

Abstract
Congenital hepatic fibrosis is part of many different malformation syndromes, of which oculo-encephalo-hepato-renal syndrome is the most common. These syndromes largely overlap, and so accurate classification of individual patients may be difficult. We present herein three syndromic siblings who were products of a consanguineous marriage. We investigated in detail at least six organ systems in these patients, namely the liver, brain, eye, kidneys, skeleton, and gonads. The common features observed in these three cases were congenital hepatic fibrosis, retinitis pigmentosa, truncal obesity, rotatory nystagmus, mental retardation, advanced myopia, and high-arched palate. The clinical dysmorphology in these patients was distinct and lacked the major features of the known syndromes associated with congenital hepatic fibrosis. Although some features of these presented cases are similar to those found in Bardet-Biedl syndrome (BBS), the absence of some major criteria of BBS (polydactyly, renal abnormality, and hypogonadism) suggests that this may be a new syndrome. All three patients remain under follow-up in the departments of Gastroenterology, Ophthalmology, and Neurology at Hacettepe University.
Keywords: Congenital hepatic fibrosis, Nystagmus, Mental retardation, Retinitis pigmentosa, High-arched palate
Core tip: Congenital hepatic fibrosis is an inherited disorder that may also accompany other congenital syndromes. Here, we present three siblings with a new variant syndrome characterized by congenital hepatic fibrosis, retinitis pigmentosa, mental retardation, nystagmus, high-arched palate, truncal obesity, and advanced myopia.
INTRODUCTION
Congenital hepatic fibrosis (CHF) is an autosomal recessive inherited malformation defined pathologically by a variable degree of periportal fibrosis and irregularly-shaped proliferating bile ducts[1,2]. The exact incidence and prevalence of CHF are not known, but it is a rare disease. By 1981, only 200 patients with CHF had been reported in the literature[3]. The first manifestations of the disease in most patients are signs or symptoms related to portal hypertension, especially splenomegaly and varices, often with gastrointestinal bleeding[4]. The clinical manifestations of CHF are non-specific, making the diagnosis of this disorder difficult. Although the onset of symptoms and signs is highly variable (ranging from early childhood to the 6th decade of life), CHF is most frequently diagnosed during adolescence or young adulthood[4]. The late appearance of symptoms and their clinical evolution suggest that CHF is a dynamic and progressive condition.
CHF occurs in association with a range of both inherited and non-inherited disorders. Described herein are three siblings from consanguineous parents, all of whom had CHF in conjunction with retinitis pigmentosa, truncal obesity, rotatory nystagmus, mental retardation, advanced myopia, and high-arched palate. The aim of this report was to evaluate the clinical findings of these three cases and to compare these findings with relevant syndromes; Joubert, Bardet-Biedl, cerebellar vermis hypoplasia, oligophrenia, ataxia, coloboma, hepatic fibrosis (COACH), Arima, and Meckel, among others.
CASE REPORT
Patient 1
The eldest child of the family is a 34-year-old female patient who presented with nystagmus, truncal obesity [body mass index (BMI): 29, waist circumference: 97 cm], and blurred vision. The family history was unremarkable, with the exception that her parents were first cousins. She reported that her liver disease and splenomegaly were discovered when she presented to the hospital for pneumonia at the age of seven, whereupon she underwent splenectomy and then cholecystectomy.
Laboratory studies revealed the following: hemoglobin 13.4 g/dL, white blood cell count 9200/mm3, platelet count 246000/mm3, international normalized ratio 1.09, partial thromboplastin time 28.3 s, aspartate aminotransferase (AST) 27 U/L, alanine aminotransferase (ALT) 25 U/L, gamma glutamyl transpeptidase (GGT) 32 U/L, total bilirubin 0.59 mg/dL, and albumin 3.4 g/dL. Serum electrolytes, renal function, and urinary examination were normal. The real time and Doppler ultrasonographic examination revealed portal vein cavernous transformation, a heterogeneous liver, and normal kidneys. A needle biopsy of the liver showed an increased number of irregularly-shaped bile ducts, with nodularity of the liver parenchyma accentuated by fibrous septa typical of CHF (Figure 1).
Figure 1.
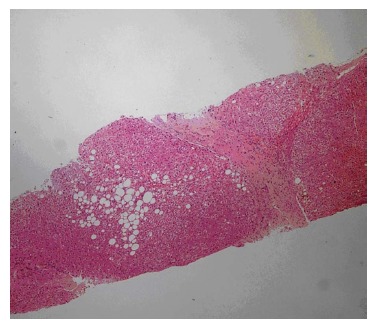
Liver biopsy showing an increased number of abnormal bile ducts, with nodularity of liver parenchyma accentuated by fibrous septa.
