time PCR using the Δ-Δ-CT method63 with 18S RNA or glyceraldehyde-3-phosphate dehydrogenase as an internal reference. 18S RNA was used for Ctx-induced regeneration experiments and for muscle development, whereas glyceraldehyde-3-phosphate dehydrogenase was used to compare all other age-matched genetic mouse strains. Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA) from frozen blocks of gastrocnemius muscle, where expression of Galgt2 protein and CT glycan had already been assessed by immunostaining. RNA was purified on a silica gel–based membrane (RNeasy; Qiagen, Germantown, MD). The integrity of RNA was verified by capillary electrophoresis using a 6000 Nano LabChip kit on a Bioanalyzer 2100 (Agilent, Santa Clara, CA). RNA content was measured using an ND-1000 spectrophotometer (Nano-Drop, Wilmington, DE). Only samples with no evidence of RNA degradation were used further. A high-capacity cDNA archive kit (Applied Biosystems, Foster City, CA) was used to reverse transcribe RNA, per manufacturer’s guidelines. Samples were subjected to real-time PCR in triplicate using a TaqMan ABI 7500 sequence detection system (Applied Biosystems, Waltham, MA). Primer sets were designed using DNASTAR Lasergene 11 software (http://www.idtdna.com/Primerquest/Home/Index) and synthesized by Integrated DNA Technologies Inc. (Coralville, IA) as described previously.64
AAV-Mediated Gene Delivery
Packaged adeno-associated virus (AAV) vector was made and purified by the Viral Vector Core at The Research Institute at Nationwide Children's Hospital, as previously described.65 For experiments involving mdx mice, mice were injected by intra-arterial injection in the femoral artery with 1 × 1011 v grAAVrh74.MCK.GALGT2, as previously described.47
Lectin Precipitation
Mouse gastrocnemius muscles were minced, and whole muscle cell lysates were prepared in lysis buffer [50 mmol/L Tris (pH 7.4) 150 mmol/L NaCl, 1 mmol/L EDTA, 1% Nonidet P-40, and proteinase inhibitor cocktail (Roche Diagnostics, Indianapolis, IN)], as previously described.49,50,53 Extracted protein (150 μg, 500 μg, 1 mg, or 3 mg) was incubated with Wisteria floribunda agglutinin (WFA) or wheat germ agglutinin (WGA) overnight at 4°C, as described before.64 Samples were spun at 3800 × g for 3 minutes and washed three times for 10 minutes each in buffer, and pellets were denatured in SDS-containing buffer as before.62 WFA binding was verified by blocking with 0.3 mol/L N-acetylgalactosamine (GalNAc) and WGA binding by blocking in 0.3 mol/L N-acetylglucosamine. Precipitated proteins were separated onto 4% to 12% SDS-PAGE linear gradient gels, transferred to nitrocellulose, and immunoblotted using antibody IIH6 for αDG (Upstate Biotechnology, Lake Placid, NY), antibody 43DAG for β dystroglycan (Leica, Buffalo Grove, IL), or antibody CT2 for the CT glycan, as previously described.62,66
Immunoblotting
Muscles were dissected, minced, and solubilized in tris-buffered saline (pH 7.4), 1% Nonidet-P40, and 1 mmol/L EDTA, with shaking overnight at 4°C. The concentration of extracted protein from muscle lysates was determined by a modified Bradford assay, after which protein was denatured by boiling the sample in buffer containing 1% SDS with 1% β-mercaptoethanol. Protein (20 μg per lane) was separated by SDS-PAGE onto 4% to 12% gradient gels and analyzed by Western blot analysis, as previously described.67 Antibodies listed for immunostaining were also used for immunoblotting, with the additional antibodies used being Plectin1 (10F6; Santa Cruz Biotechnology, Santa Cruz, CA) and actin (A5060; Sigma).
Histology and Immunofluorescence Staining
Skeletal muscles were snap frozen in liquid nitrogen–cooled isopentane and were divided into sections (8 μm thick) on a cryostat. Sections were stained with hematoxylin and eosin (H&E; Sigma) or were immunostained with antibodies against the CT carbohydrate (CT2),68 produced in our laboratory, or antibodies to dystrophin (NCL-DYS1), utrophin (NCL-DRP2), β-sarcoglycan (NCL-b-SARC), γ-sarcoglycan (NCL-g-SARC), β-dystroglycan (NCL-b-DG) (all from Leica, Buffalo Grove, IL), αDG (IIH6; Upstate Biotechnology, Lake Placid, NY), laminin α5 (from Jeffery Miner, Washington University Medical School, St. Louis, MO), laminin-α2 (L0663; from Sigma, St. Louis, MO), Galgt2 (NBP1-91229; Novus Biologicals, Littleton, CO), dystroglycan (AF6868; R&D Systems, Littleton, CO), CD68 (MCA1957GA; AbD Serotec, Kidlington, UK), CD4, and CD8 (550278 and 550281; BD Biosciences, San Jose, CA), Pax7 (from Michael Rudnicki, Ottawa Health Research Institute, Ottawa, ON, Canada), myoblast D1 (MyoD1; M3512; Dako, Carpinteria, CA), and embryonic myosin (NCL-MHCd; Leica), as previously described.49,50,53 For immunostaining with mouse monoclonal antibodies, a mouse antibody-on-mouse blocking reagent was used (Dako). All other antibodies were blocked in PBS with 10% goat or 10% donkey serum. All of the fluorophore-conjugated secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA). Rhodamine-conjugated α-bungarotoxin (Molecular Probes, Eugene, OR) was added at 50 ng/mL to identify neuromuscular junctions.
Quantification of Muscle Histopathology
Quantification of skeletal myofibers with central nuclei, myofiber diameters, and coefficients of myofiber variance was performed as previously described.49,50,53 Briefly, cross sections (8 μm thick) were cut from the midsection (belly) of the muscle and stained with H&E to assess pathology. For central nuclei measurements, all myofibers from each muscle section were counted. Measures from at least five sections were averaged per muscle, and the percentage of central nuclei was assessed using ImageJ software version 1.46r (NIH, Bethesda, MD; http://imagej.nih.gov/ij) to calculate each parameter for a given area, as previously described.50 For myofiber diameter measurements, Mini-Feret diameters were measured from a minimum of 100 myofibers randomly selected from at least five sections per muscle per mouse. These measures were then averaged using Zeiss (Jena, Germany) AxioVision software version LE 4.8, as previously described.49,50,53 To quantify CD68, CD4, and CD8 staining in heart and skeletal muscle, 10 different 1.5 × 106 μm2 images of antibody staining were captured per condition, and all positively stained cells were counted using ImageJ software, as before.64 These were then averaged from three to six animals per condition. All histopathology measures were independently assessed in a blinded manner (R.X.).
Serum Creatine Kinase Activity Assays
Blood was collected from the tail vein. Blood cells were allowed to clot, and serum was separated by centrifugation of clotted cells at 1500 × g for 10 to 20 minutes. The serum creatine kinase activity was determined in triplicate by an enzyme-coupled absorbance-based kit (CK-SL; Diagnostic Chemicals Limited, Charlottetown, PE, Canada), following the manufacturer's protocol. The enzyme activity was monitored and calculated by measuring the absorbance at 340 nm every 30 seconds for 4 minutes at 25°C.
Skeletal Muscle Physiology
Analysis of tetanic force in dissected extensor digitorum longus (EDL) muscle and strips of dissected diaphragm muscle was performed as previously described.47,64 Small linear strips of dissected diaphragm muscle were suspended between a force transducer and stimulator hook in an experimental set-up and superfused with Krebs-Henseleit solution at 37°C. Tetanic force was assessed by tetani of 600-millisecond duration, with frequencies ranging from 20 to 250 Hz and a pulse width of 1 millisecond. After measurements, muscles were weighed and cross-sectional area was calculated as previously described.47 Isolated EDL muscles were tied to a force transducer and linear servomotor. Twitch contractions at 30°C were elicited, and the muscle was stretched to optimal length. Next, one to three tetani of 500-millisecond duration, using 1-millisecond pulses at 150 Hz, were imposed on the muscle. This was followed by 10 repeats of eccentric contractions. Per repeat, the muscle was tetanized for 700 milliseconds and stretched by 5% of its initial length during the last 200 milliseconds of the tetanus. After stimulation was halted at t = 700 milliseconds, the muscle was taken back to its original length in 200 milliseconds. In between repeats, the muscle remained unstimulated for 2 minutes. After measurements, muscles were weighed and cross-sectional area was calculated as previously described.47
Echocardiography
Murine cardiac function was assessed using echocardiographic techniques that have been previously published in detail.69,70 In brief, 8-month-old male mice were anesthetized via isoflurane inhalation at a minimally effective concentration. Normothermia was maintained by heating pad. Left ventricular functional parameters were measured using the American Society for Echocardiography leading-edge technique in a blinded manner (M.J.) to measure contractile parameters, including left ventricular fractional shortening, heart rate, stroke volume, and cardiac output. In addition to baseline or non-stressed cardiac performance, cardiac response to acute stress induced was measured in anesthetized mice by i.v. injection of 2.5 mg/kg isoproterenol. The parameters described above were then remeasured at peak cardiac work after dosing.
EBD Uptake
The 5-month-old, wild-type, Galgt2−/−, mdx, and Galgt2−/−mdx mice were injected i.p. with Evans Blue Dye (EBD; E2129; Sigma) at a concentration of 50 μg/g body weight in 100 μL sterile PBS. Five hours later, mice were normalized for activity by subjecting them to 45 minutes of exercise on a horizontal treadmill at a constant speed of 12 m/minute for 15 minutes and then 24 m/minute for 30 minutes. Thirty-six hours after EBD injection, mice were sacrificed and skeletal and heart muscles were frozen and divided into sections. EBD uptake was visualized using rhodamine-specific optics on a Zeiss Axiophot epifluorescence microscope. Dye uptake was quantified using Zeiss AxioVision software, as previously described.50
Muscle Cell Isolation
Gastrocnemius, quadriceps, tibialis anterior, and triceps muscles were dissected from wild-type and Galgt2−/− mice, as previously described.62 Dissected muscles placed in ice-cold sterile PBS were minced in a laminar-flow hood using aseptic conditions. Minced muscles were digested in PBS with 1.2 U/mL dispase and 5 mg/mL collagenase IV at 37°C for 30 minutes, with titration every 10 minutes, followed by addition of Dulbecco’s modified Eagle’s medium (DMEM)/F12 media + 10% heat-inactivated horse serum + 1% penicillin-streptomycin (P/S) solution. The solution was filtered through a 70-μm cell strainer and then a 40-μm cell strainer, after which cells were centrifuged at 450 × g at 4°C for 10 minutes. The cell pellet was resuspended in 0.5% bovine serum albumin/PBS and overlayed with 100% heat-inactivated horse serum, then centrifuged again at 220 × g at 4°C for 10 minutes. After washing, the cell pellet was resuspended in growth media (DMEM/F-12 + 10% fetal bovine serum + 4% chick embryo extract + 1% P/S). Cells were preplated on uncoated tissue culture plates, with muscle cell–enriched supernatant removed after fibroblasts were allowed to adhere for 20 minutes. Purified muscle cells were then plated on 0.1% collagen-1–coated tissue culture plates. Cultures typically showed an excess of 90% positive staining for c-met, a muscle cell marker.
Muscle Cell Growth Rate and Fusion Assays
Cells extracted and purified from wild-type and Galgt2−/− skeletal muscles were grown in growth media (DMEM/F-12 + 10% fetal bovine serum + 4% chick embryo extract + 1% P/S) and were counted at three different time points (1, 2, and 3 days) after plating. For the muscle cell fusion assay, cells were grown in growth media until they reached 100% confluence and were then switched to fusion media (DMEM/F-12 + 2% horse serum + 1% P/S). Skeletal myofibers were analyzed at four time points: day 0 (just before addition of fusion media) and days 1, 3, and 6, after addition of fusion media. Sixteen equivalent fields of view were evaluated per well, with at least three wells evaluated per time point and per genotype. Data shown are averages of six experiments per condition, normalized to baseline cell number on day 0 in each instance.
Statistical Analysis
Comparisons between two groups were drawn using an unpaired t-test with equal weighting between samples. An analysis of variance with post hoc t-test was used to determine significance for comparisons involving more than two groups.
Results
Galgt2 Gene Expression and Galgt2-Dependent αDG Glycosylation Increases in Regenerating, Dystrophic, and Developing Skeletal Muscle
In adult skeletal muscle, Galgt2 protein is concentrated almost exclusively at neuromuscular and myotendinous junctions,46,62 and endogenous Galgt2 gene expression is low.51,71 To determine whether Galgt2 is highly expressed in other cases, we analyzed gene expression in regenerating, dystrophic, and developing skeletal muscles (Figure 1). To study regenerating muscle, we treated adult wild-type skeletal muscles with Ctx and assessed Galgt2 expression at 1, 4, 7, 14, and 28 days after injection, relative to uninjected day 0 muscle (Figure 1A). After acute injury, macrophages enter the muscle to clear necrotic tissue and satellite cells begin to divide on day 1.9 By days 4 and 7, satellite cells have begun to differentiate into myoblasts and fuse into immature myofibers, and this is consistent with increased expression of myogenic markers, such as MyoD, myogenin, and embryonic myosin.72 By day 14, most regenerating myofibers have formed but continue to grow and differentiate until day 21 to 28. Compared with day 0 (uninjected muscle), Galgt2 expression was significantly increased at 4 and 7 days after Ctx injection, with a maximal threefold increase found at day 4. We also found a 3.7-fold increase in Galgt2 expression in dystrophic (mdx) muscles compared with wild type (Figure 1B). The mdx muscles, having muscular dystrophy because of the dystrophin protein deficiency, are in a chronic state of muscle degeneration and regeneration.54 This increase in Galgt2 expression, however, was variable between mdx muscle samples, with only 13 of 26 muscles analyzed showing a greater than twofold elevation. We also observed increased Galgt2 expression in muscle-derived embryonal rhabdomyosarcomas (eRMSs) that occur at a low frequency in aged mdx mice73,74 and in developing muscle at postnatal day 1 wild-type muscle (Figure 1B), a time at which extrasynaptic βGalNAc and CT glycan expression levels are high.45 eRMS tumors contain regenerating skeletal myofibers in addition to rhadomyoblasts,73 and so were also of interest to study as a tissue containing several types of immature muscle cells. P1 muscle was also of interest because this is a stage of development when most muscle proteins induced by Galgt2 (eg, agrin, laminin α4, and laminin α5) are also expressed in extrasynaptic regions of skeletal myofibers.75–77 Like the CT glycan, these molecules do not show synaptic concentration until P14. This early postnatal period of muscle development (P1 to P14) also corresponds to a period when muscle growth is dramatic and driven by fusion of satellite cells into skeletal myofibers and when synapse elimination occurs, leading polyinnervated young myofibers to develop into more mature monoinnervated cells.78 Elevated Galgt2 expression correlated with increased immunostaining of Galgt2 protein and the CT glycan in all instances (Supplemental Figures S1 and S2).
Figure 1.