Her neurological examination demonstrated mild mental retardation, normal motor examination aside from hypoactive deep tendon reflexes (+1), and normal cerebellar tests. She had a high-arched palate, dystonia in her hands, and pes planus. Her vibration and position senses were decreased. Brain magnetic resonance imaging (MRI) revealed bilateral substance deposition in the globus pallidus, suggesting the presence of chronic liver disease.
She had exhibited signs of blurred vision in infancy/childhood, but her family was not concerned until she attended primary school. She had significant myopia and rotatory nystagmus with normal facial expression (Figures 2 and 3). Fundus examination revealed a pale optic disc, abundant bone spicules involving even the macula, and advanced arterial sclerosis. She also had myopia and rotatory nystagmus. Her bilateral visual acuity was restricted to hand movements only. Electroretinography yielded findings of retinitis pigmentosa (Figure 4). Examination of other cranial nerves yielded normal findings. No respiratory or cardiac abnormalities were found, and she had normal secondary sex characteristics.
Figure 2.
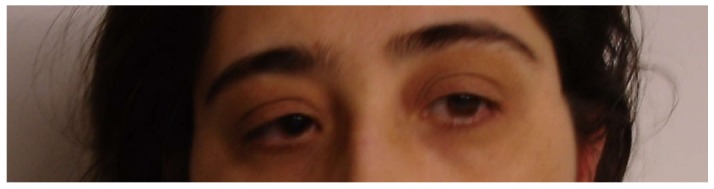
Eyes of the first patient.
Figure 3.
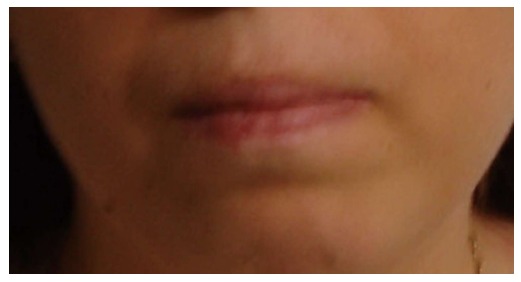
Mouth of the first patient. This appearance helps us to make a differential diagnosis of Cohen’s syndrome, in which a distinct cheerful facial expression is noted.
Figure 4.
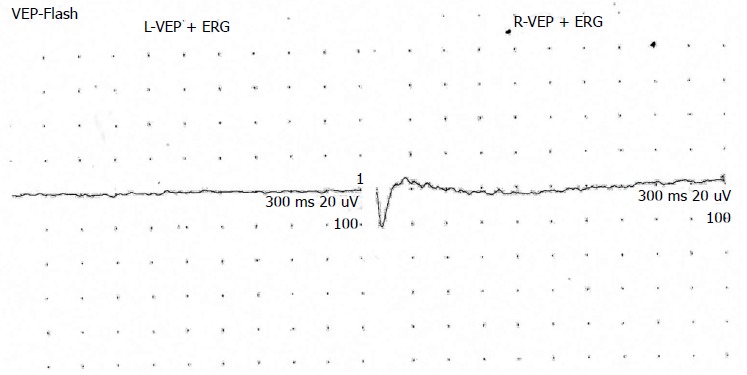
Electroretinography of case 3 showing no response in eyes bilaterally. VEP: Visual evoked potantial; ERG: Electroretinography.
Patient 2
The second patient, a 31-year-old female and the sister of the first patient, also presented with blurred vision, nystagmus, and truncal obesity (BMI: 33, waist circumference: 107 cm).
Laboratory tests revealed the following: hemoglobin 13.4 g/dL, white blood cell count 6600/mm3, platelet count 223000/mm3, ALT 19 U/L, AST 23 U/L, GGT 64 U/L, alkaline phosphatase 70 U/L, total bilirubin 0.72 mg/dL, blood urea nitrogen (BUN) 8 mg/dL, creatinine 0.69 mg/dL, and albumin 4.2 g/dL. Urinary examination was normal. Findings of a liver biopsy of this patient were also consistent with CHF (Figure 5).
Figure 5.