Increased Galgt2 gene expression and Galgt2-dependent glycosylation of α dystroglycan in regenerating, dystrophic, and developing mouse skeletal muscles. A: Fold change in Galgt2 gene expression after injection of cardiotoxin to induce skeletal muscle regeneration, relative to day 0 uninjected muscle (set at 1, line). B: Fold change in Galgt2 gene expression in mdx muscle, day 4 cardiotoxin (Ctx)-injected wild-type muscle (D4 Ctx), embryonal rhabdomyosarcoma (eRMS), and postnatal day 1 wild-type muscle (P1 WT), relative to adult WT skeletal muscle (set at 1, line). C:Wisteria floribunda agglutinin (WFA) and wheat germ agglutinin (WGA) agarose precipitation of α dystroglycan (αDG; probed with IIH6) using differing amounts of nonionic detergent-solubilized protein from adult WT, Galgt2-deficient (Galgt2−/−), dystroglycan-expressing (P3ProCreDag1lox/lox tricep), dystroglycan-deficient (P3ProCreDag1lox/lox, gastrocnemius), mdx, Galgt2-deficient mdx (Galgt2−/−mdx), postnatal day 1 WT (P1 WT), eRMS, 4 days post-Ctx injected WT muscle (D4 Ctx), 12 week-AAV-Galgt2-injected mdx (mdx + AAV-Galgt2), or Galgt2 transgenic mdx (mdx Galgt2 Tg) skeletal muscle. Errors are SEM (A and B). n = 6 per condition (A); n = 6 to 26 per group (B). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001.
To understand if increased endogenous Galgt2 gene expression led to increased glycosylation of αDG, we performed lectin precipitation on whole muscle nonionic detergent tissue lysates from developing, regenerating, and dystrophic (mdx) muscle as well as from eRMS (Figure 1C). Galgt2 transgenic muscles were used as positive controls, and Galgt2-deficient and dystroglycan (Dag1)-deficient muscles were used as negative controls. In adult wild-type muscle, αDG was precipitated by WFA only when 3 mg of total cell protein was used, whereas precipitation using 1, 0.5, or 0.15 mg protein did not generate an appreciable signal. By contrast, WGA, a control lectin that does not bind βGalNAc but binds αDG48,79 through its specificity for other glycans (sialic acid and N-acetylglucosamine), precipitated αDG from wild-type muscle when 0.15 mg of protein was used. Galgt2-deleted muscles showed highly reduced, but not absent, signal for WFA precipitates at 3 mg protein, suggesting the presence of a small amount of non–Galgt2-dependent GalNAc on αDG. WFA and WGA precipitation of P3ProCreDag1lox/lox gastrocnemius muscle, which is Dag1 deficient,57 showed no αDG, whereas precipitation from the triceps muscles of the same mice, where Dag1 is expressed,80 showed a pattern similar to wild type. In mdx muscles where Galgt2 expression was elevated, WFA precipitated αDG at protein concentrations as low as 0.5 mg, and almost all such signal was eliminated in Galgt2−/−mdx muscle (Figure 1C). In developing (P1 WT) and in regenerating (D4 Ctx) wild-type muscle, as well as in eRMS, αDG was also precipitated by WFA when lower protein amounts were used, approaching levels seen in mdx muscles 12 weeks after injection with AAV-Galgt2.47,53 WFA did not precipitate αDG from P1 or day 4 Ctx-treated Galgt2−/− muscle (data not shown), again demonstrating a requirement for endogenous Galgt2 expression. WFA and WGA precipitation showed only a 160-kDa αDG band on subsequent IIH6 immunoblots, the known molecular weight of αDG in adult skeletal muscle,79 with the exception of P1 muscle, which showed three αDG bands, at 120, 240, and 260 kDa. Thus, all conditions where endogenous Galgt2 gene expression was elevated were associated with elevated Galgt2-dependent glycosylation of αDG.
To identify the muscle cells with increased Galgt2 expression in mdx muscles, we performed costaining for Galgt2 protein with markers for regenerating myofibers (eMyosin), myoblasts (MyoD), satellite cells (Pax7), or macrophages (CD68) (Figure 2). Galgt2 is typically localized as a trans-Golgi protein,81 but it can display cytoplasmic staining when overexpressed. In regions of mdx muscle with evidence of myofiber damage and regeneration, Galgt2 protein staining was present in many cells that costained for embryonic myosin (89% of eMyosin-positive cells). The cytoplasmic pattern of Galgt2 expression in embryonic myofibers was consistent with protein overexpression, and some nuclear staining was also observed in these cells (Figure 2). There was less Galgt2 staining identified in cells costained for MyoD (37% of MyoD-positive cells) and almost no Galgt2 staining in cells stained for Pax7 (8% of Pax7-positive cells). In addition to embryonic myofibers, many CD68-positive macrophages also costained with Galgt2. Thus, with respect to muscle cells, elevated Galgt2 expression in mdx muscle occurred predominantly in regenerating myofibers, consistent with its increased gene expression at days 4 and 7 after acute muscle injury, when new myofiber formation is at its peak (Figure 1).
Figure 2.

Co-immunostaining of Galgt2 protein with Pax7, MyoD, embryonic myosin (eMyosin, also Myh3), or CD68 in mdx skeletal muscle. The mdx skeletal muscle (gastrocnemius) was costained for Galgt2 (red) and markers for regenerating myofibers (eMyosin), myoblasts (MyoD), satellite cells (Pax7), or macrophages (CD68), along with DAPI. Scale bar = 10 μm.
Skeletal and Cardiac Muscle Pathology Is Altered in Galgt2−/−mdx Mice
To assess the role of endogenous Galgt2 expression in muscular dystrophy, we generated Galgt2-deficient mdx mice (Galgt2−/−mdx). The elevated expression of Galgt2 protein in mdx muscles, which occurs primarily in regenerating myofibers, was eliminated in Galgt2−/−mdx mice, as was CT glycan expression in skeletal muscle and heart (Supplemental Figure S2). Endogenous Galgt2 expression at neuromuscular and myotendinous junctions was also eliminated in Galgt2−/− muscle (some data not shown) (Supplemental Figure S3). We stained skeletal and heart muscle sections with a modified Mason trichrome stain to assess overall muscle histopathology. Comparison of wild-type, Galgt2−/−, mdx, and Galgt2−/−mdx gastrocnemius (Figure 3A), diaphragm (Figure 3B), and heart (Figure 3C) muscles at 5 months of age showed a significant increase in collagen staining (blue) and/or increased cellular infiltrates in all Galgt2−/−mdx muscles compared with mdx. H&E staining also confirmed the presence of cellular infiltrates in skeletal and cardiac muscles (Supplemental Figure S4).
Figure 3.

Mason trichrome staining of gastrocnemius, diaphragm, and heart muscle. Thin sections of gastrocnemius muscle (A), diaphragm (B), or heart (C) from 5-month-old wild-type (WT), Galgt2−/−, mdx, and Galgt2−/−mdx mice were stained with a modified Mason trichrome stain. Scale bar = 100 μm.
We next assessed mouse (Figure 4A) and individual muscle (Figure 4B) weights, average (Mini-Feret) muscle myofiber diameters (Figure 4C), the percentage of myofibers with centrally located nuclei (Figure 4D), the percentage of nonmuscle tissue within muscle sections (Figure 4E), and average serum creatine kinase activity levels (Figure 4F). Galgt2−/−mdx hindlimb muscles (quadriceps, gastrocnemius, tibialis anterior) showed modest hypertrophy relative to mdx, but the overall weights of Galgt2−/−mdx and mdx mice were not significantly different. The average Mini-Feret diameter of myofibers and the percentage of myofibers with centrally located nuclei were not significantly different between Galgt2−/−mdx and mdx muscles, but loss of muscle cells within muscle tissue was higher for Galgt2−/−mdx compared with mdx in gastrocnemius, heart, and diaphragm at 5 months of age (Figure 4E). Thus, some Galgt2−/−mdx skeletal muscles and cardiac muscle showed evidence of increased histopathology relative to mdx, and in particular showed an increased extent of muscle fibrosis and/or inflammation. The average serum creatine kinase activity in 6- to 8-week-old animals was also elevated in Galgt2−/−mdx mice relative to mdx (Figure 4F). This is a period in mdx mice when muscle damage is severe.12 When serum creatine kinase was measured at 6 months of age, when acute muscle damage is less pronounced, levels in Galgt2−/−mdx mice were no longer higher than in mdx (data not shown). To further understand the mechanism by which Galgt2 deletion led to increased muscle histopathology, we next quantified aspects of muscle inflammation, membrane stability, and regeneration in Galgt2-deficient and control mice.
Figure 4.

Measurements of muscle size and histopathology. Wild-type (WT), Galgt2−/−, mdx, and Galgt2−/−mdx mice were compared for mouse weight (A), muscle weight (B), Mini-Feret myofiber cross-sectional diameter (C), percentage of myofibers with central nuclei (D), percentage of nonmuscle area within muscle tissue (E), and serum creatine kinase activity (F). Measures for A–E are on 5-month-old mice, and measures for F are on 6- to 8-week-old mice. Errors are SEM (A–F). n = 8 to 12 per group (A); n = 6 to 8 per group (B–F). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001. Gastroc, gastrocnemius; Quad, quadriceps; TA, tibialis anterior.
Inflammation Increases in Galgt2−/−mdx Skeletal Muscle and Heart
We stained skeletal and heart muscles from the four genotypes of mice to determine cells per constant visual field stained with CD68, a marker for macrophages, CD4, a marker for helper T lymphocytes, or CD8, a marker for cytotoxic T lymphocytes (Figure 5, A and B). Both mdx and Galgt2−/−mdx skeletal muscle and heart had more CD68-stained cells than wild-type or Galgt2−/− muscles (P < 0.01 for all such comparisons). By contrast, few CD4+ or CD8+ cells were present in any muscles from any mouse genotype examined. The number of CD68+ macrophages present in Galgt2−/−mdx muscle was significantly increased relative to mdx for both skeletal muscle and heart at 5 months of age. In addition, the number of macrophages stained with CD68, per constant unit area, was higher in both mdx and Galgt2−/−mdx skeletal muscle than in heart.
Figure 5.

Inflammation increases in Galgt2−/−mdx skeletal muscle and heart. Cells stained with CD4, CD8, or CD68 were quantified, per constant visual field, in skeletal muscle (gastrocnemius, A) and heart (B) at 5 months of age. Semiquantitative real-time PCR measures of relative increases in gene expression for markers of inflammation [CD4, CD8, CD68, IL-1β, IL-6, monocyte chemoattractant protein 1 (or chemokine C-C motif ligand 2; MCP1), macrophage inflammatory protein 1α (or chemokine C-C motif ligand 3; MIP1a), regulated on activation, normal T-cell expressed and secreted (or chemokine C-C motif ligand 5; RANTES), and tumor necrosis factor α (TNFα)] in skeletal muscle (gastrocnemius, C) and heart (D) at 5 months of age. C and D: All Galgt2−/−mdx, mdx, and Galgt2−/− measures were compared with wild-type (WT) measures for the same gene at the same age, which was set at 1 (line). Only Galgt2−/−mdx versus mdx significance comparisons are shown for clarity. Errors are SEM (A–D). n = 9 to 12 measurements per condition (A–D). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001. CD, cluster of differentiation.
We also measured the relative gene expression for markers of muscle inflammation in skeletal muscle and heart at 5 months (Figure 5, C and D). Both mdx and Galgt2−/−mdx skeletal muscle and heart had increased expression of inflammatory cytokines relative to Galgt2−/− and wild type. Expression of genes encoding CD68, Il1β, monocyte chemoattractant protein 1 (or chemokine C-C motif ligand 2), macrophage inflammatory protein 1α (or chemokine C-C motif ligand 3), regulated on activation, normal T-cell expressed and secreted (or chemokine C-C motif ligand 5), and tumor necrosis factor α was all significantly increased in Galgt2−/−mdx versus mdx skeletal muscle, whereas monocyte chemoattractant protein 1 (or chemokine C-C motif ligand 2), macrophage inflammatory protein 1α (or chemokine C-C motif ligand 3), and regulated on activation, normal T-cell expressed and secreted (or chemokine C-C motif ligand 5) were significantly elevated in heart (P < 0.05 for all). In all instances where cytokine gene expression in Galgt2−/−mdx exceeded mdx, both Galgt2−/−mdx and mdx levels were significantly increased relative to Galgt2−/− and wild type (P < 0.05 for all such comparisons). These same measures were also performed at 2 months of age, where most elevations were not yet significantly different between Galgt2−/−mdx and mdx (data not shown). These data further support the presence of increased muscle inflammation in Galgt2−/−mdx muscles with age. By contrast, expression of markers of skeletal muscle regeneration and cardiomyopathy, although elevated in some cases in both mdx and Galgt2−/−mdx, did not significantly differ between these two groups (Supplemental Figure S5).
We also measured relative gene expression changes for certain dystrophin-associated glycoprotein complex genes and integrin genes (Supplemental Figure S6, A and B) as well as extracellular matrix genes (Supplemental Figure S6, C and D) in skeletal muscle and heart. Galgt2−/−mdx skeletal muscle showed significantly increased expression of some extracellular matrix genes [laminin α2, laminin α4, laminin α5, agrin, and collagen IV (α1 and α2)], some dystroglycan-associated glycoprotein complex genes (utrophin and dystrobrevin), and several integrin genes (α7 and β1) relative to mdx (Supplemental Figure S6, A and C). In heart, Galgt2−/−mdx mice showed increased expression of markers of cardiac fibrosis [collagen I and IV(α1)] at 5 months of age relative to mdx (Supplemental Figure S6, B and D). Again, these differences were not evident between these genotypes at 2 months of age (data not shown). Immunostaining (Supplemental Figure S7A) and immunoblotting (Supplemental Figure S7B) for utrophin in Galgt2−/−mdx skeletal muscle showed higher expression than was found in mdx skeletal muscle. Expression of plectin 1 protein was also increased in Galgt2−/−mdx muscle relative to mdx (Supplemental Figure S7B). Thus, Galgt2−/−mdx muscles showed an increased pathology despite increased expression of two potential dystrophin surrogates. In addition, the increased expression of extracellular matrix genes in Galgt2-deficient mdx muscles may have contributed to increased fibrosis.
Cardiac Membrane Stability and Heart Function Is Altered in the Absence of Galgt2
To assess membrane stability, we performed EBD uptake experiments. Mice were injected with EBD and then subjected to a 45-minute period of exercise. Approximately a day later, skeletal muscle (Figure 6A) and heart muscle (Figure 6B) were assessed for dye uptake in wild-type, Galgt2−/−, mdx, and Galgt2−/−mdx mice. Both mdx and Galgt2−/−mdx skeletal muscles showed increased dye uptake relative to wild-type and Galgt2−/− muscles (P < 0.001 for all comparisons) (Figure 6A), but there was no significant difference in dye uptake between Galgt2−/−mdx and mdx. Galgt2−/−mdx dye uptake was, on average, higher. Dye uptake into skeletal muscles of both genotypes (mdx and Galgt2−/−mdx), however, was markedly increased relative to dye uptake levels found in heart muscle, consistent with previous studies.7,82 For heart, it is important to remember that although the number of cells taking up dye was low, it reflects the level of damage occurring for only 1 day after a short period of exercise, and the cumulative effects of such damage over months may, in fact, contribute to changed tissue pathology. The mdx mice at 5 months of age showed little, if any, histopathology indicative of cardiac damage (Figure 3 and Supplemental Figure S4). Consistent with this finding, we observed no increase in dye uptake in the mdx heart at this age. Galgt2−/−mdx heart, however, had increased dye uptake relative to wild-type and mdx, which may contribute to the increased cardiac muscle pathology observed with trichrome and H&E staining (Figure 3 and Supplemental Figure S4).
Figure 6.
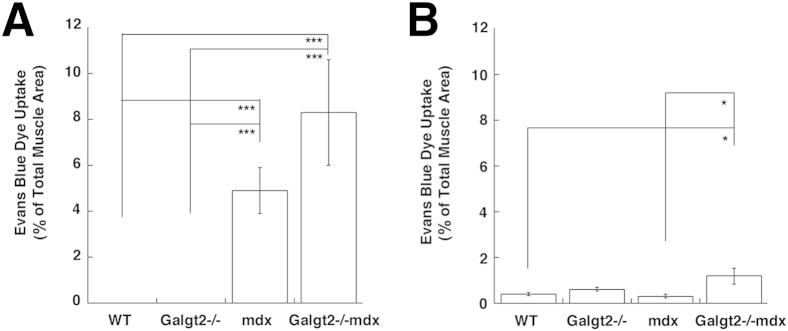
Quantification of Evans Blue Dye uptake in skeletal muscle and heart after exercise. A and B: Evans Blue Dye uptake was quantified in 2-month-old wild-type (WT), Galgt2−/−, mdx, and Galgt2−/−mdx mouse skeletal muscle (A, gastrocnemius) and heart (B) 1 day after a period of exercise. Errors are SEM (A and B). n = 6 per group (A and B). ∗P < 0.05, ∗∗∗P < 0.001.
We next assessed cardiac function by echocardiography. Heart rate, cardiac output, percentage left ventricular fractional shortening, and stroke volume were all significantly reduced in Galgt2−/−mdx mice relative to wild type after isoproterenol challenge at 8 months of age (P < 0.05) (Figure 7). Herein, we used slightly older animals where mdx cardiac dysfunction and histopathology are more pronounced than is normally observed at 5 months to get more reliable functional measures.83 After stimulation with isoproterenol, Galgt2−/−mdx mice also had reduced heart rate and percentage fractional shortening relative to mdx (P < 0.05 for both). In addition, Galgt2−/− mice showed reduced heart rate after isoproterenol challenge and reduced percentage fractional shortening and reduced cardiac output in the resting state when compared with wild type (P < 0.05 for all comparisons). Thus, several features of heart function differed between Galgt2−/−mdx and mdx mice and also between Galgt2−/− and wild-type mice.
Figure 7.