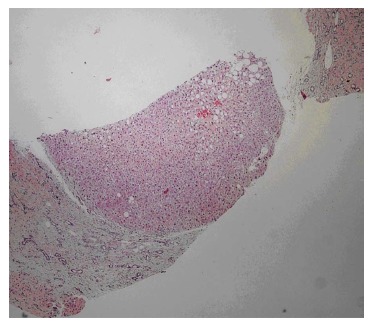
Liver showing nodular appearance due to fibrous bands in which elongated and angulated bile ducts are seen.
Her neurological examination showed mental retardation, normal motor examination aside from hypoactive deep tendon reflexes, normal cerebellar tests, and a negative Romberg test. Her sensation of vibration and position was decreased. Her brain MRI yielded normal findings. She had a high-arched palate.
The patient’s blurred vision was noticed while in primary school. Although decreased, her visual acuity was better than her elder sister; 2/20 with significant myopia bilaterally. Fundus examination revealed a pale optic disc, abundant bone spicules involving even the macula, and advanced arterial sclerosis. She also had rotatory nystagmus. Electroretinography yielded findings of retinitis pigmentosa. Examination of other cranial nerves yielded normal findings. No respiratory, cardiac, or renal abnormalities were found, and she had normal secondary sex characteristics with regular menses.
Patient 3
The third patient, a 30-year-old male and the brother of the first two patients, also presented with blurred vision, nystagmus, and truncal obesity (BMI: 28, waist circumference: 97 cm). Laboratory tests revealed the following: hemoglobin 15.3 g/dL, white blood cell count 5900/mm3, platelet count 92000/mm3, ALT 67 U/L, AST 46 U/L, GGT 122 U/L, alkaline phosphatase 107 U/L, total bilirubin 1 mg/dL, BUN 14 mg/dL, creatinine 0.85 mg/dL, and albumin 4.6 g/dL. Urinary examination was normal. Liver biopsy of this patient was also consistent with CHF (Figure 6).
Figure 6.
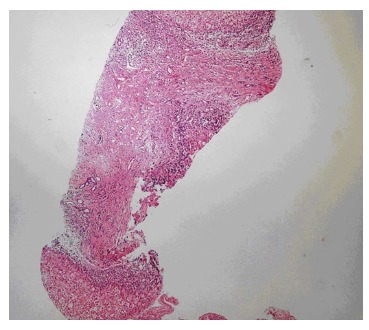
Liver showing nodular appearance due to fibrous bands in which bile ducts are elongated and periportal ductular proliferation is seen.
Patient 3 also had mental retardation, although less pronounced than his sisters. He did not have any motor deficit, aside from hypoactive deep tendon reflexes (+2). His cerebellar tests were normal and Romberg test was negative. His brain MRI revealed normal findings.
The patient’s blurred vision and night blindness were noticed while in primary school. He managed to finish primary school in a special facility for mentally retarded children. His visual acuity was 1/20 bilaterally, with significant myopia. Fundus examination revealed a pale optic disc, abundant bone spicules involving even the macula, and advanced arterial sclerosis. He also had rotatory nystagmus. Electroretinography yielded findings of retinitis pigmentosa. Examination of other cranial nerves yielded normal findings. No respiratory, cardiac, or renal abnormalities were found. He had normal secondary sex characteristics, was married, and had one child.
DISCUSSION
CHF has been described frequently in combination with other abnormalities, such as renal disease, cerebellar malformations, and mental retardation[5,6]. The term oculo-encephalo-hepato-renal syndrome is currently employed to report this association. This syndrome is not a single entity, but rather a group of disorders including COACH[7], Meckel[5], Joubert[8], and Arima syndromes[9] (Table 1). These syndromes largely overlap, and so accurate classification of individual patients may be difficult. It has been suggested that the basic defect in COACH, Joubert syndromes, and other similar conditions might be a disturbance in normal epithelial-mesenchymal interactions due to different genetic mutations[8].
Table 1.