Echocardiography measures of heart function. Measures of heart rate (A), cardiac output (B), percentage left ventricular fractional shortening (C), and stroke volume (D) were compared in 8-month-old male wild-type (WT), Galgt2−/−, mdx, and Galgt2−/−mdx mice, before (PRE-ISO) or after (POST-ISO) challenge with isoproterenol. Errors are SEM (A–D). n = 8 to 12 mice per group (A–D). ∗P < 0.05 for each condition shown versus WT and for Galgt2−/−mdx versus others, as indicated. bpm, Beats per minute.
We also compared skeletal muscle function by several measures. By using the EDL muscle, which was analyzed ex vivo, we found that both Galgt2−/−mdx and mdx muscles, but not Galgt2−/− muscles, had a significant reduction in maximal specific force relative to wild-type muscle, but that Galgt2−/−mdx did not differ significantly from mdx (Supplemental Figure S8A). Measures of force decrease during repeated eccentric contractions (of the EDL) showed that Galgt2−/−mdx muscle lost force more quickly than mdx muscles, but here again there was not a significant difference between the two genotypes (Supplemental Figure S8B). These two measures were consistent with the fact that we did not identify a significant increase in muscle wasting in Galgt2−/−mdx versus mdx for the EDL muscle at this age (data not shown). For diaphragm, ex vivo measures of maximal specific force showed again that both Galgt2−/−mdx and mdx mice had reduced specific force relative to wild type, but were not significantly different from one another (Supplemental Figure S8C). As for the EDL, force measures in Galgt2−/−mdx trended lower than in mdx.
Muscle Growth Is Delayed in Galgt2-Deficient Mice after Acute Muscle Injury
To assess the role of Galgt2 in skeletal muscle regeneration, we next compared H&E staining of thin sections after Ctx-induced regeneration in wild-type and Galgt2−/− muscles at 1, 4, 7, 14, and 28 days after injury compared with uninjected, day 0, muscle (Figure 8A). Although muscle regeneration occurred in both wild-type and Galgt2−/− mice in response to Ctx, the distribution of myofibers shifted toward smaller diameters for Galgt2−/− mice at both days 14 and 28 after injury (Figure 8, B–D). The average (Mini-Feret) diameter of regenerating muscles was reduced by 33% at day 14 and by 12% at day 28 in Galgt2−/− muscles compared with wild type. In addition, there was evidence of increased inflammation in Galgt2−/− muscles in the first week after Ctx-induced damage, with more accumulations of i.m. mononuclear cells (Figure 8A). These data are consistent with slowed muscle growth during regeneration in the absence of Galgt2. We did not, however, identify any reduction in muscle cell growth or fusion capability when cultured primary muscle cells were isolated from Galgt2−/− mice and compared with cells isolated from wild-type animals (Supplemental Figure S9). Thus, these findings appeared to be in vivo–specific effects.
Figure 8.

Deletion of Galgt2 alters muscle growth during regeneration after acute muscle injury. A: Hematoxylin and eosin staining of wild-type (WT) and Galgt2−/− muscle 1, 4, 7, 14, or 28 days after cardiotoxin-induced muscle damage compared with uninjected, day 0, control muscle. Arrow indicates region with concentrated mononuclear cell infiltrates at day 7. Distribution of Mini-Feret myofiber diameters 14 (B) or 28 (C) days after cardiotoxin (Ctx)-induced injury. D: Percentage change in average myofiber diameter 14 or 28 days after Ctx-induced injury for Galgt2−/− versus WT. Errors are SEM. n = 6 per condition (D). ∗P < 0.05, ∗∗∗P < 0.001. Scale bar = 50 μm (A).
Galgt2 Overexpression Does Not Inhibit Muscle Pathology in Dystroglycan-Deficient Muscles
Deletion of dystroglycan (Dag1) from skeletal muscles, like loss of dystrophin protein expression in mdx mice, causes muscular dystrophy.58,84 Because the primary glycoprotein glycosylated by Galgt2 when it is overexpressed in skeletal muscle is αDG,48,62,71 we next assessed whether dystroglycan was required for Galgt2 overexpression to inhibit muscle pathology in Dag1-deficient skeletal muscles. Dag1 is an essential gene in mice85; therefore, we used P3ProCreDag1lox/lox mice for these experiments, because these mice show deletion of Dag1 in caudal muscles (eg, gastrocnemius, tibialis anterior, and quadriceps) but not in more rostral muscles (eg, diaphragm, biceps, and triceps).57,80
To test the relationship between dystroglycan and the therapeutic effects of Galgt2 overexpression, we bred skeletal muscle–specific Galgt2 mCT48,62 to P3Pro-CreDag1lox/lox mice to generate mice with Galgt2 transgenic Dag1-deficient skeletal muscles (mCTDag1−/−). We observed a significant, although incomplete, decrease in the percentage of myofibers with central nuclei at 2 months in mCTDag1−/− muscles that was lost by 9 months of age (Figure 9, A and B). H&E staining of sections from 9-month-old mCTDag1−/− hind limb muscles showed severe muscle damage, with evidence of muscle tissue replacement with fat and extracellular matrix (Figure 9A). This damage was similar to damage found in Dag1−/− hind limb muscles, although these muscles were, on average, larger than mCTDag1−/− muscles because of the previously described effect of embryonic Galgt2 overexpression on myofiber growth.62,86 Similarly, unlike Galgt2 transgenic mdx mice, where serum creatine kinase activity is no higher than in wild-type mice,48 serum creatine kinase activity in mCTDag1−/− mice was significantly elevated relative to wild type, and this elevation was not significantly different from measures in Dag1−/− mice at 9 months of age (Figure 9C).
Figure 9.

Failure to sustain inhibition of muscular dystrophy in Galgt2 transgenic Dag1−/− skeletal muscles. A: Examples of hematoxylin and eosin–stained muscle sections from 2- and 9-month-old wild-type, dystroglycan-deficient (Dag1−/−), and Galgt2 transgenic dystroglycan-deficient (mCTDag1−/−) muscle. B: Quantification of percentage of myofibers with centrally located nuclei in the gastrocnemius (Gastroc), quadriceps (Quad), and tibialis anterior (TA) muscles. C: Quantification of serum creatine kinase activity. D: Semiquantitative real-time PCR measures of relative dystroglycan (Dag1) gene expression at 2 and 9 months, normalized to 1 for 2-month-old wild-type muscle. E: Semiquantitative real-time PCR measures of relative Galgt2 gene expression normalized to 1 for 2-month-old wild-type muscle. Errors are SEM (B and C) or SD (D and E). n = 6 per group (B); n = 4 to 9 per group (C); n = 3 to 6 per group (D and E). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001. Scale bar = 200 μm (A).
We measured both Dag1 gene (Figure 9D) and Galgt2 gene (Figure 9E) expression in wild-type, mCT, Dag1−/−, and mCTDag1−/− muscles and found that the reduction in Dag1 gene expression was not significantly different between Dag1−/− and mCTDag1−/− muscles at 2 or 9 months of age. Similarly, Galgt2 gene expression was equivalently elevated in mCT and mCTDag1−/− muscles at 2 or 9 months of age. We observed a 25% reduction in Galgt2 gene expression between 2 and 9 months in both mCT and mCTDag1−/− muscles, likely the result of reduced transgene promoter activity at the older age. The level of Galgt2 overexpression at 9 months in mCTDag1−/− muscle, however, was still much higher than levels needed for therapeutic effects in AAV-Galgt2–treated mdx,53 dyW49, or Sgca−/−50 muscles, which typically can saturate therapeutic effects at a 1000-fold level of gene overexpression (data not shown). The high level of Galgt2 overexpression observed in mCT and mCTDag1−/− muscles largely results from the low level of endogenous Galgt2 gene expression in wild-type muscle, because of its normal confinement to neuromuscular and myotendinous junctions. The small, but insignificant, apparent increase in Dag1 gene expression at 2 months in mCTDag1−/− muscle compared with Dag1−/− did not correlate with an increased percentage of myofibers expressing dystroglycan protein (Supplemental Figure S10); thus, this change was also not likely to have any significant biological effect. Thus, Galgt2 overexpression ultimately failed to inhibit muscular pathology from occurring in dystroglycan-deficient muscles, but did show a partial inhibition of dystrophic pathology in young Dag1-deficient muscles.
Discussion
Several surrogate gene therapies currently targeted for DMD, including utrophin, integrin α7, sarcospan, and follistatin, can inhibit muscle pathology when overexpressed and exacerbate muscle pathology when deleted in mdx mice.35–44,87,88 The experiments presented herein suggest that Galgt2 is similar to these other genes. When overexpressed in mdx skeletal muscles, Galgt2 can inhibit the development of muscle damage and pathology associated with disease.47,48,53 Herein, we show that endogenous Galgt2 gene and protein expression levels are elevated in mdx muscles, particularly in regenerating skeletal myofibers, and that deletion of endogenous Galgt2 expression in mdx mice leads to more severe disease pathology. Moreover, elevation of Galgt2 expression in mdx muscle, as well as in developing and regenerating wild-type muscle, is associated with Galgt2-dependent glycosylation of αDG. The fact that the level of Galgt2 elevation in mdx muscles is modest, 3.7-fold as observed herein and twofold as reported in another study,56 further suggests that even relatively low levels of Galgt2 induction in mdx muscles may have an impact on muscle pathology. Strikingly, deletion of Galgt2 in mdx skeletal muscles exacerbates disease, even though both utrophin and plectin1 protein levels are up-regulated. Further work will be required to better understand the relationship between these modifiers, but this may suggest an epistatic relationship between membrane glycosylation and the function of certain dystrophin surrogates.
We report herein four disease-relevant findings that occur on deletion of the Galgt2 gene in mice: i) reduced muscle growth in response to acute muscle injury in wild-type muscle, suggesting a role for endogenous Galgt2 expression in muscle regeneration, ii) increased inflammation, particularly invasion of macrophages, in dystrophin-deficient mdx heart and skeletal muscles, iii) increased muscle histopathology, particularly increased wasting in skeletal muscle and increased necrotic foci in cardiac muscle in mdx mice, and iv) reduced cardiac function, both in wild-type and mdx mice.
With regard to a potential role for Galgt2 in muscle regeneration, we found reduced muscle growth at 14 and 28 days after acute muscle injury induced with Ctx. Increased Galgt2 gene expression peaks at 4 to 7 days after acute muscle injury, a period when newly made myofibers are first being formed in abundance, and then expression is reduced back to near basal levels at 14 to 28 days after injury. This is consistent with our finding of muscle Galgt2 protein expression predominantly occurring in embryonic myosin-positive myofibers in regions with regenerating muscle, whereas myoblasts, evidenced by costaining with MyoD, or satellite cells, evidenced by costaining with Pax7, showed much less Galgt2 protein co-expression. This is similar to what occurs in early postnatal development, where Galgt2 expression peaks in early postnatal myofibers at birth and then is dramatically reduced in expression over the first 2 postnatal weeks, becoming confined to the neuromuscular and myotendinous junctions.45 When Galgt2 is transgenically overexpressed in skeletal myofibers from embryonic time points onward, so that high myofiber expression is maintained into adulthood, satellite cells that normally would fuse into developing postnatal myofibers to contribute to their robust postnatal growth often fail to do so.62 It is possible that a similar function occurs during acute injury, whereby high Galgt2 expression in early developing myofibers prevents premature fusion of satellite cells into regenerating myofibers, allowing for increased myoblast formation and increased muscle growth. This stands in contrast to what occurs with deletion of Galgt1, another member of the β1,4GalNAc transferase family. Galgt1, which is responsible for the biosynthesis of complex gangliosides,89 peaks in expression 1 day after acute injury and is almost exclusively expressed in satellite cells and/or myoblasts.72 Deletion of Galgt1 causes premature differentiation of satellite cells into myoblasts and also increases satellite cell apoptosis, with decreased expression of satellite cell markers (Pax7) and increased expression of myoblast markers (MyoD and Myog) after acute muscle injury.72 This, too, leads to impaired myofiber growth during regeneration after acute injury, but likely for entirely different reasons than occurs with Galgt2 deletion.72
Another changed disease phenotype resulting from deletion of Galgt2 in mdx mice was increased muscle inflammation. The number of CD68-positive macrophages, per unit area, was increased in both heart and in skeletal muscle in Galgt2−/−mdx muscles relative to mdx, whereas numbers of CD4+ helper and CD8+ cytotoxic T lymphocytes did not differ significantly between the two genotypes. The increased numbers of macrophages in Galgt2−/−mdx muscle were consistent with the finding of increased gene expression for cytokines known to stimulate macrophage muscle recruitment and infiltration, including monocyte chemoattractant protein 1 (or chemokine C-C motif ligand 2), macrophage inflammatory protein 1α (or chemokine C-C motif ligand 3), and regulated on activation, normal T-cell expressed and secreted (or chemokine C-C motif ligand 5). Because Galgt2 protein expression was also found to be present in CD68 macrophages in regenerating muscle, it is not clear if this inflammation phenotype results from an autocrine or paracrine function in myofibers or macrophages, or if indeed both cell types are involved. The response of macrophages to acute injury involves multiple macrophage cell populations, including M1 macrophages, which invade the muscle immediately after injury and are responsible for clearance of necrotic tissue, and M2 macrophages, which invade muscle only several days after injury and secrete cytokines that can stimulate the regenerative response.9 Further studies would be required to delineate the relative positive and negative influences of an increased muscle inflammatory response. However, given that muscle pathology was generally more severe in Galgt2−/−mdx muscles than in mdx, the increased macrophage response may reflect the presence of increased muscle necrosis.
The third disease-related finding was the presence of increased skeletal and cardiac muscle damage. This was evident on staining of thin sections with modified Mason trichrome and with H&E. In both instances, increased collagen staining and increased evidence of inflammatory infiltrates were present. Heart and skeletal fibrosis was further confirmed by significantly increased collagen and laminin gene expression using semiquantitative real-time PCR. The modest hypertrophy of Galgt2−/−mdx hind limb muscles, relative to mdx, may also reflect increased fibrosis. Dye uptake was also altered in Galgt2−/−mdx heart relative to mdx, and trended higher in skeletal muscle, suggesting increased membrane damage in the absence of Galgt2 expression. Although the levels of dye uptake were low in heart, this level of damage, which is assayed for only 1 day in time, may contribute to the increased muscle pathology observed in adult animals, which occurs over many months. As a group, these findings suggest that deletion of Galgt2, in the context of already existing dystrophy, increases the severity of muscle histopathology. To some degree, this may merely reflect increased kinetics of muscle damage as opposed to an overall change in level, and future work will be required to delineate between these possibilities.
Our fourth finding in this study was that deletion of Galgt2 altered cardiac function in both wild-type and mdx mice. We chose to study mice at 8 months of age for cardiac function, because mdx mice begin to show significant reductions, relative to wild-type mice, in ejection fraction and percentage fractional shortening as well as increases in left ventricular diameter and overall cardiac inflammation, fibrosis, and damage at this age.90–94 Deletion of other disease-modifying genes that can alter cardiac pathology in mdx mice (eg, matrix metalloproteinase-995 or IL-1096) also demonstrates phenotypes at or near this age. After isoproterenol challenge, Galgt2−/−mdx mice had reduced heart rate, cardiac output, percentage fractional shortening, and stroke volume relative to wild-type mice and also reduced heart rate and percentage fractional shortening relative to mdx mice. Furthermore, resting measures for Galgt2−/− mice for cardiac output and percentage fractional shortening were reduced relative to wild-type mice. These findings suggest that the increased inflammation and increased overall muscle damage found in Galgt2−/−mdx heart, relative to wild-type and mdx, likely have consequences for heart function, which is important, because cardiomyopathy is a major factor in DMD mortality. The presence of functional changes in Galgt2-deleted wild-type heart, however, may also suggest that Galgt2 is essential for overall cardiac function, independent of the presence of muscular dystrophy.
Our final finding in this study was that Galgt2 overexpression required dystroglycan to fully inhibit muscular dystrophy. Galgt2 overexpression ultimately has no impact on the extent of skeletal muscle pathology in dystroglycan (Dag1)-deficient skeletal muscles by 9 months of age, although clearly some effect was evident in younger (2-month-old) mice. More modest levels of pathology are also seen in mdx at 2 months of age compared with later time points,48,53 and this reduced extent of muscle damage may allow for a partial therapeutic response.
αDG is the major glycoprotein target for Galgt2 glycosylation with the CT glycan in skeletal muscle.48,62 In our first study of AAV-Galgt2 overexpression in mdx skeletal muscle,53 we did not observe induction of αDG glycosylation, as we had previously observed in Galgt2 transgenic mdx skeletal muscles.48,62 The work presented herein, coupled with our recent study of AAV-Galgt2 overexpression in rhesus macaques,71 demonstrates that Galgt2 does induce the glycosylation of αDG when the gene is overexpressed in adult skeletal muscle. Our failure to demonstrate this in our first study in 2007 was likely because of the relatively low dose of virus used and the fact that we only chose a 6-week time point for analysis. The slow turnover time of muscle membrane proteins (which can take weeks), coupled with the 3- to 4-week requirement for AAV to induce maximal muscle gene expression,49 and the general down-regulation of αDG protein expression in mdx muscles,97 likely made detecting αDG glycosylation at this short time point more difficult. One additional target of Galgt2 glycosylation is a glycolipid, which in other systems has been identified as sialopentosylceramide.98,99 The glycosylation of this membrane glycolipid has allowed us to observe Galgt2 overexpression in skeletal muscle, even in the absence of dystroglycan. Because Galgt2 overexpression showed a partial inhibition of muscle pathology in Dag1−/− muscles at 2 months (but that was lost by 9 months), it is possible that this Galgt2-glycosylated glycolipid, or other factors induced by Galgt2 overexpression, other than dystroglycan, may also affect skeletal muscle pathology.
The combination of the findings presented herein, increased inflammation, histopathology, and slowed muscle growth during regeneration in skeletal muscle, and increased inflammation, histopathology, and membrane permeability in cardiac muscle, point to potential mechanisms by which loss of Galgt2 could increase muscle pathology in mdx mice. Further experiments will be required to understand the cell or cells of origin that produce these various loss-of-function phenotypes, but as a group, these studies support a role for endogenous Galgt2 expression as a modifier of muscle pathology phenotypes in mdx mice and suggest that Galgt2 glycosylation of αDG is important for these biological effects.
Acknowledgments
Galgt2−/− mice were obtained from the Consortium for Functional Glycomics. We thank John Lowe (Genentech) for generating and allowing us to use Galgt2−/− mice; Jeffery Miner (Washington University, St. Louis, MO) for laminin α5 antibody and P3ProCreDag1lox/lox mice; Kevin Campbell (The University of Iowa, Iowa City, IA) for generating and allowing us to use Dag1lox/lox mice; John Epstein for generating and allowing us to use P3Pro-Cre mice; Reed Clark (Nationwide Children's Hospital, Columbus, OH) for AAV viral vector production; and Bethannie Golden, Guohong Shao, Marybeth Camboni, Sarah deVries, Ying Jia, and Randall Evans (The Research Institute at Nationwide Children's Hospital, Columbus, OH) for technical support. Pax7 antibodies were provided by Michael Rudnicki (Ottawa Health Research Institute, Ottawa, ON, Canada).
Footnotes
Supported by NIH grant R01 AR049722 (P.T.M.). Muscle physiology experiments were partially supported by NIH grant P30 NS045758 (Core D support to P.M.L.J.).
Disclosures: None declared.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2015.06.008.
Supplemental Data
Supplemental Figure S1.

Expression of Galgt2 protein and CT glycan in developing and regenerating skeletal muscle and in embryonal rhabdomyosarcoma. CT2 staining of CT glycan and Galgt2 immunostaining increase in day 4 regenerating muscle after cardiotoxin injection (P4 Ctx) and in embryonal rhabdomyosarcoma (eRMS). Developing normal muscle shows high CT glycan expression (stained with CT2) at postnatal day P1, with primarily synaptic concentration by P14 and P42. P42 staining serves as a primary antibody control for P4 Ctx staining in a normal muscle. Control shows staining with secondary antibody alone. Scale bar = 100 μm.
Supplemental Figure S2.
CT glycan staining is reduced in Galgt2−/−mdx skeletal muscle and heart. A: Longitudinal sections of the left ventricular wall of the heart from wild-type (WT), Galgt2−/−, mdx, and Galgt2−/−mdx mice stained with CT2 at 2 months of age. B: Cross sections of gastrocnemius muscle from WT, Galgt2−/−, mdx, and Galgt2−/−mdx mice stained with CT2 at 2 months of age. Scale bar = 50 μm (A and B).
Supplemental Figure S3.

Galgt2 and dystrophin protein expression in wild-type (WT), Galgt2−/−, mdx, and Galgt2−/−mdx mice. A: Nonidet-P40–extracted mouse protein (40 μg) from WT, Galgt2−/−, mdx, and Galgt2−/−mdx gastrocnemius muscle was separated by SDS-PAGE and immunoblotted for Galgt2 or dystrophin protein, with actin as a loading control. B: Immunostaining for Galgt2 and dystrophin protein in 2-month-old gastrocnemius muscle. Arrows indicate a myotendinous junction. Scale bar = 50 μm (B).
Supplemental Figure S4.

Hematoxylin and eosin (H&E) staining of gastrocnemius, diaphragm, and heart muscle. Gastrocnemius (A), diaphragm (B), and heart (C) muscle sections were stained with H&E. Sections from 5-month-old wild-type (WT), Galgt2−/−, mdx, and Galgt2−/−mdx muscles are shown. Scale bar = 200 μm (A–C).
Supplemental Figure S5.
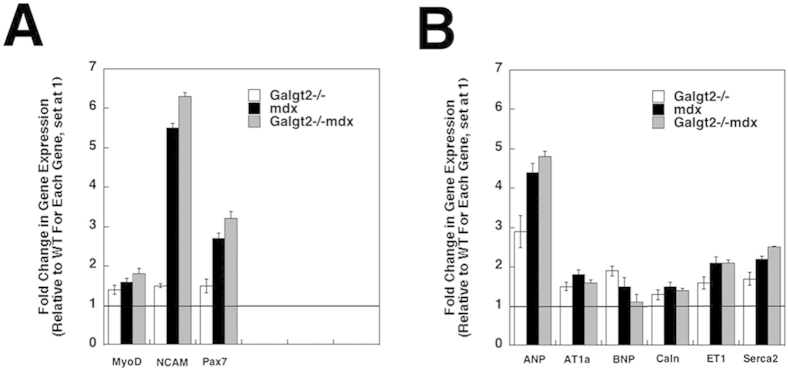
Expression of markers of muscle regeneration and cardiac hypertrophy or cardiomyopathy in skeletal muscle and heart. Semiquantitative real-time PCR measurements of gene expression were made in wild-type (WT), Galgt2−/−, mdx, and Galgt2−/−mdx skeletal muscle (gastrocnemius, A) and heart (B) at 5 months of age, comparing relative gene expression for genes related to muscle regeneration (A) or cardiac hypertrophy or cardiomyopathy (B). All Galgt2−/−mdx, mdx, and Galgt2−/− measures are compared with wild-type (WT) measures for the same gene at the same age, which was set at 1 (line). Errors are SEM (A and B). n = 6 to 9 measurements per condition (A and B). ANP, atrial natriuretic peptide; AT1a, angiotensin II receptor; BNP, B-type natriuretic peptide; Caln, calcineurin; ET1, endothelin 1; MyoD, myogenic differentiation factor D; NCAM, neural cell adhesion molecule; Pax7, paired box protein 7; Serca2, sarcoplasmic reticulum calcium ATPase 2.
Supplemental Figure S6.