Comparison of our cases with other related syndromes
| Joubert syndrome | Bardet-Biedl syndrome | COACH syndrome | Arima syndrome | Meckel syndrome | Our cases | |
| Cerebellar vermis hypoplasia | +1 | +1 | +1 | |||
| Ataxia | +1 | +1 | ||||
| Abnormal breathing pattern | +1 | |||||
| Abnormal eye movements | +1 | |||||
| Hypotonia | +1 | |||||
| Retinitis pigmentosa | +3 | +1 | + | |||
| Polydactyly | +1 | +3 | +1 | |||
| Truncal obesity | +1 | + | ||||
| Mental retardation | +1 | + | ||||
| Psychomotor retardation | +1 | |||||
| Hypogonadism/genital abnormalities | +1 | |||||
| Renal abnormalities | +1 | +1 | +1 | |||
| Speech disorder/delay | +2 | |||||
| Strabismus/cataract/astigmatism | +2 | |||||
| Brachydactyly/syndactyly | +2 | |||||
| Mild hypertonia | +2 | |||||
| Dental abnormalities | +2 | |||||
| High-arched palate | +2 | + | ||||
| Cardiovascular abnormalities | +2 | +3 | ||||
| Encephalocele | +1 | |||||
| Diabetes mellitus | +2 | |||||
| Oligophrenia | +1 | |||||
| Ocular coloboma | +1 | |||||
| Nystagmus | +3 | +1 | + | |||
| Hepatic fibrosis | +3 | +3 | +1 | +1 | +3 | + |
| Advanced myopia | + |
One of the primary features of this syndrome;
One of the secondary features of this syndrome;
Not a constant feature but has been reported in the literature.
A special subgroup of CHF is COACH syndrome, which is characterized by hypoplasia of the cerebellar vermis, oligophrenia, congenital ataxia, coloboma, and hepatic fibrosis[7]. The abnormalities observed in this syndrome appear to be variable. Numerous congenital anomalies were reported to accompany this syndrome, including slender long bones, postaxial polydactyly, pulmonary stenosis, and atrial septal defect. We found no anomalies involving the kidneys, lungs, or heart in any of our patients, and they did not have ataxia. Furthermore, the absence of the primary features of COACH syndrome (i.e., oligophrenia and ocular coloboma, polydactyly, ataxia, and cerebellar vermis hypoplasia) in our patients excludes that diagnosis.
Another member of this group of disorders, Arima syndrome, is characterized by cerebellar vermis hypoplasia, psychomotor retardation, ocular abnormalities including nystagmus, and polycystic kidneys. Of those, only mental retardation and nystagmus was evident in our patients. Moreover, death in infancy is common in this syndrome, usually due to respiratory failure, and survivors usually have severe mental retardation[10].
Joubert syndrome is an autosomal recessive condition distinguished by hypoplasia of the cerebellar vermis, hypotonia, retinal dystrophy characterized by abnormal eye movements, and impaired psychomotor development together with an abnormal respiratory pattern[11]. This syndrome is genetically heterogeneous with mutations in two genes (AHI-1 and CEP290) identified to date[12]. Although not a constant feature, CHF has also been reported to co-exist with Joubert syndrome. Molar tooth sign (MRI appearance of hypoplasia of the cerebellar vermis and accompanying brainstem abnormalities in an axial plane through the junction of the midbrain and pons) is nearly a pathognomonic finding for this syndrome. In our previous study, we reported two sisters with Joubert syndrome and CHF who presented with abnormal eye movements, speech disorder, and mental motor retardation. Their MRIs were suggestive of Joubert syndrome[6]. However, none of the current three patients had molar tooth sign on MRI, essentially excluding Joubert syndrome. The overlapping features of our patients with Joubert syndrome included poor vision, nystagmus, retinitis pigmentosa, and CHF. These are not constant findings for Joubert syndrome, but have been reported in the literature as co-existent.
Cohen’s syndrome is one of the rare autosomal recessive disorders that are over-represented in the Finnish population[13]. The phenotype in Finnish patients is highly homogenous, consisting of non-progressive mild to severe psychomotor retardation, motor clumsiness, microcephaly, characteristic facial features, childhood hypotonia and joint laxity, progressive retinochoroidal dystrophy, myopia, intermittent isolated neutropenia, and a cheerful disposition. The characteristic facial features include high-arched or wave-shaped eyelids, a short philtrum, thick hair, and low hairline. Truncal obesity appearing during or after mid-childhood can be seen in a minority of individuals with the syndrome[14]. Our cases share some features of Cohen’s syndrome, such as retinitis pigmentosa, myopia, mental retardation, and obesity, but facial dysmorphism and the other aforementioned features which are highly typical for Cohen’s syndrome were absent in our cases.