Expression of dystrophin-associated glycoprotein (DAG) genes and extracellular matrix (ECM) genes in skeletal muscle and heart. Semiquantitative real-time PCR measurements of gene expression were made in wild-type (WT), Galgt2−/−, mdx, and Galgt2−/−mdx skeletal muscle (gastrocnemius, A and C) and heart (B and D) at 5 months of age, comparing relative gene expression for genes related to the dystrophin-associated glycoprotein complex (A and B) or extracellular matrix genes (C and D). All Galgt2−/−mdx, mdx, and Galgt2−/− measures are compared with wild-type (WT) measures for the same gene at the same age, which was set at 1 (line). Errors are SEM (A–D). n = 6 to 9 measurements per condition (A–D). ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001. Agrn, agrin; Col1α1, collagen 1(α1); Col3α1, collagen 3(α1); Col4α1, collagen 4(α1); Col4α2, collagen 4(α2); αDB, α dystrobrevin; DG, dystroglycan; Dys, dystrophin; Intα7, integrin α7; Intβ1, integrin β1; LNα2, laminin α2; LNα4, laminin α4; LNα5, laminin α5; αSG, α sarcoglycan; βSG, β sarcoglycan; γSG, γ sarcoglycan; δSG, δ sarcoglycan; Utrn, utrophin.
Supplemental Figure S7.

Expression of dystrophin-associated glycoproteins in skeletal muscle and heart. A: Co-immunostaining of utrophin (Utrn) and acetylcholine receptors (AChRs; stained with α bungarotoxin) in wild-type (WT), Galgt2−/−, mdx, and Galgt2−/−mdx skeletal muscle. 2nd Only shows staining with secondary antibody alone along with positive AChR staining. B: Western blots of whole muscle protein from wild-type (Dmd+) and mdx (Dmd−) mice, with or without deletion of Galgt2, analyzed with antibodies to α dystroglycan (αDG), β dystroglycan (βDG), γ sarcoglycan (γSG), utrophin (Utrn), plectin 1 (Plec1), or actin. Scale bar = 50 μm (A).
Supplemental Figure S8.

Skeletal muscle physiology measures comparing wild-type, Galgt2−/−, mdx, and Galgt2−/−mdx mice. A: Maximal tetanic-specific force in the extensor digitorum longus (EDL) muscle. B: Force decrease during eccentric contractions in the EDL muscle. C: Specific force in diaphragm muscle measured at 180 Hz. Errors are SEM (A–C). n = 5 to 10 per group. ∗P < 0.05, ∗∗P < 0.01.
Supplemental Figure S9.
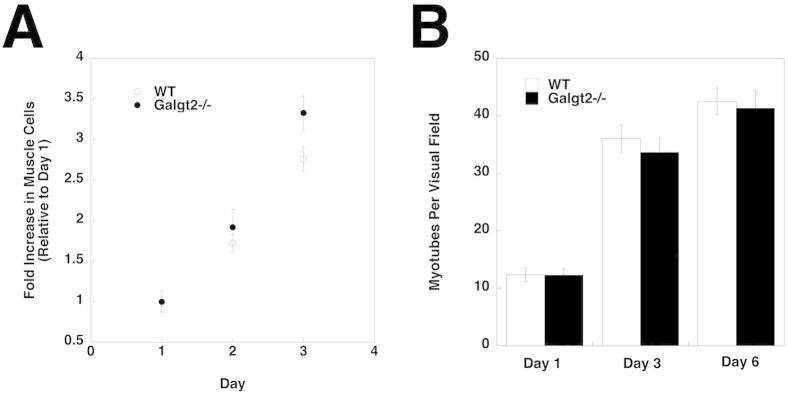
Growth and fusion of Galgt2−/− primary muscle cells in culture. A: Growth rate of Galgt2-deficient (Galgt2−/−) cultured primary muscle cells does not change relative to primary muscle cells from wild-type (WT) mice. B: Muscle cells grown to confluence were placed in fusion medium, and myotubes were counted at 1, 3, and 6 days after fusion. Galgt2−/− muscle cells are not significantly changed in their fusion capability compared with WT muscle cells. Errors are SEM (A and B). n = 6 mice per condition, with n = 3 replicates per experiment (A and B).
Supplemental Figure S10.

Percentage of myofibers expressing dystroglycan protein in wild-type, Galgt2 transgenic (mCT), dystroglycan-deficient (Dag1−/−), and mCTDag1−/− muscles. Muscle cross sections were stained for dystroglycan protein, with a laminin α2-counterstain, to identify all dystroglycan-expressing skeletal myofibers. Errors are SD. n = 3 per condition.
References
- 1.Mendell J.R., Rodino-Klapac L.R., Sahenk Z., Roush K., Bird L., Lowes L.P., Alfano L., Gomez A.M., Lewis S., Kota J., Malik V., Shontz K., Walker C.M., Flanigan K.M., Corridore M., Kean J.R., Allen H.D., Shilling C., Melia K.R., Sazani P., Saoud J.B., Kaye E.M. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. 2013;74:637–647. doi: 10.1002/ana.23982. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman E.P., Brown R.H., Jr., Kunkel L.M. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 3.Koenig M., Hoffman E.P., Bertelson C.J., Monaco A.P., Feener C., Kunkel L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- 4.Mann C.J., Honeyman K., Cheng A.J., Ly T., Lloyd F., Fletcher S., Morgan J.E., Partridge T.A., Wilton S.D. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc Natl Acad Sci U S A. 2001;98:42–47. doi: 10.1073/pnas.011408598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Head S.I., Williams D.A., Stephenson D.G. Abnormalities in structure and function of limb skeletal muscle fibres of dystrophic mdx mice. Proc Biol Sci. 1992;248:163–169. doi: 10.1098/rspb.1992.0058. [DOI] [PubMed] [Google Scholar]
- 6.Sacco P., Jones D.A., Dick J.R., Vrbova G. Contractile properties and susceptibility to exercise-induced damage of normal and mdx mouse tibialis anterior muscle. Clin Sci (Lond) 1992;82:227–236. doi: 10.1042/cs0820227. [DOI] [PubMed] [Google Scholar]
- 7.Bridges L.R. The association of cardiac muscle necrosis and inflammation with the degenerative and persistent myopathy of MDX mice. J Neurol Sci. 1986;72:147–157. doi: 10.1016/0022-510x(86)90003-1. [DOI] [PubMed] [Google Scholar]
- 8.Spencer M.J., Tidball J.G. Do immune cells promote the pathology of dystrophin-deficient myopathies? Neuromuscul Disord. 2001;11:556–564. doi: 10.1016/s0960-8966(01)00198-5. [DOI] [PubMed] [Google Scholar]
- 9.Tidball J.G., Villalta S.A. Regulatory interactions between muscle and the immune system during muscle regeneration. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1173–R1187. doi: 10.1152/ajpregu.00735.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y.X., Rudnicki M.A. Satellite cells, the engines of muscle repair. Nat Rev Mol Cell Biol. 2012;13:127–133. doi: 10.1038/nrm3265. [DOI] [PubMed] [Google Scholar]
- 11.Verdijk L.B., Snijders T., Drost M., Delhaas T., Kadi F., van Loon L.J. Satellite cells in human skeletal muscle: from birth to old age. Age (Dordr) 2014;36:545–547. doi: 10.1007/s11357-013-9583-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De la Porte S., Morin S., Koenig J. Characteristics of skeletal muscle in mdx mutant mice. Int Rev Cytol. 1999;191:99–148. doi: 10.1016/s0074-7696(08)60158-8. [DOI] [PubMed] [Google Scholar]
- 13.Cardasis C.A., Cooper G.W. An analysis of nuclear numbers in individual muscle fibers during differentiation and growth: a satellite cell-muscle fiber growth unit. J Exp Zool. 1975;191:347–358. doi: 10.1002/jez.1401910305. [DOI] [PubMed] [Google Scholar]
- 14.Stedman H.H., Sweeney H.L., Shrager J.B., Maguire H.C., Panettieri R.A., Petrof B., Narusawa M., Leferovich J.M., Sladky J.T., Kelly A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature. 1991;352:536–539. doi: 10.1038/352536a0. [DOI] [PubMed] [Google Scholar]
- 15.Pastoret C., Sebille A. mdx mice show progressive weakness and muscle deterioration with age. J Neurol Sci. 1995;129:97–105. doi: 10.1016/0022-510x(94)00276-t. [DOI] [PubMed] [Google Scholar]
- 16.McNally E.M. New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annu Rev Med. 2007;58:75–88. doi: 10.1146/annurev.med.58.011706.144703. [DOI] [PubMed] [Google Scholar]
- 17.Mendell J.R., Moxley R.T., Griggs R.C., Brooke M.H., Fenichel G.M., Miller J.P., King W., Signore L., Pandya S., Florence J., Schierbecker J., Robison J., Kaiser K., Mandel S., Arfken C., Gilder B. Randomized, double-blind six-month trial of prednisone in Duchenne's muscular dystrophy. N Engl J Med. 1989;320:1592–1597. doi: 10.1056/NEJM198906153202405. [DOI] [PubMed] [Google Scholar]
- 18.Biggar W.D., Harris V.A., Eliasoph L., Alman B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord. 2006;16:249–255. doi: 10.1016/j.nmd.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 19.Manzur A.Y., Muntoni F. Diagnosis and new treatments in muscular dystrophies. Postgrad Med J. 2009;85:622–630. doi: 10.1136/jnnp.2008.158329. [DOI] [PubMed] [Google Scholar]
- 20.Chicoine L.G., Montgomery C.L., Bremer W.G., Shontz K.M., Griffin D.A., Heller K.N., Lewis S., Malik V., Grose W.E., Shilling C.J., Campbell K.J., Preston T.J., Coley B.D., Martin P.T., Walker C.M., Clark K.R., Sahenk Z., Mendell J.R., Rodino-Klapac L.R. Plasmapheresis eliminates the negative impact of AAV antibodies on microdystrophin gene expression following vascular delivery. Mol Ther. 2014;22:338–347. doi: 10.1038/mt.2013.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yue Y., Ghosh A., Long C., Bostick B., Smith B.F., Kornegay J.N., Duan D. A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs. Mol Ther. 2008;16:1944–1952. doi: 10.1038/mt.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Judge L.M., Chamberlain J.S. Gene therapy for Duchenne muscular dystrophy: AAV leads the way. Acta Myol. 2005;24:184–193. [PubMed] [Google Scholar]
- 23.Harper S.Q., Hauser M.A., DelloRusso C., Duan D., Crawford R.W., Phelps S.F., Harper H.A., Robinson A.S., Engelhardt J.F., Brooks S.V., Chamberlain J.S. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 24.Scott J.M., Li S., Harper S.Q., Welikson R., Bourque D., DelloRusso C., Hauschka S.D., Chamberlain J.S. Viral vectors for gene transfer of micro-, mini-, or full-length dystrophin. Neuromuscul Disord. 2002;12(Suppl 1):S23–S29. doi: 10.1016/s0960-8966(02)00078-0. [DOI] [PubMed] [Google Scholar]
- 25.Cirak S., Arechavala-Gomeza V., Guglieri M., Feng L., Torelli S., Anthony K., Abbs S., Garralda M.E., Bourke J., Wells D.J., Dickson G., Wood M.J., Wilton S.D., Straub V., Kole R., Shrewsbury S.B., Sewry C., Morgan J.E., Bushby K., Muntoni F. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rinaldi F., Perlingeiro R.C. Stem cells for skeletal muscle regeneration: therapeutic potential and roadblocks. Transl Res. 2014;163:409–417. doi: 10.1016/j.trsl.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karpati G., Ajdukovic D., Arnold D., Gledhill R.B., Guttmann R., Holland P. Myoblast transfer in Duchenne muscular dystrophy. Ann Neurol. 1993;34:8–17. doi: 10.1002/ana.410340105. [DOI] [PubMed] [Google Scholar]
- 28.Bonuccelli G., Sotgia F., Schubert W., Park D.S., Frank P.G., Woodman S.E., Insabato L., Cammer M., Minetti C., Lisanti M.P. Proteasome inhibitor (MG-132) treatment of mdx mice rescues the expression and membrane localization of dystrophin and dystrophin-associated proteins. Am J Pathol. 2003;163:1663–1675. doi: 10.1016/S0002-9440(10)63523-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Assereto S., Stringara S., Sotgia F., Bonuccelli G., Broccolini A., Pedemonte M., Traverso M., Biancheri R., Zara F., Bruno C., Lisanti M.P., Minetti C. Pharmacological rescue of the dystrophin-glycoprotein complex in Duchenne and Becker skeletal muscle explants by proteasome inhibitor treatment. Am J Physiol Cell Physiol. 2006;290:C577–C582. doi: 10.1152/ajpcell.00434.2005. [DOI] [PubMed] [Google Scholar]
- 30.Wagner K.R., McPherron A.C., Winik N., Lee S.J. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol. 2002;52:832–836. doi: 10.1002/ana.10385. [DOI] [PubMed] [Google Scholar]
- 31.Haidet A.M., Rizo L., Handy C., Umapathi P., Eagle A., Shilling C., Boue D., Martin P.T., Sahenk Z., Mendell J.R., Kaspar B.K. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci U S A. 2008;105:4318–4322. doi: 10.1073/pnas.0709144105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mendell J.R., Sahenk Z., Malik V., Gomez A.M., Flanigan K.M., Lowes L.P., Alfano L.N., Berry K., Meadows E., Lewis S., Braun L., Shontz K., Rouhana M., Clark K.R., Rosales X.Q., Al-Zaidy S., Govoni A., Rodino-Klapac L.R., Hogan M.J., Kaspar B.K. A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol Ther. 2015;23:192–201. doi: 10.1038/mt.2014.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flanigan K.M., Campbell K., Viollet L., Wang W., Gomez A.M., Walker C.M., Mendell J.R. Anti-dystrophin T cell responses in Duchenne muscular dystrophy: prevalence and a glucocorticoid treatment effect. Hum Gene Ther. 2013;24:797–806. doi: 10.1089/hum.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sonnemann K.J., Heun-Johnson H., Turner A.J., Baltgalvis K.A., Lowe D.A., Ervasti J.M. Functional substitution by TAT-utrophin in dystrophin-deficient mice. PLoS Med. 2009;6:e1000083. doi: 10.1371/journal.pmed.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deconinck N., Tinsley J., De Backer F., Fisher R., Kahn D., Phelps S., Davies K., Gillis J.M. Expression of truncated utrophin leads to major functional improvements in dystrophin-deficient muscles of mice. Nat Med. 1997;3:1216–1221. doi: 10.1038/nm1197-1216. [DOI] [PubMed] [Google Scholar]
- 36.Deconinck A.E., Rafael J.A., Skinner J.A., Brown S.C., Potter A.C., Metzinger L., Watt D.J., Dickson J.G., Tinsley J.M., Davies K.E. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- 37.Grady R.M., Teng H., Nichol M.C., Cunningham J.C., Wilkinson R.S., Sanes J.R. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- 38.Tinsley J., Deconinck N., Fisher R., Kahn D., Phelps S., Gillis J.M., Davies K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med. 1998;4:1441–1444. doi: 10.1038/4033. [DOI] [PubMed] [Google Scholar]
- 39.Burkin D.J., Wallace G.Q., Nicol K.J., Kaufman D.J., Kaufman S.J. Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J Cell Biol. 2001;152:1207–1218. doi: 10.1083/jcb.152.6.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rooney J.E., Welser J.V., Dechert M.A., Flintoff-Dye N.L., Kaufman S.J., Burkin D.J. Severe muscular dystrophy in mice that lack dystrophin and alpha7 integrin. J Cell Sci. 2006;119:2185–2195. doi: 10.1242/jcs.02952. [DOI] [PubMed] [Google Scholar]
- 41.Nakatani M., Takehara Y., Sugino H., Matsumoto M., Hashimoto O., Hasegawa Y., Murakami T., Uezumi A., Takeda S., Noji S., Sunada Y., Tsuchida K. Transgenic expression of a myostatin inhibitor derived from follistatin increases skeletal muscle mass and ameliorates dystrophic pathology in mdx mice. FASEB J. 2008;22:477–487. doi: 10.1096/fj.07-8673com. [DOI] [PubMed] [Google Scholar]
- 42.Tsuchida K. Myostatin inhibition by a follistatin-derived peptide ameliorates the pathophysiology of muscular dystrophy model mice. Acta Myol. 2008;27:14–18. [PMC free article] [PubMed] [Google Scholar]
- 43.Bogdanovich S., Krag T.O., Barton E.R., Morris L.D., Whittemore L.A., Ahima R.S., Khurana T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. doi: 10.1038/nature01154. [DOI] [PubMed] [Google Scholar]
- 44.Bogdanovich S., Perkins K.J., Krag T.O., Whittemore L.A., Khurana T.S. Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J. 2005;19:543–549. doi: 10.1096/fj.04-2796com. [DOI] [PubMed] [Google Scholar]
- 45.Martin P.T., Scott L.J., Porter B.E., Sanes J.R. Distinct structures and functions of related pre- and postsynaptic carbohydrates at the mammalian neuromuscular junction. Mol Cell Neurosci. 1999;13:105–118. doi: 10.1006/mcne.1999.0737. [DOI] [PubMed] [Google Scholar]
- 46.Hoyte K., Kang C., Martin P.T. Definition of pre- and postsynaptic forms of the CT carbohydrate antigen at the neuromuscular junction: ubiquitous expression of the CT antigens and the CT GalNAc transferase in mouse tissues. Brain Res Mol Brain Res. 2002;109:146–160. doi: 10.1016/s0169-328x(02)00551-x. [DOI] [PubMed] [Google Scholar]
- 47.Martin P.T., Xu R., Rodino-Klapac L.R., Oglesbay E., Camboni M., Montgomery C.L., Shontz K., Chicoine L.G., Clark K.R., Sahenk Z., Mendell J.R., Janssen P.M. Overexpression of Galgt2 in skeletal muscle prevents injury resulting from eccentric contractions in both mdx and wild-type mice. Am J Physiol Cell Physiol. 2009;296:C476–C478. doi: 10.1152/ajpcell.00456.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nguyen H.H., Jayasinha V., Xia B., Hoyte K., Martin P.T. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc Natl Acad Sci U S A. 2002;99:5616–5621. doi: 10.1073/pnas.082613599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu R., Chandrasekharan K., Yoon J.H., Camboni M., Martin P.T. Overexpression of the cytotoxic T cell (CT) carbohydrate inhibits muscular dystrophy in the dyW mouse model of congenital muscular dystrophy 1A. Am J Pathol. 2007;171:181–199. doi: 10.2353/ajpath.2007.060927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu R., DeVries S., Camboni M., Martin P.T. Overexpression of Galgt2 reduces dystrophic pathology in the skeletal muscles of alpha sarcoglycan-deficient mice. Am J Pathol. 2009;175:235–247. doi: 10.2353/ajpath.2009.080967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoon J.H., Chandrasekharan K., Xu R., Glass M., Singhal N., Martin P.T. The synaptic CT carbohydrate modulates binding and expression of extracellular matrix proteins in skeletal muscle: partial dependence on utrophin. Mol Cell Neurosci. 2009;41:448–463. doi: 10.1016/j.mcn.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jayasinha V., Hoyte K., Xia B., Martin P.T. Overexpression of the CT GalNAc transferase inhibits muscular dystrophy in a cleavage-resistant dystroglycan mutant mouse. Biochem Biophys Res Commun. 2003;302:831–836. doi: 10.1016/s0006-291x(03)00271-7. [DOI] [PubMed] [Google Scholar]