Retinitis pigmentosa is the term given to a set of hereditary retinal diseases that feature degeneration of rod and cone photoreceptors[15]. A major form of syndromic retinitis pigmentosa, Bardet-Biedl syndrome (BBS), is variably associated with obesity, cognitive impairment, polydactyly, hypogenitalism, and renal disease[16]. BBS has also been found to be associated with CHF. Three families with BBS mapped to the BBS2, BBS3, and BBS4 loci of 2q31 were recruited in one study for a comprehensive eye exam and, in selected cases, electroretinography testing. The results of that study suggested that BBS3 and BBS4 mutations may play a role in the development of myopia[17]. Our patients share primary (truncal obesity and retinitis pigmentosa) and secondary (high-arched palate and CHF) features of BBS. Truncal obesity was defined in our patients according to the International Diabetes Federation 2005 criteria[18]. However, the primary features of BBS, such as postaxial polydactyly, hypogonadism, and renal abnormalities, were absent in the presented cases. Different mutations in unknown genes could possibly be responsible for the advanced myopia in our patients, similar to the situation in BBS. Although myopia has been rarely reported in BBS, advanced myopia was noted in our cases.
In forming the diagnostic criteria for each syndrome, it is important to consider anatomical malformations in conjunction with the clinical signs and symptoms. We investigated six organs in detail, namely the liver, brain, eye, kidneys, skeleton, and gonads. There are major anatomical malformations of the kidney, hands (polydactyly), and gonads in BBS, and of the brain in Joubert, Arima, COACH, and Meckel syndromes. As shown in Table 1, our cases, who presented with CHF, advanced myopia, rotatory nystagmus, truncal obesity, retinitis pigmentosa, mental retardation, and high-arched palate, did not have all or even at least three major components of any listed syndrome. The authors entertain the possibility that our cases may represent a new syndrome. As shown in Table 1, the presented cases most closely resemble BBS. Beales et al[16] reviewed the diagnostic criteria of BBS after evaluating 112 cases based on their clinical findings and they added new criteria; however, liver fibrosis was not included as part of BBS. The predominance of liver fibrosis and the absence of polydactyly, renal abnormality, and hypogonadism in our cases distinguish them from BBS. These clinical findings suggest that our patients might represent a new syndrome.
Our report may contribute to a better delineation of the variable clinical expression of cases within the spectrum of oculo-encephalo-hepato-renal syndromes.
COMMENTS
Case characteristics
Three patients presented with blurred vision and truncal obesity.
Clinical diagnosis
Abnormal signs on physical examination were mental retardation, high-arched palate, pes planus, myopia, rotatory nystagmus, and retinitis pigmentosa on electroretinography.
Differential diagnosis
Cerebellar vermis hypoplasia, oligophrenia, ataxia, coloboma, hepatic fibrosis (COACH), Meckel, Joubert, Arima, Cohen, Bardet-Biedl syndromes.
Laboratory diagnosis
Laboratory test results were essentially within normal limits, with the exception of mildly-elevated alanine aminotransferase in two patients and mild thrombocytopenia in one patient.
Imaging diagnosis
Doppler examination in one patient revealed portal cavernous transformation, while magnetic resonance imaging of the brain was within normal limits in all patients.
Pathological diagnosis
Liver biopsy revealed an increased number of irregularly-shaped bile ducts, with nodularity of the liver parenchyma accentuated by fibrous septa typical of congenital hepatic fibrosis (CHF) in all patients.
Treatment
The patients did not require immediate treatment, but were treated for their ophthalmological disturbances.
Related reports
The co-existence of CHF, retinitis pigmentosa, mental retardation, nystagmus, high-arched palate, truncal obesity, and advanced myopia in the patients may indicate a new variant syndrome different from the known oculo-encephalo-hepato-renal syndromes (i.e., COACH, Meckel and Joubert).
Term explanation
CHF has been described frequently in combination with other abnormalities, such as renal diseases, cerebellar malformations, and mental retardation. The term oculo-encephalo-hepato-renal syndrome is currently employed to report this association.
Experiences and lessons
CHF may present as part of a syndrome affecting the central nervous system and eyes.
Peer-review
The authors have described three cases of CHF associated with ophthalmological and neurological findings which could represent a new syndrome.
Footnotes
Institutional review board statement: The study was reviewed and approved by the Hacettepe University Institutional Review Board.
Informed consent statement: All study participants, or their legal guardian, provided informed written consent prior to study enrollment.