- 53.Xu R., Camboni M., Martin P.T. Postnatal overexpression of the CT GalNAc transferase inhibits muscular dystrophy in mdx mice without altering muscle growth or neuromuscular development: evidence for a utrophin-independent mechanism. Neuromuscul Disord. 2007;17:209–220. doi: 10.1016/j.nmd.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sicinski P., Geng Y., Ryder-Cook A.S., Barnard E.A., Darlison M.G., Barnard P.J. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 55.Bulfield G., Siller W.G., Wight P.A., Moore K.J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc Natl Acad Sci U S A. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marshall J.L., Holmberg J., Chou E., Ocampo A.C., Oh J., Lee J., Peter A.K., Martin P.T., Crosbie-Watson R.H. Sarcospan-dependent Akt activation is required for utrophin expression and muscle regeneration. J Cell Biol. 2012;197:1009–1027. doi: 10.1083/jcb.201110032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jarad G., Miner J.H. The Pax3-Cre transgene exhibits a rostrocaudal gradient of expression in the skeletal muscle lineage. Genesis. 2009;47:1–6. doi: 10.1002/dvg.20447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cohn R.D., Henry M.D., Michele D.E., Barresi R., Saito F., Moore S.A., Flanagan J.D., Skwarchuk M.W., Robbins M.E., Mendell J.R., Williamson R.A., Campbell K.P. Disruption of DAG1 in differentiated skeletal muscle reveals a role for dystroglycan in muscle regeneration. Cell. 2002;110:639–648. doi: 10.1016/s0092-8674(02)00907-8. [DOI] [PubMed] [Google Scholar]
- 59.Li J., Chen F., Epstein J.A. Neural crest expression of Cre recombinase directed by the proximal Pax3 promoter in transgenic mice. Genesis. 2000;26:162–164. doi: 10.1002/(sici)1526-968x(200002)26:2<162::aid-gene21>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 60.Muscat G.E., Kedes L. Multiple 5′-flanking regions of the human alpha-skeletal actin gene synergistically modulate muscle-specific expression. Mol Cell Biol. 1987;7:4089–4099. doi: 10.1128/mcb.7.11.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brennan K.J., Hardeman E.C. Quantitative analysis of the human alpha-skeletal actin gene in transgenic mice. J Biol Chem. 1993;268:719–725. [PubMed] [Google Scholar]
- 62.Xia B., Hoyte K., Kammesheidt A., Deerinck T., Ellisman M., Martin P.T. Overexpression of the CT GalNAc transferase in skeletal muscle alters myofiber growth, neuromuscular structure, and laminin expression. Dev Biol. 2002;242:58–73. doi: 10.1006/dbio.2001.0530. [DOI] [PubMed] [Google Scholar]
- 63.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 64.Chandrasekharan K., Yoon J.H., Xu Y., deVries S., Camboni M., Janssen P.M., Varki A., Martin P.T. A human-specific deletion in mouse Cmah increases disease severity in the mdx model of Duchenne muscular dystrophy. Sci Transl Med. 2010;2:42ra54. doi: 10.1126/scitranslmed.3000692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clark K.R., Liu X., McGrath J.P., Johnson P.R. Highly purified recombinant adeno-associated virus vectors are biologically active and free of detectable helper and wild-type viruses. Hum Gene Ther. 1999;10:1031–1039. doi: 10.1089/10430349950018427. [DOI] [PubMed] [Google Scholar]
- 66.Xia B., Martin P.T. Modulation of agrin binding and activity by the CT and related carbohydrate antigens. Mol Cell Neurosci. 2002;19:539–551. doi: 10.1006/mcne.2001.1095. [DOI] [PubMed] [Google Scholar]
- 67.Hoyte K., Jayasinha V., Xia B., Martin P.T. Transgenic overexpression of dystroglycan does not inhibit muscular dystrophy in mdx mice. Am J Pathol. 2004;164:711–718. doi: 10.1016/S0002-9440(10)63158-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Conzelmann A., Lefrancois L. Monoclonal antibodies specific for T cell-associated carbohydrate determinants react with human blood group antigens CAD and SDA. J Exp Med. 1988;167:119–131. doi: 10.1084/jem.167.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang T., Lu X., Arnold P., Liu Y., Baliga R., Huang H., Bauer J.A., Liu Y., Feng Q. Mitogen-activated protein kinase phosphatase-1 inhibits myocardial TNF-alpha expression and improves cardiac function during endotoxemia. Cardiovasc Res. 2012;93:471–479. doi: 10.1093/cvr/cvr346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Han B., Baliga R., Huang H., Giannone P.J., Bauer J.A. Decreased cardiac expression of vascular endothelial growth factor and redox imbalance in murine diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2009;297:H829–H835. doi: 10.1152/ajpheart.00222.2009. [DOI] [PubMed] [Google Scholar]
- 71.Chicoine L.G., Rodino-Klapac L.R., Shao G., Xu R., Bremer W.G., Camboni M., Golden B., Montgomery C.L., Shontz K., Heller K.N., Griffin D.A., Lewis S., Coley B.D., Walker C.M., Clark K.R., Sahenk Z., Mendell J.R., Martin P.T. Vascular delivery of rAAVrh74.MCK.GALGT2 to the gastrocnemius muscle of the rhesus macaque stimulates the expression of dystrophin and laminin alpha2 surrogates. Mol Ther. 2014;22:713–724. doi: 10.1038/mt.2013.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Singhal N., Martin P.T. A role for Galgt1 in skeletal muscle regeneration. Skelet Muscle. 2015;5:3. doi: 10.1186/s13395-014-0028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fernandez K., Serinagaoglu Y., Hammond S., Martin L.T., Martin P.T. Mice lacking dystrophin or alpha sarcoglycan spontaneously develop embryonal rhabdomyosarcoma with cancer-associated p53 mutations and alternatively spliced or mutant Mdm2 transcripts. Am J Pathol. 2010;176:416–434. doi: 10.2353/ajpath.2010.090405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Camboni M., Hammond S., Martin L.T., Martin P.T. Induction of a regenerative microenvironment in skeletal muscle is sufficient to induce embryonal rhabdomyosarcoma in p53-deficient mice. J Pathol. 2012;226:40–49. doi: 10.1002/path.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patton B.L., Miner J.H., Chiu A.Y., Sanes J.R. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol. 1997;139:1507–1521. doi: 10.1083/jcb.139.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chiu A.Y., Sanes J.R. Development of basal lamina in synaptic and extrasynaptic portions of embryonic rat muscle. Dev Biol. 1984;103:456–467. doi: 10.1016/0012-1606(84)90333-6. [DOI] [PubMed] [Google Scholar]
- 77.Hoch W., Ferns M., Campanelli J.T., Hall Z.W., Scheller R.H. Developmental regulation of highly active alternatively spliced forms of agrin. Neuron. 1993;11:479–490. doi: 10.1016/0896-6273(93)90152-h. [DOI] [PubMed] [Google Scholar]
- 78.Singhal N., Martin P.T. Role of extracellular matrix proteins and their receptors in the development of the vertebrate neuromuscular junction. Dev Neurobiol. 2011;71:982–1005. doi: 10.1002/dneu.20953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ervasti J.M., Campbell K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell. 1991;66:1121–1131. doi: 10.1016/0092-8674(91)90035-w. [DOI] [PubMed] [Google Scholar]
- 80.Singhal N., Xu R., Martin P.T. Distinct contributions of Galgt1 and Galgt2 to carbohydrate expression and function at the mouse neuromuscular junction. Mol Cell Neurosci. 2012;51:112–126. doi: 10.1016/j.mcn.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smith P.L., Lowe J.B. Molecular cloning of a murine N-acetylgalactosamine transferase cDNA that determines expression of the T lymphocyte-specific CT oligosaccharide differentiation antigen. J Biol Chem. 1994;269:15162–15171. [PubMed] [Google Scholar]
- 82.Yue Y., Li Z., Harper S.Q., Davisson R.L., Chamberlain J.S., Duan D. Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation. 2003;108:1626–1632. doi: 10.1161/01.CIR.0000089371.11664.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Janssen P.M., Hiranandani N., Mays T.A., Rafael-Fortney J.A. Utrophin deficiency worsens cardiac contractile dysfunction present in dystrophin-deficient mdx mice. Am J Physiol Heart Circ Physiol. 2005;289:H2373–H2378. doi: 10.1152/ajpheart.00448.2005. [DOI] [PubMed] [Google Scholar]
- 84.Cote P.D., Moukhles H., Lindenbaum M., Carbonetto S. Chimaeric mice deficient in dystroglycans develop muscular dystrophy and have disrupted myoneural synapses. Nat Genet. 1999;23:338–342. doi: 10.1038/15519. [DOI] [PubMed] [Google Scholar]
- 85.Williamson R.A., Henry M.D., Daniels K.J., Hrstka R.F., Lee J.C., Sunada Y., Ibraghimov-Beskrovnaya O., Campbell K.P. Dystroglycan is essential for early embryonic development: disruption of Reichert's membrane in Dag1-null mice. Hum Mol Genet. 1997;6:831–841. doi: 10.1093/hmg/6.6.831. [DOI] [PubMed] [Google Scholar]
- 86.Chandraskeharan K., Martin P.T. Embryonic overexpression of Galgt2 inhibits skeletal muscle growth via activation of myostatin signaling. Muscle Nerve. 2009;39:25–41. doi: 10.1002/mus.21198. [DOI] [PubMed] [Google Scholar]
- 87.Duclos F., Straub V., Moore S.A., Venzke D.P., Hrstka R.F., Crosbie R.H., Durbeej M., Lebakken C.S., Ettinger A.J., van der Meulen J., Holt K.H., Lim L.E., Sanes J.R., Davidson B.L., Faulkner J.A., Williamson R., Campbell K.P. Progressive muscular dystrophy in alpha-sarcoglycan-deficient mice. J Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peter A.K., Marshall J.L., Crosbie R.H. Sarcospan reduces dystrophic pathology: stabilization of the utrophin-glycoprotein complex. J Cell Biol. 2008;183:419–427. doi: 10.1083/jcb.200808027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Takamiya K., Yamamoto A., Furukawa K., Yamashiro S., Shin M., Okada M., Fukumoto S., Haraguchi M., Takeda N., Fujimura K., Sakae M., Kishikawa M., Shiku H., Aizawa S. Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc Natl Acad Sci U S A. 1996;93:10662–10667. doi: 10.1073/pnas.93.20.10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vianello S., Bouyon S., Benoit E., Sebrie C., Boerio D., Herbin M., Roulot M., Fromes Y., de la Porte S. Arginine butyrate per os protects mdx mice against cardiomyopathy, kyphosis and changes in axonal excitability. Neurobiol Dis. 2014;71:325–333. doi: 10.1016/j.nbd.2014.08.023. [DOI] [PubMed] [Google Scholar]
- 91.Sciorati C., Staszewsky L., Zambelli V., Russo I., Salio M., Novelli D., Di Grigoli G., Moresco R.M., Clementi E., Latini R. Ibuprofen plus isosorbide dinitrate treatment in the mdx mice ameliorates dystrophic heart structure. Pharmacol Res. 2013;73:35–43. doi: 10.1016/j.phrs.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 92.Uaesoontrachoon K., Quinn J.L., Tatem K.S., Van Der Meulen J.H., Yu Q., Phadke A., Miller B.K., Gordish-Dressman H., Ongini E., Miglietta D., Nagaraju K. Long-term treatment with naproxcinod significantly improves skeletal and cardiac disease phenotype in the mdx mouse model of dystrophy. Hum Mol Genet. 2014;23:3239–3249. doi: 10.1093/hmg/ddu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Quinlan J.G., Hahn H.S., Wong B.L., Lorenz J.N., Wenisch A.S., Levin L.S. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord. 2004;14:491–496. doi: 10.1016/j.nmd.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 94.Spurney C.F., Knoblach S., Pistilli E.E., Nagaraju K., Martin G.R., Hoffman E.P. Dystrophin-deficient cardiomyopathy in mouse: expression of Nox4 and Lox are associated with fibrosis and altered functional parameters in the heart. Neuromuscul Disord. 2008;18:371–381. doi: 10.1016/j.nmd.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dahiya S., Givvimani S., Bhatnagar S., Qipshidze N., Tyagi S.C., Kumar A. Osteopontin-stimulated expression of matrix metalloproteinase-9 causes cardiomyopathy in the mdx model of Duchenne muscular dystrophy. J Immunol. 2011;187:2723–2731. doi: 10.4049/jimmunol.1101342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nitahara-Kasahara Y., Hayashita-Kinoh H., Chiyo T., Nishiyama A., Okada H., Takeda S., Okada T. Dystrophic mdx mice develop severe cardiac and respiratory dysfunction following genetic ablation of the anti-inflammatory cytokine IL-10. Hum Mol Genet. 2014;23:3990–4000. doi: 10.1093/hmg/ddu113. [DOI] [PubMed] [Google Scholar]
- 97.Matsumura K., Ervasti J.M., Ohlendieck K., Kahl S.D., Campbell K.P. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- 98.Kawamura Y.I., Kawashima R., Fukunaga R., Hirai K., Toyama-Sorimachi N., Tokuhara M., Shimizu T., Dohi T. Introduction of Sd(a) carbohydrate antigen in gastrointestinal cancer cells eliminates selectin ligands and inhibits metastasis. Cancer Res. 2005;65:6220–6227. doi: 10.1158/0008-5472.CAN-05-0639. [DOI] [PubMed] [Google Scholar]
- 99.Dohi T., Ohta S., Hanai N., Yamaguchi K., Oshima M. Sialylpentaosylceramide detected with anti-GM2 monoclonal antibody: structural characterization and complementary expression with GM2 in gastric cancer and normal gastric mucosa. J Biol Chem. 1990;265:7880–7885. [PubMed] [Google Scholar]