Conflict-of-interest statement: The authors declare no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non-commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: March 1, 2015
First decision: April 23, 2015
Article in press: July 22, 2015
P- Reviewer: Gao RP S- Editor: Yu J L- Editor: Rutherford A E- Editor: Jiao XK
References
- 1.Dobbs RH. Congenital hepatic fibrosis with portal hypertension. Proc R Soc Med. 1960;53:327–328. [PubMed] [Google Scholar]
- 2.Kerr DN, Harrison CV, Sherlock S, Walker RM. Congenital hepatic fibrosis. Q J Med. 1961;30:91–117. [PubMed] [Google Scholar]
- 3.De Vos M, Barbier F, Cuvelier C. Congenital hepatic fibrosis. J Hepatol. 1988;6:222–228. doi: 10.1016/s0168-8278(88)80036-9. [DOI] [PubMed] [Google Scholar]
- 4.Di Bisceglie A, Befeler A. Cystic and nodular diseases on the liver. In: Schiff ER, Madderey W, Sorrell M, editors. Schiff’s Diseases of the Liver. 10th ed. Philadelphia: Lippincott Williams and Wilkins; 2007. pp. 1231–1251. [Google Scholar]
- 5.Salonen R. The Meckel syndrome: clinicopathological findings in 67 patients. Am J Med Genet. 1984;18:671–689. doi: 10.1002/ajmg.1320180414. [DOI] [PubMed] [Google Scholar]
- 6.Yönem O, Ozkayar N, Balkanci F, Harmanci O, Sökmensüer C, Ersoy O, Bayraktar Y. Is congenital hepatic fibrosis a pure liver disease? Am J Gastroenterol. 2006;101:1253–1259. doi: 10.1111/j.1572-0241.2006.00642.x. [DOI] [PubMed] [Google Scholar]
- 7.Kirchner GI, Wagner S, Flemming P, Bleck JS, Gebel M, Schedel I, Schüler A, Galanski M, Manns MP. COACH syndrome associated with multifocal liver tumors. Am J Gastroenterol. 2002;97:2664–2669. doi: 10.1111/j.1572-0241.2002.06051.x. [DOI] [PubMed] [Google Scholar]
- 8.Saraiva JM, Baraitser M. Joubert syndrome: a review. Am J Med Genet. 1992;43:726–731. doi: 10.1002/ajmg.1320430415. [DOI] [PubMed] [Google Scholar]
- 9.Matsuzaka T, Sakuragawa N, Nakayama H, Sugai K, Kohno Y, Arima M. Cerebro-oculo-hepato-renal syndrome (Arima’ syndrome): a distinct clinicopathological entity. J Child Neurol. 1986;1:338–346. doi: 10.1177/088307388600100404. [DOI] [PubMed] [Google Scholar]
- 10.Satran D, Pierpont ME, Dobyns WB. Cerebello-oculo-renal syndromes including Arima, Senior-Löken and COACH syndromes: more than just variants of Joubert syndrome. Am J Med Genet. 1999;86:459–469. [PubMed] [Google Scholar]
- 11.Silverstein DM, Zacharowicz L, Edelman M, Lee SC, Greifer I, Rapin I. Joubert syndrome associated with multicystic kidney disease and hepatic fibrosis. Pediatr Nephrol. 1997;11:746–749. doi: 10.1007/s004670050381. [DOI] [PubMed] [Google Scholar]
- 12.Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, Ozilou C, Faivre L, Laurent N, Foliguet B, et al. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007;80:186–194. doi: 10.1086/510499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen MM, Hall BD, Smith DW, Graham CB, Lampert KJ. A new syndrome with hypotonia, obesity, mental deficiency, and facial, oral, ocular, and limb anomalies. J Pediatr. 1973;83:280–284. doi: 10.1016/s0022-3476(73)80493-7. [DOI] [PubMed] [Google Scholar]
- 14.Kolehmainen J, Black GC, Saarinen A, Chandler K, Clayton-Smith J, Träskelin AL, Perveen R, Kivitie-Kallio S, Norio R, Warburg M, et al. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet. 2003;72:1359–1369. doi: 10.1086/375454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 16.Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36:437–446. [PMC free article] [PubMed] [Google Scholar]
- 17.Héon E, Westall C, Carmi R, Elbedour K, Panton C, Mackeen L, Stone EM, Sheffield VC. Ocular phenotypes of three genetic variants of Bardet-Biedl syndrome. Am J Med Genet A. 2005;132A:283–287. doi: 10.1002/ajmg.a.30466. [DOI] [PubMed] [Google Scholar]
- 18.Holt RI. International Diabetes Federation re-defines the metabolic syndrome. Diabetes Obes Metab. 2005;7:618–620. doi: 10.1111/j.1463-1326.2005.00519.x. [DOI] [PubMed] [Google Scholar]